
A compartmentalized microfluidic chip with crisscross microgrooves and electrophysiological electrodes for modeling the blood–retinal barrier†
Jose
Yeste
 *abc,
Marta
García-Ramírez
d,
Xavi
Illa
ab,
Anton
Guimerà
ab,
Cristina
Hernández
d,
Rafael
Simó
d and
Rosa
Villa
ab
*abc,
Marta
García-Ramírez
d,
Xavi
Illa
ab,
Anton
Guimerà
ab,
Cristina
Hernández
d,
Rafael
Simó
d and
Rosa
Villa
ab
aInstitut de Microelectrònica de Barcelona, IMB-CNM (CSIC), 08193, Bellaterra, Barcelona, Spain. E-mail: jose.yeste@csic.es
bCIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, Barcelona, Spain
cDepartamento de Microelectrónica y Sistemas Electrónicos, Universitat Autònoma de Barcelona, Spain
dDiabetes and Metabolism Research Unit, Vall d'Hebron Research Institute, Barcelona, and CIBERDEM-Instituto de Salud Carlos III, Madrid, Spain
First published on 14th November 2017
Abstract
The interconnection of different tissue–tissue interfaces may extend organ-on-chips to a new generation of sophisticated models capable of recapitulating more complex organ-level functions. Single interfaces are largely recreated in organ-on-chips by culturing the cells on opposite sides of a porous membrane that splits a chamber in two or by connecting the cells of two adjacent compartments through microchannels. However, it is difficult to interconnect more than one interface using these approaches. To address this challenge, we present a novel microfluidic device where cells are arranged in parallel compartments and are highly interconnected through a grid of microgrooves, which facilitates paracrine signaling and heterotypic cell–cell contact between multiple tissues. In addition, the device includes electrodes on the substrate for the measurement of transepithelial electrical resistance (TEER). Unlike conventional methods for measuring the TEER where electrodes are on each side of the cell barrier, a method with only electrodes on the substrate has been validated. As a proof-of-concept, we have used the device to mimic the structure of the blood–retinal barrier by co-culturing primary human retinal endothelial cells (HREC), a human neuroblastoma cell line (SH-SY5Y), and a human retinal pigment epithelial cell line (ARPE-19). Cell barrier formations were assessed by a permeability assay, TEER measurements, and ZO-1 expression. These results validate the proposed microfluidic device with microgrooves as a promising in vitro tool for the compartmentalization and monitoring of barrier tissues.
Introduction
Organ-on-a-chip is an emerging technology addressed to reduce animal testing and accelerate clinical trials. Devices based on this technology enable an in vitro microenvironment and microarchitecture that closely mimics in vivo conditions.1,2 These factors stimulate a physiological response of the cells allowing the recapitulation of tissue- and organ-level functions. Organ-on-chips offer the advantage of accurately controlling the microenvironment of different cells by placing them in different compartments.3 Therefore, each cell type can be exposed to particular mechanical and biochemical conditions. However, this separation is detrimental to heterotypic cell–cell interactions, especially when dealing with many different cells and compartments. In fact, to reproduce more than one tissue–tissue interface while maintaining a particular microenvironment for each cell type is a microengineering challenge.Tissue–tissue interfaces in organ-on-chips have mimicked the interface between microvascular endothelium and brain parenchyma in the central nervous system4 or the interface between the alveolar epithelium and the pulmonary microvascular endothelium in the lung,5 among others. A common strategy to simulate a tissue–tissue interface is by using a polymeric porous membrane to separate two cell types in different compartments of a microfluidic device.5–8 The membrane offers close contact between tissues (∼10 μm in thickness) and the possibility to apply mechanical stretches if using a flexible membrane.5 However, there are few works based on multi-layered membranes to deal with several interfaces.9 Indeed, the stacking of porous membranes hampers the application of live cell imaging techniques. An alternative strategy is the creation of microchannels to link two adjacent compartments to permit side-by-side interconnections. Typically, these planar microfluidic devices are built using a flat glass substrate which is bonded to a polydimethylsiloxane (PDMS) slab containing the compartments and the microchannels. This technology is commercially available (SynVivo Inc., US) and has been used to model different tissue barrier functions.10–13 Since compartments are arranged in parallel over a flat surface, these devices are suitable for real-time imaging of the whole cell culture. In addition, they are easy to fabricate and only requires the bonding of the PDMS slab once the mold is built. A disadvantage of this strategy is the small interface area, i.e., a barrier function model will be associated with the few cells occupying the entrance of the microchannels.
To reproduce more than one tissue–tissue interface in a single device, we have developed a novel microfluidic device where cells are arranged in parallel compartments but are highly interconnected through a grid of microgrooves under the cells (Fig. 1), which facilitates paracrine signaling and heterotypic cell–cell contact between multiple tissues. Therefore, it is possible to build multiple interconnected interfaces maintaining a particular microenvironment for each tissue. The device comprises a PDMS slab that defines the compartments and a glass chip including a grid of microgrooves and metal electrodes. Its planar disposition facilitates real-time imaging and the integration of electrodes. In addition, permeability studies can be performed on epithelium and endothelium tissues covering the grid. Since the whole extent of the compartments is located on the grid, the measured permeability is representative of a large culture area unlike microchannel approaches.
 | ||
| Fig. 1 Recapitulating the cell structure of the retina. (A) Cell layers of the retina. From the anterior part of the retina to the posterior part of the retina: retinal endothelial cells (REC), ganglion cell layer (GCL), inner nuclear layer (INL), outer nuclear layer (ONL), and retinal pigment epithelium (RPE). Double arrows indicate the transport crossing the inner and outer BRB. (B) Schematic representation of the presented microfluidic cell culture device, including cell arrangement used in the experimental setup. Endothelial (HREC) and epithelial (ARPE-19) cells are located at the end compartments where there are TEER measurement electrodes, and SH-SY5Y cells are located in the middle compartments where there are extracellular potential recording electrodes. | ||
Microfluidic cell culture devices are commonly built using microfabrication processes such as photolithography, deposition, and etching. With this technology it is also possible to integrate metal electrodes in such devices.14 In fact, our microfluidic device includes metal electrodes on the glass substrate that permit the measurement of transepithelial electrical resistance (TEER). TEER measurement allows us to quantify tissue barrier function, with the advantages of being non-destructive, label-free and easily applicable in real time.15 Several authors have developed different electrode configurations and strategies to measure TEER in microfluidic systems16–19 and have integrated electrodes in microfluidic barrier models for its online monitoring.6,7,16–21 Unlike methods aimed at measuring the TEER based on electrodes located on each side of the cell barrier, we present in this work a method—also compatible with transport studies—that only requires electrodes to be on the basal side similar to the work of Sun et al.22 Thus, TEER can be measured in multiple compartments avoiding the laborious task of integrating electrodes in the PDMS block. With the commercially available electric cell-substrate impedance sensing (ECIS) technology23,24 (Applied Biophysics Inc, US), it is also possible to assess cell proliferation and barrier function using only electrodes in a substrate. However, ECIS systems lack a basal compartment since cells are attached to a surface that contains the electrodes. Thus, permeability and transport studies cannot be performed in such systems, which is a significant limitation in a barrier organ-on-a-chip system. In addition to the TEER measurement electrodes, the device includes an array of electrodes to be used in future applications for the recording of extracellular field potential. Recording electrodes allow evaluation of the electrical activity of neuronal tissues through local field potentials (LFPs) and extracellular action potentials (EAPs); additionally they could also be used to electrically stimulate neuronal activity.25
To show the potential applicability of the structure of multi-compartments and microgrooves in organ-on–a-chip technology as well as its monitoring capabilities, we have used the device to mimic the structure of the blood–retinal barrier (BRB). The BRB maintains the homeostasis of the neuroretina by regulating the transport of compounds from systemic blood circulation26 (Fig. 1A). This is composed of two barriers: 1) the inner barrier at the retinal microvascular endothelium forming capillaries (anterior part of the retina), and 2) the outer barrier formed by the retinal pigment epithelium (RPE) (posterior part).27 Between both barriers there is the neuroretina, which is formed by well-defined neuronal layers. Failure of the BRB leads to the accumulation of fluid in the area of the retina of the highest visual acuity (macula edema).28 This occurs in early stages of diabetic retinopathy (DR) and in particular diabetic macular edema (DME), which are common complications of diabetes that remain the leading causes of blindness in working-age populations in developed countries.29
Disease modeling in organ-on-chips has the potential to accelerate the identification of clinical targets since animal models often fail to represent specific human diseases.30,31 Some research studies have been focused on diseases associated with BRB breakdown.32,33 For example, Chung et al.32 managed to recapitulate choroidal neovascularization that resulted in outer BRB breakdown using a microfluidic cell culture consisting of a RPE and a blood vessel network.
To validate the herein proposed microfluidic device, we developed a co-culture of primary human retinal endothelial cells (HREC), the human neuroblastoma cell line (SH-SY5Y), and the human retinal pigment epithelial cell line (ARPE-19). The formation of endothelial and epithelial barriers was assessed by a permeability assay, TEER measurements, and ZO-1 expression. With this compartmentalization approach, it is possible to expose the endothelium to flow-induced shear stress, and the neuronal tissue can be subjected to a particular damage in order to evaluate the neurovascular coupling. In addition, it is possible to examine not only the particular response of one isolate type of cell in front to several stimuli but also its repercussion on the other retinal cell types, thus reproducing what really happens in the human retina.
Materials and methods
Microfluidic device design
The microfluidic device is structured in side-to-side compartments that are interconnected through a grid of microgrooves. It comprises a PDMS slab containing 7 compartments and a glass chip with microgrooves and electrodes for TEER measurements and recording of extracellular field potential. The compartments are 500 μm in width, 230 μm in height, and spaced by 50 μm. Two arrays of perpendicular microgrooves are etched in the glass and arranged in an area of 4 × 4 mm in the middle of the chip. Microgrooves are 2 μm in width, 4 μm in depth, and spaced by 10 μm. The total dimensions of the device are 22 × 22 × 4 mm.Platinum electrodes are located at the bottom of the microgrooves. The three compartments in the middle have an array of 24 recording electrodes regularly distributed. The area of each electrode is 50 × 50 μm, although the effective area that is exposed to the surface is 30 × 30 μm due to the microgrooves. Each of the other two compartments at both ends has a pair of straight electrodes of 40 μm in width and 4 mm in length (effective area of 0.58 × 10−3 cm2) to measure the TEER. All electrodes have accessible pads at the edges of the chip. A custom-made holder with spring contacts enables the electrical connection between the chip and an impedance analyzer, thereby facilitating the assembly.
Microfluidic device fabrication
The building of the device was divided into three steps: 1) fabrication of the glass chip with microgrooves and electrodes, 2) replica molding to make the PDMS slab with compartments, and 3) the final alignment and bonding of these two parts.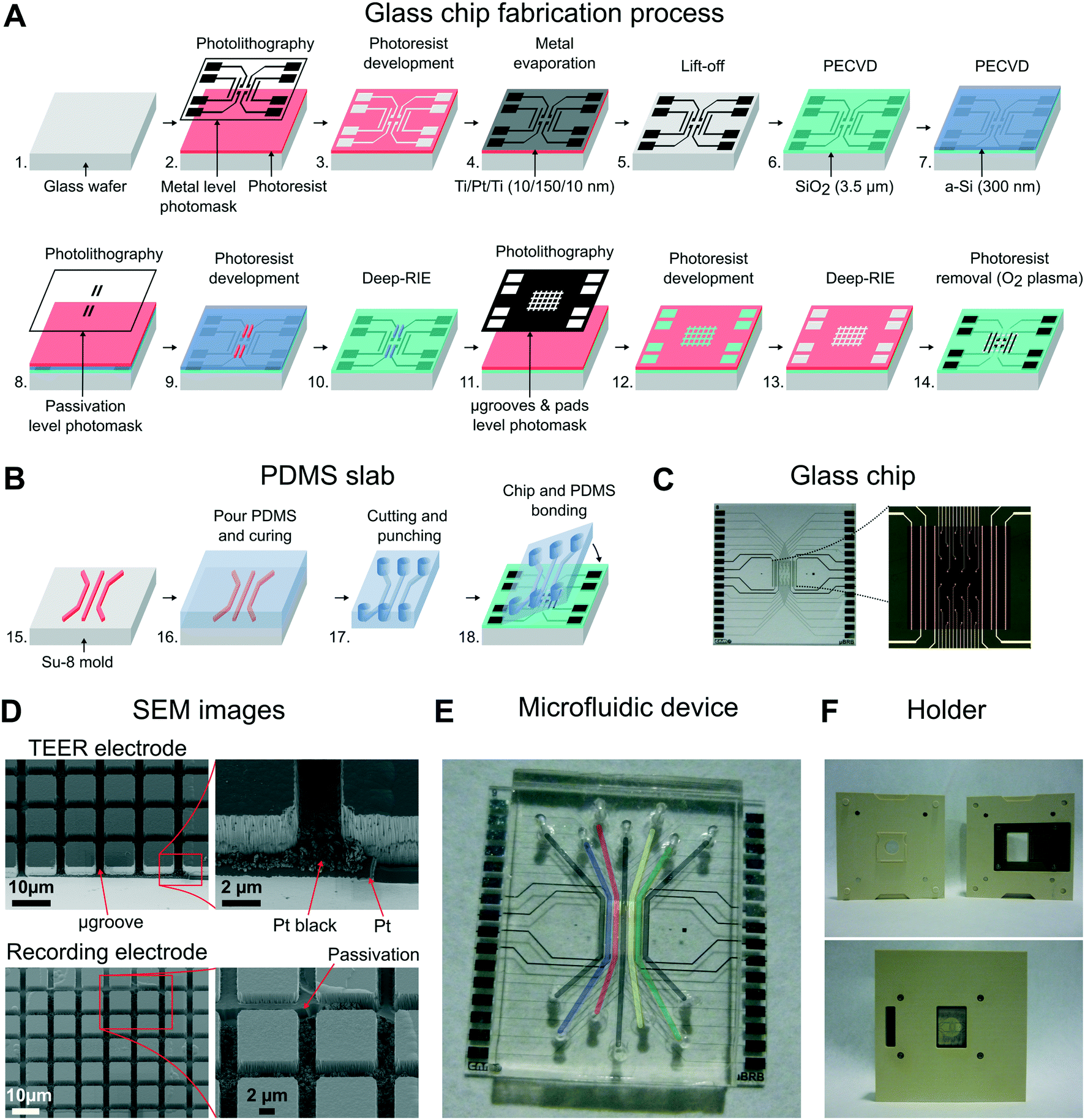 | ||
| Fig. 2 Microfluidic device fabrication process. (A) Fabrication steps of the glass chip. Proceeding from the beginning to the end are the photolithography process with the metal level photomask, Ti/Pt/Ti (10/150/10 nm) evaporation, lift-off process, plasma enhanced chemical vapor deposition (PECVD) of silicon dioxide (SiO2) (3.5 μm), PECVD of amorphous silicon (a-Si) (300 nm), photolithography process with the passivation level photomask, deep reactive-ion etching (deep-RIE) to etch the a-Si, photolithography process with the microgrooves and pad level photomask, and deep-RIE to etch SiO2. (B) Fabrication steps of the PDMS stamp consisted of pouring prepolymer PDMS on the SU-8 mold, curing of the PDMS, cutting of pieces, punching fluidic holes, and bonding of the PDMS slab with the glass chip. (C) Microscopy images of the glass chip, including an image of the central zone where the grid of microgrooves is located. (D) Scanning electron microscopy (SEM) images of the glass chip detailing the microgrooves and metal electrodes. (E) Full assembly of the device. Light color lines are superimposed on the image for clarity of the fluidic channels. (F) Custom-made holder to handle the device and to bring the electric pads into contact. | ||
A standard photolithography process using 1.2 μm-thick positive photoresist (HIPR 6512 Fujifilm Electronic Materials, US) was performed to define the passivation layer (steps 8, 9). The a-Si layer was etched using a deep reactive ion etching (DRIE) process (Alcatel AMS-110DE), and the remaining photoresist was stripped using O2 plasma (TEPLA 300-E, Technics Plasma, US) (step 10). Microgrooves were defined using the μ-grooves & pad level photomask in a second standard photolithography procedure (steps 11, 12) and etched using a DRIE process (steps 13, 14). The etch profile resulted in two levels because of the patterned a-Si layer: the one that reached the surface of the platinum layer (4 μm in depth), and a second one (2.5 μm in depth) that insulated the metal tracks of the recording electrodes. Finally, the wafer was then cut into chips of 22 × 22 mm, obtaining 9 chips per wafer.
To decrease the electrode polarization impedance of the TEER and recording electrodes, these were electrochemically coated with a layer of platinum black.34 Details about platinization are provided in Fig. S1 in the ESI†. In addition, apart from their use in future applications, the recording electrodes were electrically characterized by measuring a simulated signal of 10 Hz ranging from 10 to 1000 μVp (Fig. S3†). The impedance of the recording electrodes was 10 kΩ at 1 kHz, and the obtained average noise was lower than 5 μVrms.
The PDMS prepolymer (Sylgard 184, Dow corning Europe SA, BE) was prepared in a ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (base:curing agent, w/w) and degassed in a vacuum desiccator. The prepolymer was cast on a Petri dish containing the SU-8 mold and left on a flat surface for 15 min. The PDMS was cured at 75 °C for 2 h and left overnight at room temperature (step 16). The PDMS was then peeled off from the mold, and pieces of 17 × 22 mm were cut and punched for fluid access (step 17). The resulting height of the compartments was 230 μm, and the thickness of the PDMS slab was 4 mm.
:1 (base:curing agent, w/w) and degassed in a vacuum desiccator. The prepolymer was cast on a Petri dish containing the SU-8 mold and left on a flat surface for 15 min. The PDMS was cured at 75 °C for 2 h and left overnight at room temperature (step 16). The PDMS was then peeled off from the mold, and pieces of 17 × 22 mm were cut and punched for fluid access (step 17). The resulting height of the compartments was 230 μm, and the thickness of the PDMS slab was 4 mm.
Permeability assay
The endothelial and epithelial permeabilities were quantified by the passage of labelled tracers. This assay was carried out after the cells were perfused for 3 days in the device. Afterwards, pipette tips were connected at the inlets and outlets to fill the compartments with the tracers. The compartment with epithelial cells was carefully filled with medium containing fluorescein isothiocyanate–dextran (FITC-dextran) (70 kDa, Sigma Aldrich) at a concentration of 50 μg mL−1. The compartment with endothelial cells was filled with medium containing Rhodamine B isothiocyanate–dextran (RITC-dextran) (70 kDa, Sigma Aldrich) at a concentration of 5 mg mL−1. The device was then placed in an incubator at 37 °C and 5% CO2. At 30 min, fluorescence microscopy images were taken from the device using an Olympus 1X71 inverted microscope at an exposure time of 0.33 (RITC) and 0.67 ms (FITC). In the same way, a compartment of a device without cells was filled with both labelled dextrans, and fluorescence microscopy images were taken every 5 min for 30 min. Fluorescence images were analyzed with ImageJ software to quantify the intensity in the compartments. Normalization was performed by dividing the intensity in the receiving compartment by the intensity in the donor compartment. According to Fick's First Law and provided that there is a lineal relation between intensity and dextran amount, the initial slope of the normalized intensity will be related to the permeability as11,35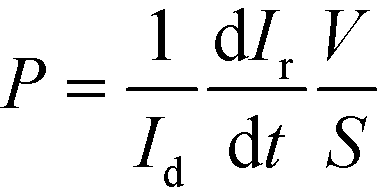 | (1) |
Transepithelial electrical resistance
TEER is conventionally obtained using electrodes in apical and basal sides, so that the electrical current flows across the cell barrier. In our approach, there are two electrodes placed at the bottom of the microgrooves, and the current flows from one electrode to another crossing the cell barrier twice. A schematic representation of this measurement system and the equivalent electric circuits with lumped elements are shown in Fig. 3. Unlike the conventional measurement system, this approach includes the parasitic resistance of the solution in the basal side (the medium in the microgrooves, Rμg). At relatively middle frequencies (∼1–10 kHz), the measured impedance is purely resistive (phase = 0°) and associated with the equivalent electric circuit shown in Fig. 3C. This is because the electrode polarization impedances (CPEe) and the cell layer capacitances (Ccl) do not contribute significantly to the measured impedance in this range of frequencies. To convert TEER values in units of Ω cm2, a numerical study according to our previous work18 is provided in the ESI† (Fig. S5). Normalizing the TEER to the area of the electrodes does not take into account that the effective area through which the current passes the cell layer depends on the TEER value.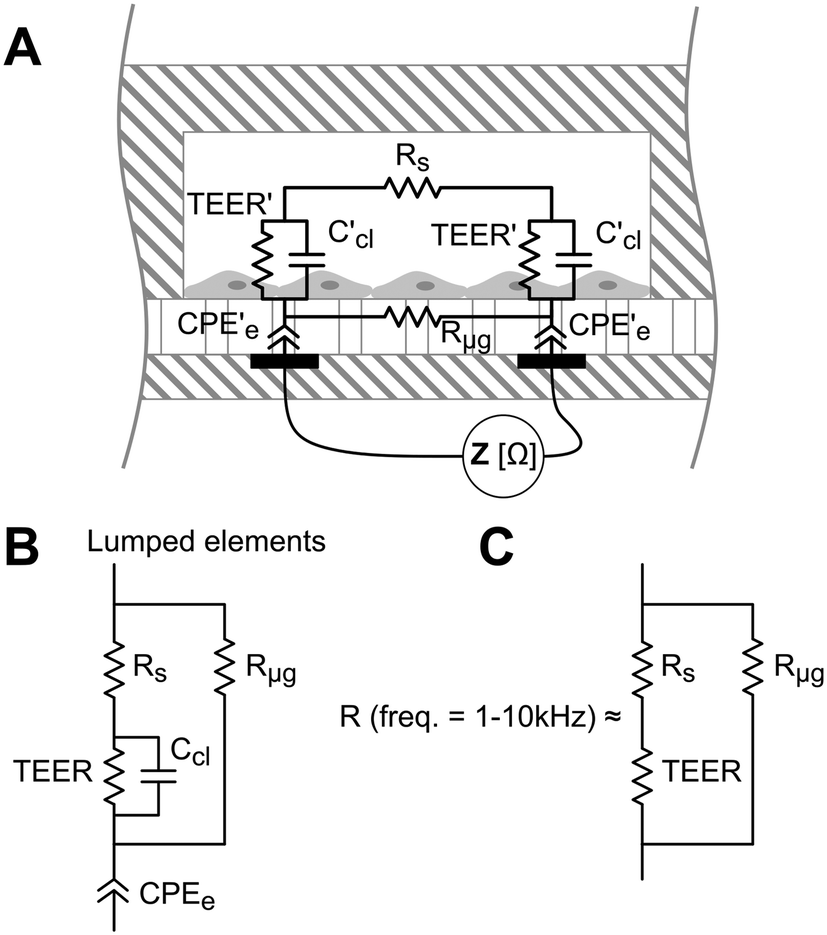 | ||
| Fig. 3 (A) Schematic representation of the measurement system consisting of two electrodes under the microgrooves. The equivalent electric circuit includes constant phase element (CPEe) that represents the electrode polarization impedance, cell layer capacitance (Ccl), luminal medium resistance (Rs), basolateral medium resistance (Rμg), and transepithelial electrical resistance (TEER). (B) Lumped element model of the equivalent electric circuit. (C) Equivalent electric circuit of the resistance measured at relatively middle frequencies (from 1 to 10 kHz). | ||
Electrical impedance between the two electrodes was acquired with an impedance analyzer36 and measured at 20 frequencies ranging from 100 Hz to 1 MHz. The analyzer was adapted to apply a voltage of 10 mVp with an output resistance of 1 kΩ, limiting the generated perturbation in voltage and current by 10 mVp and 10 μAp, respectively. The resistance at middle frequencies (Fig. 3C) was obtained from fitting the impedance spectra using the least-squares method in Matlab.
Cell culture
Epithelial monolayers were obtained with ARPE-19 cells (ATCC, US), an immortalized human retinal pigment epithelial cell line. These epithelial cells form stable monolayers, which exhibit morphological and functional polarity under specific culture conditions. ARPE-19 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) and Ham's F-12 nutrient mixture (1:1), 10% fetal bovine serum (FBS), and 2 mmol L−1 penicillin/streptomycin. Experiments were performed at passage 23. Endothelial monolayers were obtained with primary HREC (Innoprot, ES). Endothelial cells were cultured in Endothelial Medium (EM, Innoprot, ES), (5%) FBS, 2 mmol L−1 penicillin/streptomycin, and experiments were performed at passage 3. Human neural cells (SH-SY5Y cell line) were obtained from ECACC (Sigma-Aldrich, ES), and cultured in DMEM, 5% FBS, 2 mmol L−1 penicillin/streptomycin.
Compartments of the device were coated with fibronectin (Millipore, ES) at a concentration of 100 μg mL−1 (2.3 μg cm−2) and incubated at 37 °C for 1 h. The device with fibronectin was rinsed with culture medium and incubated for 30 min. SH-SY5Y, HREC, and ARPE-19 cells were then seeded at a concentration of 2 × 106 cells cm−2 and incubated at 37 °C and 5% of CO2 until the end of the experiment.
Experimental setup
Neuronal cells were cultured in the three middle compartments of the device containing the recording electrodes. Otherwise, endothelial and epithelial cells were cultured in the compartments adjacent to those with the neurons as shown in Fig. 1B.After seeding, cells were incubated under static conditions for 1.5–3 h to ensure cell attachment. Then, the cells were perfused with their respective culture media at a rate of 0.1 mL min−1 for 3 days. For the case of the neuronal cells, the medium was only flowed through the central compartment to simplify the fluidic connections. Culture medium was injected using a syringe pump (NE-4000, New Era Pump Systems, Inc., US) in infusion mode. Polytetrafluoroethylene (PTFE) tubes (0.5 mm internal diameter (ID) and 1 mm outer diameter (OD)) were cut and inserted into the inlets and outlets of the device. Inlet tubes were connected to syringes containing culture media, and the outlet tubes were connected to reservoirs (silicone tube of 0.8 mm ID and 2.4 mm OD). To ensure proper equilibration of the medium with the incubator, reservoirs were vented and placed near the outlets.
Immunocytofluorescence
Cells were cultured under the conditions previously described. At the end of each experiment, cells were washed with Dulbecco's phosphate-buffered saline (DPBS) and blocked with DPBS containing 2% bovine serum albumin (BSA) and 0.05% Tween for 1 h at room temperature. Then, the cells were incubated with mouse anti-human ZO-1 antibody (Zymed, Invitrogen, ES) diluted 1/200 in the same buffer overnight at 4 °C. After washing two times with DPBS, the cells were further incubated with a fluorescent secondary antibody (Alexa 594, Invitrogen, ES) for 1 h at room temperature. Nuclei were stained in blue with 4′-6-diamidino-2-phenylindole (DAPI) (Vector Laboratory, US). Images were acquired with a confocal laser scanning microscope (FV1000, Olympus, DE) (ARPE-19 cells) and an Olympus 1X71 inverted microscope (SH-SY5Y cells).Results and discussion
We validated the developed microfluidic device with a co-culture of primary HREC, the SH-SY5Y human neuroblastoma cell line, and the ARPE-19 human retinal pigment epithelial cell line. Cells were cultured at confluence, and maintained with a continuous supply of culture medium for 3 days (Fig. 4). The formation of endothelial and epithelial barriers was assessed by their permeability to dextran 70 kDa, TEER measurements, and ZO-1 expression. In addition, SH-SY5Y cells exhibited a neuronal morphology. | ||
| Fig. 4 Phase contrast images of the cells in the zone of the grid of microgrooves of the device. Images were taken (A) at cell seeding and (B) at the third day. Cells in the compartments pointed with arrows were perfused with their respective culture media at a rate of 0.1 mL min−1 for 3 days. | ||
Permeability assay and immunocytofluorescence
Reduced permeability to large dextran molecules was found in both endothelial and epithelial cell monolayers (Fig. 5). Fluorescence images taken from the blank control (without cells) show that the devices were properly sealed. The PDMS successfully bonded on top of the microgroove grid, letting the tracer pass only through the grooves. In Fig. 5D, a zoomed view of the PDMS wall between two adjacent compartments is shown. In addition, cells did not cross between compartments, and those compartments without cells still remained empty at the end of the experiment. The size of the microgrooves are small enough (2 μm width and 4 μm depth) to prevent cell migration between compartments, but they are large enough to allow neuritic processes to extend between compartments through the microgrooves under the wall.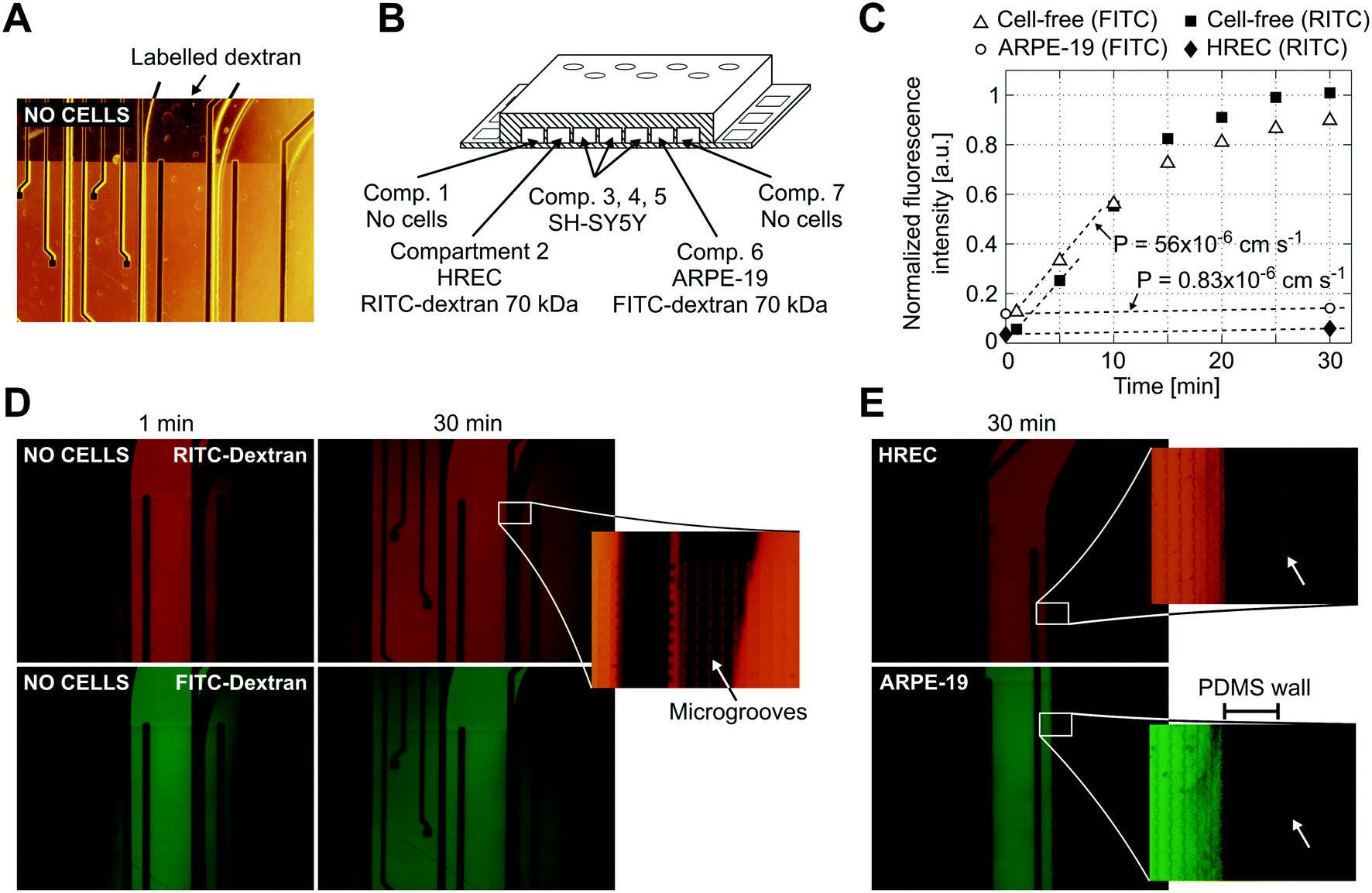 | ||
| Fig. 5 Permeability to labelled dextran 70 kDa. (A) Contrast phase image of the compartments without cells. Note that the metal electrodes and electric tracks are opaque. (B) Schematic drawing (not to scale) of the arrangement of the cells and labelled dextrans. (C) Normalized fluorescence intensity in the receiving compartment that is adjacent to the donor compartment. Intensity was quantified from the mean of an area in the middle of the compartment with ImageJ software and normalized by dividing by the intensity at the donor compartment. Permeability was calculated according to eqn (1) using the initial slope of the normalized fluorescence ((in cm s−1) Pcell-free(FITC) = 56 × 10−6, Pcell-free(RITC) = 53 × 10−6, PARPE-19(FITC) = 0.83 × 10−6, and PHREC(RITC) = 0.88 × 10−6). (D) Fluorescence microscopy images without cells at 1 and 30 min after the administration of RITC and FITC dextrans in one of the compartments. In the zoomed view, a PDMS wall that separates two compartments and a TEER measurement electrode (left side) is shown. Indicated with an arrow is the location of the microgrooves under the PDMS wall. (E) Fluorescence microscopy image at 30 min after administration of RITC or FITC dextran in a compartment with a monolayer of HREC or ARPE-19. Images were taken once the cells have stayed in the device for 3 days. The arrow in the zoomed view points to a little fluorescence in the adjacent compartment after the PDMS wall. | ||
We used RITC and FITC dextrans to simultaneously quantify the permeability of both endothelial and epithelial barriers. Fluorescence intensities in the receiving compartments, adjacent to the donor compartments, and calculated permeabilities are shown in Fig. 5C. In the blank control, both labelled dextrans were rapidly distributed throughout the device when they were injected into one compartment as evidenced by the fluorescence images taken at 1 and 30 min. Otherwise, the endothelial and epithelial compartments exhibited little permeation to dextran, at least for 30 min. Only a little fluorescence was observed in the adjacent compartments as shown in the zoomed view in Fig. 5E. Permeability was shown to decrease by more than 1 order of magnitude. However, further studies using longer assay times are needed. The planar disposition of the device allows real-time imaging of the whole cell culture. Furthermore, transport studies can be conducted with high spatiotemporal resolution which is not possible with conventional methods based on collecting samples. Indeed, some authors have utilized similar planar approaches to perform time course monitoring of labelled tracers.11
In addition to permeability analysis and TEER, immunocytofluorescence staining for ZO-1 was performed after 3 days for ARPE-19 cells. It demonstrated barrier formation on the grid of microgrooves. The ZO-1 protein was localized at the peripheral membrane and continuously distributed alongside the epithelial monolayer (Fig. 6B). SH-SY5Y cells also proliferated on the grid. At the end of the experiment, there were some cells forming clusters with neuronal phenotypes and extended neuritic processes (Fig. 6D).
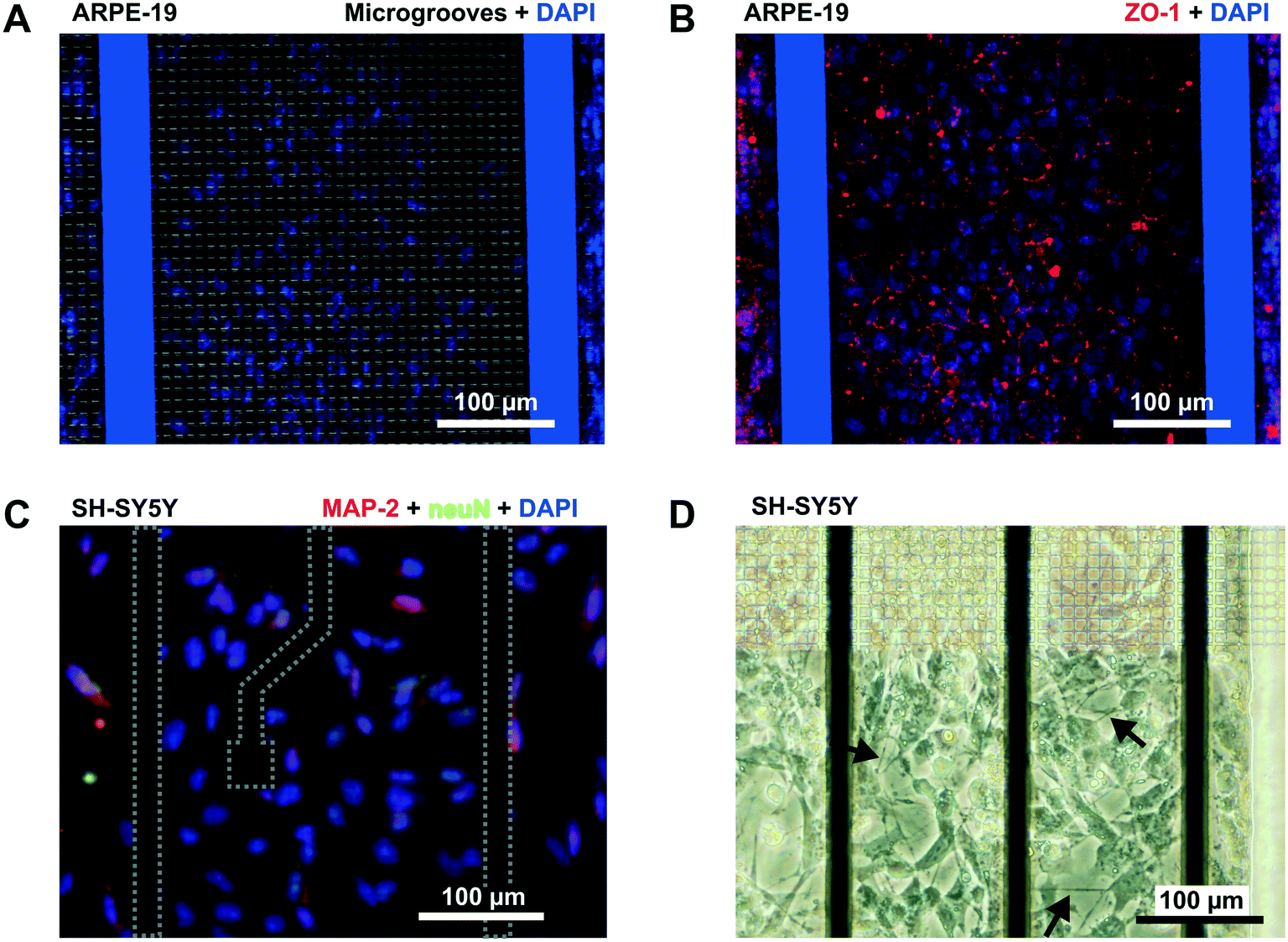 | ||
| Fig. 6 Immunofluorescence. (A and B) Confocal microscopy image (×10) of the human retinal epithelial monolayer lining the grid of microgrooves in the device. (A) The image is an overlap of the grid of microgrooves and cell nuclei stained with DAPI (blue). (B) Overlap of the ZO-1 protein (red) and DAPI. (C) Fluorescence image of SH-SY5Y cells following staining with DAPI (blue), anti-NeuN antibody [EPR12763] – neuronal marker with Alexa Fluor 488 (green), and anti-Map2 (ab32454)-neuronal marker with Alexa Fluor 568 (red). (D) Light microscopy of the SH-SY5Y taken at the edge of the grid of microgrooves. Arrows point to occasional extensions of neuritic processes. | ||
Transepithelial electrical resistance
To validate the TEER measurement system that uses two straight parallel electrodes at the bottom of the microgrooves, we compared the resistance measured using this two basal electrodes to the conventional TEER measurement using electrodes in apical and basal sides. For that, the PDMS slab of the device was perforated as in Fig. 7A to introduce an additional Pt wire electrode in the apical side. In Fig. 7B, the time course of the resistances is shown during the administration and removal of ethylenediaminetetraacetic acid (EDTA) in ARPE-19 cells cultured for 1 day for both configurations. The application of the calcium chelating agent EDTA induced in a few minutes rapid barrier breakdown, whereas its removal caused the recovery of the barrier to its initial values. EDTA removes extracellular Ca2+ of the medium, which in turn opens the paracellular pathway presumably through stimulation of protein kinase C37,38 and upregulation of intracellular Ca2+. As a result, Ca2+ depletion induces disruption of intercellular junctions, disruption of actin filaments, and event delocalization of tight junctions when depletion is prolonged.39 During transient Ca2+ removal (approximately less than 1 h), tight junctions remain localized in the region of cell–cell contacts, and TEER alternatively drops and is recovered in a process that usually takes several hours; this transient disruption has been exploited to enhance drug permeability.40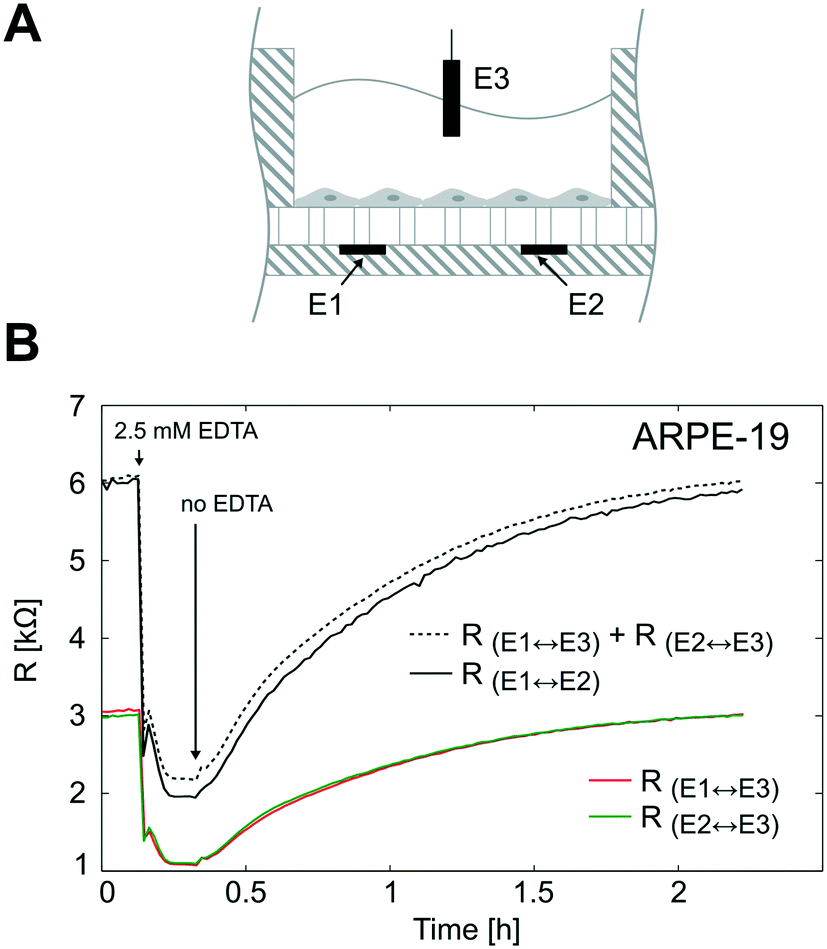 | ||
| Fig. 7 TEER measurement system validation. (A) Schematic representation of the electrode configuration used to validate the measurement system based on two electrodes under the microgrooves (E1 and E2). For this purpose, the device was perforated and an additional Pt wire electrode (E3) was introduced in the apical side. This electrode configuration allowed us to measure three resistances; 1) the resistance between the electrodes under the microgrooves (RE1↔E2), 2) the resistance between E1 and E3 (RE1↔E3), and 3) the resistance between E2 and E3 (RE2↔E3). (B) ARPE-19 cells were cultured for 1 day, and the three resistances were measured during a calcium switch procedure. The sum of RE1↔E3 and RE2↔E3 is shown in dashed lines and fits the same time course variations of RE1↔E2. | ||
It can be observed that the time courses of the resistance measured between the apical electrode and each one of the basal electrodes (RE1↔E3 and RE2↔E3) were equal as expected. In addition, the sum of these two resistances (RE1↔E3 + RE2↔E3) fitted the same time course of the resistance measured between basal electrodes in our method (RE1↔E2). This demonstrates that the proposed measurement system is feasible to determine the TEER. It offers the advantage of measuring the TEER in many compartments without integrating electrodes in the PDMS slab, which is time-consuming and has poor reproducibility. However, some considerations have to be addressed with this methodology.
Depending on the microgroove dimensions, there may be a limitation when measuring high TEER values because the resistance of the solution in the basal side (Rμg) is in parallel with the TEER. Therefore, there will be an underestimation of the TEER as its value approaches Rμg. For example, if TEER plus the resistance of the solution in the apical side (Rs) is smaller than Rμg (TEER + Rs < Rμg) most of the current will flow across the cell barrier, and the measured resistance will be the TEER plus the Rs. Otherwise (TEER + Rs > Rμg), most of the current will flow through the solution contained in the microgrooves without crossing the cell barrier. For our electrodes and microgroove dimensions, this Rμg (40 kΩ, details are provided in Fig. S2†) is much higher than the maximum measured resistance (R < 6 kΩ). Thus, the measured resistance (R) was mainly contributed by the TEER in series with Rs. Although the measurement system is suitable to measure a large range of TEER values neglecting Rμg, it would be possible to incorporate the calculated Rμg value when measuring high TEER values.
In Fig. 8A, the impedance spectra measured with and without a monolayer (ARPE-19 or HREC) covering the grid are shown. In the Bode representation, the change that causes the cell barrier in the impedance spectra is clearly observed. Herein, the rise in resistance in the middle frequencies is associated with the increase of the TEER value. The time course of R is shown in Fig. 8B. After 1 h, cells were successfully attached to the microgroove grid, and during the first day, the R increased supposedly due to cell spreading until it reached a plateau. This coincided with the monolayer formation observed under the microscope.
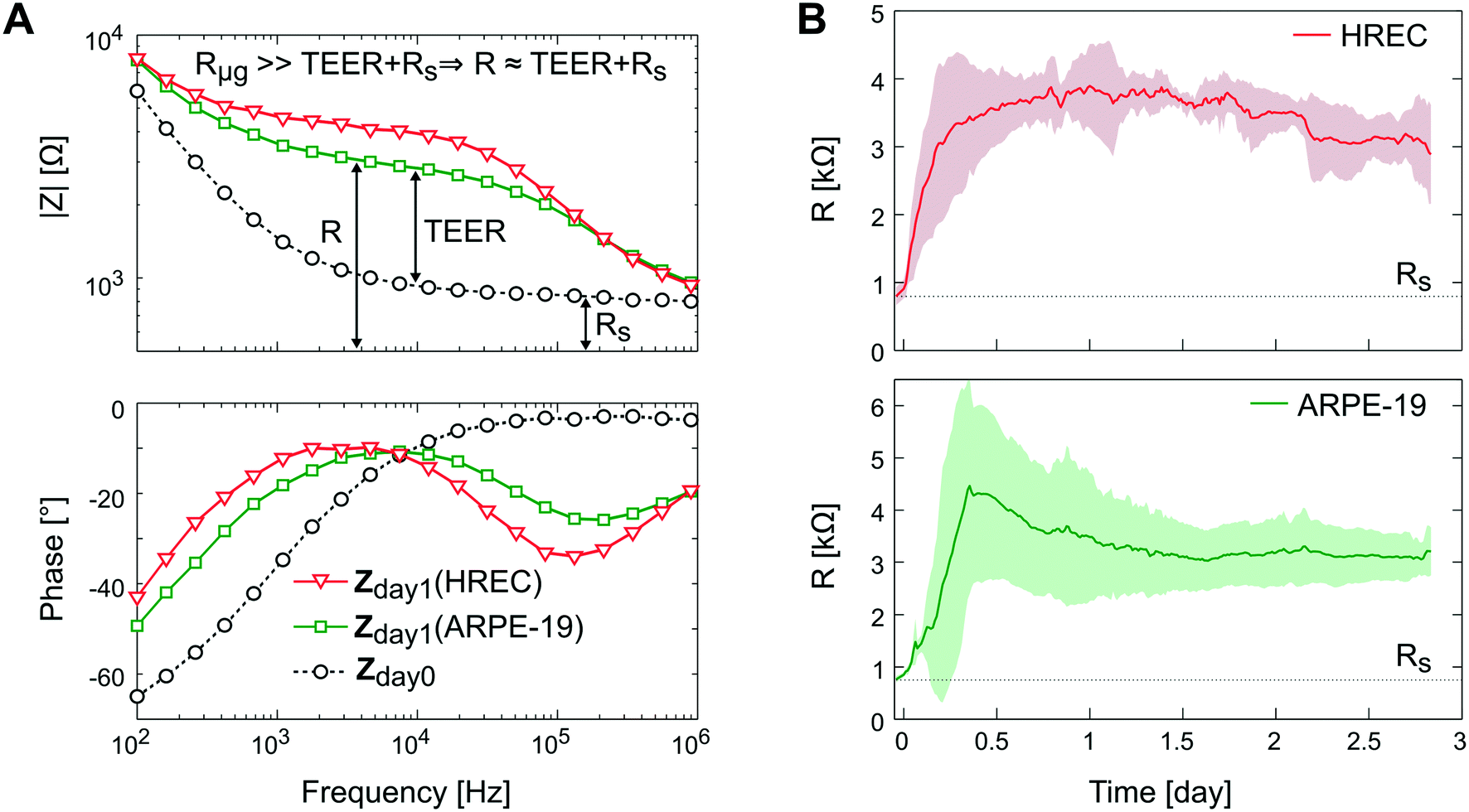 | ||
| Fig. 8 Transepithelial electrical resistance measurement. (A) Bode representation of the impedance spectra measured before cell seeding (dashed line and circle symbol) and after 1 day for HREC (green square symbol) and ARPE-19 (red inverted triangle symbol) cultures. (B) Time course of the resistance measured for HREC (red line) (n = 4) and ARPE-19 (green line) (n = 3) cultures. Data were acquired every 15 min for 3 days. Note that there are data before cell seeding (time 0) that correspond to the measurement without cells. | ||
Although there was no significant difference in R between ARPE-19 and HREC at the 3rd day (3.2 ± 0.46 kΩ (4.9 Ω cm2) and 2.9 ± 0.73 kΩ (4.2 Ω cm2), respectively), the real-time functionality allowed us to observe some different behaviors in cell attachment and spreading. For instance, the R of HREC cells experienced an exponential increase after cell seeding. Otherwise, the R of ARPE-19 cells increased with a constant slope, and made a noticeable overpeak. Herein, it has to be noted that the variability between experiments was observed when we connected the flow.
The formation of the tight junctions depends on the experimental conditions and in particular on the number of cells plated to reach confluence. In cultures in which a high amount of cells has been seeded, the tight junction ZO-1 appears by western blot as soon as 4–8 h but the protein forming an efficient barrier between cells is not visible by immunohistochemistry until 2–3 days. This has been reported in both ARPE-19 (ref. 41 and 42) and HREC.43–46 In our hands this has also been reproduced as is shown in Fig. S6.†
Limiting factors
Herein, we have summarized several limiting factors of the device as well as some issues of the microfluidic cell culture that is important to discuss:Conclusions
The interconnection of different tissue–tissue interfaces together with cell exposure to constant flow may expand organ-on-chips to a new generation of sophisticated models capable of recapitulating more complex organ-level functions. We have developed a microfluidic cell culture device where more than one tissue barrier function can be emulated. It is based on a grid of microgrooves that allows heterotypic cell–cell interactions and the formation of several tissues. Microfabrication techniques were used to construct the device, allowing the integration of TEER electrodes for continuous monitoring. Moreover, its planar disposition enables real-time imaging with high spatiotemporal resolution.As a proof-of-concept, we used the fabricated device to mimic the structure of the BRB by co-culturing HREC (inner BRB), SH-SY5Y cells (neuroretina), and ARPE-19 cells (outer BRB). Tissues on the microgrooves were assessed by a permeability assay, TEER measurements, and ZO-1 expression. To measure the TEER in real-time, we validated a method based on electrodes placed at the basal side (situated under the microgrooves) of both cell monolayers.
In conclusion, the presented multi-compartment microfluidic device with integrated electrophysiological monitoring electrodes will permit us to make more accurate measurements of cell behavior than can be obtained in the currently static models. Thus, it can be envisaged as a promising new approach for recapitulating multi-barrier models. In particular, it will facilitate the study of retinal diseases affecting BRB integrity and neurovascular coupling such as DR, which remains the leading cause of blindness in working-age individuals in the western world.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the requirements to achieve the PhD degree in Electrical and Telecommunication Engineering at the Universitat Autònoma de Barcelona, and it was supported by grants from Ministerio de Economía y Competitividad (MINECO) (SAF2014-62114-EXP and SAF2016-77784-R) and Fundació La Marató de TV3 (Exp. 201629-10). This work has made use of the Spanish ICTS Network MICRONANOFABS partially supported by MINECO and the ICTS ‘NANBIOSIS’, more specifically by the Micro-Nano Technology Unit of the CIBER in Bioengineering, Biomaterials & Nanomedicine (CIBER-BBN) at the IMB-CNM.References
- K. Yum, S. G. Hong, K. E. Healy and L. P. Lee, Biotechnol. J., 2014, 9, 16–27 CrossRef CAS PubMed.
- S. N. Bhatia and D. E. Ingber, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS PubMed.
- C. Moraes, G. Mehta, S. C. Lesher-Perez and S. Takayama, Ann. Biomed. Eng., 2012, 40, 1211–1227 CrossRef PubMed.
- A. K. H. Achyuta, A. J. Conway, R. B. Crouse, E. C. Bannister, R. N. Lee, C. P. Katnik, A. A. Behensky, J. Cuevas and S. S. Sundaram, Lab Chip, 2013, 13, 542–553 RSC.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- L. M. Griep, F. Wolbers, B. de Wagenaar, P. M. ter Braak, B. B. Weksler, I. A. Romero, P. O. Couraud, I. Vermes, A. D. van der Meer and A. van den Berg, Biomed. Microdevices, 2013, 15, 145–150 CrossRef CAS PubMed.
- R. Booth and H. Kim, Lab Chip, 2012, 12, 1784–1792 RSC.
- A. K. H. Achyuta, A. J. Conway, R. B. Crouse, E. C. Bannister, R. N. Lee, C. P. Katnik, A. A. Behensky, J. Cuevas and S. S. Sundaram, Lab Chip, 2013, 13, 542–553 RSC.
- D. Sticker, M. Rothbauer, S. Lechner, M.-T. Hehenberger and P. Ertl, Lab Chip, 2015, 15, 4542–4554 RSC.
- B. Prabhakarpandian, M.-C. Shen, J. B. Nichols, I. R. Mills, M. Sidoryk-Wegrzynowicz, M. Aschner and K. Pant, Lab Chip, 2013, 13, 1093 RSC.
- S. P. Deosarkar, B. Prabhakarpandian, B. Wang, J. B. Sheffield, B. Krynska and M. F. Kiani, PLoS One, 2015, 10, e0142725 Search PubMed.
- J. H. Yeon, D. Na, K. Choi, S.-W. Ryu, C. Choi and J.-K. Park, Biomed. Microdevices, 2012, 14, 1141–1148 CrossRef CAS PubMed.
- T. B. Terrell-Hall, A. G. Ammer, J. I. G. Griffith and P. R. Lockman, Fluids Barriers CNS, 2017, 14, 3 CrossRef PubMed.
- R. Booth, S. Noh and H. Kim, Lab Chip, 2014, 14, 1880–1890 RSC.
- B. Srinivasan, A. R. Kolli, M. B. Esch, H. E. Abaci, M. L. Shuler and J. J. Hickman, J. Lab. Autom., 2015, 20, 107–126 CrossRef CAS PubMed.
- O. Y. F. Henry, R. Villenave, M. J. Cronce, W. D. Leineweber, M. A. Benz and D. E. Ingber, Lab Chip, 2017, 17, 2264–2271 RSC.
- M. W. van der Helm, M. Odijk, J.-P. Frimat, A. D. van der Meer, J. C. T. Eijkel, A. van den Berg and L. I. Segerink, Biosens. Bioelectron., 2016, 85, 924–929 CrossRef CAS PubMed.
- J. Yeste, X. Illa, C. Gutiérrez, M. Solé, A. Guimerà and R. Villa, J. Phys. D: Appl. Phys., 2016, 49, 375401 CrossRef.
- M. Odijk, A. D. van der Meer, D. Levner, H. J. Kim, M. W. van der Helm, L. I. Segerink, J.-P. Frimat, G. A. Hamilton, D. E. Ingber and A. van den Berg, Lab Chip, 2015, 15, 745–752 RSC.
- Y. I. Wang, H. E. Abaci and M. L. Shuler, Biotechnol. Bioeng., 2017, 114, 184–194 CrossRef CAS PubMed.
- B. M. Maoz, A. Herland, O. Y. F. Henry, W. D. Leineweber, M. Yadid, J. Doyle, R. Mannix, V. J. Kujala, E. A. FitzGerald, K. K. Parker and D. E. Ingber, Lab Chip, 2017, 17, 2294–2302 RSC.
- T. Sun, E. J. Swindle, J. E. Collins, J. A. Holloway, D. E. Davies and H. Morgan, Lab Chip, 2010, 10, 1611–1617 RSC.
- J. Wegener, C. R. Keese and I. Giaever, Exp. Cell Res., 2000, 259, 158–166 CrossRef CAS PubMed.
- I. Giaever and C. R. Keese, IEEE Trans. Biomed. Eng., 1986, BME-33, 242–247 CrossRef PubMed.
- M. E. J. Obien, K. Deligkaris, T. Bullmann, D. J. Bakkum and U. Frey, Front. Neurosci., 2015, 8, 423 Search PubMed.
- J. G. Cunha-Vaz, in Retinal Pigment Epithelium and Macular Diseases, ed. G. Coscas and F. C. Piccolino, Springer, Netherlands, Dordrecht, 1998, pp. 5–11 Search PubMed.
- M. Campbell and P. Humphries, in Biology and Regulation of Blood-Tissue Barriers, ed. C. Y. Cheng, Springer New York, New York, NY, 2013, pp. 70–84 Search PubMed.
- I. Klaassen, C. J. F. Van Noorden and R. O. Schlingemann, Prog. Retinal Eye Res., 2013, 34, 19–48 CrossRef CAS PubMed.
- T. Y. Wong, C. M. G. Cheung, M. Larsen, S. Sharma and R. Simó, Nat. Rev. Dis. Primers, 2016, 2, 16012 CrossRef PubMed.
- A. van de Stolpe and J. den Toonder, Lab Chip, 2013, 13, 3449 RSC.
- A. P. Haring, H. Sontheimer and B. N. Johnson, Stem Cell Rev. Rep., 2017, 13, 381–406 CrossRef CAS PubMed.
- M. Chung, S. Lee, B. J. Lee, K. Son, N. L. Jeon and J. H. Kim, Adv. Healthcare Mater., 2017, 1700028 CrossRef PubMed.
- L.-J. Chen, S. Ito, H. Kai, K. Nagamine, N. Nagai, M. Nishizawa, T. Abe and H. Kaji, Sci. Rep., 2017, 7, 3538 CrossRef PubMed.
- G. Gabriel, I. Erill, J. Caro, R. Gómez, D. Riera, R. Villa and P. Godignon, Microelectron. J., 2007, 38, 406–415 CrossRef CAS.
- V. H. Huxley, F. E. Curry and R. H. Adamson, Am. J. Physiol., 1987, 252, H188 CAS.
- A. Guimerà, G. Gabriel, D. Parramon, E. Calderón and R. Villa, in World Congress on Medical Physics and Biomedical Engineering (Munich, DE, 7–12 September 2009), ed. O. Dössel, W. C. Schlegel and R. Magjarevic, Springer, Berlin, 2009, vol. 25/7, pp. 868–871 Search PubMed.
- S. Citi, J. Cell Biol., 1992, 117, 169–178 CrossRef CAS PubMed.
- M. Tomita, M. Hayashi and S. Awazu, J. Pharm. Sci., 1996, 85, 608–611 CrossRef CAS PubMed.
- J. D. Siliciano, J. Cell Biol., 1988, 107, 2389–2399 CrossRef CAS PubMed.
- M. A. Deli, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 892–910 CrossRef CAS PubMed.
- T. Yoshikawa, N. Ogata, H. Izuta, M. Shimazawa, H. Hara and K. Takahashi, Curr. Eye Res., 2011, 36, 1153–1163 CrossRef CAS PubMed.
- J. Hirata, J.-A. Ko, H. Mochizuki, K. Funaishi, K. Yamane, K.-H. Sonoda and Y. Kiuchi, Open Life Sci., 2014, 9, 461–468 CAS.
- T. W. Gardner, T. Lesher, S. Khin, C. Vu, A. J. Barber and W. A. Brennan, Biochem. J., 1996, 320, 717–721 CrossRef CAS PubMed.
- J.-H. Kim, J. H. Kim, H.-O. Jun, Y. S. Yu and K.-W. Kim, Am. J. Pathol., 2010, 176, 1517–1524 CrossRef CAS PubMed.
- Y. Jiang, L. Liu and J. J. Steinle, Invest. Ophthalmol. Vis. Sci., 2017, 58, 185 Search PubMed.
- E.-A. Ye, L. Liu and J. J. Steinle, Vision Res. DOI:10.1016/j.visres.2017.07.007.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7lc00795g |
| This journal is © The Royal Society of Chemistry 2018 |