DOI:
10.1039/C6LC01208F
(Paper)
Lab Chip, 2017,
17, 178-185
Selection of aptamers based on a protein microarray integrated with a microfluidic chip†
Received
26th September 2016
, Accepted 17th November 2016
First published on 17th November 2016
Abstract
We developed an efficient and fast method based on a protein microarray integrated with a microfluidic chip for the process of SELEX (systematic evolution of ligands by exponential enrichment). Lactoferrin from bovine milk was used as a target protein, while bovine serum albumin (BSA), α-lactalbumin, β-lactoglobulin and casein were used as negative proteins. They were separately dotted and immobilized to prepare the protein microarray and the resulting microarray was further integrated into a microfluidic chip for the SELEX (PMM–SELEX) process. The interaction between aptamer candidates and targets could be monitored using a fluorescence microarray scanner and the whole PMM–SELEX process was performed through seven-round selection. As a result, five aptamers (Lac-14, Lac-6a, Lac-9, Lac-5, Lac-3a) with high specificity and affinity can be repeatedly obtained during three times of independent repeated selection. Surface plasmon resonance (SPR) was used to calculate the dissociation constants (Kd). The aptamer Lac-6a was then used for detection of lactoferrin by fluorescence polarization. A linear response was observed for lactoferrin concentrations in the range of 0.78–50 μg mL−1 and the detection limit was 0.39 μg mL−1. Thus, the innovative PMM–SELEX presented shows stability, accuracy and high efficiency for aptamer screening.
Introduction
Since the invention of systematic evolution of ligands by exponential enrichment (SELEX) for selection of aptamers,1–3 great efforts have been made to develop innovative methods for rapid, efficient and high-throughput production of aptamers.4–6 The most critical steps in SELEX are separation efficiency, PCR and clone. Magnetic bead-based SELEX in tubes was usually used to isolate aptamers for proteins with high affinity and selectivity.7–11 Nanomaterial-assisted non-immobilized SELEX in tubes was explored to avoid immobilization of proteins on solid surfaces and reduce the rounds of selection.12–14 More recently, a wide variety of microfluidic techniques have been explored to increase the separation efficiency, including capillary electrophoresis (CE) microfluidic SELEX,15–17 sol–gel microfluidic SELEX,18–20 surface plasmon resonance (SPR) microfluidic SELEX,21,22 magnetic bead-based microfluidic SELEX,23–26 and bead-based microfluidic SELEX.27,28 For improvement of PCR efficiency, agarose droplet PCR29 and emulsion PCR (ePCR)30–32 were explored and the positive clones were directly picked out without an extra process of clone detection which was usually used in conventional SELEX. All the developed methods aim to make the SELEX more time-saving, automated, efficient and inexpensive. Therefore, it is considered to be still of critical importance to improve the stability, reproducibility and quality control of SELEX as well as simplify and standardize the whole procedure.
Microfluidic technology is undoubtedly one of the most popular methods in SELEX because of its improved speed, decreased costs, increased resolving power, and automation.5 The magnetic bead-based microfluidic SELEX could not provide the one-line monitoring method for the enrichment of aptamers.23–26 The bead-based microfluidic SELEX28 could be monitored by fluorescence microscopy; however, the observed region is limited and the uniform collection of fluorescence intensity is also difficult. Taking advantage of the microfluidic system, we developed the PMM–SELEX method for rapid, efficient and quantitative selection of aptamers. Compared with the current microfluidic SELEX, the PMM–SELEX had the following advantages. Firstly, the immobilization of targets in the microfluidic chip is free of solid beads such as magnetic beads and microbeads. Instead, the targets could be dotted in the form of a microarray with good reproducibility by physical adsorption in the PMM chips. Secondly, the enrichment of the whole PMM–SELEX can be quantitatively monitored by scanning the microarray. Thus, an effective evaluation on the efficiency of enrichment was achieved. Thirdly, PMM–SELEX is time-saving and efficient, the repeated sequences can be found in the fifth round of selection and the whole process can be finished at the seventh round. Fourthly, the PMM–SELEX was highly reproducible and high-throughput. The PMM–SELEX was firstly used for the selection of important proteins in milk. Lactoferrin is a nonheme iron-binding glycoprotein that is strongly expressed in bovine and human milk and it plays important functions during infancy.33,34 Some methods have been used for detection of lactoferrin, such as liquid chromatography–tandem mass spectrometry,35,36 paper-based analytical devices with low sensitivity37,38 and sandwich immunoassay.39 It is urgent to develop a sensitive, simple and rapid method to improve the abovementioned methods. As a result, we established a new aptamer-based method benefiting from our SELEX results for sensitive and rapid detection of lactoferrin.
Experimental
Materials and reagents
SG-2506 borosilicate glass was purchased from Changsha Shaoguang Chrome Blank Co., Ltd. Sylgard 184 elastomer base and curing agent for polydimethylsiloxane (PDMS) were purchased from Dow Corning (Midland, MI). The dechroming liquid was a mixture of 20% (m/v) Ce(NH4)2(NO3)6 and 3.5% (v/v) CH3COOH. The etching solution contained 1.86% (m/v) NH4F, 4.64% (v/v) HNO3 and 5.00% (v/v) HF. Aldehyde-modified slides (Shanghai Biotechnology Co., Ltd.), lactoferrin, casein, α-lactalbumin, β-lactoglobulin from bovine milk and bovine serum albumin (BSA) were purchased from Sigma Aldrich (St. Louis, MO, USA). Streptavidin magnesphere® paramagnetic particles (SA-MPs) were purchased from Promega Corporation (Madison, USA). 2× Taq Master Mix (Tiangen Biotech. Co., Ltd.), 2× SYBR® Premix Ex TaqTM II (Takara Bio Inc., Japan) was used for PCR amplification. The following phosphate buffers were used in the experiment: 1× PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4·12H2O, 2 mM KH2PO4), 1× PBSM (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, 1 mM MgCl2, pH 7.4), 1× PBST (1× PBS + 0.05% Tween-20); 1× PBS was purchased from Invitrogen (Carlsbad, CA, USA). 1× TBE (89 mmol L−1 Tris, 89 mmol L−1 boric acid, 2 mmol L−1 EDTA) was used for 30% polyacrylamide gel electrophoresis.
Apparatus
A microfluidic pump (Model LSP04-1A, Longer Pump Corp., Baoding, China) was used for liquid manipulation. A plasma cleaner (Model PDC-MG, Chengdu MINGHENG Science & Technology Co., Ltd., China) was used to bond the glass slide with the PDMS piece. The fluorescence intensity was measured using a BioTek Synergy H1 (USA). The protein microarrays were obtained using a Smart Arrayer and scanned using a Luxscan-10K/A microarray scanner (532 nm laser source for TAMRA, Beijing CapitalBio Co., China). An Applied Biosystems® SimpliAmp™ thermal cycler (Thermo Fisher Scientific Inc., USA) was used for PCR amplification. A GEN4T thermal cycler (Hangzhou Bio-gener Technology Co., Ltd., China) was used for heating the microarray chip at 95 °C. SPR measurements were performed using Biacore X100 apparatus (GE Healthcare, Sweden) and the data were fitted by date using BIAcore X100 evaluation software.
SELEX library and primers
All the ssDNA used in this study were synthesized according to Yang's group28 by Sangon Biotech (Shanghai, China). The selection ssDNA library consisted of 40 nucleotide (nt) randomized sequences and 20 nt primer sequences. Lib: 5′-TAMRA-GACAGGCAGGACACCGTAAC-N40-CTGCTACCTCCCTCCTCTTC-3′. The primers for PCR amplification are the TAMRA-labeled forward primer (TAMRA-FP) and biotinylated reverse primer (Bio-RP). TAMRA-FP: 5′-TAMRA-GACAGGCAGGACACCGTAAC-3′, Bio-RP: 5′-GAAGAGGAGGGAGGTAGCAG-biotin-3′.
Design and fabrication of protein microarrays in microfluidic chips
The microfluidic device was prepared on borosilicate glass substrates using photolithographic and wet chemical etching techniques. It consisted of two serpentine microfluidic channels which were connected by a silicone pipe (700 μm wide, 100 μm deep). One channel was dotted with target proteins for positive selection while another channel was dotted with counter proteins for negative selection. The procedures for fabrication of a photomask and microchannels were described below. Briefly, a pattern design on a photomask with microchannels was firstly transferred onto the borosilicate glass substrate following UV exposure of 35 seconds. Then, they are rinsed with NaOH (0.5% (w/v)) for 1 min and dechroming liquid for 5 min. For etching, the glass with micropatterns was etched with HF/NH4F/HNO3 solution at 37 °C for 40 min. The resulting glass substrate was then washed with ultrapure water and dried at 80 °C for 1 h. The PDMS pre-polymer and curing agent were mixed, degassed and heated to make a PDMS layer. The procedures for fabrication of a PMM chip were described below. First, a glass slide was treated with NaOH and thoroughly washed using ultrapure water to increase the hydrophilicity and physical adsorption. Second, the target protein (lactoferrin) and negative proteins (BSA, α-lactalbumin, β-lactoglobulin, casein) were dotted on the glass slide using a Smart Arrayer. Lastly, the PDMS layer was treated with plasma and immediately bound the glass slide to fabricate the PMM chip.
Aptamer selection and image capture of microarrays
The target protein (lactoferrin) and negative proteins (BSA, α-lactalbumin, β-lactoglobulin, casein) were dotted on aldehyde-modified slides. For the first round of selection, only lactoferrin was immobilized. After standing for 2 h at 37 °C, the slide was blocked by 20 mg mL−1 BSA with 0.01 mM random short ssDNA (20 nt) for 1 h and washed with 1× PBST (150 μL) for 10 min. Then, the ssDNA library (1 nmol) dissolved in 125 μL of 1× PBSM (1× PBS + 1 mM MgCl2, pH 7.4) was injected into the channel at 2.5 μL min−1 for 50 min, and 150 μL of 1× PBS were pumped into the channel at 15 μL min−1 to wash the unreacted aptamers. Finally, PDMS was torn off and the slide was scanned using a Luxscan-10K/A microarray scanner. After scanning, the microarray was washed with 500 μL of DEPC water at 95 °C for 5 min, and the solution was blown dry to 92 μL using high purity nitrogen for PCR amplification.
PCR amplification
The collected products are divided into four parts. 23 μL of the collected products was mixed with 2× Taq Master Mix (25 μL), 20 μM TAMRA-FP (1 μL), and 20 μM Bio-RP (1 μL). The thermal cycling conditions were as follows: 94 °C for 5 min, 10 cycles of 94 °C for 30 s, 60.5 °C for 30 s, and 72 °C for 30 s, followed by an extra extension at 72 °C for 5 min. Then, the amplified PCR products were used as templates, and 5 μL of PCR products were mixed with 25 μL of 2× SYBR® Premix Ex TaqTM II, 20 μM TAMRA-FP (1 μL), and 20 μM Bio-RP (1 μL) and 18 μL of DEPC water. The mixtures were reamplified and 10 μL of the PCR mixture was collected during the extension step of each cycle. The fluorescence of the PCR mixture was detected using a BioTek Synergy H1 to find the optimal cycle number. Next, the remaining collected products were all amplified using 2× Taq Master Mix at the optimized cycle number.
ssDNA generation
The PCR product was incubated with SA-MPs for 1 h with strong shaking at 37 °C to separate the dsDNA from the PCR mixture. ssDNA was then generated in solution by incubating the beads with 50 mM NaOH (25 μL) for 5 min at room temperature. The supernatant was collected and neutralized with 100 mM HCl (12.5 μL). Finally, 25 μL of DEPC water and 62.5 μL of 2× PBSM (NaCl 254 mM, KCl 5.4 mM, Na2HPO4 20 mM, KH2PO4 4 mM, MgCl2 2 mM) were added to the solution to achieve a final salt concentration which is the same as that of 1× PBSM. The amount of ssDNA in solution was detected using a BioTek Synergy H1. The purity of the ssDNA was examined by 30% polyacrylamide gel electrophoresis.
Cloning and sequencing of selected aptamers
One part of the fifth, sixth and seventh selected products were PCR amplified with unlabeled primers, respectively. The three PCR products were cloned by Sangon Bio-technology Co., Ltd. A hundred and twenty colonies of every round were randomly picked and sequenced at Sangon (Shanghai, China). The secondary structure analysis of selected aptamers was performed using the IDT (http://sg.idtdna.com/calc/analyzer).
Specificity of selected aptamers toward different proteins
A 6 × 6 microarray was fabricated on aldehyde-modified slides (2 × 4 = 8 reaction pond) using a Smart Arrayer. First, 5 mg mL−1 BSA, α-lactalbumin, β-lactoglobulin, casein and lactoferrin were immobilized on the slide and left to stand for 2 h at 37 °C. Second, the slide was blocked by reacting with 10 mg mL−1 BSA for 1 h and washed with 1× PBST 3 times, for 5 min for each washing. Thirdly, 50 μL of 1 μM aptamers (Lac-14, Lac-6a, Lac-9, Lac-6b, Lac-3a, Lib) were added and allowed to react for 60 min at 37 °C followed by the washing step as described before. The array was scanned and the data were collected using a Luxscan-10K/A microarray scanner.
K
d determination using SPR
A Biacore X100 instrument was used to characterize the binding features of selected aptamers to lactoferrin. The CM5 chip was activated with 400 mM N-ethyl-N′-(dimethylaminopropyl)carbodiimide (EDC) and 100 mM N-hydroxysuccinimide (NHS) for 7 min, followed by injection of proteins into two channels, i.e. FC1 and FC2. 50 μg mL−1 lactoferrin was dissolved in 1× PBS (pH 7.4) and injected into the FC2 channel. A mixture of counter proteins (50 μg mL−1 α-lactalbumin, 50 μg mL−1 β-lactoglobulin, 5 μg mL−1 casein, and 100 μg mL−1 BSA) was dissolved in 10 mM sodium acetate (pH 5.0) and injected into the FC1 channel. Lactoferrin and counter proteins were immobilized by a target level model at 4900 RU and 4500 RU (1 RU ≈ 1 pg mm−2), respectively. To block the remaining unreacted functional groups on the CM5 chip, 1 M ethanolamine hydrochloride (pH 8.5) was injected into the flow cell for 7 min. Then the aptamers (Lac-14, Lac-6a, Lac-9, Lac-3a, Lac-5 and Lib) were respectively injected and 1× PBSM was used as the running buffer. The concentrations of the aptamers are 1 nM, 2 nM, 4 nM, 8 nM, and 16 nM; in addition, different concentrations of aptamers are sequentially injected. The flow rate (10 μL min−1) was maintained constant during the whole process, the association time was kept at 120 s and the dissociation time was kept at 600 s, respectively. Regeneration was carried out with 2 M NaCl for 15 s. Data analysis was carried out using X100 evaluation software, and the data were fitted to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model.
:1 binding model.
Detection of lactoferrin using fluorescence polarization
For specificity, Lac-6a-FITC (10 μL, 250 nM) was separately added to 25 μg mL−1 lactoferrin and 500 μg mL−1 α-lactalbumin, β-lactoglobulin, casein, and BSA. For lactoferrin detection, different concentrations of lactoferrin in the range of 0–50 μg mL−1 (100 μL) were added to the Lac-6a-FITC (10 μL, 250 nM) in a 96 black micro-well plate for 15 min at 37 °C. Then the plate was directly scanned using a BioTek Synergy H1 and the data were collected (excitation: 485/20, emission: 528/20).
Results and discussion
Selection of lactoferrin aptamers
The principle of PMM–SELEX for aptamer screening is shown in Scheme 1. The target protein (lactoferrin) and the negative proteins (BSA, α-lactalbumin, β-lactoglobulin, casein) were dotted into the microarray integrated in the microfluidic chip. For the first round of selection, only the target protein was dotted. Firstly, a library of ssDNA (Lib) was pumped into the PMM chip for 50 minutes (min), and then 1× PBS was used for washing nonspecific Lib. After scanning using the Luxscan-10K/A microarray scanner, the microarray was eluted with DEPC water at 95 °C for 5 min to collect the specific Lib for PCR amplification. The collected Lib was amplified and separated by SA-MPs to produce dsDNA, and then 50 mM NaOH was used to transform dsDNA into ssDNA for the next round of selection. The concentration of the newly generated Lib was detected and calculated by fluorescence assay. In the experiment, the fifth, sixth and seventh rounds of PCR products were cloned and sequenced. The sequences which are repeated more than three times are chosen for further analysis.
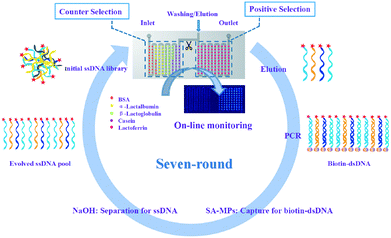 |
| | Scheme 1 The preparation of the PMM chip and the principle of PMM–SELEX for aptamer screening. | |
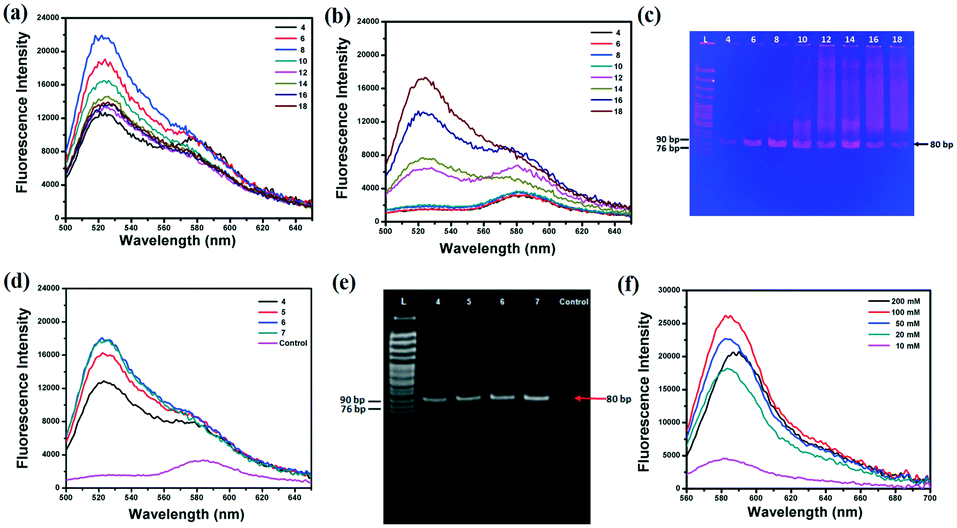
For efficient SELEX, quantitative analysis of every parameter is very important. PCR and separation efficiencies are undoubtedly the two most vital parameters. To shorten the time of the whole PCR process, monitoring the fluorescence intensity of SYBR Green I during the PCR process substitute for traditional polyacrylamide gel electrophoresis (PAGE)24–26 or agarose gel electrophoresis27,28 to find the optimal PCR amplification cycle. The Lib (10 fM and 0 fM) was firstly pre-amplified with 10 cycles by 2× Taq Master Mix and then one-tenth of products were re-amplified with 2× SYBR® Premix Ex TaqTM. The cycles of PCR should be optimized as by-products will be produced with the increased cycles of PCR. To investigate the effect of PCR cycles on the generation of by-products, the primary Lib was firstly used as a model. The results showed that the fluorescence intensity reached a maximum (Fig. 1a), and the background signal was greatly increased after 8 cycles of PCR (Fig. 1b). And the PAGE results also showed that the by-products were formed after 8 cycles of PCR (Fig. 1c). In addition, the collected Lib from the first round of selection was pre-amplified with 10 cycles and then re-amplified; the fluorescence intensity and PAGE image of the re-amplified PCR products are shown in Fig. 3d and e. The optimal PCR cycle produced by fluorescence spectrum and gel electrophoresis was consistent, 6 cycles was chosen as the optimal cycle. In the next round of selection, the optimal PCR was also monitored by the fluorescence spectrum of SYBR. The optimal PCR cycle could be deduced just by the fluorescence spectrum which greatly reduced the time for the whole PCR process. After PCR amplification, the PCR products were separated and purified by SA-MPs, and finally NaOH was used to transform dsDNA into ssDNA. A high concentration of NaOH was found to shift the fluorescence spectrum of TAMRA, and the optimal concentration of NaOH should be smaller than 100 mM. Thus, 50 mM was chosen for the next experiments (the separation efficiency is 83.3%).
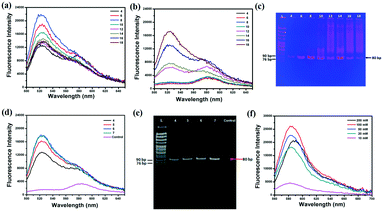 |
| | Fig. 1 The fluorescence intensity of the re-amplified PCR products in different PCR cycles, (a) 10 fM Lib, (b) 0 fM Lib. (c) The gel electrophoretogram of the re-amplified PCR products. The fluorescence spectrum (d) and gel electrophoretogram (e) of the re-amplified PCR products of the collected Lib in 4 cycles, 5 cycles, 6 cycles, 7 cycles and the negative control. (f) The fluorescence spectrum of the generated Lib in different NaOH concentrations. | |
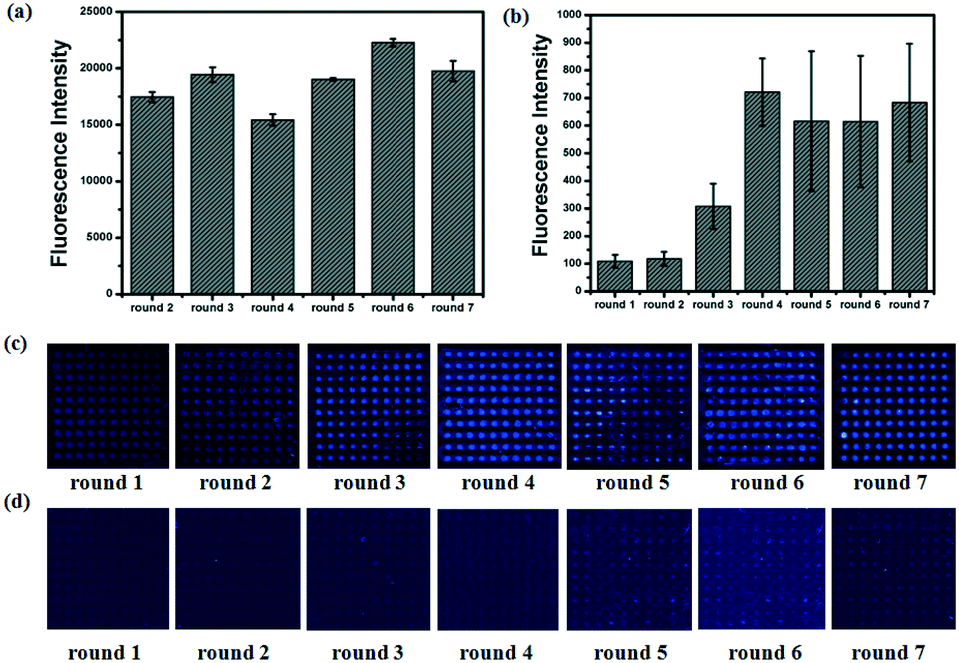
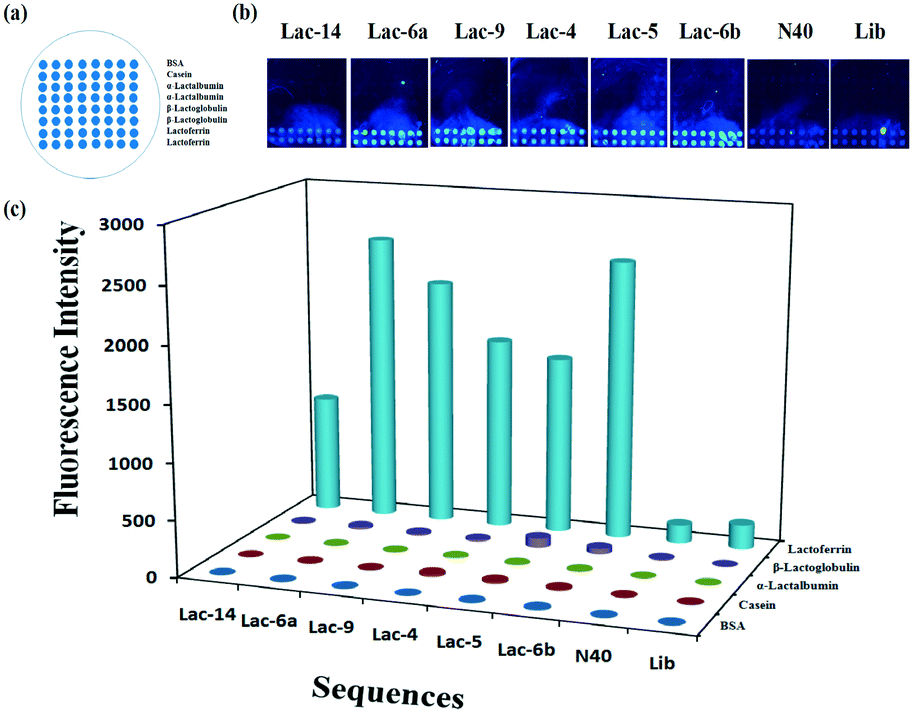
To calculate the concentration of Lib generated by NaOH, the fluorescence intensity of the primary Lib in different concentrations was collected (Fig. S1†). The concentration of the generated Lib in every round of selection was calculated as 48.6 pmol, 53.13 pmol, 43.86 pmol, 52.19 pmol, 59.51 pmol and 53.85 pmol respectively, according to the fluorescence intensity in Fig. 2a. The enrichment of specific aptamers on lactoferrin was monitored using a Luxscan-10K/A microarray scanner (Fig. 2c). In addition, the enriched aptamers were totally eluted and the fluorescence images after elution with water are shown in Fig. 2d. The fluorescence intensity of Lib specific to lactoferrin was significantly increased with the selected rounds. From round 1 to round 5 of selection, the fluorescence intensity was increased gradually. The signal surged in round 5 and reached a plateau in the next four rounds, so the fifth, sixth, and seventh PCR products with unlabeled primers were cloned and sequenced. 120 colonies of every round were randomly picked and sequenced at Sangon (Shanghai, China). The products from the fifth, sixth, and seventh rounds of aptamer selection were sequenced to reveal the selected lactoferrin-binding aptamers. Among the fifth colonies, three sequences were repeated twice, and a sequence was repeated 3-fold. Among the sixth colonies, two sequences were repeated twice and two sequences were repeated 3-fold. Among the seventh colonies, a sequence was repeated 11-fold, 5-fold, 4-fold, 4-fold respectively, two sequences were repeated 3-fold, and a sequence was repeated 2-fold. It demonstrated that the selected products of round 7 showed a relatively good enrichment. From the PMM–SELEX, 11 sequences which were repeated more than two times are selected, and the sequences and repeated times are shown in Table 1. Six sequences (Lac-14, Lac-6a, Lac-9, Lac-4, Lac-5, Lac-6b) which were repeated more than three times are used for the study of specificity of selected aptamers towards other proteins. To study the specificity of selected aptamers to lactoferrin, a fluorescence method based on a microarray chip was used. 5 mg mL−1 of different proteins are dotted as microarrays on aldehyde-modified slides and the schematic is shown in Fig. 3a. After immobilization, blocking and washing steps, 1 μM TMARA, labeled Lac-14, Lac-6a, Lac-9, Lac-4, and Lac-5, was added onto the slides for 1 h at 37 °C. The sequences and ΔG of the aptamers are shown in Table 1, ΔG indicates the stability of the selected aptamers in buffers, and the website of IDT was used to calculate ΔG and simulate the secondary structures of the aptamers. The selected sequences of N40 could not form stable secondary structures (Table 1). Thus, N40 with primers of selected aptamers was simulated and the truncated sequences are shown in Table 1. When the irrelevant bases which did not affect the secondary structures of the aptamers were truncated, a lower ΔG of the aptamers is generated. So the truncated sequences are used to test specificity towards other proteins. The fluorescence images and intensities of selected aptamers on different proteins are shown in Fig. 3b and c. As shown in Fig. 3b and c, the proteins of BSA, α-lactalbumin, β-lactoglobulin and casein showed almost no binding force with selected aptamers. The ssDNA of Lib and N40 also showed a much lower fluorescence intensity to lactoferrin indicating the high enrichment of selected aptamers in PMM–SELEX. The aptamers of Lac-14, Lac-6a, Lac-9, Lac-4, Lac-5, and Lac-6b showed a high fluorescence intensity which indicated their high specificity and good selectivity towards lactoferrin. Therefore, our PMM–SELEX is highly efficient.
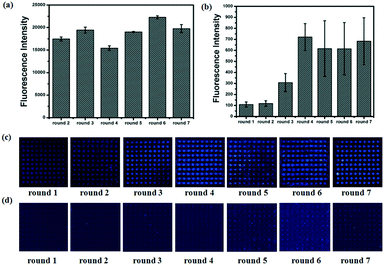 |
| | Fig. 2 During each round of selection, from round 1 to round 7. (a) The fluorescence intensity of the evolved Lib. (b) The fluorescence intensity of the enriched aptamers on lactoferrin microarrays. The fluorescence images of the enriched aptamers on lactoferrin microarrays (c) before and (d) after elution with DEPC water. | |
Table 1 The information about the selected aptamers
| Aptamer ID |
Sequences of N40 from 5′ to 3′ |
ΔG (kcal mol−1) |
Truncated sequences of Lib from 5′ to 3′ |
ΔG (kcal mol−1) |
Repeated times |
| The fifth round |
The sixth round |
The seventh round |
| Lac-14 |
CCTAACACGTACGGGGCATTTATGGCATAGCTCTTCCTCC |
−0.77 |
GCAGGACACCGTAACCCTAACACGTACGGGGCATTTATGGCATAGCTCTTCCTCCCTGC |
−5.92 |
2 |
1 |
11 |
| Lac-6a |
CGGTGCATCTATGGCTACTAGCTTTTCCTGCCTATACTAC |
−2.27 |
AGGCAGGACACCGTAACCGGTGCATCTATGGCTACTAGCTTTTCCTGCCT |
−11.21 |
2 |
3 |
1 |
| Lac-9 |
GGGCAATCTTGCTCTTATTTTCACACTTGATAAATATGTC |
−4.27 |
GCAGGACACCGTAACGGGCAATCTTGCTCTTATTTTCACACTTGATAAATATGTCCTGC |
−10.25 |
2 |
2 |
5 |
| Lac-4 |
GGGCGAACGCTCTAAATAAATGGCATCAATTTCTTTTGCC |
−4.29 |
GCAGGACACCGTAACGGGCGAACGCTCTAAATAAATGGCATCAATTTCTTTTGCCCTGC |
−5.82 |
0 |
0 |
4 |
| Lac-5 |
GGGCTTATGCTCTTAAAAATCCTGAGCGACTTTTTATGTA |
−3.08 |
GCAGGACACCGTAACGGGCTTATGCTCTTAAAAATCCTGAGCGACTTTTTATGTACTGC |
−5.46 |
1 |
3 |
1 |
| Lac-6b |
TCGGGCAAAGCTCTGAATAATGTTCAACCAATATTCTGTC |
−2.56 |
GCAGGACACCGTAACTCGGGCAAAGCTCTGAATAATGTTCAACCAATATTCTGTCCTGC |
−9.49 |
1 |
1 |
4 |
| Lac-3a |
AATACTCCTGTTACCGTGCATCTATGGCCATTGGCTTTTC |
−2.26 |
GCAGGACACCGTAACAATACTCCTGTTACCGTGCATCTATGGCCATTGGCTTTTCCTGC |
−8.12 |
0 |
0 |
3 |
| Lac-3b |
ACGGGCTCTGCTTTTCCAATAATGACAAGTATTGGATGGC |
−4.91 |
GCAGGACACCGTAACACGGGCTCTGCTTTTCCAATAATGACAAGTATTGGATGGCCTGC |
−9.39 |
0 |
1 |
2 |
| Lac-3c |
ACTCTCAACCATGACGGTGCAATATTGGCGCTAGCTCTTC |
−3.29 |
GCAGGACACCGTAACACTCTCAACCATGACGGTGCAATATTGGCGCTAGCTCTTCCTGC |
−8.06 |
0 |
2 |
1 |
| Lac-3d |
GTATTAACTAGTACCAGTGCATCTATGGCTATTGCTTTTC |
−1.62 |
GCAGGACACCGTAACGTATTAACTAGTACCAGTGCATCTATGGCTATTGCTTTTCCTGC |
−2.76 |
0 |
0 |
3 |
| Lac-3e |
GTTCGTAACAACACGGCATCTATGGCTTTAGCTCTTGTAC |
−3.1 |
GCAGGACACCGTAACGTTCGTAACAACACGGCATCTATGGCTTTAGCTCTTGTACCTGC |
−5.32 |
3 |
0 |
0 |
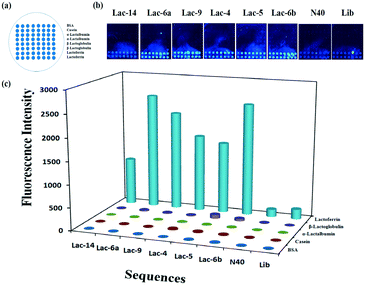 |
| | Fig. 3 (a) Schematic of the microarray chip for protein immobilization, (b) fluorescence images and (c) the intensity of selected aptamers to lactoferrin. | |
To test the reproducibility and stability of the PMM–SELEX method, we repeated the whole process of PMM–SELEX for another two times. Some parameters were changed to prove our assumption on PMM–SELEX. Firstly, the selected number of aptamers is related to the fluorescence intensity on the microarrays. Secondly, increasing the fluorescence intensity of the microarray in each round could shorten the round of selection. For the second round of PMM–SELEX, the concentration of lactoferrin was 2.5 mg mL−1, and the concentration of Lib in every round of selection was about 90 pmol at the second round of PMM–SELEX (Table S1†). As the concentration of Lib in each round was increased, the fluorescence intensities of each round are obviously increased (Fig. S2†). In addition, the selected sequences and repeated times are shown in Table S2.† Compared to Table 1, we could conclude that some aptamers were well enriched just in the fifth round.
What's more, six sequences (Lac-14, Lac-6a, Lac-9, Lac-5, Lac-3a, Lac-6b) were repeatedly selected in the second round of selection indicating the good reproducibility and stability of PMM–SELEX. In the third round of PMM–SELEX, the concentration of lactoferrin was 5 mg mL−1, and the concentration of Lib in every round of selection was about 70 pmol (Table S1†). The fluorescence intensities and images of each round are shown in Fig. S3.† And the selected sequences and repeated times are shown in Table S3.† The number of selected aptamers is increased and five sequences (Lac-14, Lac-6a, Lac-9, Lac-5, Lac-3a) were still repeatedly selected. The repeated PMM–SELEX process proved our assumption and also indicated the good reproducibility and stability of PMM–SELEX. In addition, the number of selected aptamers could be tuned by the amount of Lib and the fluorescence intensity of the microarrays. Therefore, the PMM–SELEX is highly reproducible and stable. The selected aptamers in each cycle of PMM–SELEX are shown in Fig. 4a, and five aptamers (Lac-14, Lac-6a, Lac-9, Lac-5, Lac-3a) were chosen for further examination including the structure and affinity constant. Firstly, the structures of the aptamers are simulated using OligoAnalyzer 3.1 and the loop–stem is found in five aptamers. Therefore, we believe that the loop–stem may be necessary for binding with lactoferrin. Secondly, the structures with minimum ΔG were analyzed and the tails at the end of the loop–stem were deleted. Thirdly, the aptamers with truncated bases were simulated again and the final simulated structures of Lac-14, Lac-6a, Lac-9, Lac-5, and Lac-3a are shown in Fig. 4. As shown in Fig. 4, the aptamers have very similar secondary structures which are characteristic of stems and loops. And Lac-14 had more loops which may indicate its different affinity to lactoferrin.
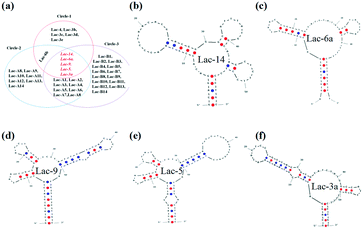 |
| | Fig. 4 (a) The selected aptamers from three times of PMM–SELEX with 7 rounds of each time. The simulated structure of selected aptamers using OligoAnalyzer 3.1 at the minimum ΔG. (b) Lac-14, (c) Lac-6a, (d) Lac-9, (e) Lac-5, and (f) Lac-3a. | |
Determination of aptamer binding affinity
SPR binding studies were performed for five representative aptamers. The immobilization level of lactoferrin and counter proteins were 4900 RU and 4500 RU, respectively, indicating successful immobilization of proteins on the CM5 chip. For SPR measurement, aptamers were sequentially injected into the FC1 channel and then into FC2. The resonance units (RUs) were collected and ΔRU (RUtarget protein − RUnegative proteins) was used to calculate the binding affinity of the different aptamers. Dose-dependent binding sensorgrams for aptamers to lactoferrin are shown in Fig. 5. The SPR sensorgrams obtained for each aptamer illustrated the concentration-dependent binding kinetics in the range of 1–16 nM in which Lib shows no detectable response. The Lib was tested for its binding but did show little response to lactoferrin when the concentration of Lib is in the range of 100–1600 nM. However, the selected aptamers of Lac-14, Lac-6a, Lac-9, Lac-3a, and Lac-5 showed high binding affinity. The calculated Kon (association rate constant) was from 1.20 × 106 to 11.66 × 106 (M−1 s−1) and the Koff (dissociation rate constant) was from 3.43 × 10−3 to 1.04 × 10−2 (s−1) (Table S4†). As a result, the Kd values of Lac-14, Lac-6a, Lac-9, Lac-3a, and Lac-5 were 5.48 ± 1.79 nM, 1.04 ± 0.50 nM, 1.07 ± 0.34 nM, 0.63 ± 0.06 nM, and 0.92 ± 0.02 nM, respectively. The Kd value of Lac-14 is higher than those of the other aptamers, which may be related to its structure having more loops than the other aptamers. In a word, the selected aptamers had high affinity and the SPR results are directly supportive of the high efficiency of the PMM–SELEX method.
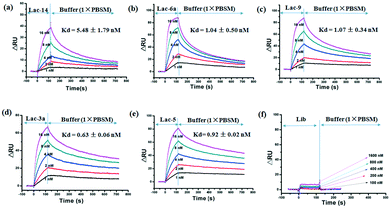 |
| | Fig. 5 SPR studies for binding kinetics of selected aptamers: (a) Lac-14, (b) Lac-6a, (c) Lac-9, (d) Lac-3a, (e) Lac-5, (f) Lib. The concentrations of aptamers and Kd are indicated in the overlay of the sensorgrams. | |
Detection of lactoferrin using fluorescence polarization
Fluorescence polarization is a simple and reliable method for rapid analysis of the interaction between fluorophore-labeled aptamers and targets. Fluorescence polarization has many advantages.40,41 First, polarization assays are homogeneous without a separation process. Second, polarization assays are reproducible and facilely automated. Third, polarization assays are rapid and insensitive to photobleaching and environmental fluctuation. Among aptamers of Lac-14, Lac-6a, Lac-9, Lac-3a, and Lac-5, the bases of Lac-6a was fewest, so Lac-6a was chosen to decrease the background signal of fluorescence polarization. The selected aptamer Lac-6a was labeled with fluorescein isothiocyanate (FITC), and polarization was used to evaluate the interaction between Lac-6a-FITC and lactoferrin. Firstly, the incubation time between lactoferrin and Lac-6a-FITC was examined and the interaction speed was very fast (Fig. 6a). Second, the concentration of Lac-6a-FITC was optimized and the signal of blank increased when the concentration of Lac-6a-FITC was lower than 250 nM (Fig. 6b). The aptamer Lac-6a-FITC showed high specificity towards lactoferrin, because the polarization of Lac-6a-FITC in other proteins was much lower than that in lactoferrin (Fig. 6c). In addition, a linear response was observed for lactoferrin concentrations in the range of 0.78–50 μg mL−1 (r = 0.98) (Fig. 6d). The interaction between selected aptamers and proteins could be easily detected by this assay. Compared to the current methods for detection of lactoferrin,35–39 this assay is simple, rapid and efficient. As shown in Fig. 4a, there are 39 sequences, for a further decrease of the detection limit for fluorescence polarization. Aptamers (Lac-A1–Lac-A14 and Lac-B1–Lac-B14) in Fig. 4a can be further truncated and analyzed to find more suitable aptamers with minimum bases for fluorescence polarization. We offer an efficient and sensitive way to detect lactoferrin based on our selected aptamers.
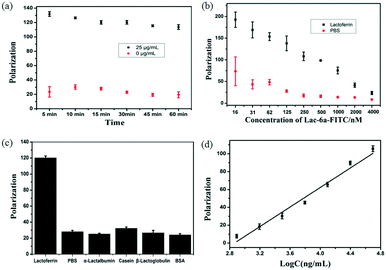 |
| | Fig. 6 The polarization of Lac-6a-FITC (a) at different incubation times and (b) at different concentrations when the concentration of lactoferrin was 50 μg mL−1. (c) In different proteins, lactoferrin (25 μg mL−1), α-lactalbumin (500 μg mL−1), β-lactoglobulin (500 μg mL−1), casein (500 μg mL−1), and BSA (500 μg mL−1). (d) The concentration of lactoferrin was in the range of 0.78–50 μg mL−1. | |
Conclusions
To conclude, taking advantage of a protein microarray and a microfluidic chip, we have established a new PMM–SELEX method that allowed rapid, repetitive and efficient selection of lactoferrin. The PMM allowed for rapid immobilization of proteins, separation of unbinding aptamers and imaging of enriched aptamers on microarrays. This method demonstrated fast selection, taking only 7 days to finish 7 rounds to obtain aptamers with binding constants at the 10−9 level. In addition, the PMM–SELEX is highly reproducible and efficient, and the selected aptamers are highly specific and selective towards the target protein. Moreover, this method is available for selection of any proteins. The presented approach may provide a standard method for rapid and stable selection of aptamers.
Author contributions
D. Xu and H. Li conceived the idea. H. Li, X. Liu and D. Xu designed the experiments. X. Liu and H. Li carried out most of the experiments and analyzed the results, and W. Jia carried out the SPR experiment. Z. Chen analyzed the data of the clone. H. Li, X. Liu and D. Xu interpreted the results and wrote of the paper.
Acknowledgements
We acknowledge financial support of the National Natural Science Foundation of China (21227009, 21475060 and 21405077), Natural Science Foundation of Jiangsu Province (BK20140591). Research Foundation of Jiangsu Province Environmental Monitoring (1116), Special Fund for Agro-scientific research in the Public interest (201403071) and the National Science Fund for Creative Research Groups (21121091).
References
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- D. L. Robertson and G. F. Joyce, Nature, 1990, 344, 467–468 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- M. Darmostuk, S. Rimpelova, H. Gbelcova and T. RumL, Biotechnol. Adv., 2015, 33, 1141–1161 CrossRef CAS PubMed.
- H. Lin, W. Zhang, S. Jia, Z. Guan, C. J. Yang and Z. Zhu, Biomicrofluidics, 2014, 8, 041501 CrossRef PubMed.
- M. Yüce, N. Ullah and H. Budak, Analyst, 2015, 140, 5379–5399 RSC.
- D. Barash, R. Stoltenburg, T. Schubert and B. Strehlitz, PLoS One, 2015, 10, e0134403 Search PubMed.
- L. Chen, F. Rashid, A. Shah, H. M. Awan, M. Wu, A. Liu, J. Wang, T. Zhu, Z. Luo and G. Shan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10002–10007 CrossRef CAS PubMed.
- Q. Gong, J. Wang, K. M. Ahmad, A. T. Csordas, J. Zhou, J. Nie, R. Stewart, J. A. Thomson, J. J. Rossi and H. T. Soh, Anal. Chem., 2012, 84, 5365–5371 CrossRef CAS PubMed.
- J.-C. Lai and C.-Y. Hong, J. Mater. Chem. B, 2014, 2, 4114 RSC.
- Z. Xi, R. Huang, Z. Li, N. He, T. Wang, E. Su and Y. Deng, ACS Appl. Mater. Interfaces, 2015, 7, 11215–11223 CAS.
- H. Gu, N. Duan, S. Wu, L. Hao, Y. Xia, X. Ma and Z. Wang, Sci. Rep., 2016, 6, 21665 CrossRef CAS PubMed.
- J.-W. Park, R. Tatavarty, D. W. Kim, H.-T. Jung and M. B. Gu, Chem. Commun., 2012, 48, 2071–2073 RSC.
- Y. Tan, Q. Guo, Q. Xie, K. Wang, B. Yuan, Y. Zhou, J. Liu, J. Huang, X. He, X. Yang, C. He and X. Zhao, Anal. Chem., 2014, 86, 9466–9472 CrossRef CAS PubMed.
- L. Dong, Q. Tan, W. Ye, D. Liu, H. Chen, H. Hu, D. Wen, Y. Liu, Y. Cao, J. Kang, J. Fan, W. Guo and W. Wu, Sci. Rep., 2015, 5, 15552 CrossRef CAS PubMed.
- S. D. Mendonsa and M. T. Bowser, J. Am. Chem. Soc., 2004, 126, 20–21 CrossRef CAS PubMed.
- H. Nie, Y. Chen, C. Lü and Z. Liu, Anal. Chem., 2013, 85, 8277–8283 CrossRef CAS PubMed.
- J.-Y. Ahn, S. Lee, M. Jo, J. Kang, E. Kim, O. C. Jeong, T. Laurell and S. Kim, Anal. Chem., 2012, 84, 2647–2653 CrossRef CAS PubMed.
- H. Bae, S. Ren, J. Kang, M. Kim, Y. Jiang, M. M. Jin, I. M. Min and S. Kim, Nucleic Acid Ther., 2013, 23, 443–449 CrossRef PubMed.
- S.-m. Park, J.-Y. Ahn, M. Jo, D.-k. Lee, J. T. Lis, H. G. Craighead and S. Kim, Lab Chip, 2009, 9, 1206 RSC.
- M. Khati, M. Schuman, J. Ibrahim, Q. Sattentau, S. Gordon and W. James, J. Virol., 2003, 77, 12692–12698 CrossRef CAS PubMed.
- T. S. Misono and P. K. R. Kumar, Anal. Biochem., 2005, 342, 312–317 CrossRef CAS PubMed.
- C.-J. Huang, H.-I. Lin, S.-C. Shiesh and G.-B. Lee, Biosens. Bioelectron., 2012, 35, 50–55 CrossRef CAS PubMed.
- H.-I. Lin, C.-C. Wu, C.-H. Yang, K.-W. Chang, G.-B. Lee and S.-C. Shiesh, Lab Chip, 2015, 15, 486–494 RSC.
- X. Lou, J. Qian, Y. Xiao, L. Viel, A. E. Gerdon, E. T. Lagally, P. Atzberger, T. M. Tarasow, A. J. Heeger and H. T. Soh, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2989–2994 CrossRef CAS PubMed.
- S. S. Oh, K. M. Ahmad, M. Cho, S. Kim, Y. Xiao and H. T. Soh, Anal. Chem., 2011, 83, 6883–6889 CrossRef CAS PubMed.
- J.-W. Park, S. J. Lee, S. Ren, S. Lee, S. Kim and T. Laurell, Sci. Rep., 2016, 6, 27121 CrossRef CAS PubMed.
- Q. Wang, W. Liu, Y. Xing, X. Yang, K. Wang, R. Jiang, P. Wang and Q. Zhao, Anal. Chem., 2014, 86, 6572–6579 CrossRef CAS PubMed.
- W. Y. Zhang, W. Zhang, Z. Liu, C. Li, Z. Zhu and C. J. Yang, Anal. Chem., 2012, 84, 350–355 CrossRef CAS PubMed.
- J. Wang, Q. Gong, N. Maheshwari, M. Eisenstein, M. L. Arcila, K. S. Kosik and H. T. Soh, Angew. Chem., Int. Ed., 2014, 53, 4796–4801 CrossRef CAS PubMed.
- R. Yufa, S. M. Krylova, C. Bruce, E. A. Bagg, C. J. Schofield and S. N. Krylov, Anal. Chem., 2015, 87, 1411–1419 CrossRef CAS PubMed.
- Z. Zhu, Y. Song, C. Li, Y. Zou, L. Zhu, Y. An and C. J. Yang, Anal. Chem., 2014, 86, 5881–5888 CrossRef CAS PubMed.
- S. Mayeur, S. Spahis, Y. Pouliot and E. Levy, Antioxid. Redox Signaling, 2016, 24, 813–836 CrossRef CAS PubMed.
- P. Pomastowski, M. Sprynskyy, P. Zuvela, K. Rafinska, M. Milanowski, J. J. Liu, M. Yi and B. Buszewski, J. Am. Chem. Soc., 2016, 138, 7899–7909 CrossRef CAS PubMed.
- X. Ke, Q. Chen, X. D. Pan, J. S. Zhang, W. M. Mo and Y. P. Ren, RSC Adv., 2016, 6, 12280–12285 RSC.
- J. Zhang, S. Lai, Z. Cai, Q. Chen, B. Huang and Y. Ren, Anal. Chim. Acta, 2014, 829, 33–39 CrossRef CAS PubMed.
- K. Yamada, T. G. Henares, K. Suzuki and D. Citterio, ACS Appl. Mater. Interfaces, 2015, 7, 24864–24875 CAS.
- K. Yamada, S. Takaki, N. Komuro, K. Suzuki and D. Citterio, Analyst, 2014, 139, 1637 RSC.
- L. Liu, D. Kong, C. Xing, X. Zhang, H. Kuang and C. Xu, Anal. Methods, 2014, 6, 4742 RSC.
- D. M. Jameson and J. A. Ross, Chem. Rev., 2010, 110, 2685–2708 CrossRef CAS PubMed.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6lc01208f |
|
| This journal is © The Royal Society of Chemistry 2017 |