DOI:
10.1039/C7GC02383A
(Communication)
Green Chem., 2017,
19, 4792-4797
Greener synthesis of nanofibrillated cellulose using magnetically separable TEMPO nanocatalyst†
Received
4th August 2017
, Accepted 5th September 2017
First published on 29th September 2017
Abstract
Nanofibrillated cellulose was synthesized by magnetically separable 4-oxo-2,2,6,6 tetramethylpiperidine-1 oxyl (TEMPO) mediated oxidation and mechanical disintegration of wood pulp. 4-oxo-TEMPO was covalently tethered to the surface of magnetic nanoparticles, which enabled reuse of the catalyst making the oxidation process cost effective through catalyst recovery and eliminating the catalyst release into waste water streams. This is a first report on use of solid catalyst to synthesize nanofibrillated cellulose using water as a medium.
Introduction
Cellulose is the most abundant renewable resource for making chemicals and functional materials. On the other hand, cellulose is one of the most inert organic materials on the planet. To mitigate this issue, synthetic catalysts have an important role in the conversion of cellulose into derivatives with various substitution patterns. Regioselective oxidation of cellulose pulp converts accessible C6 surface hydroxyls on microfibril surfaces into carboxylic acid groups which serve as reactive “handles” that can be modified with a number of chemistries to enable functionalization of cellulose for many applications.1–4 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO), with sodium hypochlorite as oxidant, is used as an efficient catalytic system for oxidation of native cellulose.5 Further, nanofibrillated cellulose (NFC) in particular, is synthesized by successive oxidation and mechanical disintegration of cellulose pulp.6 In fact, successive TEMPO mediated oxidation and homogenization for fibrillation is reported to have the lowest energy demand to produce NFC.7 However, TEMPO is a toxic chemical to aquatic life that cannot be released into waste effluent after oxidation, as it can accumulate in the environment.8,9 This issue creates a need to recycle TEMPO in order for this process to be a sustainable technology to create value added materials from plant-based resources.10,11 The reuse of TEMPO is thus critical to develop a greener and cost effective process for the synthesis of NFC.12
There have been efforts to recycle TEMPO by tethering it covalently on nanoparticles or polymers.13,14 Using magnetic nanoparticles as support enabled easy separation of TEMPO after oxidation while polymers can be precipitated by non-solvents.15 Tethered nanoparticles have been used for oxidation of primary alcohols to carboxylic acids.16–19 However, there has been no report on use of such heterogeneous magnetic catalyst to modify solid biopolymer substrates such as wood pulp fibre. We report the synthesis of heterogeneous TEMPO catalyst (Fe@MagTEMPO) wherein, TEMPO was tethered to magnetic nanoparticles through covalent bonding and used to oxidize cellulose pulp and synthesize NFC. The oxidation was carried out using water as the medium, sodium bromide as promoter and sodium hypochlorite as oxidant. At the end of the oxidation, TEMPO was recycled and sodium hypochlorite was reduced to sodium chloride, leading to nanocellulose production with greener chemistry principles with renewable starting material, low energy processing, and no toxic by-product formation.
Results and discussion
Pulp oxidation
Carboxylic acid content of 0.17 mmol g−1 was obtained on bleached cellulose kraft pulp when it was allowed to react with sodium hypochlorite for 6 h without any TEMPO catalyst (Table 1). This level of oxidation provided the fibre with an overall negative charge, albeit weak. Use of homogeneous TEMPO as catalyst dramatically increased the carboxyl content of pulps up to 1 to 1.2 mmol g−1 in time periods of only 2 hours to create highly negatively charged fibres.20 Hence, the effectiveness of homogeneous TEMPO to oxidize a heterogeneous substrate such as wood pulp was demonstrated in the conversion of primary hydroxyls into carboxylic acids and aldehydes. A solid Fe@MagTEMPO catalyst was reacted with a solid substrate in a water medium that produced an oxidized fibre with a carboxyl content of 0.5 to 0.9 mmol g−1 under controlled conditions at room temperature (Table 1). These numbers indicated the heterogeneous TEMPO was effective at oxidizing the pulp fibre; however, an extended reaction time was required to obtain higher levels of oxidation.
Table 1 Effect of reaction conditions on oxidation level of bleached cellulose kraft pulp
| Catalyst |
Carboxyl content (mmol g−1) |
Time (h) |
Temperature (°C) |
Rate of oxidation (mmol g−1 h−1) |
| 2 g never dried bleached cellulose kraft pulp, 200 cm3 water, 0.1 mmol NaBr, 5 mmol g−1 NaClO, NaOH (0.5 M) to maintain pH 10 in all scenarios. |
| No catalyst |
0.17 |
6 |
25 |
0.028 |
| Homogeneous TEMPO |
1 |
2 |
25 |
0.48 |
| Fe@MagTEMPO |
0.54 |
6 |
25 |
0.085 |
| Fe@MagTEMPO |
0.9 |
3.5 |
65 |
0.25 |
In addition to the time factor, catalyst loading per volume of reaction mixture was investigated to determine the impact on the rate of oxidation (Fig. ES1†). Nearly tripling the loading of the catalyst did not change the reaction rate. This result indicated a diffusion controlled reaction; the catalyst had to penetrate through the pores of cellulose fibres to oxidize cellulose. These pores were noted to be up to 80 nm in size.21 Compared to the Fe@MagTEMPO dimensions of 30 nm (Fig. ES2† & Fig. 4), the heterogeneous catalyst was able to diffuse within the walls of the fibre and was able to catalyze the oxidation by virtue of its size. Further, the rate of oxidation was influenced by the addition time of the hypochlorite with increased rates when the oxidant was added slowly to the reaction mixture (Fig. ES3†). The oxidant had two roles during the reaction: it activated the free radical on TEMPO and activated the NaBr–NaBrO cycle, so that TEMPO remained active for oxidization of the hydroxyl groups (Fig. ES4†). If the oxidant was added only at beginning of reaction, it would primarily oxidize the promoter (NaBr), as activating sterically hindered heterogeneous TEMPO is difficult.22–24
The other parameter that had a significant impact on the reaction was the temperature, as the rate of oxidation increased with increased temperature (Fig. ES5†). The reaction rate increased three fold to 0.25 mmol g−1 h−1 when used at 65 °C. The increase in the rate of reaction was attributed to increased number of vibrations due to thermal currents within the medium. When homogeneous TEMPO was used as catalyst, oxidation rate of 0.48 mmol g−1 h−1 was obtained at room temperature. Hence, for this heterogeneous reaction that involved the solid catalyst and fibre substrate, the reaction was less efficient, yet the heterogeneous catalyst still produced oxidized fibre up to levels of 0.9 mmol g−1 (Table 1).
Further, it was reported that TEMPO oxidation of primary alcohols into carboxylic acids occurs via an initial oxidation of the alcohol into an aldehyde. Typical values for TEMPO oxidized fibre had a total carbonyl content of 1 mmol g−1 with approximately 70% that occurred as carboxylic acids and the remainder were aldehydes.25 The aldehydes were converted into carboxylic acids, post reaction, via oxidation with sodium chlorite. In the present study, the initial carboxylic acid content of the fibre was 0.9 mmol g−1 and the content remained the same after sodium chlorite treatment. This data suggested that the TEMPO catalyst was effective to create a cycle where neighbouring activated TEMPO groups oxidized accessible cellulose hydroxyls to a final product of carboxylic acids.
Nanocellulose production
Oxidation levels with the heterogeneous catalyst were sufficient to fibrillate the oxidized cellulose pulp and generate NFC. After sonication, the oxidized fibre suspension was transparent, while the sonicated sampled contained suspended fibre (Fig. 1). The fibrillation was effective for the creation of completely transparent films after solvent casting. The films demonstrated slight iridescence that is not always seen for nanofibrillated pulp. SEM images of fractured film showed the layers of NFC which the lamellar structure would provide a mechanism towards iridescence (Fig. 2). However, this layered structure was not as regular as cellulose nanocrystalline films that demonstrated structural colour.26,27 AFM analysis of NFC showed individualised fibrils of approximately 5 nm (±2 nm) in diameter along with a few larger “macrofibrils” that were on the scale of 25 nm (Fig. 3). The thickness of nanofibrils were similar to that obtained by homogeneous TEMPO mediated oxidation yet larger in average diameter for previous reported sonicated TEMPO oxidized nanocellulose.7,20
 |
| | Fig. 1 Evidence of nanofibrillation achieved after oxidation of cellulose pulp using Fe@MagTEMPO as catalyst. | |
 |
| | Fig. 2 SEM images of fractured nanofibrillated cellulose film obtained by oxidation of cellulose pulp using Fe@MagTEMPO as catalyst. | |
 |
| | Fig. 3 AFM image of nanofibrillated cellulose obtained by oxidation of cellulose pulp using Fe@MagTEMPO as catalyst. | |
With the use of solid catalyst for oxidation of solid reactant yielding a solid product, there was a possibility of catalyst being entrapped in the product. A typical separation process included filtering the fibre slurry at the end of oxidation across 11 μm pore opening membrane and then rinsing the fibre with excess water in the presence of a magnetic field (bar magnet) to recover the particles prior to fibrillation. To confirm that the catalyst was completely separated from the product, the NFC film was analyzed with energy dispersive X-ray analysis (EDX) which has a detection limit of 300 ppm for silica.28 The analysis of micron thick NFC films did not detect Fe@MagTEMPO catalyst elements of iron or silica, indicating complete separation of catalyst from oxidized pulp fibre (Table 2). ATR-FTIR has been used as complementary analysis technique to SEM-EDX for investigation of organic fouling on porous organic composite membranes, for analysis of rubber diaphragms and for analysis of zinc oxide nanoparticles coated paper.29–31 ATR-FTIR spectrum of NFC film made using Fe@MagTEMPO catalyst showed no peaks associated with Fe–O (640 cm−1) or silica hydroxide (800, 900 cm−1) indicating the lack of detection of residual catalyst in the NFC film (Fig. ES6†). Moreover, the spectra for NFC films made from homogeneous TEMPO and Fe@MagTEMPO were similar except for the peak associated with carboxylate groups in sodium salt form (1615 cm−1). This result was from the lower oxidation level obtained with Fe@MagTEMPO catalyst. Peaks associated with C–O–C stretching vibration of pyranose ring skeleton (1059 cm−1), sp3 hybridized C–H stretching (2900 cm−1), hydrogen bonded O–H stretching (3350 cm−1), C–H deformation (1372–1429 cm−1) and O–H out of plane bending (670 cm−1) owing to sonication were seen in both films.32,33
Table 2 EDX analysis of Fe@MagTEMPO and nanofibrillated cellulose film
| Sample |
Elemental composition (wt%) |
| |
Fe |
Si |
C |
O |
| Fe@MagTEMPO |
18.97 |
17.50 |
14.86 |
48.67 |
| NFC_Fe@MagTEMPO |
Not detected |
Not detected |
51.36 |
48.64 |
Catalyst evaluation and stability
The loading of TEMPO on the Fe3O4 nanoparticle anchor was measured using UV-Visible light spectroscopy at 461 nm wavelength.34 The TEMPO loading was found to be 0.35 mmol g−1catalyst. The catalyst nanoparticle size was ∼30 nm giving an area of 2875 nm2 per nanoparticle for binding TEMPO. Considering the 0.35 mmol g−1catalyst loading, there was an average of 200 TEMPO groups per catalyst nanoparticle, providing enormous oxidizing power for the catalyst as it diffuses through the swollen fibre cell wall. TEMPO tethered on iron oxide nanoparticles has been used previously for oxidation of alcohols and hence it was necessary to determine the stability of Fe@MagTEMPO in alkaline reaction environment. The catalyst sample was dispersed in sodium hydroxide solution (pH 10) for 15 days. Qualitative comparison of TEM images of fresh and aged catalyst samples confirmed the structural fidelity of the catalyst with significant coating of silica on the particles after 15 days (Fig. 4). The images contain aggregated particles with iron oxide cores along with silica surfaces. However, during synthesis, not all the silica surface was modified with APTES and TEMPO, as there were peaks associated with silica hydroxide (800 cm−1, 935 cm−1) (Fig. 5).35 After soaking in alkali for 15 days, these functional groups decreased in intensity relative to the silica peaks (∼1090, ∼460 cm−1). Additionally, the broad stretching peak of the hydroxyl groups was observed to decrease at 3400 cm−1 as well indicating a change to the surface hydroxyls. This data suggested a slightly altered surface chemistry, still dominated in Si–O–Si bonds but existing in an altered environment after alkali treatment; this change was seen with the formation of a new peak at 1511 cm−1 relating to the interaction of the unreacted amino groups on APTES with the Si–OH/Si–O− groups on the particle surface.36 Changes to TEMPO related peaks were not observed in the spectra due to the low concentrations.
 |
| | Fig. 4 TEM images of fresh Fe@MagTEMPO and 15 days aged Fe@MagTEMPO in sodium hydroxide solution (pH 10). | |
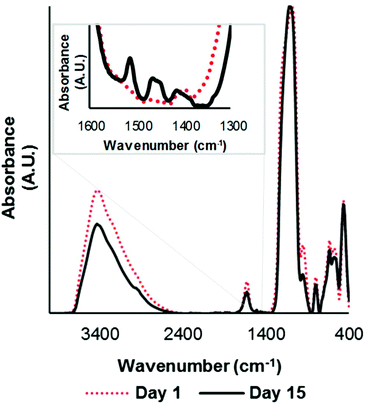 |
| | Fig. 5 FTIR analysis of fresh Fe@MagTEMPO and 15 days aged Fe@MagTEMPO in sodium hydroxide solution (pH 10). | |
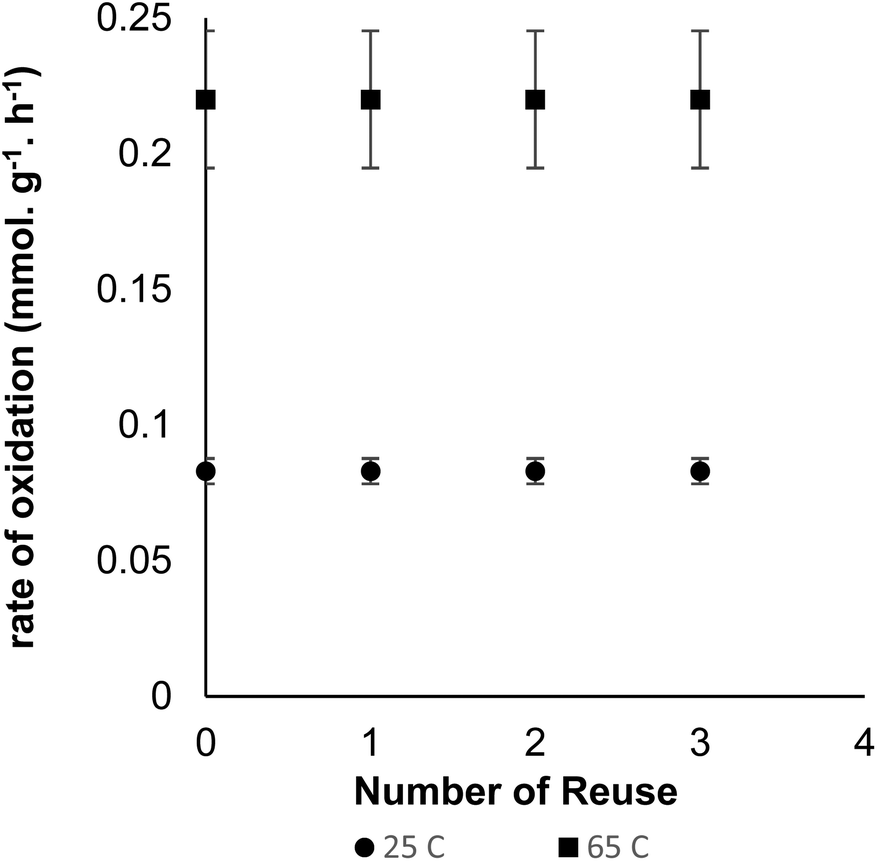
A further stability test was performed on the catalyst in a multi-step oxidation experiment, where the heterogeneous catalyst was removed, based on the steps indicated above, and the water solution captured and pulp fibre resuspended in the same batch without the heterogeneous catalyst. Continued addition of oxidant did not cause further oxidation of the pulp fibre, as indicated by the in situ pH probe. However, when the heterogeneous catalyst was reintroduced into the system, oxidation of the fibre continued until completion. This experiment indicated that there were no free TEMPO molecules that were not tethered to the catalyst as well as the catalyst could be recovered from the reaction medium. The reusability of the catalyst was further studied by reusing the same catalyst at 25 °C for four cycles with each cycle running for 6 h (Fig. 6). The catalyst was collected at the end of each oxidation reaction with an external magnet and used “as-is” for the next oxidation cycle. More pertinent reusability studies were carried out at 65 °C at similar reaction conditions (Fig. 6). For every reuse, the reaction was allowed to proceed for 3.5 h to obtain carboxyl content of at least 0.78 mmol g−1 on pulp fibre. There was no loss of catalyst activity at end of four cycles (Fig. 6) as well as loss of magnetic capabilities in both scenarios.
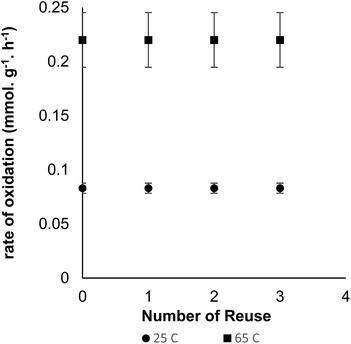 |
| | Fig. 6 Reusability of Fe@MagTEMPO catalyst for oxidation of cellulose pulp: 2 g never dried bleached softwood pulp, 200 cm3 water, 0.1 mmol NaBr, 0.175 mg cm−3 Fe@MagTEMPO, 5 mmol g−1 NaClO, NaOH (0.5 M) to maintain pH 10. | |
Conclusion
Nanoparticle (solid) catalyst was used to oxidize cellulose pulp fibre (solid) with water as medium. Oxidation levels and rate of oxidation with heterogeneous catalyst were slightly lower than the homogeneous TEMPO mediated oxidation but were sufficient to achieve effective levels of oxidation that led to fibrillation and nanocellulose production. Subsequently, TEMPO heterogeneous catalyst was recovered from the reaction mixture and reused which prevented the release of the catalyst into wastewater streams during nanocellulose synthesis.
Experimental
Synthesis of Fe@MagTEMPO catalyst
The catalyst was synthesized in four stages of precipitation, condensation, silanization and reductive amination. 5 g Iron(III) chloride and 3 g iron(II) chloride tetrahydrate were dissolved in 2 M HCl solution using a bath sonicator. The dissolved salts were precipitated with ammonium hydroxide solution. Fe3O4 nanoparticles with 5–10 nm diameter were obtained (Fig. ES2†). The nanoparticles were suspended in mixture of ethanol (35 cm3) and water (6 cm3) under sonication. TEOS (1.5 cm3) was added dropwise. Silica nanoparticles were obtained as a uniform coating on Fe3O4 nanoparticles by hydrolysis and condensation using ammonium hydroxide solution as catalyst (Fig. ES2†). The silica coated Fe3O4 nanoparticles (10 g) were suspended in toluene (200 cm3) using a bath sonicator and APTES (2.5 cm3) was added to it. The mixture was heated at 105 °C for 20 h to allow completion of silanization reaction. Fe3O4 nanoparticles with silica coating and APTES (3 g) were suspended in ethanol (100 ml) and 4-Oxo-TEMPO (1.5 mmol) was added to it. The suspension was kept under continuous agitation at 40 °C using shaker-incubator. 5-Ethyl-2-methylpyridine borane complex (2 mmol) was used as green catalyst for reductive amination reaction.37 The reductive amination catalyst was added to the reaction mixture in three equal parts after 3 h, 24 h and 48 h of agitation at 40 °C. The final product was separated with an external magnet (Fig. ES7†), washed with ethanol and dried under vacuum. The final catalyst was referred as Fe@MagTEMPO and stored in vacuum oven at 40 °C.
Oxidation of cellulose pulp using Fe@MagTEMPO38
The oxidation of cellulose pulp was carried out in a 1 L round bottom flask. Temperature was controlled using heating mantle equipped with PID controller (±2 °C). An overhead stirrer was used to disperse the catalyst in reaction mixture. pH was monitored using a Fischer Scientific AR 15 meter. Oxidation tests were done using never dried pulp dispersed in distilled water such that the concentration of pulp was 1% (w/w). Required quantities of sodium bromide and Fe@MagTEMPO were added to the dispersion of cellulose pulp. The mixture was heated to the desired temperature and oxidation was started by addition of sodium hypochlorite. The progress in oxidation was evident with decrease in pH of the reaction mixture. The pH was maintained at 10 using 0.5 M NaOH solution. The reaction was stopped when no further drop in pH was observed. The catalyst was separated from the reaction mixture by filtration and collected using an external magnet for reuse. The carboxyl content on oxidized pulp was calculated by conductometric titration. 0.25 g of oxidized pulp was soaked in 0.01 M HCl solution for 1 h. The soaked pulp was washed and resuspended in 0.001 M NaCl solution. 200 μl of 0.1 M HCl was added just prior to titration. Conductivity was measured after adding 50 μl aliquots of 0.05 M NaOH solution when the meter (Fischer Scientific Accumet AR50 with two-cell conductivity probe) showed stable reading. The carboxyl content was calculated from results as:
where, CNaOH is concentration of titrant (mmol cm−3), VNaOH is volume of titratnt consumed at equivalence point (cm3) and Wpulp is oven dry weight of cellulose pulp used for titration (g). The oxidised pulp was blended for 15 min and sonicated for 30 min to get NFC.
Abbreviation
| SEM | Scanning electron microscopy |
| AFM | Atomic force microscopy |
| TEM | Transmission electron microscopy |
| FTIR | Fourier transform infrared spectroscopy |
| ATR-FTIR | Attenuated total reflectance-Fourier transform infrared spectroscopy |
| EDX | Energy dispersive X-ray spectroscopy |
| TEOS | Tetra-ethyl-orthosilicate |
| APTES | 3-Aminopropyltriethoxysilane |
| NFC_Fe@MagTEMPO | Nanofibrillated cellulose film generated using Fe@MagTEMPO catalyst |
Conflicts of interest
Authors declare no conflict of interest.
Acknowledgements
S C Patankar acknowledges Rideau Hall Foundation for Queen Elizabeth Scholarship. Scott Renneckar acknowledges NSERC Discovery Grants Program and Canada Research Chair Program. Authors acknowledge help of Mijung Cho for SEM analysis and Muzaffer Karaaslan for AFM analysis.
References
- W. Zhang, R. K. Jhonson, Z. Lin, C. Chandoha-Lee, A. Zink-Sharp and S. Renneckar, Cellulose, 2013, 20, 2935–2945 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- S. R. Collinson and W. Thielemans, Coord. Chem. Rev., 2010, 254, 1854–1870 CrossRef CAS.
- A. Gandini and N. M. Belgacem, Polym. Int., 1998, 47, 267–276 CrossRef CAS.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- Q. Li, S. McGinnis, C. Sydnor, A. Wong and S. Renneckar, ACS Sustainable Chem. Eng., 2013, 1, 919–928 CrossRef CAS.
- TEMPO, MSDS No. 214000 [online], Sigma-Aldrich Corporation: St. Louis, Missouri, Nov 19, 2016, http://www.sigmaaldrich.com/safetycenter.html (accessed Dec 17, 2016).
- 4-Oxo-TEMPO, MSDS No. 179485 [online], Sigma-Aldrich Corporation: St. Louis, Missouri, Nov 19, 2016, http://www.sigmaaldrich.com/safetycenter.html (accessed Dec 17, 2016).
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- A. Dijksman, I. W. C. E. Arends and R. A. Sheldon, Chem. Commun., 2000, 271–272 RSC.
- Y. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 CAS.
- B. Karimi and E. Farhangi, Chem. – Eur. J., 2011, 17, 6056–6060 CrossRef CAS PubMed.
- C. W. Lim and I. S. Lee, Nano Today, 2010, 5, 412–434 CrossRef CAS.
- A. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- A. Schatz, R. N. Grass, W. J. Stark and O. Reiser, Chem. – Eur. J., 2008, 14, 8262–8266 CrossRef PubMed.
- A. K. Tucker-Schwartz and R. L. Garrell, Chem. – Eur. J., 2010, 16, 12718–12726 CrossRef CAS PubMed.
- D. Brunel, F. Fajula, J. B. Nagy, B. Deroide, M. J. Verhoef, L. Veum, J. A. Peters and H. van Bekkum, Appl. Catal., A, 2001, 213, 73–82 CrossRef CAS.
- Y. Sun, C. Cao, F. Wei, P. Huang, S. Yang and W. Song, Sci. Bull., 2016, 61(10), 772–777 CrossRef CAS.
- R. Shinoda, T. Saito, Y. Okita and A. Isogai, Biomacromolecules, 2012, 13, 842–849 CrossRef CAS PubMed.
- S. Park, R. A. Venditti, H. Jameel and J. J. Pawlak, Carbohydr. Polym., 2006, 66, 97–103 CrossRef CAS.
-
G. Togo and M. I. Fernandez, Oxidation of Primary alcohols to Carboxylic acids- A guide to current common practice, Springer-Verlag, New York, 2007, pp. 79–103 Search PubMed.
- F. W. Brodin and H. Theliander, BioResources, 2013, 8(4), 5925–5946 CrossRef.
- M. Krystof, M. Perez-Sanchez and P. Dominguez de Maria, ChemSusChem, 2013, 6, 826–830 CrossRef CAS PubMed.
- T. Saito and A. Isogai, Biomacromolecules, 2004, 5, 1983–1989 CrossRef CAS PubMed.
- P. X. Wang, W. Y. Hamad and M. J. MacLachlan, Nat. Commun., 2016, 7, 11515 CrossRef PubMed.
- D. G. Gray, Nanomaterials, 2016, 6, 213, DOI:10.3390/nano6110213.
-
J. Goldstein, D. Newbury, D. Joy, C. Lyman, P. Echlin, E. Lifshin, L. Sawyer and J. Michael, Scanning Electron Microscopy and X-Ray Microanalysis, Springer Science+Business Media, New York, 2003 Search PubMed.
-
M. Rabiller-Baudry, F. Gouttefangeas, J. Le Lannic and P. Rabiller, in Current Microscopy Contributions to Advances in Science and Technology, ed. A. Mendez-Vilas, Formatex Microscopy Series Number 5, 2012, vol. 2, pp. 1066–1076 Search PubMed.
- Shimadzu Analytical and Measuring Instruments, http://www.shimadzu.com/an/industry/petrochemicalchemical/chem0201010.htm (accessed Sep 1, 2017).
- K. Ghule, A. V. Ghule, B. Chen and Y. Ling, Green Chem., 2006, 8, 1034–1041 RSC.
- S. Coseri, G. Biliuta, L. F. Zemljic, J. S. Srndovic, P. T. Larsson, S. Strnad, T. Kreze, A. Naderi and T. Lindstrom, RSC Adv., 2015, 5, 85889–85897 RSC.
- Q. Li and S. Renneckar, Biomacromolecules, 2011, 12, 650–659 CrossRef CAS PubMed.
- S. Liu, Y. Xing, J. Han and E. Tang, Cellulose, 2017, 24, 3635–3644 CrossRef CAS.
- C. H. Yu, A. Al-Saadi, S. J. Shih, L. Qiu, K. Y. Tam and S. C. Tsang, J. Phys. Chem. C, 2009, 113, 537–543 CAS.
- J. Kim, P. Seidler, L. S. Wan and C. Fill, J. Colloid Interface Sci., 2009, 329, 114–119 CrossRef CAS PubMed.
- E. R. Burkhardt and B. M. Coleridge, Tetrahedron Lett., 2008, 49, 5152–5155 CrossRef CAS.
-
S. Renneckar and S. Patankar, US Pat, 62479542, 2017 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7gc02383a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Scott
Renneckar
and
Scott
Renneckar