
Biomimetic and bioinspired approaches for wiring enzymes to electrode interfaces
Patrick O.
Saboe
a,
Emelia
Conte
a,
Megan
Farell
a,
Guillermo C.
Bazan
b and
Manish
Kumar
*a
aDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: manish.kumar@psu.edu
bCenter for Polymers and Organic Solids, University of California at Santa Barbara, Santa Barbara, CA 93106, USA
First published on 8th November 2016
Abstract
Biomimetic and bioinspired approaches to redox enzyme wiring involve borrowing structures and strategies found in biological electron transfer systems for use in engineered devices. Redox protein–electrode systems are evolving for several applications, including energy, biomedical and environmental purposes. This review is intended to be both “tutorial” and comprehensive in that we provide a guide to understand, design, and improve electrode interfaces for redox enzyme electron transfer processes in devices. The review examines electrode interfaces by directly comparing them with biological electron transfer systems. First, the mechanisms, theory, and structures for electron transfer in biological systems are provided, followed by analysis of the strategies and structures engineered in redox-protein devices. The review describes the challenges of constructing and applying redox enzyme devices, including the poor electrical contact between electrodes and enzymes and low lifetime and scalability of devices.
 Patrick O. Saboe | Patrick O. Saboe is a researcher at the National Renewable Energy Lab (NREL) in Golden, Colorado. He received his BS in Chemical Engineering from the University of New Haven in 2011 and his PhD in Chemical Engineering from the Pennsylvania State University in 2016 under PhD advisor, Manish Kumar. He has been a visiting researcher at Harvard Medical School in the Tom Walz' Lab in 2012, at ETH Zurich in the Hyung Gyu Park Lab in 2015 and in the Gregg Beckham group at NREL in 2016. He has served as the chair of the Gordon Research Seminar on Membranes: Materials & Processes in 2016 and won the Environmental Protection Agency (EPA) Science to Achieve Results (STAR) fellowship from 2014–2016. His research interests include enzyme electrodes, artificial photosynthesis, membrane separations, biofuels, and biomimetic and bioinspired membranes. |
 Emelia Conte | Emelia Conte is an undergraduate student in Chemical Engineering (Junior) at Penn State. She conducts research into biocompatible electrical interfaces and membrane proteins in Manish Kumar's group. |
 Megan Farell | Megan Farell is a PhD student in the Department of Chemical Engineering at the Pennsylvania State University. She received her Bachelor's in Chemical Engineering at the University of Tennessee in 2015. Currently, she works in Manish Kumar's group, and her research focuses on creating innovative bioelectrochemical platforms for various applications. |
 Guillermo C. Bazan | Professor Bazan obtained his BSc from the University of Ottawa in 1986 and his PhD from MIT in Inorganic Chemistry working under the guidance of Professor Richard R. Schrock. After a postdoctoral appointment at Caltech with John Bercaw, he joined the Department of Chemistry at the University of Rochester in 1992. His move to UCSB came in 1998, where he is now Professor in the Department of Chemistry & Biochemistry and in the Department of Materials. His research interests are in the area of preparative macromolecular chemistry and the design and application of conjugated materials for applications in optoelectronic and bioelectronics devices. His awards include the Humboldt Foundation Bessel Award, the American Chemical Society Cope Scholar Award, and a Fellowship to the American Association for the Advancement of Science. |
 Manish Kumar | Manish Kumar is an assistant professor of Chemical Engineering, Biomedical Engineering, and Environmental Engineering at Penn State. He received his bachelors from the National Institute of Technology in Trichy, India in Chemical Engineering. He completed a masters in environmental engineering at the University of Illinois and then worked for approximately seven years in the consulting industry on applied research projects. He returned to Illinois to complete a PhD in the area of biomimetic membranes and then conducted postdoctoral research at the Harvard Medical School on the structure of water channel proteins, aquaporins. He works in the area of membrane protein biophysics, membrane protein enhanced synthetic membranes and on developing artificial membrane proteins and biocompatible electrical interfaces. He has received the NSF CAREER award. |
Broader contextThe interfacing of reduction–oxidation (redox) enzymes to synthetic electrode materials is useful in the fields of energy, biomedical, and environmental research. Although redox enzyme devices show promise through their specific redox potentials and flexibility, their effectiveness is challenged by their lack of long term stability and generally poor electrical sensitivity. Biomimetic and bioinspired approaches build on structures and concepts found in biological electron transfer systems and are applied to engineered devices for improved current density, stability and electron transfer efficiency. In this work, we discuss the challenges currently facing redox protein based bioelectronics and highlight how they can be solved using biomimetic approaches. We identify equivalent biological examples for existing electrode interfaces and wiring techniques, and focus on the use of the biological systems to improve existing electrode interfaces. For the first time, examples from biology are used to directly address the optimization of engineered devices for better stability, protein loading, and electrical performance. |
Introduction to bioelectrochemical electrode interfaces for energy, environmental and biomedical applications
Engineered combinations of biotic and abiotic materials for various applications have become a prominent field of research in recent years.1–4 Many of these engineered constructs utilize one or more enzymes to exploit the diverse set of functions offered by proteins that have been finely tuned over billions of years of evolution. Reduction–oxidation (redox) enzymes have received considerable attention due to their ability to catalyze numerous reactions that are potentially applicable for energy, biomedical, and environmental purposes. We provide a brief summary of the state-of-the-art devices for each of these applications below, including a focus on glucose biosensors, a multi-billion dollar industry for diabetes management.5 A variety of engineered electrodes are used to immobilize enzymes for specific applications while maintaining their stability, activity, proper orientation, and effective electrical connection. These emerging materials include graphene,6 hydrogels,7,8 conductive9 and redox polymers10 and carbon nanotubes.11 Such innovative materials present attractive properties, such as flexibility,12 high surface areas,13 and specific redox potentials.10,14 The stability and electronic coupling of redox proteins on electrodes remain fundamental challenges for many redox protein based bioelectronics.15 This review highlights how these challenges have been addressed through biomimetic and bioinspired approaches. Biomimetic approaches incorporate biological or comparable structures and concepts into a design. A bioinspired approach uses tailored or engineered biological structures and concepts.16 We place these approaches in context of biological electron transfer processes and the chemical environments supporting these processes (Fig. 1). A number of reviews have been published on the various aspects of bioelectrochemical systems, including on biosensors,5,17 microbial1 and enzymatic fuel cells,18 photo-bioelectronics,3,19,20 interface materials,2,9 and protein-film voltammetry.21 These reviews address the goal of effective electron transfer from an electrode to a bio-derived material. However, there is less attention on how engineered systems are related to biological electron transfer processes. Our review is distinct from previous works in that we use examples from biology as a guide to understand, design and improve electrode interfaces. We find an equivalent biological example for every electrode interface or wiring technique introduced. These biological examples provide insight into electron transfer mechanisms and kinetics, biocompatibility and scalability of redox protein based bioelectronics.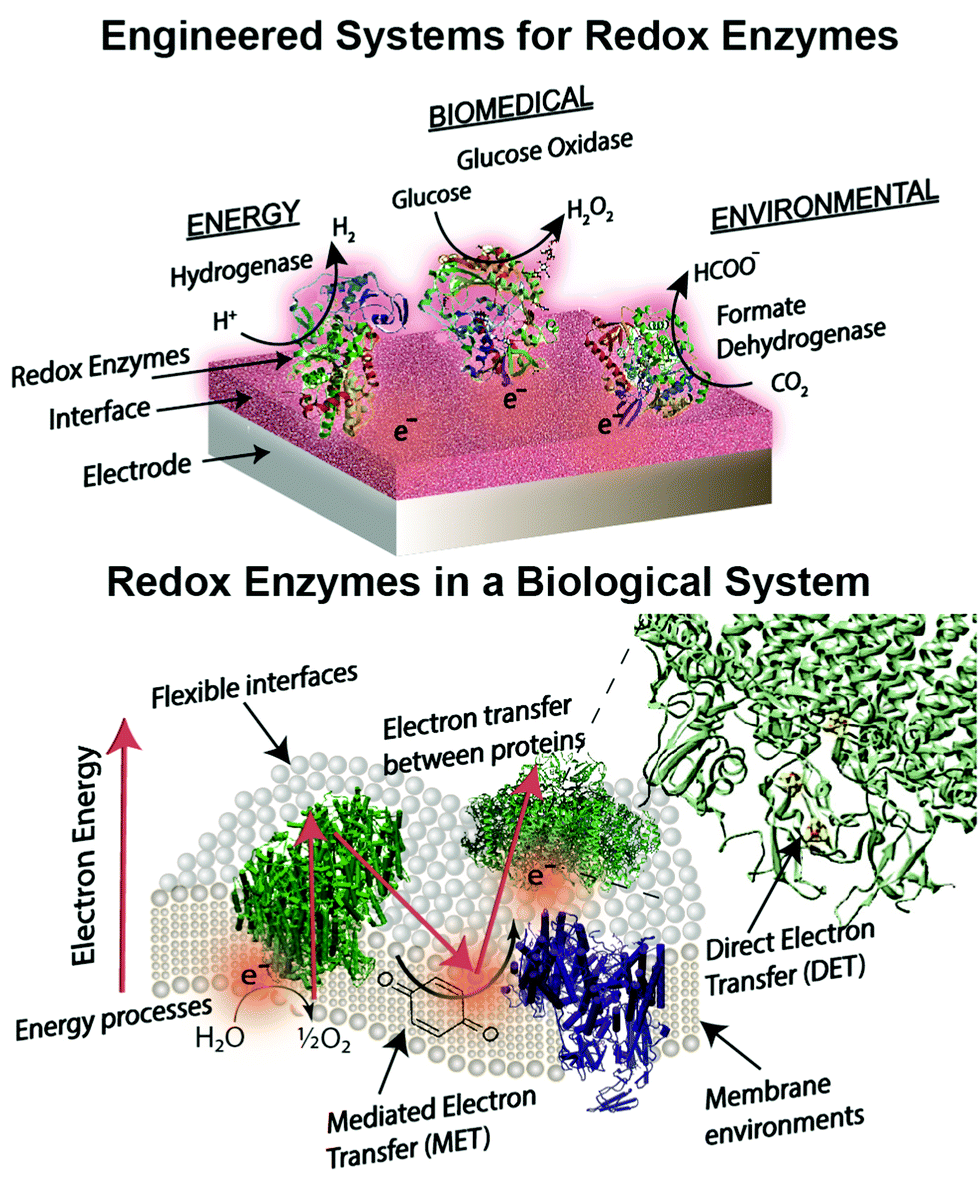 | ||
| Fig. 1 Biological features of redox enzymes important for bioelectrochemical devices. Applications in energy, biomedical, and environmental fields require an efficient method of electron transfer, a driving force for electron transfer, interactions of interfaces and enzymes for wiring, and optimizing bioelectrochemical devices for use on a larger scale. Biological examples of these processes are given within the figure. In this review we find useful information in biological designs as a framework to review the current literature on redox protein based bioelectronics. | ||
Redox proteins are essential to many cellular energy conversion processes including respiration,22 photosynthesis,23 extracellular electron transfer,24 and biodegradation.25 Redox proteins have one or more electrochemically active centers that serve to provide a route for electrons to and from a catalytic site.26 Oxidoreductases with multiple electron centers have redox site “chains”, which are spatially separated assemblies of single electron redox sites, to aid in transporting electrons within their structure and with other proteins.27 There are many types of redox sites, including hemes, chlorins, quinones, favins, Fe–S clusters, tyrosine and tryptophan residues,28,29 and copper, molybdenum and manganese ions.27,30 Redox enzymes may have a variety of these components in a redox cluster or redox chain, for example the nitrogenase has a redox cluster consisting of molybdenum, iron and sulfur.31Table 1 lists relevant examples and the reactions they catalyze. Many redox protein mediated reactions are targeted for various energy applications including hydrogen generation,32,33 enzymatic fuel cells (EFCs),18,34,35 and photo-electrochemical systems.20 For example, hydrogenase is a widely researched redox enzyme that catalyzes the reversible oxidation of molecular hydrogen into two protons and two electrons. Hydrogenase and photosystem I (PSI) have been wired together with a dithiol hydrocarbon (for example 1,6-hexanedithiol) to create a tethered nanoconstruct capable of catalyzing the photoreduction of protons into hydrogen gas.33 Another significant area of research focusing on exploiting redox enzymes for energy conversion is EFCs. EFCs generate an external electrical current between an enzyme modified anode and cathode. Enzymes used as an anode electrocatalyst include glucose dehydrogenase, catalase and glucose oxidase. Enzymes including laccase and bilirubin oxidase are used on cathodes in EFCs.18,36 EFCs have already displayed their potential by powering implantable small-scale devices.37 When multiple EFC cells were stacked together, enough power was generated to drive a toy car for two hours.38 EFCs operating with direct electron transfer (DET) have achieved peak power outputs of 1.3 mW cm−2 and have maintained stability for a maximum of 168 hours.39 Current research for EFCs focuses on generating greater power density and increasing the lifetime of enzyme-powered devices by using different techniques and strategies for connecting enzymes to electrodes.
| Protein | Reaction | Redox potential (vs. SHE) |
|---|---|---|
| Photosystem II (PSII) | P680+ + e− ↔ P680 | ∼1.1 V40 |
| Laccase | Laccasered + O2 → H2O + laccaseox | 0.82 V14 |
| Photosystem I (PSI) | P700+ + e− ↔ P700 | 0.49 V40 |
| Cytochrome a | Cytochrome a (Fe3+) + e− ↔ cytochrome a (Fe2+) | 0.29 V41 |
| Glucose oxidase | Glucose + O2 → gluconic acid + H2O2 | −0.080 V at pH 7.042 |
| Formate dehydrogenase | HCOO− → CO2 + H+ + e− | −0.420 V43 |
| Hydrogenase | 2H+ + 2e− ↔ H2 | −0.075 to −0.450 V44 |
Biomedical applications for redox enzymes include biosensors,45 implantable biofuel cells,46 and electrochemical immunoassays.4 Redox enzymes used in biosensors include cytrochrome c, hemoglobin, catalase and glucose oxidase.47 As an example, hemoglobin is used as a hydrogen peroxide biosensor.48 One of the most commonly used enzymes for biomedical applications is glucose oxidase. Glucose oxidase-powered enzymatic fuel cells have shown their ability to fuel a variety of biosensors, glucose monitors, and pacemakers.49 In the sixty years since Clark and Lyons introduced the idea of glucose oxidase electrodes, there has been immense progress in glucose biosensors for monitoring diabetes.5 Glucose biosensors comprise about 85% of the biosensor market, and glucose oxidase is the major recognition element for these sensors.45 However, current glucose oxidase biosensors cannot be implemented for continuous monitoring in humans due to time dependent sensitivity loss due to biofouling and enzyme instability. Several emerging interfaces are more conducive to human implantation compared to traditional materials, such as metals, because they offer greater physical flexibility, enhanced biocompatibility, and useful redox properties. For instance, synthetic hydrogels provide a flexible and biocompatible interface for potential use in biomedical applications.50 Another engineered interface appealing to the biomedical industry are conductive polymers. Conductive polymers have been studied for drug delivery and sensing due to their unique electrical properties including electrochemical triggered chemical release.51 Their redox properties promote controlled ionic transport though their polymeric membranes, allowing different anions to be electrostatically entangled in conductive polymer membranes and freed upon reduction. A recent study combined glucose oxidase and a modified graphene surface to create a glucose sensor.52 The polyvinylpyrrolidone-protected graphene and polyethyleimine-functionalized ionic liquid allowed direct electron transfer to glucose oxidase and supported glucose oxidase activity. This setup could act as a biosensor up to 14 mM glucose and maintained over 95% of its functionality after a week.
Redox enzymes such as laccase, alkyl sulfatase, and horseradish peroxidases have demonstrated their use in environmental applications, including as chemical biosensors53 and in organic54 and inorganic55 contaminant removal.56 Laccase has been extensively studied due to its potential to detoxify wastewater containing a variety of contaminants including dyes,57 toxins,57 hormones,54 cresols,58 and chlorophenols.58 A study focused on removing estrogen from synthetic and municipal wastewater showed that a laccase activity of 20 U mL−1 was able to entirely remove steroid estrogens.54 Furthermore, the incorporation of 1-hydroxy-benzotriazole as a mediator increased laccase's efficiency for estrogen removal, making this hormone-removal system more economically feasible and effective. Other enzymes exploited for pollutant removal include hydrogenase, Mn-dependent hydrogenase, cellobiose dehydrogenase, and lignin peroxidase.59,60
The utilization of bioelectrochemical systems for electrosynthesis has become an emerging research area in recent years. Enzymatic electrosynthesis uses one or more enzymes to transform low-cost chemicals into value-added products at low overpotentials.61,62 Currently, the majority of studies on bio-electrosynthesis utilize microbes as the biocatalyst, although enzyme electrode systems are gaining momentum in the field considering the recent developments in bioelectrochemistry and a focus on highly selective and inexpensive catalysts.61 An example of enzymatic electrosynthesis used an enzyme cascade, including formate, aldehyde, and alcohol dehydrogenase, to convert carbon dioxide into methanol.63 The addition of carbonic anhydrase to the cascade greatly augmented the effectiveness of the system by increasing the rate of CO2 hydration to bicarbonate, a substrate for formate dehydrogenase. In addition, the search for carbon neutral energy has instigated increased research in carbon sequestration. One method researched for carbon dioxide removal is the enzymatic electrocatalysis of formate using formate dehydrogenase to catalyze the reduction of carbon dioxide.64 Formic acid production of 413.8 mg mL−1 was achieved at −1 V, and the recycling of NAD+ allows for the continuous production of formate. These studies demonstrated the potential of using an enzymatic construct to couple carbon dioxide sequestration to biofuel generation and for chemical production. Although enzyme electrosynthesis is a rapidly evolving field that has many applications,62 enzyme electrode systems including enzyme fuel cells, sensors and photosynthetic devices are well-studied and provide examples to contextualize future research in the enzyme electrosynthesis field.61
The following review describes biological systems with integrated electron transfer and compares them with current bioelectrochemical systems (Table 2). The review focuses on the design of the electrode interfaces that mimic or could be improved by mimicking biological materials, systems or strategies. Four considerations are highlighted as effective engineering building blocks of interfaces for energy, environmental, and biomedical applications of redox enzymes. These considerations are aimed at improving the lifetime of the enzyme and device, and the sensitivity of the electrode (from improved performance on a per protein basis). We organize these goals into the following four considerations (1) routes of electron transfer from electrode to enzyme – toward effective wiring, (2) driving forces for electron transfer – redox proteins and electrodes within the energy landscape, (3) compatible interfaces for immobilization and stabilization of redox enzymes, (4) optimizing the performance of a bioelectrochemical device toward scalable designs. In each section we find a relevant biological system to compare with and contrast to current approaches to engineered devices.
| Design consideration (section) | Electrode interface or wiring technique | Equivalent biological example | Similarity between engineering and biological approach |
|---|---|---|---|
| Routes of electron transfer from electrodes to enzymes – toward direct wiring (1.0) | – MET in enzymatic devices (1.2) | – Laccase mediated system (LMS) (1.2) | – Electron transfer to sterically hindered substrates and redox enzymes. |
| – Direct electron transfer (DET) with carbon nanotubes (1.3) | – Bacterial nanowires (1.3) | – Long range electron transfer without electron mediators. | |
| Driving forces and kinetics of electron transfer – redox enzymes and electrodes within an energy landscape (2.0) | – Tuning the redox potential of osmium redox polymers (2.2) | – Redox potential of protein cofactors (2.2) | – Electron transfer rate depends on the difference in redox potential of donor acceptor. |
| – Photoswitchable devices (2.3) | – Photolyase and cryptochrome proteins (2.3) | – Light activated function | |
| – Semi-conductor electrodes (2.4) | – Photosystem I (PSI) (2.5) | – Band gap properties | |
| Compatible, biomimetic and bioinspired interfaces for immobilization of redox enzymes (3.0) | – Electrostatic and energy-based enzyme immobilization (3.2) | – Electron transfer between proteins (3.2) | – Electrostatic interactions facilitate electron transfer between donor and acceptor. |
| – Covalent binding approaches (3.3) | – Redox cofactors bound to protein framework (3.3) | – Covalent bonds help stabilize and orient electron transfer components. | |
| – Enzyme wiring with nanoparticles and electrode substrates – metals, semiconductor and carbon materials as electrodes (3.4) | – Shewanella and Geobacter use mineral oxides including Fe(III) and Mn(III/IV) as electron acceptors (1.3) | – Solid electrodes do not need extensive surface modifications to interface with biological systems. | |
| – Soluble proteins in porous or hydrogel polymer interface (3.5) | – Soluble proteins are stable in the intracellular fluids (3.5) | – Soluble redox proteins are stable in a buffered and hydrophilic environment. | |
| – Tethered lipid bilayer membranes (tBLMs) (3.6) | – Lipid membranes stabilize membrane proteins (3.6) | – Membrane proteins are functional in a hydrophobic membrane environment. | |
| – Synthetic bilayers and conjugated oligoelectrolytes (COEs) (3.7) | – Mediated electron transport in biological electron transport chains (3.7) | – Membrane intercalated mediators provide an electron pathway through hydrophobic membranes. | |
| Optimizing the performance of bioelectrochemical devices – toward scalable designs (4.0) | – Protein loading on electrodes (4.2) | – Multi-layered membranes in chloroplast (4.2) | – Layered structures create a high protein density (protein loading) |
| – Long-term stabilization of proteins on interfaces (4.3) | – Lifetime of enzymes in biological systems: enzymes are replaceable (4.3.1) | – Bioelectrochemical systems may benefit by implementing recycling of active interfaces with new enzymes. | |
| – Protein orientation in interfaces (4.4) | – Electron transfer between proteins (3.2) | – Directional electron transfer | |
1.0 Routes of electron transfer from electrodes to enzymes – toward direct wiring
1.1 Overview of mechanisms of electron transfer
Electron transfer between the electrode surface and the enzyme active site is the most obvious requirement for engineering bioelectrochemical devices. There are two methods to obtain electrical contact between a redox enzyme and an electrode: (1) mediated electron transfer (MET), and (2) direct electron transfer (DET).65 In MET, chemical mediators, bound or unbound to the electrode, are used to transport electrons to or from redox enzymes. Mediators include low molecular weight molecules, redox polymers, and conductive polymers.66,67 DET occurs via electron tunneling from electrodes to enzymes and occurs over short distances.67,68 The primary difference between MET and DET is that in DET, electrons are transferred in a one-step electron transfer process from the electrode material to the enzyme through the solution medium. In MET, two electron transfer steps take place: (1) from the electrode to the chemical mediator and (2) from the mediator to the enzyme.The many electron transfer methods used in biology are similar to those used to couple enzymes to electrodes. Underlying both biological processes and electrochemical systems are the two distinct methods, DET and MET. Engineered systems use chemically stable small mediators such as ferrocene and osmium complexes, and conductive polymers such as polyaniline, as substitutes for natural mediators such as flavins, soluble proteins, and quinones.67 In biological systems, MET is used primarily to transport electrons over longer distances (Section 1.2) or within membranes (see Section 3.6). In the enzymatic fuel cell literature, electron transfer through MET has produced the highest power from an enzymatic fuel cell.36 This may be because chemical mediators have access to many proteins packed on an electrode, even if there are multiple layers of enzymes. This situation is in contrast to DET, which is effective only for a catalytic monolayer proximal to the electrode surface. DET is more difficult to achieve than MET in engineered systems, especially if the redox center is embedded in the protein matrix.39 However, the theoretical kinetic gains from DET (Section 2.1) and the biological DET within and between proteins has inspired many wiring techniques. We next compare engineered electrochemical examples of MET and DET with biological examples.
1.2 MET in enzymatic devices
One of the redox enzymes utilized in a multitude of EFCs through MET is glucose oxidase (GOx). Due to glucose oxidase's catalytic site being around 13 Å from the enzyme's surface, DET is challenging to achieve without creating direct connections to the electrode. Methods that can overcome the distance to the active site of glucose oxidase are the use of osmium redox polymers and carbon nanotubes.69 An EFC study utilized glucose oxidase from Aspergillus niger at the anode and laccase from Trametes versicolor at the cathode with osmium redox polymers as mediators.70 Osmium redox polymers have an osmium center within an aromatic core coordinated by a nitrogen linkage to a polymer backbone, and are typically hydrophilic and maintain good contact with water-soluble enzymes (Fig. 2).71 The osmium redox polymer, [Os(4,4-diamino-2,2-bipyridine)2(poly(N-vinylimidazole))-(poly(N-vinylimidazole))9Cl]Cl (E0 = −0.110 V vs. Ag/AgCl) was crosslinked with GOx for use at the anode.70 Using a phosphate-based buffer, the performance of the entire cell, anode, and cathode were examined through a range of pH values. At pH 4.7, the maximum current density at the anode and cathode was close to 0.08 mA cm−2 and 0.28 mA cm−2 respectively. Increasing the pH to 7.4 caused a decrease in the anode current density to 0.065 mA cm−2 and to 0.02 mA cm−2 for the cathode. The maximum power density from this EFC was 0.04 mW cm−2 at 0.4 V and pH 5.5.70 Recently, Milton et al. (2016) used cobaltocene as an electron mediator with the nitrogenase redox protein for the production of ammonia and hydrogen with the nitrogenase redox protein (Fig. 3).72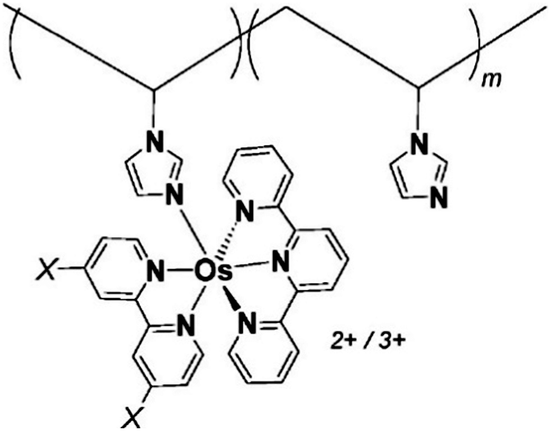 | ||
| Fig. 2 Osmium redox centers are commonly covalently bond to polymer backbones for immobilization on electrodes. Osmium redox polymers facilitate MET by wiring the redox centers of incorporated enzymes to electrode surfaces and transporting electrons close to enzyme active sites. Such wiring allows “electron hopping” from the enzyme active site through osmium centers until the electrons are passed on to the electrode surface.73,74 X = Cl, H, CH3, or OCH3. Each X group produces a different redox potential of the osmium redox center. Copyright (2008) American Chemical Society. | ||
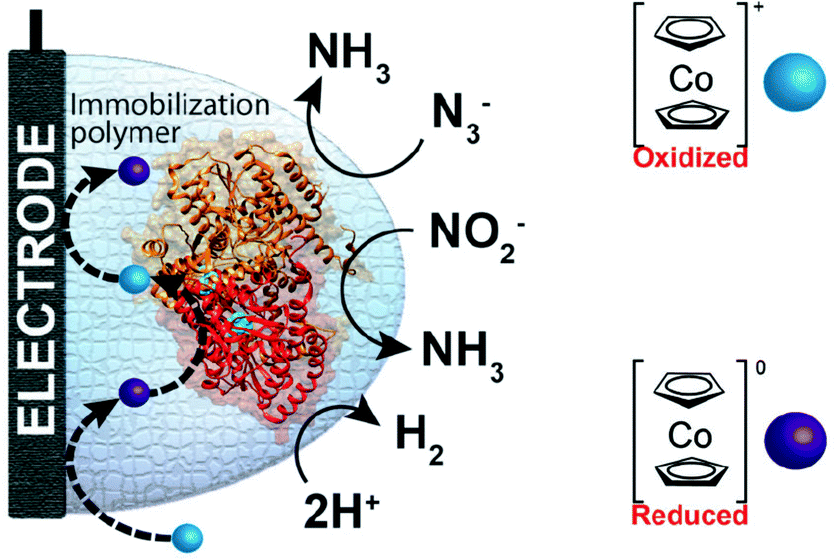 | ||
| Fig. 3 Milton et al. (2016)72 immobilized MoFe protein of Azotobacter vinelandii nitrogenase. The MoFe enzyme catalyzes a sustainable pathway to produce hydrogen and reduce azide (N3−) and nitrite (NO2−) to ammonia (NH3). The electron mediator cobaltocene was used to transfer electrons between the electrode and the MoFe protein. Copyright (2016) The Royal Society of Chemistry. | ||
Biological systems also use MET to achieve sterically hindered redox reactions. For example, in the laccase mediator system, electrons are transferred from lignin through low molecular weight compounds to the laccase enzymes. This system allows two important processes: accessibility to lignin molecules and degradation of non-phenolic lignin.25 Laccase, an oxidase widely distributed in higher plants, fungi, and bacteria, couples the reduction of oxygen with the oxidation of a broad range of organic substrates, including phenols, polyphenols, methoxy-substituted monophenols, aromatic and aliphatic amines, lignin, and even certain inorganic compounds.75 In some cases, however, the organic compounds of interest may not be oxidized directly because the molecules are too large to access the active site of laccases, or have a relatively high redox potential. The laccase mediator system (LMS) is used to degrade substrates that laccase cannot directly oxidize, such as non-phenolic lignin. Mediators that have a high reduction potential are able to rapidly oxidize lignin, however the regeneration of the mediator may be slow and controlled by the endergonic reaction with laccase.76 Naturally occurring mediators include 3-hydroxyanthranilic acid and 4-hydroxybenzoic acid (Fig. 4).77 A typical synthetic mediator used for laboratory experiments is 2,2′-azinobis-[3-ethylthiazoline-6-sulfonate] (ABTS).75
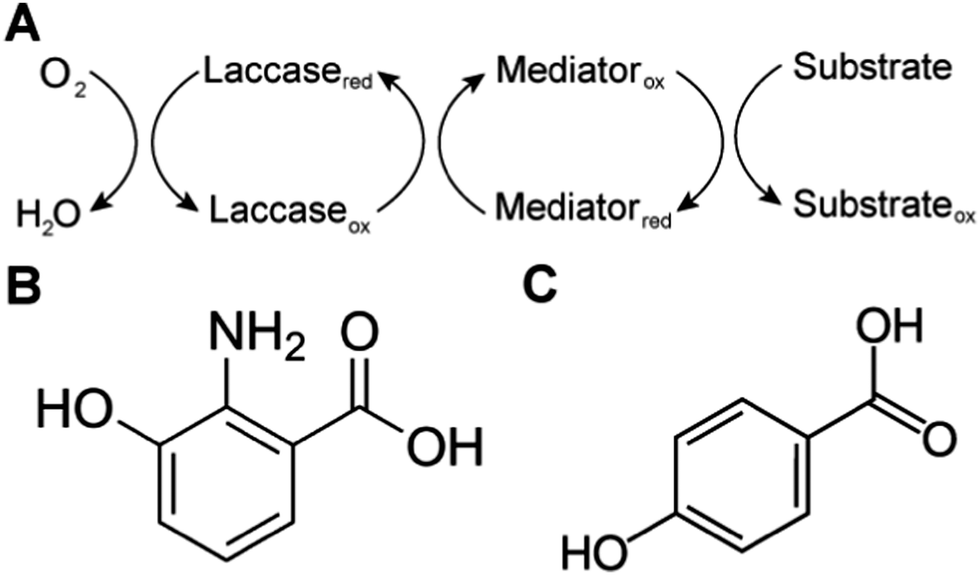 | ||
| Fig. 4 (A) Laccase mediated system (LMS) redox reactions. Substrates are oxidized by mediators. Laccase oxidizes these mediators and reduces oxygen to water. (B and C) Natural mediators include (B) 3-hydroxyanthranilic acid and (C) 4-hydroxybenzoic acid. | ||
Common synthetic mediators used in electrochemical systems include ferri/ferrocyanide, ferrocene and derivatives,78 organic dyes,79 osmium complexes, quinones, conductive polymers such as polyaniline and polypyrrole,9 and even small redox proteins.66,78,80 An ideal electron mediator should be an oxygen stable molecule, with oxidized and reduced forms that react rapidly with the desired enzyme.66 Several small molecule mediators have been crosslinked with polymers to form composites that minimize leaching of mediators and to increase electron transfer rates.81–85 For example, ferrocene and its derivatives have been used in biochemical devices for many years due to their advantageous properties including low molecular weight, high stability and reversibility,66,83 and have been linked to enzymes or materials such as CNTs,86 ferrocene-doped silica nanoparticles,87 and thermo-responsive poly-N-isopropylacrylamide-ferrocene. A full review of synthetic mediators has been provided by Chaubey and Malhotra (2002).66
1.3 DET in enzymatic devices: carbon nanotube (CNT) enzyme composite electrodes for glucose biofuel cells (GBFCs)
Diffusion limited reactions are a common challenge in enzymatic devices, especially for fuel cells.88 Biofuel cells that eliminate diffusing mediators have been developed for DET to redox proteins. Biofuel cells use biological catalysts and offer opportunity for replacement of noble metals used in traditional fuel cells. Enzymatic catalysts of glucose, glycerol and ethanol have the advantage of ambient operating temperature and pressure, neutral pH, and high selectivity. For industrial applications, GBFCs need stability, longevity, and low operating costs. One commonly used electrode material in GBRCs is carbon nanotubes (CNTs). CNTs are popular for enzymatic bioelectronics because they have high electrical conductivities and stability, and are compatible with enzymes.17,86,89–91 Zebda et al. (2010)36 fabricated a GBFC with electrodes composed of enzymes on CNTs (Fig. 5).36 The electrode material was made by compacting laccase and glucose oxidase with CNTs. This system produced a high current of 1.3 mW cm−2 with an open circuit voltage of 0.95 V, remaining stable for at least one month.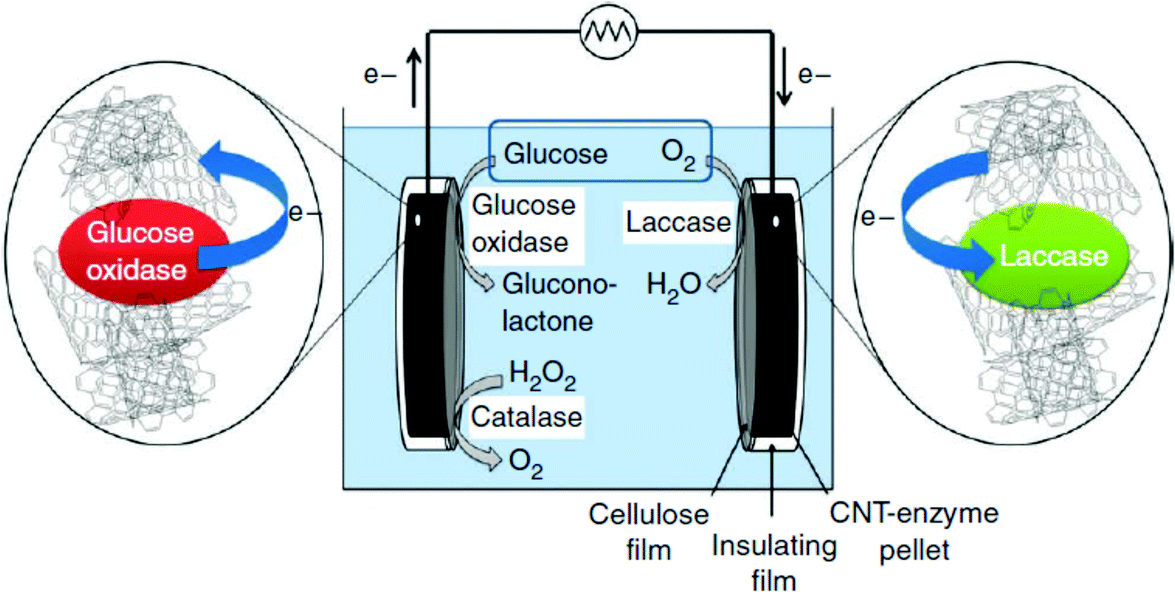 | ||
| Fig. 5 Enzymatic fuel cells use glucose, oxygen or hydrogen peroxide as fuel to produce an external power source. The difference in redox potential of glucose oxidase or catalase and laccase drives an external current. When glucose oxidase and laccase enzymes were immobilized on carbon nanotube (CNT) electrodes, the systems produced a high current of 1.3 mW cm−2 with an open voltage of 0.95 V. Copyright (2011) Nature Publishing Group. | ||
Perhaps an excellent model for synthetic systems to emulate is DET through the bacterial nanowires of Shewanella and Geobacter. These microorganism use bacterial nanowires to facilitate respiration by transferring electrons to mineral oxides over an impressive distance of up to centimeters at rates comparable to doped organic semiconductors (5 mS cm−1).24,92,93Shewanella nanowires are outer membrane extensions containing multi-heme cytochrome proteins.94 A second proposed mechanism of EET is through pili filaments in Geobacter. Electron transfer through the filaments may be possibly due to the redox proteins within the pili and from π–π stacking of aromatic residues (Fig. 6).
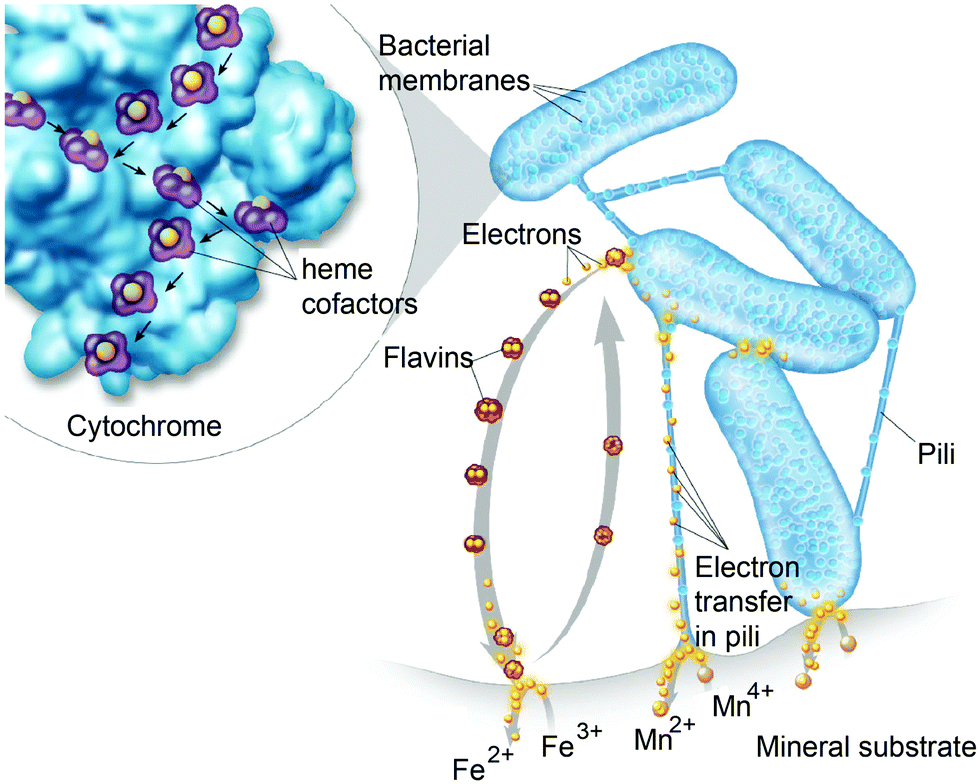 | ||
| Fig. 6 Electrons are shuttled to a mineral substrate from bacterial cells. Cytochrome proteins are present in Shewanella appendages and participate in extracellular electron transfer (EET). Direct electron transfer may occur along cytochrome proteins in pili due to closely spaced heme redox centers. Flavins are also produced by the bacterial cells to shuttle electrons to the mineral substrate. Copyright (2013) Thom Graves. | ||
Several homology modelling approaches have been used to explain the conductances measured by experiment.93,95 Xiao et al. (2016) recently showed that Geobacter sulfurreducens has a separation distance of 3.5–3.6 Å between the closest atoms in aromatic rings within the pili, which is consistent with X-ray diffraction data and may explain the metallic-like electron transport of the pili.96 It is speculated that the short length of the Geobacter sulfurreducens piliA monomer permits tight and ideal positioning of amino acids for enhanced conductivity.97
Although bacterial nanowires have excellent electron transfer capability, modifications can further increase their conductivity to within the range of CNTs. Tan et al. (2016) recently replaced the phenylalanine and tyrosine amino acids on the carboxyl terminus of Geobacter sulfurreducens with tryptophan, an amino acid known to promote enhanced electron transfer in enzymes and peptides.98 The incorporation of tryptophan produced a strain with a pili diameter reduced by 0.5 to 1.5 nm due to the increased hydrophobicity of tryptophan that encourages the association of the carboxyl-terminus with the pilus core. Enhanced conductivities of up to 977 ± 41 S cm−1, a 2000-fold increase from that of the wild-type pili, were reported as a result of the mutations. Although this performance is slightly less than that of traditional CNTs, these modified bacterial nanowires are much more conductive than DNA or PEDOT nanowires.98
1.4 DET vs. MET in enzymatic devices
In contrast to glucose oxidase and amine oxidase (AO) of Arthrobacter globiformis which have their active redox site buried (20 Å deep in a hydrophobic channel for AO),99 the structure of cellobiose dehydrogenase (CDH) can easily facilitate direct electron transfer in addition to mediated electron transfer. The presence of a protruding cytochrome domain allows electrons to be directly transferred between the active site of CDH and the electrode surface.100 The structure of CDH is depicted in Fig. 7. CDH, a fungal enzyme, can oxidize a broad range of fuels, including glucose, cellobiose, and lactose, and is also useful for the detection of several chemicals.101 Multiple research studies have compared MET and DET of CDH for the same system.102,103 One study investigated the different modes of electron transfer of CDH from Phanerochaete sordida at modified anodes.104 The electrodes were prepared with single wall carbon nanotubes (SWCNTs) on pyrolytic graphite electrodes. For MET, osmium redox polymers were used to facilitate electron transport. The optimum pH range for DET was between 4 and 4.5 whereas pH 5–6 improved performance for MET. Compared to CDHs from other organisms, CDH from P. sordida had high rates of DET. However, MET still achieved an order of magnitude greater maximum current and maximum power density compared to a CDH anode operating with DET. A CDH anode using MET generated a maximum power density of 0.3 mW cm−2 at 350 mV while the highest power density generated through DET was 0.032 mW cm−2. Although DET produced lower power density compared to MET, this type of electron coordination is favored for amperometric biosensors due to rapid response and lower costs.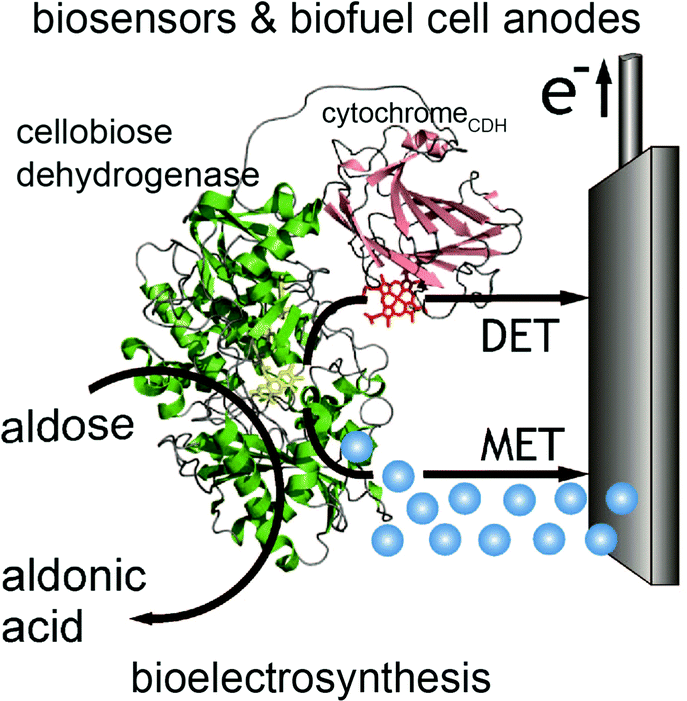 | ||
| Fig. 7 Structure of cellobiose dehydrogenase (CDH) and its connection to an electrode is shown. In addition to mediated electron transfer (MET) from CDH, direct electron transfer (DET) to the electrode occurs through a cytochrome attached to the CDH. The CDH system is proposed for bioelectrosynthesis, biosenors and for biofuel cells as an anode electrode.100 Copyright (2010) John Wiley and Sons. | ||
Both MET and DET mechanisms are used by Shewanella and Geobacter to reduce mineral oxides including Fe(III) and Mn(III/IV). For MET, Shewanella secretes flavins as electron mediators that reduce minerals. Flavins then diffuse back into the cell and can be re-reduced.105 An alternative to the flavin MET are bacterial nanowires (Fig. 8),92,93 which may use DET through multi-heme cytochrome proteins to reduce minerals.
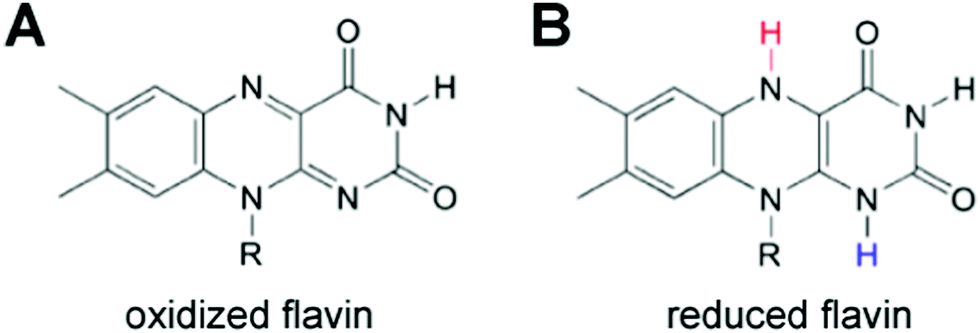 | ||
| Fig. 8 (A and B) Flavin electron shuttles are used to transfer electrons to mineral substrates from Shewanella and Geobacter bacterial cells. Flavins have both an (A) oxidized and (B) reduced form and can be recycled as electron mediators. | ||
2.0 Driving forces and kinetics of electron transfer – redox enzymes and electrodes within an energy landscape
2.1 Introduction to the theory of biological electron transfer and kinetic control at the nano-engineered bioelectrochemical interface
The kinetics of electron transfer from electrode to enzyme may limit maximum current generated in bioelectrochemical devices.88,106,107 In this section we present an overview of the theory of electrochemical driving forces for biological electron transfer as tools to optimize the kinetics at an electrode interface. In Sections 2.2–2.4 we review engineered systems that use specifically designed driving forces for controlled or optimized current generation. In each of the following sections, we present biological electron transport structures and processes where driving forces between redox centers have been tuned to optimize protein function.The relationship between kinetics of electron transfer and electrochemical driving force, developed by Marcus,108 shows that the rate of electron transfer between proteins depends on the difference in redox potential of donor and acceptor (G), the reorganization energy, and the distance between the donor and acceptor sites.109 The reorganization energy is the energy needed for the change in molecular structure of the molecules and environment associated with an electron transfer process, and is usually in the range of 0.7–1 eV.110
A simplified empirical expression relating the rate of exergonic (downhill) electron transfer (ket) to free energy of electron transfer (ΔG), edge-to-edge distance between redox cofactors (R, in Å) and reorganization energy (λ) is given by eqn (1):110
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kexeret = 15 − 0.6(R − 3.6) − 3.1(ΔG + λ)2/λ kexeret = 15 − 0.6(R − 3.6) − 3.1(ΔG + λ)2/λ | (1) |
| logkenderet = logkexeret − ΔG/0.06 | (2) |
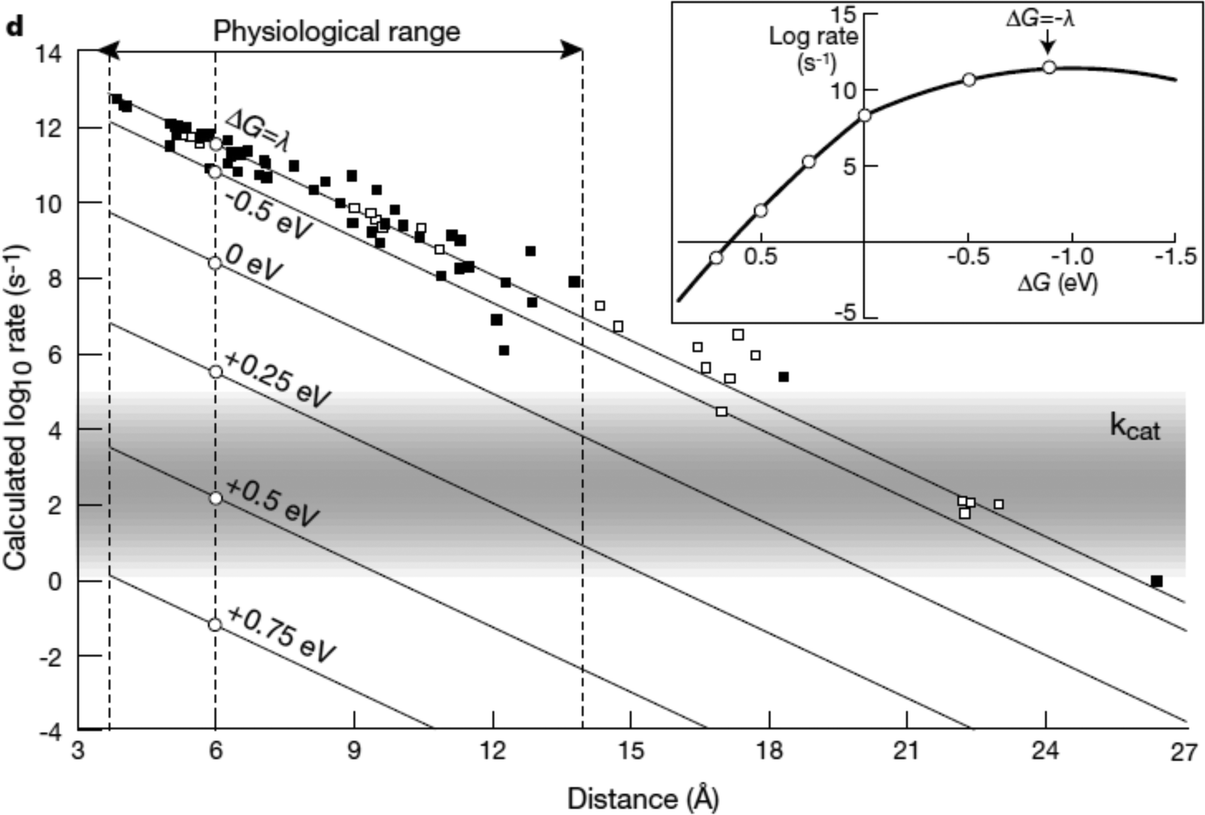 | ||
| Fig. 9 Biological enzymes can transfer an electron from redox at speeds comparable to metal conductors (10−16 s). The closer the distance between cofactors, the higher the rate of electron transfer. Page et al. (1999)26 used Marcus theory to plot predicted electron transfer rates against the distance between redox centers determined for 31 redox proteins. The rate of electron transfer falls off exponentially with increasing distance between cofactors. Inset: The rate of electron transfer is a function of the difference in redox potential between the redox centers (ΔG). When ΔG = −λ, the electron transfer rate is the greatest.26 Copyright (1999) Nature Publishing Group. | ||
Similar to biological electron transfer between cofactors, the difference in redox potential between a synthetic mediator and the protein's donor or acceptor redox center determines both direction of electron flow and kinetics. The biological redox center redox potential may be mimicked in engineered systems by redox polymers, semiconductor materials, conductive polymers, or freely diffusing mediators. Synthetic mediators mimic biological redox centers by having a similar redox potential range.66 For example, ferrocene derivatives have redox potentials in the range of ∼0.35 V vs. Ag/AgCl (∼0.15 V vs. SHE).113 The following section highlights how the redox potential of osmium mediators influence the power density of the enzymatic fuel cell.
2.2 Tuning the redox potential of osmium redox polymers
Relative redox potentials of proteins and electrons impact the kinetics of electron transfer, so it is important to be able to tune the redox potentials of elements of any artificial or hybrid redox “chain” for enhanced kinetics. Bioinspired osmium redox polymers10,14 have redox potentials that can be tuned to fit these needs. In an investigation of laccase biofuel cells by Gallaway and Barton (2008),14 the redox potentials of various osmium redox polymers were tuned by binding ligands of different Lewis base characters.14 By testing polymers with redox potentials between 0.11 and 0.80 V, they discovered that the polymers with the highest redox potentials generated the lowest current densities, and that redox mediators with potentials near that of laccase showed a dramatic drop in catalytic performance. By balancing the tradeoffs between current density and power density, it was possible to determine the optimum mediator potential for their laccase biofuel system, which was 0.66 V (Fig. 10). Typically, mediators' redox potentials are tuned close to that of the target enzyme14,74 and small changes in redox potential (∼50 mV) can cause significant changes in the overall rate of electron transfer between electrode and enzyme.14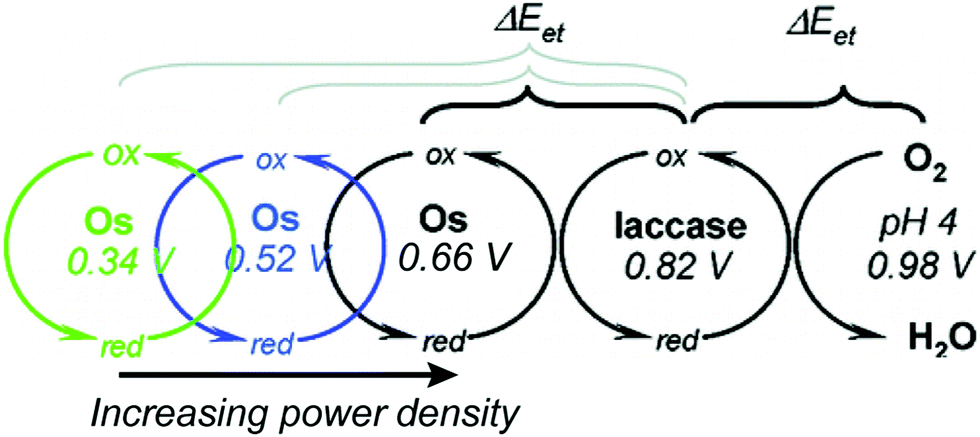 | ||
| Fig. 10 The redox potential of osmium redox polymers can be varied from 0.11 to 0.8 V. The rate of electron transfer between mediators and redox proteins depends on their difference in redox potential. This difference is non-linear and large driving forces can actually lead to a decrease in electron transfer rates (manifested in current density) due to mismatch with the reorganization energy of the system. An increasing power density is produced from the system as the driving force is decreased. Copyright (2008) American Chemical Society. | ||
Similarly to tuned osmium mediators, biological redox centers are tuned to a certain redox potential within a protein. The redox potentials of redox centers can span the entire biological range of −0.5 to 1.0 V vs. SHE.114 To achieve this range, the redox potential is tuned by the environment created by the protein scaffold through the use of electrostatic interactions, solvent accessibility, and hydrogen bonding. For instance, iron sulfur proteins usually have multiple FeS clusters used for electron transport. Although the structure of the clusters is the same, the redox potential varies from cluster to cluster due to the nature and arrangement of surrounding residues. Using the Protein Dipoles Langevin Dipoles (PDLD) model and density functional theory (DFT), Jensen et al. (1994)115 was able to predict redox potentials for the [4Fe–4S] clusters of three different redox enzymes and compare them to experimentally determined redox values.115 In the PDLD model, the area near the enzyme's redox center is simulated as a matrix of partially charged, polarizable atoms, and the solvent is modeled as an array of permanent dipoles encompassed in a continuum dielectric. The redox potentials calculated based on the PDLD model concurred with the experimental redox potentials of the clusters. This investigation concluded that the main contributor to redox potential of [4Fe–4S] clusters is the configuration of amide groups in the primary chains proximal to the clusters, while hydrogen bonding is not a major factor.
2.3 Photoswitchable devices
In engineered systems, electron transfer can be influenced by non-invasive external forces. For example, glucose oxidase (GOx) was incorporated into a photoswitchable device with ferrocene as the electron mediator.19 These photoswitchable devices use light to control the redox state or current of the devices, leading to “On” and “Off” states (Fig. 11). GOx was functionalized with ferrocene bound to the exposed lysine residues. The device was turned “On” using an external light source with a wavelength between 360 and 380 nm and off at wavelengths longer than 475 nm. The “On” and “Off” states are basically controlled by the charge of the chemical modifier on the electrode. In the ‘On’ state, the nitrospiropyran tethered to the electrode has a positive charge and attracts GOx to the surface by electrostatics, increasing the current. In the off state, the nitrospiropyran is protonated through photoisomerization to nitromerocyanine which does not attract GOx to the electrode. Molecular wires can also exhibit two states, allowing or blocking electron transfer. The voltage and light responsive molecular wire, 1,2-bis[2-methyl-5-(4-pyridinium-3-thienyl]cyclopentene-N,N′-diacetic acid was linked to an electrode. The molecule becomes redox inactive at an applied voltage of 0.35 V or higher. A full review of photoswitchable devices was recently provided by Wang, Willner et al. (2013).19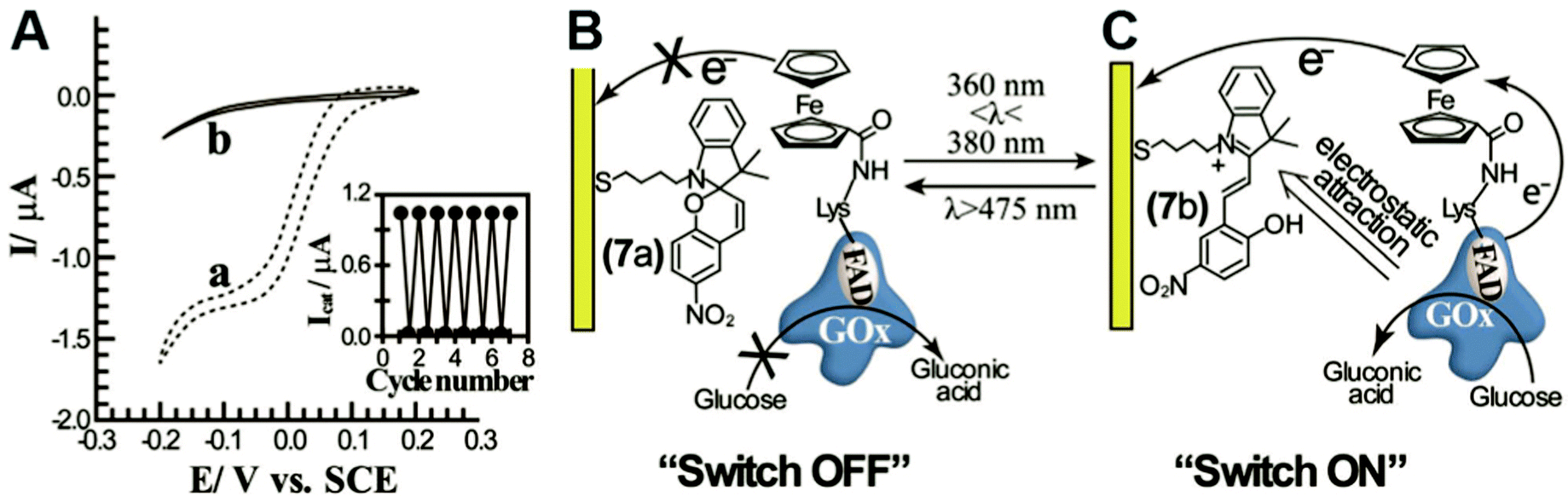 | ||
| Fig. 11 A photoswitchable enzyme device was built by using ferrocene modified glucose oxidase (GOx) and electrodes modified with the light activated chemical nitromerocyanine. (A) Depending on the wavelength, current generation can be switched ‘On’ or ‘Off.’ A low current is produced in the ‘Off’ state (trace b). (B) In the ‘Off’ state the nitromerocyanine blocks electron transfer from the ferrocene mediator. (C) In the ‘On’ state, GOx is attracted to the electrode and nitromerocyanine is allows electron transfer. Copyright (2012) John Wiley and Sons. | ||
The biological equivalent example of photoswitchable devices are proteins that are activated by external forces such as light and magnetic fields. For example, photolyase enzymes catalyze DNA repair reactions using electron transfer reactions following light activation of a FAD cofactor.116 Evolutionarily related to photolyase, cryptochrome proteins may produce a light induced radical pair that is influenced by magnetic fields and linked to long-range navigation of animals such as migratory birds.117
2.4 Enzymes on semiconductor materials
Redox proteins can be described as wide band gap doped semiconductors with redox centers as the dopants.118 For example, photosystem I (PSI) acts a photo-induced semiconductor required for directional electron transfer and conversion of light energy (see Section 2.5). The photo-induced zinc-substituted cytochrome b562 and c can act as an n-type and p-type semiconductor respectively.119 Composite systems of typical semiconductor materials and redox proteins are investigated as photo-bioelectrochemical devices due to their photosensitivity. Semiconductor materials have high chemical stability, and can form nanostructures including nanoparticles and nanowires.120–122 Cadmium sulfide (CdS), indium tin oxide (ITO), zinc oxide (ZnO), titanium dioxide (TiO2)122 and silicon semiconductors have been linked to redox proteins.123 In these devices, the semiconductor material's valance or conductive band can act as an electron donor or acceptor during photoexcitation. Curri et al. (2002)120 used CdS (cadmium sulfide) nanoparticles as photocatalysts linked to formaldehyde dehydrogenase (FDH) enzymes to oxidize formaldehyde. Additionally, photosynthetic proteins have been immobilized on semi-conductor materials as composite materials with enhanced photocurrent generation (Fig. 12A). The driving force for electron transfer from a semiconductor to an enzyme is the electrochemical energy difference between the conductive band of the semiconductor and the redox potential of the redox protein (Fig. 12B).124 We propose that the kinetics of electron transfer from semiconductors could be optimized by using matching the electrochemical driving force between semiconductor and enzyme to the reorganization energy of the enzyme. Semiconductor materials with various Fermi levels and band gaps could be tested with redox enzymes to develop these systems.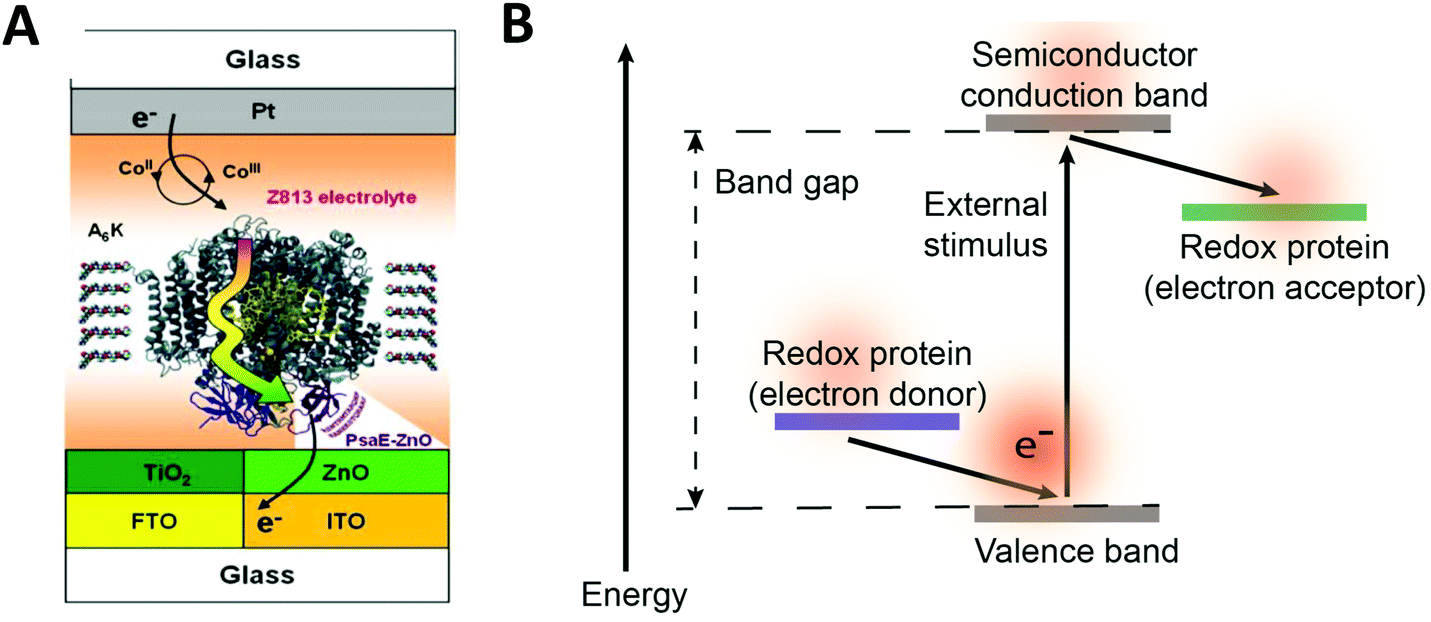 | ||
| Fig. 12 (A) Mershin et al. (2012)122 immobilized photosystem I (PSI) on semiconductor materials to generate high photocurrents. The PSI protein was immobilized with small peptides which stabilize the hydrophobic band of the protein. PSI was immobilized on TiO2 nanocrystals and ZnO nanowires to produce photocurrents of up to 362 μA cm−2. (B) The driving force for electron from a redox protein to a semiconductor is the energy difference between the valence band and the redox potential of the redox protein. Similarly, the driving force for electron transfer from a semiconductor to a redox protein is the energy difference between the conduction band and the redox potential of the redox protein. (A) Copyright (2012) Nature Publishing Group. | ||
2.5 Distance and redox potentials help photosynthetic reaction centers stabilize photo-induced reactions
Many of the concepts of biological electron transfer theory can be illustrated by examining electron transfer in photosynthetic reaction centers. Specific tuning of the distances between biological redox centers and their redox potentials provides a means for these proteins to convert the energy of a photon of light into a stable low potential reductant. For example, the photosynthetic membrane protein photosystem I (PSI) uses organic molecules such as chlorophyll to absorb light and convert the energy of the photon into chemical potential in the form of a reduced iron–sulfur cluster. Fig. 13 illustrates the energy landscape of the redox cofactors within PSI, including their edge-to-edge distance and electron transfer lifetimes.128 Following light absorption, a dimer of chlorophyll a molecules denoted P700 is promoted to its excited singlet state (P700*) which is rapidly oxidized to P700+ and the electron transferred sequentially across the RC through a number of redox cofactors (A0, A1, FX, FA, and FB) at the expense of free energy.40 Ultimately, the distance between P700+ and the final electron acceptor (FB) is large (∼5 nm) and therefore direct electron transfer between these species occurs at slow rates (100 ms)125 and is easily outcompeted by forward electron transfer from cytochrome (τ1/2 = ∼4 μs)126 which reduces P700+ to P700, thus further stabilizing the reduced terminal acceptor FB−.127 Therefore by increasing the distance between the donor and acceptor pair in the electron transfer chain at the expense of free energy, the lifetime of the charge separated state is extended long enough to allow the downstream diffusion-limited electron transfer processes of the cellular metabolism to capitalize on the reductive potential of the reduced terminal acceptor within the reaction center (RC).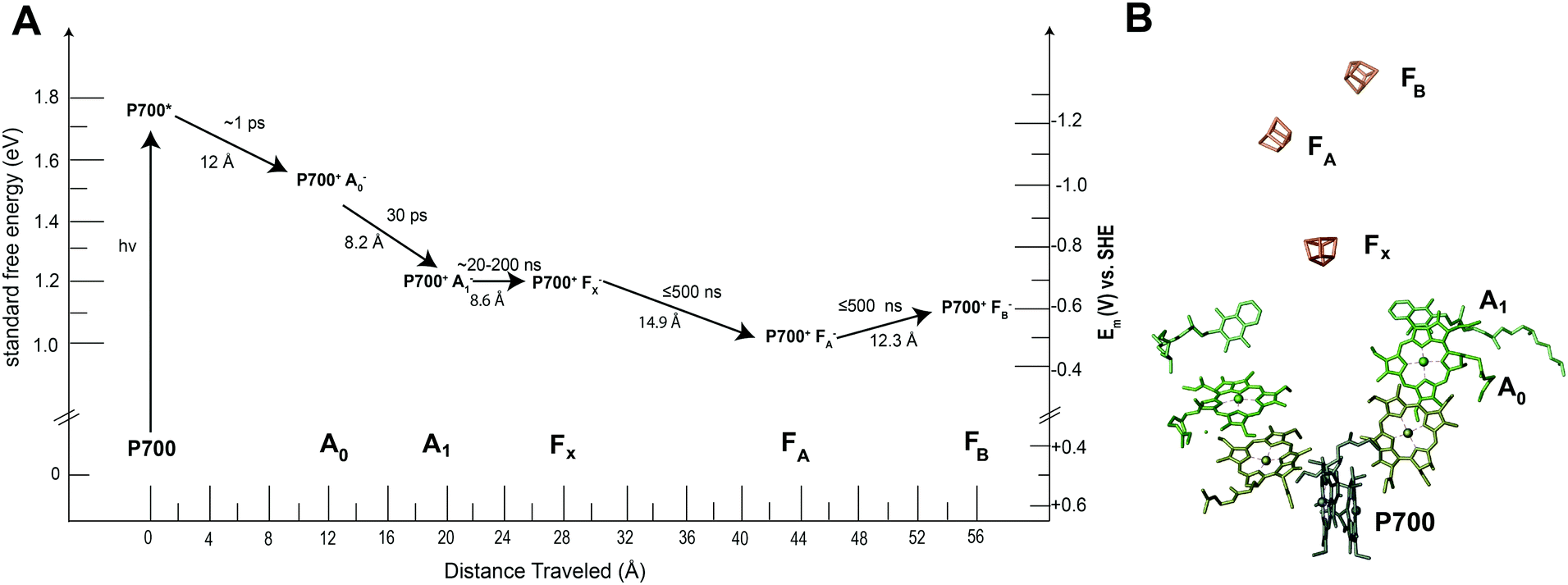 | ||
| Fig. 13 Photosystem I has quinone (A1), iron sulfur clusters (FX, FA, FB) and chlorophyll cofactors (A0 and P700) that form the electron transport chain within the protein. When photosystem I absorbs a photon of light, P700 is promoted to an excited state (P700*). This electron is quickly transferred to neighboring redox centers to stabilize its energy. (A) Each electron transfer reaction requires a loss of free energy. In return, a long-lived charge separated state is produced.129 (B) The X-ray structure of each cofactor in the electron transport chain is shown. There are two pathways present in PSI to the FX cluster (A and B branch) since the reaction center is a protein dimer. | ||
3.0 Compatible, biomimetic and bioinspired interfaces for immobilization of redox enzymes
3.1 Introduction to a biocompatible environment for electron transfer
In this section, we examine biocompatibility of electrode interfaces from the perspective of the enzyme's native biological environment and review the similarities and differences of engineered electrode environments and naturally-evolved optimized stabilizing environments. We investigate various types of engineered electrode interfaces and their modifications (summarized in Table 3). Immobilization or stabilization of proteins at the interface is a critical step in the development of a workable enzymatic device. Immobilization can be accomplished in four major ways: (1) electrostatic binding, (2) binding induced by surface energy, (3) covalent binding, or by (4) binding to metal nanoparticles. Immobilization is also influenced by (1) substrate material – what type of electrode proteins are immobilized on, and (2) environment – whether proteins are immobilized on or within an interface (such as a membrane or a hydrogel). We discuss these considerations below, noting how well each interface and technique mimics biological electron transfer processes.| Interface/electrode | MET vs. DET | Advantages | Disadvantages |
|---|---|---|---|
| Self assembled monolayers (SAMs) |
DET
and MET |
– Selective protein adsorption130–132
– Mimics protein environment131,134 – Adjustable chain packing density134, chain length132,135, and terminal endgroup130–132 |
– Not stable in nonpolar solvent134
– Challenging to synthesize uniform layers135 – Require proper protein orientation for electron transfer130,132 |
| Osmium redox polymers | MET |
– Can be cross-linked for stability136
– Adjustable redox potential14 – Tunable mediator density, film thickness, and electron diffusivity136 |
– Prone to inconsistent mediator loading14
– Use of an expensive, rare earth transition metal – High cross-linking restricts electron transfer to immobilized proteins136 |
| Conductive polymer (CPs) | MET |
– Low impendence137
– Conductivity close to semiconductors138 – Can control protein distribution and activity139 – Can be tailored for greater biocompatibility and conductivity140,141 – Easily synthesized142,143 |
– Brittle with high rigidity and poor water solubility141,142,144
– Can be cytotoxic at high concentrations138,145,146 – Loss of dopant leads to loss of electronic activity147 |
| Tethered bilayer lipid membranes (tBLMs) | MET |
– Properties adjusted by addition of spacer molecules, change in lipid composition, or functional groups148–151
– Quick, easy assembly149,152 |
– Tethering molecule can induce conformational constraints148,149,153,154
– Inconsistent electrode coverage153 – Lipid membrane stability limited to days or months148,152,155 |
| Conjugated oligoelectrolytes (COEs) | MET |
– Decrease internal resistance of lipid membranes156
– Spontaneously align in lipid membranes156–158 – Variety of existing COEs with unique properties including length and redox potential |
– Toxicity at high concentrations156,158,159
– Require a membrane to orient |
| Composite hydrogels – (conductive polymer in hydrogels) | MET |
– High stiffness and tensile strength160–162
– Incorporate proteins with high biological stability160,161,163 – Adjustable mechanical strength and crosslink density161,162,164 |
– CPs conjugate toward electrode surface143
– Poor adherence of CPs and hydrogels in the long-term143 – Poor mechanical properties of PVA hydrogels165 |
| Graphene | DET |
– Single atomic thickness166
– High mechanical strength and flexibility166,167 – Chemical modification possible168–171 – Can be used to generate selected carbon interfaces166,167 – High conductivity properties167 |
– Can be expensive to produce using CVD172
– Cannot generate large-scale, pure amounts of graphene for industrial use167,172 – Low biocompatibility173 |
| Graphene oxide (GO) | DET |
– Less expensive alternative to graphene172
– Oxygen-containing functional groups provides good biocompatibility174 – Disperses well in water175 |
– Hydrophilicity changes with changing size175
– Electrically insulating167 |
| Nanoparticles (NPs) | DET |
– Easily functionalized174
– Can act dually as an interface and as an immobilization technique on other electrodes174,176 – High conductivity achieved by packing177 – Binding can change properties of enzyme178 |
– Some bound proteins have conformational changes and loss of biological activity178
– Difficult to control structure formation processes179 |
| Carbon nanotubes (CNTs) | DET |
– High surface area17
– Good biocompatibility180 – High stability17 – Functionalization can improve solubility167 |
– Difficult to disentangle modified CNT bundles167
– Sonication can shorten CNTs167 – Moderate toxicity, especially with surface modifications181 |
3.2 Electrostatic and energy-based enzyme immobilization
Some biomimetic and bioinspired polymeric interfaces bind proteins using mechanisms similar to those of charge-based protein–protein binding. This electrostatic method involves tailoring protein binding to interfaces based on the opposite charges between the interface and the enzyme. Enzymes can be positively or negatively changed, as reflected by their isoelectric point182 (Fig. 14). Electrostatic binding has been used to immobilize proteins on a variety of interfaces, including self-assembled monolayers (SAMs) and hydrogels.183,184 Löfås et al. (1990)183 used a negatively charged carboxy-methyl-modified dextran hydrogel for protein immobilization.183 Developed atop a glass substrate coated with a gold film, the negatively charged hydrogel bound positively charged proteins in solutions of low ionic strength after activation with reactive groups. Other studies of charge immobilization include work by Narváez et al. (2000),185 Calvo et al. (2000),186 Wang et al. (2006),187 and Lee and Tsai (2009).188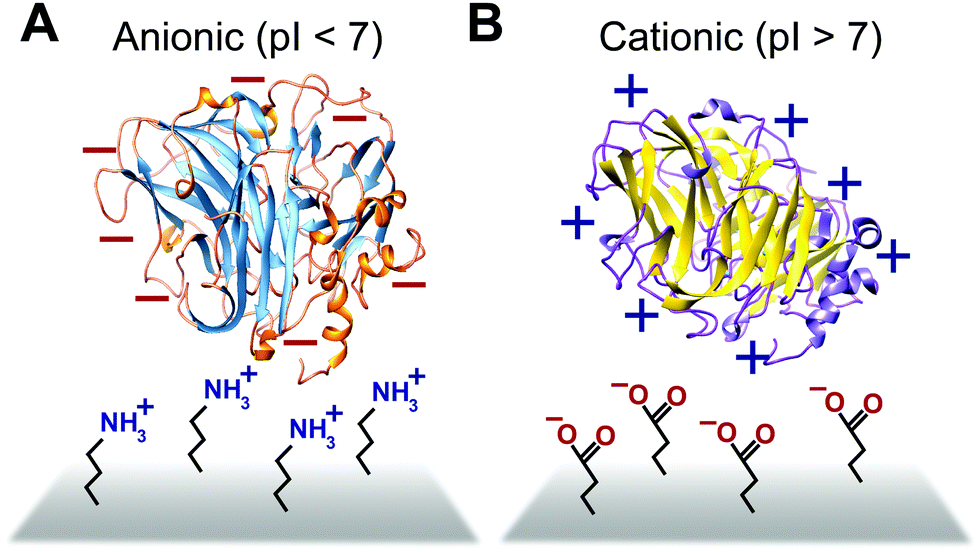 | ||
| Fig. 14 Proteins are anionic or cationic depending on their isoelectric point (pI). (A) Positively charged electrode interfaces such as amine functionalized interfaces are used to immobilize anionic (pI < 7) proteins. (B) Negatively charged electrode interfaces such as those containing carboxylate functionality are used to immobilize cationic proteins (pI > 7). | ||
The electrostatic immobilization approach mimics the structure and chemical nature of protein binding sites. The formation of a donor–acceptor complex enables protein–protein electron transfer. Redox proteins have one or more protein binding sites189 that attract and align the solvent accessible redox sites (∼1000 to 2000 Å2).190 Binding sites have three common characteristics; (1) the binding sites on each protein have opposite charged areas that attract each other, (2) the exposed redox centers in the binding site are often surrounded by hydrophobic residues and (3) the binding site has loose geometric requirements.190 In some cases, such as electron transfer between Fe protein and the MoFe protein or nitrogenase, conformation charges of the binding site are needed to form a donor–acceptor complex.191,192 In addition to distance, the rate of electron transfer between proteins is influenced by the structure and viscosity of the solvent between the proteins189,193 and the structure of the donor–acceptor complex.194
Binding by chemical modification of interfaces also acts similarly to electrostatic-based protein–protein binding in biological systems, but with one caveat; instead of binding due to electrostatic interactions, binding occurs based on the protein's attraction to high-energy chemical groups.131 This type of energy-based binding is characteristically used with SAM interfaces.130–132 The chemical mechanism is similar to the biological charge-based technique because this phenomenon occurs partly as a result of protein affinity to high energy surfaces.131 Modifying SAMs with high energy end-groups, such as hydroxyl (–OH) and carboxyl (–COOH) functionalities, results in the selective adsorption of proteins to these high energy regions.130 Conversely, decorating SAMs with low energy end-groups, such as tetrafluoro (–CF3) and methyl (–CH3) functionalities, results in minimized protein binding in those areas.130 For example, carboxyl groups have a higher energy than hydroxyl groups, which can provide an additional gradient of protein adsorption on the interface.132 Increasing chain length is also known to increase adsorption of proteins, specifically LHI–RC complexes, to SAM interfaces, aiding as a supplementary effect to the chemical modification of SAM end-groups.132 These examples both show an effective chemical approach to the immobilization of proteins on a bioelectrochemical device.
3.3 Covalent binding approaches
Much like in biological systems where heme redox cofactors are bound to cytochromes (Fig. 15), bioelectrochemical devices may utilize covalently bound enzymes at an interface. A common way to achieve improved stability and enhanced binding of enzymes to polymeric interfaces is through cross-linking on electrodes. Cross-linking is a popular method of enzyme incorporation in a variety of polymers, including osmium redox polymers,136 tethered bilayer lipid membranes (tBLMs),195 and conductive organic polymers.196 Specifically, poly(ethylene glycol)diglycidyl ether (PEGDGE) cross-linking has proven to be an effective means of stabilizing conductive polymer interfaces.196 The two epoxy groups present in PEGDGE can react with amino, hydroxyl, and carboxyl groups.197 In a recent study by Mano et al. (2007),196 polyaniline (PANI) was incorporated into a hydrogel with glucose oxidase by cross-linking.196 In trials of PANI systems in the absence of a cross-linker, the weakly bound films slowly dissolved in solution. After cross-linking with PEGDGE, the PANI hydrogel remained stable through multiple conductivity tests. Most importantly, glucose oxidase (GOx) was able to be co-crosslinked within the hydrogel to make electrical contact between the electrode and enzyme. This crosslinking stabilized the GOx within the polymer hydrogel, resulting in a maximum current density of 200 μA cm−2 at 15% weight GOx. Such studies attest to the effectiveness of covalently binding proteins to interfaces in a variety of bioelectrochemical devices. Osmium redox mediators can be crosslinked for integration onto polymers and hydrogel networks.74 In a study by Chakraborty et al. (2014),198 laccase enzymes and osmium redox polymers were co-immobilized within a hydrogel film using a chemical cross-linker to evaluate the electron transfer properties of the system.198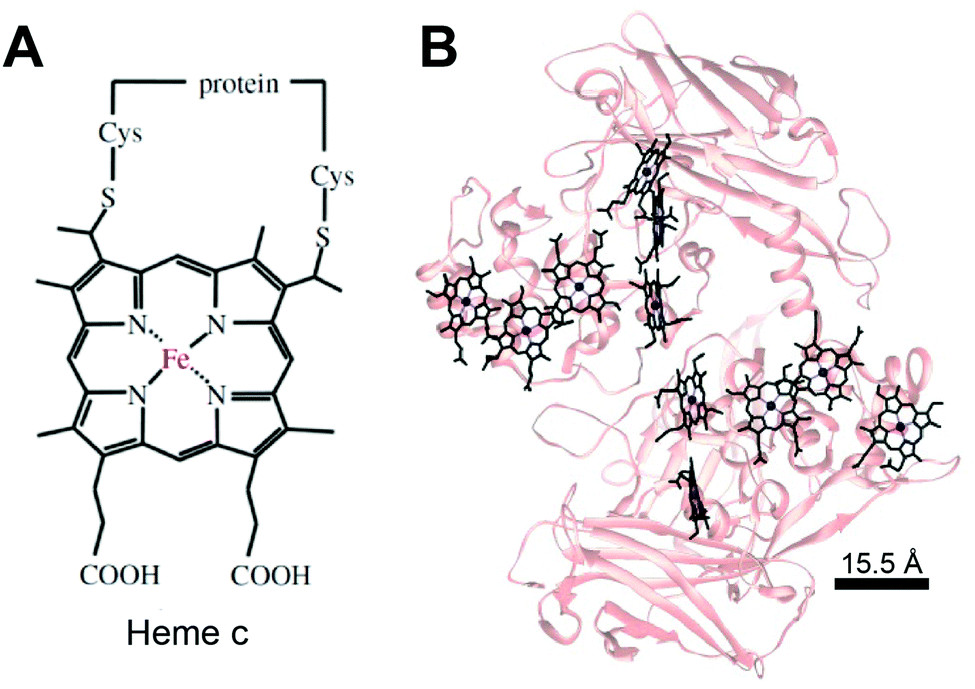 | ||
| Fig. 15 (A) Chemical structure of heme c (or heam) redox center.199 Thioether bonds link the heme structure to a protein (B) heme cofactors are commonly found in redox proteins including cytochromes. The spacing between multi-heme cofactors is 15.5 Å or less within cytochrome proteins. The heme redox centers are arranged in chains to enable direct electron transfer across the entire protein. | ||
Despite crosslinking's effectiveness with stabilization and protein binding, in some cases it may work too well and can restrict the electron transport properties of the material. A study by Aoki and Heller (1993) reports that, as opposed to a 5% wt cross-linker, a 25% wt cross-linked osmium hydrogel restricts the motion of the electron transport chain.136 It can therefore be important to weigh the potential for protein binding with the desired electron transport properties of the material.
The biological equivalent system of crosslinked enzymes at the electrode interface is bound cofactors in protein structures. For example, multi-heme cytochrome proteins, (Fig. 15) have two or more heme cofactors covalently stabilized within the protein by a thioether bond.199 Each heme molecule is spaced 15.5 Å apart within multi-heme cytochromes and form hopping sites that transfer electrons across distances spanning 10's of nanometers.199 Electrochemical systems may benefit from specific, biomimetic covalent bounds, similarly to those used for heme's in cytochrome, to stabilize and orient enzymes and mediators.
3.4 Enzyme wiring with nanoparticles and electrode substrates – metals, semiconductor and carbon materials as electrodes
In addition to the many biomimetic and bioinspired approaches of soft materials as electrode interfaces (mimicking the protein interface), research efforts are also focused on the electrode material. The biological inspiration of redox proteins immobilized directly on electrodes is the bacterial nanowires from Shewanella and Geobacter (Section 1.3), where cytochromes reduce mineral substrates through DET and MET. These microorganisms use mineral oxides including Fe(III) and Mn(III/IV) as electron acceptors to facilitate respiration (Fig. 6).24,200 A similar approach can be used for redox enzymes on electrodes. Electrode materials without extensive surface modifications (SAMs, polymers or mediators) have advantages including high conductivity, increased DET possibility, large surface areas, and high structural stability under changes in solvent and pH.201 Many electrodes, like metal-oxide materials, semiconductors and carbon electrodes, can be modified with simple surface chemistries including hydroxyl, carboxyl, and carbonyl to facilitate binding of enzymes.4 Carboxylic acid and amine groups on the electrode (biomimetic approach) allow the material to covalently couple complementary amino or carboxyl groups on the immobilized enzyme.4 On carbon materials, it is especially easy to develop carboxylic acid functionalities, as oxidation of the surface provides a means of generating these functional groups.4 Carbon nanotubes (CNTs) and graphene electrodes have also gained attention as electrode materials. In the subsequent subsections we discuss graphene electrodes and nanoparticles as substrates for redox enzymes.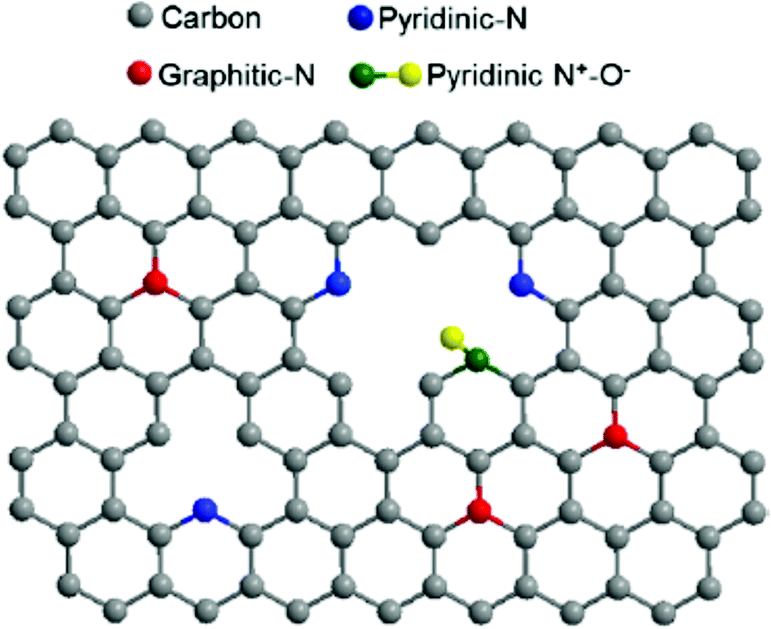 | ||
| Fig. 16 The structure of N-doped graphene. Doping the interface with nitrogen causes enhanced ability for electron transfer between redox enzymes and the graphene electrode surface.211 Copyright (2012) American Chemical Society. | ||
GO is a graphene sheet with carboxylic acids at its edges and phenol, hydroxyl and epoxide groups along the basal plane.167 GO has been investigated as an alternative to graphene due to its ability to be dispersed in water167 and its unique chemical and electronic structures, brought out by the collection of chemical groups on its surface.172,212 Graphene production by conventional methods, including chemical vapor deposition, exfoliation from bulk graphite with Scotch tape, epitaxial growth, and “unzipping” of CNTs, are currently incapable of inexpensively producing large, pure graphene monolayers,172 making graphene oxide's use in research applications more prevalent. Although GO has proven to be a fair electrode for applications with redox-enzymes, it is an electrically insulating material with high thermal instability, making it poorly conductive in its natural state.167 Thus, a partially reduced sheet of GO must be used in electrochemical applications.167 In a study by Wang et al. 2009, GOx was covalently bound to reduced GO adsorbed on an amino-modified electrode for application in DET.213 By ensuring that cyclic voltammetry readings were constant with those of the native GOx enzyme and assessing the electrocatalysis towards glucose at concentrations between 0 and 24 mM, the group confirmed that GOx was still active after immobilization on the GO interface.
These particles can also be used as wires or electrodes for biomolecule applications123 due to their ability to achieve a high conductivity in electrochemical devices by closely packing nanoparticles on a substrate surface.177 In order to facilitate the biomolecule–NP binding required for such devices, positively charged proteins can be electrostatically bound to anionic ligands on NPs, covalently tethered to NPs that are functionalized with thiolated molecules, or bound to affinity sites functionalized on NPs.178 Although such covalent attachment can help to overcome instability and inactivation of enzymes, some proteins and enzymes directly adsorbed on NPs show conformational changes and loss of biological activity upon incorporation.178 However, such binding can also have the added effect of stabilization of the biomolecule–NP structure, and can change the properties of NPs and biomolecules to have such advantages as increased solubility and increased stability of hydrogen bonds formed by the biomolecule.178
Xiao et al. (2003)176 constructed a glucose oxidase biosensor using NP–enzyme binding, where the metallic NP adjacent to the enzyme redox center would enhance redox properties by acting as an electron relay.176 In their most effective experimental set-up, functionalized Au-NP was reacted with flavin adenine dinucleotide (FAD) (Fig. 17), resulting in a FAD functionalized NP, with which Aspergillus niger apoglucose oxidase (apo-GOx) could be reconstituted. Upon assembly on a gold electrode and functionalization with a dimercaptoxylene monolayer, the interface produced a 60% random, densely packed coverage of GOx on the electrode surface. More impressively, the electron transfer rate was calculated to be seven times higher than that of native GOx with oxygen as an electron acceptor, far surpassing the capabilities of the analogous biological system. This system showed the NP was effective as an electrical relay element between the FAD redox site and the electrode surface, and the high current density achieved shows the effectiveness of NP tethering of biomaterials to electrode surfaces.
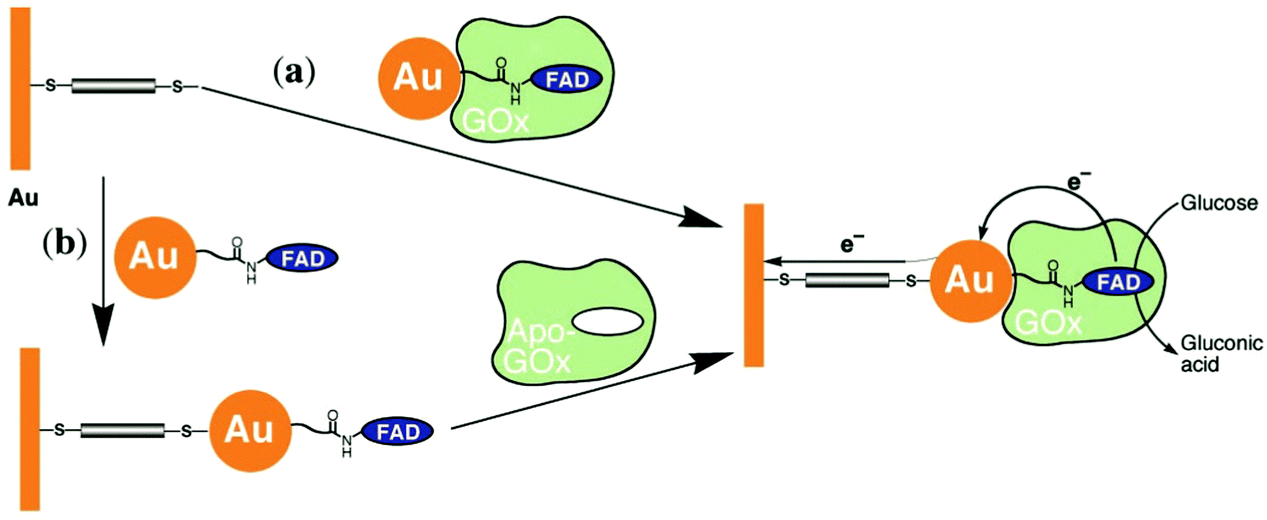 | ||
| Fig. 17 Possible pathways for creating a biomolecule–NP interface using GOx and Au-NPs. (a) Apo-GOx is reconstituted with FAD functionalized Au-NP. The complex is assembled on a gold electrode functionalized with a 1,4-dimercaptoxylene monolayer. (b) A FAD-functionalized Au-NP is assembled on a functionalized gold electrode. The exposed FAD on the electrode–NP interface provides a means of assembly for GOx. Copyright (2003) The American Association for the Advancement of Science. | ||
3.5 Soluble proteins in conductive porous and hydrogel interfaces
Soluble enzymes are typically hydrophilic, and their native environment is the intracellular fluid. The cellular environment may be mimicked at the electrode interface with porous or hydrogel materials. Mesoporous structures have a high surface area and allow immobilized protein access to a buffered solution, which can stabilize the protein. In addition, porous interfaces are ideal for mediated electron transfer and the removal of reactive products.61 Porous interfaces include silica sol–gel materials,216,217 metal oxides including SnO2, TiO2, and ITO.218,219 and hydrogel materials including DNA,220 protein and peptides,221,222 chitosan223 and conductive polymer hydrogels.224Zhai et al. (2013)163 used GOx incorporation within a hydrogel for use as an effective glucose sensor with a sensitivity of 96.1 μA mM−1 cm−2 and a low detection limit of 0.7 μM (Fig. 18).163 By first creating a platinum nanoparticle/PANI hybrid hydrogel system, GOx was incorporated through the open channel 3D matrix of the hydrogel. Because of the porosity of the hydrogel, a larger effective surface area was available for GOx incorporation, and the concentration of immobilized GOx on the hydrogel surface was greatly increased compared to simply depositing the enzyme onto an electrode surface. Packing immobilized GOx on the hydrogel in this way allows the enzyme to better facilitate the transport of molecules and electrons, which led to a high sensitivity and performance of the resultant glucose sensor. Although hydrogels are just one way of incorporating soluble proteins to electrochemical interfaces, their examples show that this type of immobilization can be an effective means of increasing the performance of a bioelectrochemical device.
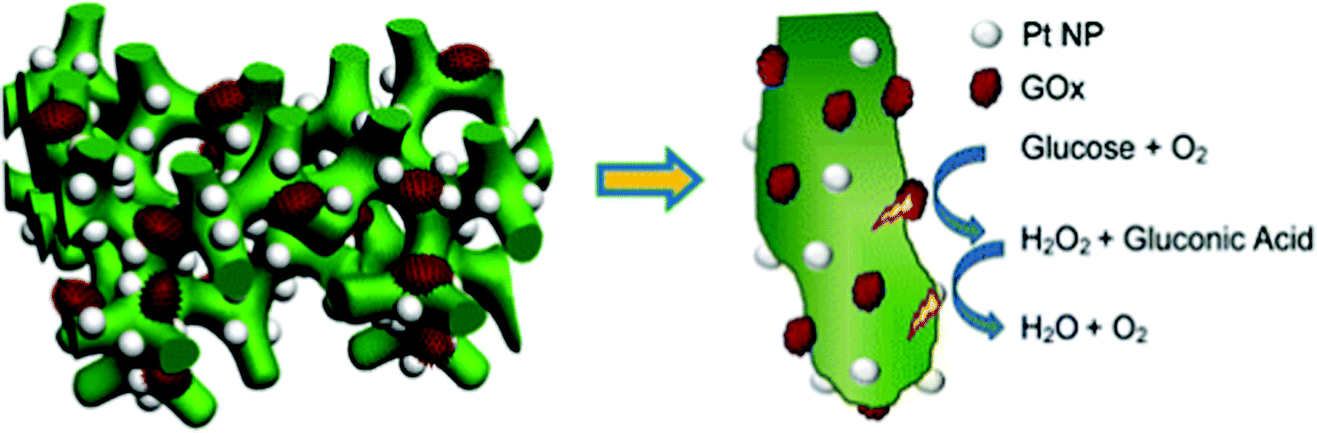 | ||
| Fig. 18 Structure of Pt-NP/PANI hydrogel with incorporated GOx. On this interface, GOx performs oxidation of glucose with high efficiency leading to high sensitivity. The PANI hydrogel provides high surface area for immobilization of GOx leading to high loading capacity. Pt-NPs enhance electron transfer by providing even more surface area for immobilization. The NPs are similar in size to the GOx to mimic electron transfer between proteins. Copyright (2013) American Chemical Society. | ||
Hydrogenase is a commonly used redox enzyme that needs a tailored environment to function properly. Hydrogenases catalyze the reversible reaction of molecular hydrogen to protons and electrons. Since hydrogenase is inactivated on oxygen exposure, several studies have focused on increasing hydrogenase's tolerance to oxygen or limiting oxygen exposure. Recently, Plumeré et al. (2014)225 utilized a redox hydrogel to shield hydrogenase from high-potential and oxygen deactivation. Polyethylenimine modified with viologen moieties acted as a buffer between the electrode surface containing hydrogenase and the air, insulating the enzyme from the negative effects of both high potentials and oxygen.225
3.6 Tethered lipid bilayer membranes (tBLMs)
In some applications of osmium redox polymers,198 SAMs,131,226 and tBLMs,153 non-ionic surfactants are used to stabilize membrane proteins for such purposes as improved spreading of protein-containing solutions over the polymer. Some studies using such detergents as Triton X-100 report no negative effects on protein activity of specific proteins.198 However, a number of studies have shown that short chain non-ionic detergents can inactivate proteins,227 which could negate the redox effects of proteins meant to be incorporated within an interface. These types of phenomena draw attention to the challenges of detergents and solvents when considering the maintenance of a protein-rich interface.Much research on developing devices with membrane proteins focuses on their compatibility within biomimetic tethered lipid bilayer membranes (tBLMs). tBLMs, made through self-assembly of tethering lipids in solution,149,155 are closely analogous to biological cell membranes, in which channels and other proteins are embedded. In an application of tBLMs, cytochrome bo3 was co-adsorbed into a phospholipid bilayer membrane with cholesterol tethers, which attached the lower leaflet of the bilayer to the electrode surface (Fig. 19).228 Spacer molecules were used to prevent the formation of a monolayer from the cholesterol tethers. The resulting membrane provided sufficient space for the integration of the cytochrome, which was co-adsorbed and randomly distributed throughout the tBLM. Most importantly, no large defects in the membrane were detected by electrochemical impedance spectroscopy (EIS), and a large part of the surface was shown to be active, proving that the integrity and functionality of the enzyme was preserved during its incorporation into the tBLM. Such interfaces that mimic the natural environment of enzymes and preserve their functionality are relevant for the incorporation of membrane proteins into a functional bioelectrochemical device.
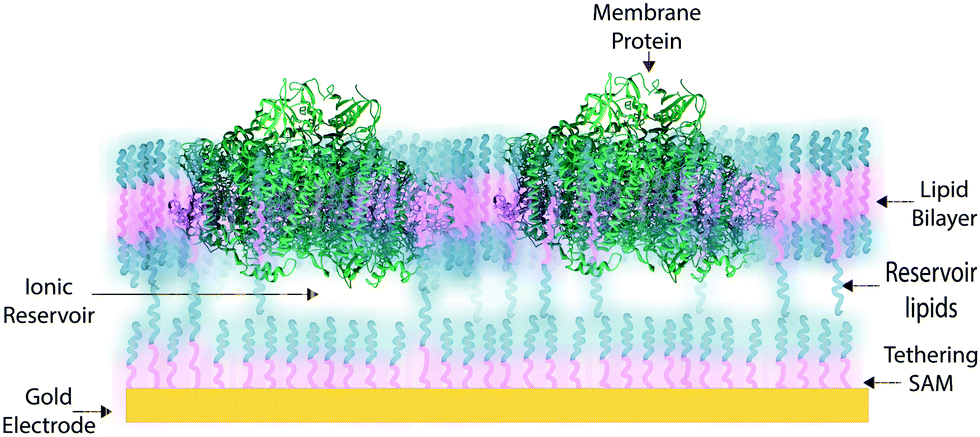 | ||
| Fig. 19 Example illustration of a membrane protein in a tethered lipid or synthetic bilayer. A gold electrode functionalized with thiol-tethers that bind to the lipid bilayer and stabilize the membrane system. The membrane protein is then near the electrode surface. An ionic reservoir is formed between the thiol-monolayer and the protein membrane. | ||
Artificial tethered lipid bilayers are inspired by biological lipid membranes (Fig. 20). These lipid membranes are essential for compartmentalization of cellular components. Lipid bilayer membranes are 4–5 nm in thickness, provide a barrier to both solute passage and electron passage, and enable ionic gradients to build up across membranes.229,230 Lipid membranes also provide a means to arrange proteins in close proximity within the membrane plane including into highly ordered structures, such as 2D crystals.231 Photosynthetic membrane proteins of the purple bacteria pack closely in a lipid bilayer.232 Membrane proteins have external hydrophobic residues, which stabilize the proteins in a lipid bilayer, in the absence of which proteins can aggregate and lose activity. There are three approaches to stabilizing membrane proteins on electrodes: (1) with detergent micelles, (2) with small peptides,122 or (3) with a membrane-modified electrode.
 | ||
| Fig. 20 (A) Purple bacterial membranes are packed with light harvesting membrane proteins. Light harvesting proteins (I and II) pack with reaction centers (RCs) to absorb light as part of the photosynthetic pathway. The lipid bilayer provides an environment to densely pack proteins within the cell. Copyright (2009) Springer. | ||
3.7 Synthetic bilayers and conjugated oligoelectrolytes
Despite the successes of biomimetic tBLMs in incorporating membrane proteins on interfaces, there are some drawbacks with using tBLMs in bioelectrochemical devices. Several tBLMs use reservoir lipids, which are lipids incorporated within a tBLM to create a space (reservoir) between the mobile lipid bilayer and electrode surface. This space is intended to increase the ability of electrons to travel through the lipid bilayer to the electrode surface, facilitated by ionic carriers within the reservoir region.149 tBLMs with idealized reservoir lipids for enhanced conductivity can be difficult to synthesize, and bulky protein domains could interfere with the tethering lipids.149,154 Tethering can also introduce constraints on bilayer diffusion and protein conformation, preventing redox proteins from undergoing conformational changes which allow their functionality within these membranes.149,154 When coupled with the tendency of tBLMs to only partially cover an electrode, resulting in decreased performance,153 these properties decrease the reliability of tBLM interfaces.Bioinspired artificial bilayers that mimic the functionality, bio-compatibility, and structure of tBLMs are being explored as more effective, long-lasting membranes for incorporating membrane proteins interfaced with a bioelectrochemical device. One type of artificial bilayer uses synthetic block copolymers (BCPs) to replicate the bilayer structure of tBLMs while providing enhanced packing and protection of proteins from surface interactions.150 By cross-linking,233 BCPs can provide a stable, compatible environment for membrane proteins. Although there are currently limited studies regarding integration of redox proteins within BCP membranes in devices,234,235 Jia et al. (2009)235 developed a diblock copolymer film that mimicked hemoglobin's native environment to immobilize redox-active hemoglobin proteins.235 The successful immobilization resulted in good direct electron transfer between the redox protein and electrode, and exhibited the biocompatibility of BCP artificial bilayers for redox applications. More recently Saboe et al. (2016) used a diblock copolymer to stabilize the photosystem I (PSI) proteins into large membranes and integrated these proteins into a long-lived photocurrent producing device (Fig. 21).
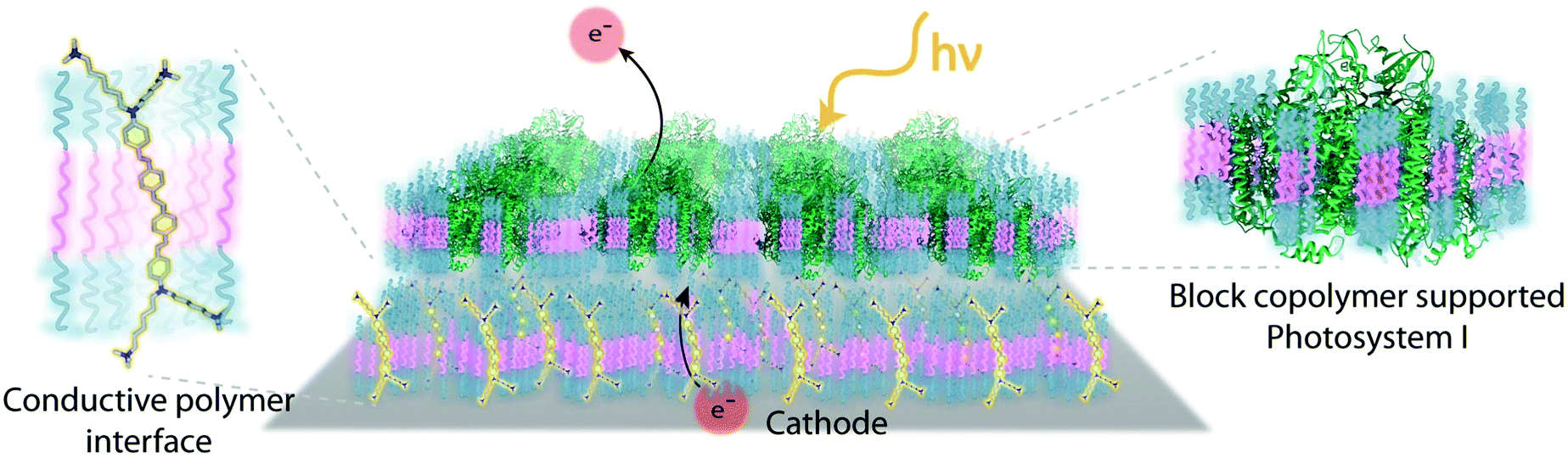 | ||
| Fig. 21 A block copolymer membrane integrated with DSSN+ (a COE) as an interface for PSI-block copolymer membranes. Upon exposure to light, electrons travel through the COE-integrated membrane from a gold electrode, generating high photocurrents on a per protein basis. This system showed good longevity over 30 days. COEs are ∼4 nm in length and provide excellent charge transfer through a lipid bilayer. The diblock copolymer interface was used as an immobilization cushion for the PSI protein membranes. Copyright (2016) The Royal Society of Chemistry. | ||
Although tBLMs and synthetic membranes provide a means to stabilize membrane proteins, these membranes have a low dielectric constant, which may prevent multiple layers of membrane proteins from gaining electrical contact with the electrode through mediators.230 The integration of conjugated oligoelectrolytes (COEs) within membranes creates a bioinspired pathway for electrons through these membranes that could further increase electron transfer between redox proteins and an electrode surface.
COEs have been used as cell membrane-spanning electron transfer molecules to increase current output in microbial fuel cells (MFCs) and recently in photosynthetic devices.234,236,237 COEs have a specific orientation in a lipid bilayer – perpendicular to the plane of the membrane – allowing enhanced electron transfer across the membrane.140,156,158,238 Using COEs, Saboe et al. (2016)234 produced an interface to immobilize PSI on a diblock copolymer membrane intercalated with DSSN+ that produced four times more photocurrent than the same device without the integrated COE component (Fig. 21).234,236 Such effective modifications to the traditional tBLM structure provide an indication of their long-term promise.
The biological equivalent of COEs in a membrane is the quinones within mitochondrial and thylakoid membrane. In the mitochondrial membrane is a series of membrane proteins that use oxygen, nicotinamide adenine dinucleotide (NADH) and a proton gradient to make the high energy molecule, adenosine triphosphate (ATP).239,240 Electrons are transferred between these membrane proteins by using membrane intercalated, freely diffusing molecules. For example, quinone molecules such as ubiquinone are used in cyclic electron transfer within the hydrophobic region of the lipid membrane.241 Oxidization of NADH and succinate by complexes I and II generate protonated quinone, which transfers electrons to complex III (Fig. 22A).242,243 Photosynthetic membranes utilize a similar electron shuttling mechanism. In the thylakoid membranes of cyanobacteria, membrane intercalated plastoquinone shuttles electrons between photosystem II and cytochrome b6f (Fig. 22B).23
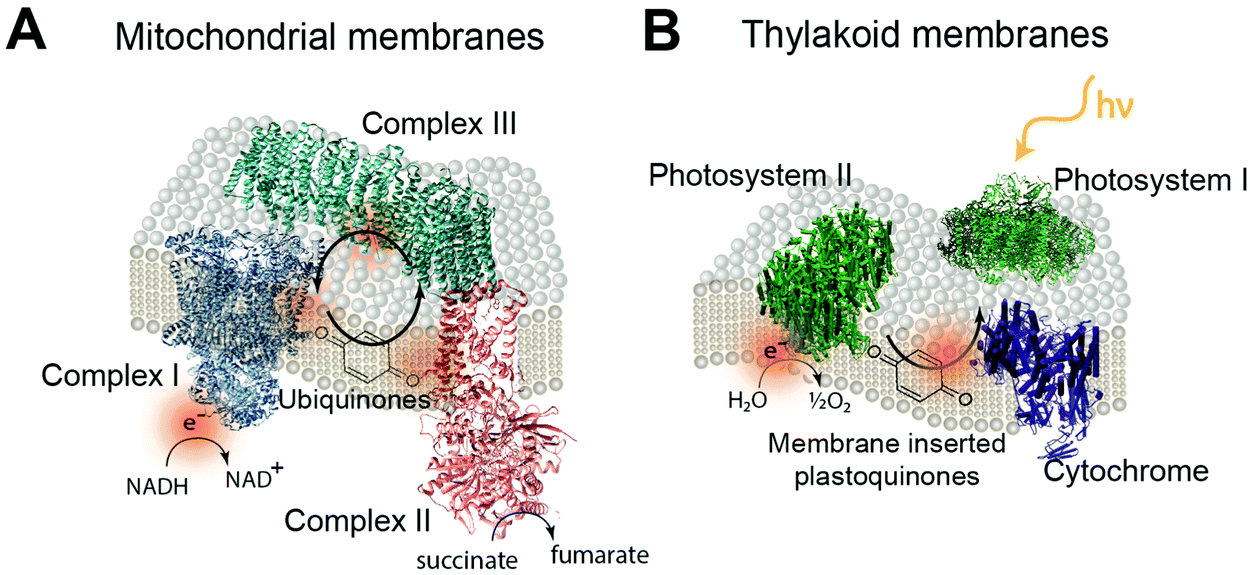 | ||
| Fig. 22 Biological electron transport chains are important for energy production within the mitochondria (A) and in photosynthesis (B). Both processes are centered around a membrane structure. A lipid bilayer supports and orients redox membrane proteins in biological systems. Small redox mediators including ubiquinone (in mitochondrial membranes) and plastoquinones (in thylakoid membranes) intercalate in the lipid bilayer and provide an electron pathway between redox membrane proteins. | ||
4.0 Optimizing the performance of bioelectrochemical devices – toward scalable designs
4.1 Introduction to optimized and scalable designs
Enzymes have contributed to industrial processes for over a century. Over this time, the industrial and personal use of enzymes have expanded including in yeast fermentation, pharmaceuticals, food, and textiles.244 The scale up of many bioprocesses, such as fermentation and other bioreactors, are well established and may be optimized by using mathematical models. Even though these catalytic proteins have been utilized in a broad range of large scale manufacturing scenarios, it remains challenging to scale up emerging enzyme-based applications, such as enzymatic fuel cells or even simple sensors (beyond glucose sensors) with good sensitivity and low production cost.5 Factors that limit the expansion of bioelectrochemical devices include enzyme longevity, cost, and device performance (sensitivity or power output). Since there are existing technologies that can execute similar functions as enzyme-driven devices, it is vital that bioelectrochemical devices are competitive in both economics and performance. In order for enzymatic constructs to be utilized on production scale or even as personal devices, redox proteins and their devices must be durable or have easily replaceable components while still maintaining high activity. For instance, an implantable continuous glucose monitor powered by glucose oxidase must have a long catalytic lifetime or the enzyme should be noninvasively renewed as needed for the device to be practical. Another primary factor in design scale up is cost. The techniques used to obtain high enzyme purity could be expensive. For industrial and therapeutic applications, downstream purification can constitute up to 80% of the entire production cost, making enzyme yield and purification techniques huge factors in the sale price.215 For example, Biogen's Interferon, a drug generated in a small volume, sells at about $3000 per g, whereas intravenous immune globulin (IVIG) is produced with greater yield and sells at around $50 per g.245 Considerations of enzyme lifetime, device power output and durability, and economic feasibility are crucial for transitioning bioelectrochemical devices from a research lab scale to production for widespread usage.In this section, we review the potential of enzymatic devices in industry and address fundamental challenges toward scalable technologies, including stability, protein loading and orientation challenges. We analyze how these challenges are overcome in biological systems. In the following sections (Sections 4.2–4.5) we comment on (1) increasing protein loading on electrodes, (2) methods to control the orientation of proteins on electrodes, and (3) the long-term stability of enzymatic devices.
4.2 Protein loading on electrodes
Enzyme loading is an important property to consider in the design of enzymatic devices since a high density of enzyme on the electrode is desirable for high current density. Jeuken et al. (2006)228 produced an active enzyme coverage between 3 and 8.5 × 10−15 mol cm−2 of cbo3 proteins on a tBLM surface.228 The theoretical maximum loading for a single monolayer of protein that has an area of 102 Å2 is ∼2.2 × 10−12 mol cm−2.246Enzyme loading has also been a point of investigation with SAMs. An extensive study of the properties of SAMs by Gooding et al. (1999)184 brings to light important information about enzyme loading.184 In an initial study of enzyme loading capacity on SAMs, the loading was calculated at 1 × 10−12 mol cm−2. However, this value came in contrast to the calculated theoretical loading of 2.6–3.8 × 10−12 mol cm−2, and thus does not correspond to the close packing of enzymes on the SAM. Instead, enzymes were arranged on the SAM surface in rings of five or six enzymes in clusters. Therefore, limitations for enzyme loading are not just based on the amount of area available to the enzymes, but also on the unique way that they pack onto provided interfaces.
The type of interface used for a bioelectrochemical application can also have an effect on enzyme loading. Typically, interface materials with high surface areas, including graphene, provide an increased enzyme loading on the surface of a device.247 A study by Kang et al. (2009)247 reported an increased glucose oxidase loading of 1.12 × 10−9 mol cm−2 for a graphene–chitosan composite interface on a glassy carbon electrode compared to using only the glassy carbon electrode for protein immobilization.247 Similarly, Ekanayake et al. (2007)248 use platinum coated, nano-porous alumina electrodes to create polypyrrole nanotube arrays that lead to a high level of glucose oxidase loading, which can be further improved by varying the combination of substrate, polymer, and dopant used.248
Zhang et al. (2010)249 investigated electrostatic factors that can influence protein loading on interfaces.249 Immobilization of both horseradish peroxidase (HRP) and lysozyme were investigated on unmodified graphene oxide (GO) sheets. When immobilizing HRP, enzyme loading on GO decreased with increasing pH due to changes in the surface charge of HRP; for buffers with pH below 7.2, HRP retained a net positive charge and were favorably attracted to the negatively charged GO sheets. At pH's above 7.2, HRP developed a net negative charge, and was thus repelled by the negatively charged GO sheets, resulting in poor immobilization. Furthermore, when HRP and lysozyme in buffers of pH 7.0 were compared, lysozyme had a loading of 700 μg mg−1 on GO sheets while HRP reached a maximum loading of 100 μg mg−1. This large discrepancy was likely due to lysozyme's surface charge interacting more favorably with GO than HRP, showing that substrate–enzyme interactions informing enzyme loading are largely dependent on surface charge interactions. The group also noticed that increasing enzyme loading causes an increase in surface coverage of the enzyme, and that increasing enzyme loading did not affect the efficiency of the enzyme.
Enzyme loading can play a large, direct role in device performance. Gooding and coworkers have showed quantitatively that enzyme loading can affect current production in interfaces.184 They were able to control the loading of enzymes on SAMs by altering the time the activated SAM is exposed to the enzyme solution. By changing the enzyme loading, the group observed an almost linear increase in current response with increased enzyme loading. Similarly, enzyme loading can be tuned to produce a device that is functional over a specific set of environmental or experimental parameters. Brahim et al. (2002)250 tested a variety of glucose oxidase loadings for use in a polypyrrole–hydrogel composite glucose biosensor.250 The group found that loadings of 760 and 1520 U cm−2 resulted in sensors that detected glucose within the range of physiological glucose concentrations in patients without diabetes (between 4.5–6.0 mM glucose). A correlation between response time and enzyme loadings was observed, where response time of the biosensor decreased with increasing enzyme loadings until reaching 1520 U cm−2, when an increased response rate was observed. These applications show the direct relevance of protein loading on the production of bioelectrochemical devices.
High protein loading is effective in generating large current responses in enzymatic devices. In these systems, enzymes are usually randomly mixed with immobilized electron mediator polymers. Although this type of design effectively increases the electrode area, direct electron transfer between all the loaded enzymes and the electrode is not possible. On the other hand, biological systems use precise nano-structures, including multi-layered membrane structures, to maximize function per unit volume. An excellent example is multi-layered membranes in chloroplast (Fig. 23). Thylakoid membranes are folded into a unique layered structure which enhances light absorption per unit area.251
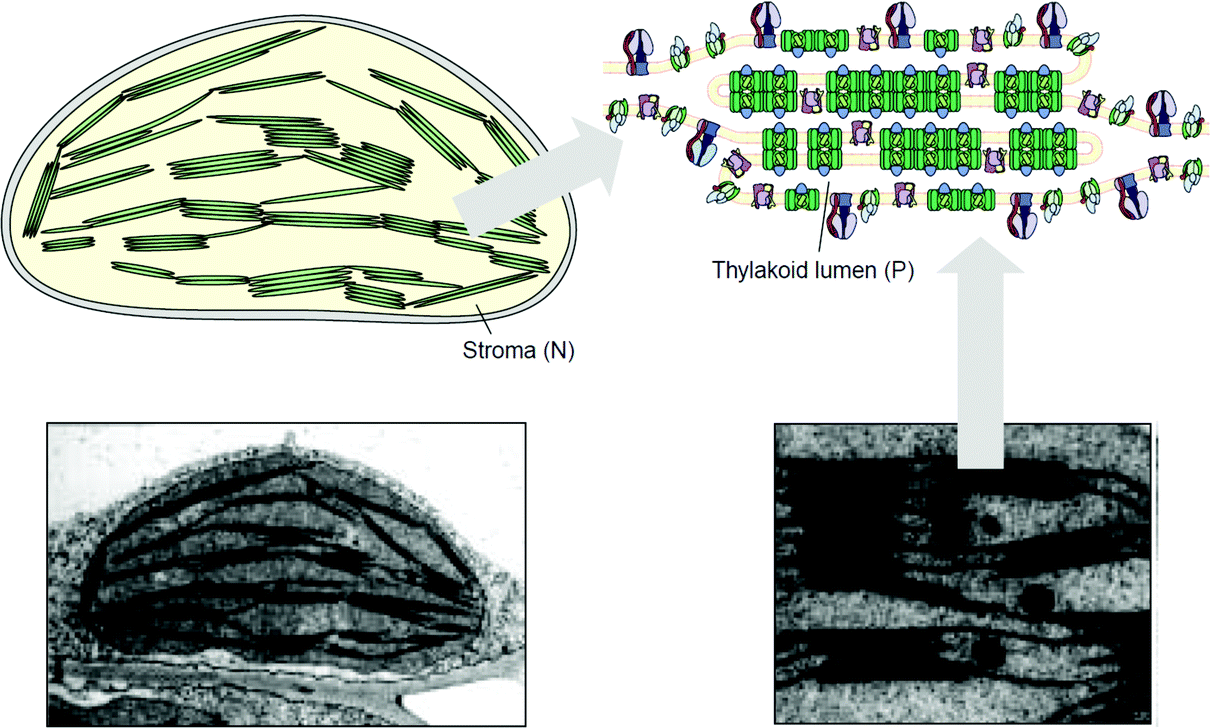 | ||
| Fig. 23 Thylakoid membranes have a three-dimensional design to increase light absorption. The thylakoid membranes are folded into densely packed structures in chloroplast. Within the thylakoid membrane, photosystem II, photosystem I, cytochrome and ATPase membrane proteins are present which in total convert light energy into chemical energy. Copyright (2001) Elsevier. | ||
4.3 Long-term stabilization of proteins on interfaces
In biological systems, proteins can be degraded and manufactured when needed to keep the cell alive. Some proteins can even last for years and there are other proteins that function to reduce oxidized proteins.252 However, in bioelectronic devices, enzymes are not replaced and the maximize lifetime of the device may depend directly on the enzyme stability. Stability can be achieved by integrating redox proteins onto interfaces that mimic the biological cell environment. Several studies point to the long-term stability of proteins on electrode interfaces and while integrated into lipid membranes (Table 4). Blanford et al. (2007)253 report a laccase electrode system stabilized on a chemically modified pyrolytic graphite ‘edge’ electrode. Bound to the electrode by a diazonium salt complex, the laccase system retained 57% of its original current density after an eight week period.254 Similarly, Rüdiger et al. (2005)254 covalently bound hydrogenase to an amine-functionalized carbon electrode and produced an interface that maintained 90% of its original current after one week.253 This stability can also be achieved in photoelectrochemical cells of PSI proteins. A majority of studies that have measured the stability of enzymes show decreases in activity over time (Table 4). The research field should focus on improving the long-term stability of redox proteins on electrodes through protein or electrode modifications or alternately, a renewal strategy may need to be devised.| Protein | Electrode interface | Stability (days) | Activity retentionRef. |
|---|---|---|---|
| a Continuous operation at −0.1 V vs. Ag/AgCl and 1.4 mW cm−2. b Construct not tested in an electrode setting. | |||
| Photosystem I | – Alkanethiol SAM with Os(bpy)2Cl2 mediator, 0.03% DDM detergent | >0.125a | N/A255 |
| – 1,4-Benzenedimethanethiol SAM on gold with vitamin K1 linked to a Au-NP (∼1.6 nm) | 365 | ∼100%256 | |
| – Tethered nanoconstruct of PSI and H2ase using an alkane dithiol molecular wire to Fe clustersb | 64 | ∼100%33 | |
| – PSI multilayer on unmodified gold electrode, 0.05% Triton-X 100 surfactant | 280 | N/A257 | |
| Glucose oxidase | – CNT in chitosan matrix on glassy carbon | 15 at 4 °C | ∼100%258 |
| – Os(bpy)2Cl2 crosslinked to poly(vinylpyridine) on gold | 21 | N/A259 | |
| – Crosslinked-GOx to CNTs on carbon felt electrode | 0.67 | ∼100%260 | |
| – Ferrocene modified graphite electrodes | 90 | 96%261 | |
| – 7 μm diameter, 2 cm long carbon fiber with PAA-PVI-Os(bpy)2Cl on carbon electrode | 2 | 60%262 | |
| Laccase | – CNT in chitosan composite on glassy carbon | 15 | 99%263 |
| – 3-Aminopropyltriethoxysilane-modified Pt electrode | 60 | 80%264 | |
| – AuNPs on poly(amidoamine) linked to a conductive polymer (3′,4′-diamine-2,2′;5′2′′-terthiophene), with poly(amidoamine) on glass carbon electrodes | 60 | 92%265 | |
| – Anthracene-2-diazonium to pyrolytic graphite ‘edge’ electrode | 56 | 57%253 | |
| Bilirubin oxidase (BOD) | – Microcrystalline cellulose–MWCNT modified electrode on glassy carbon | 40 | 50%266 |
| – 15 nm diameter Au-NP modified Au electrode | 2 | 90%267 | |
| – 7 μm diameter, 2 cm long carbon fiber with PAA-PVI-Os(bpy)2Cl on carbon electrode | 2 | 60%262 | |
| – Polycationic polymer with Os complex, PEGDE crosslinked | 1 | 78%268 | |
| D-Fructose dehydrogenase (FDH) | – Microcrystalline cellulose–MWCNT modified electrode on glassy carbon | 40 | 93%266 |
| – 15 nm diameter Au-NP modified Au electrode | 2 | 76%267 | |
| – Carbon paste electrode with Os(bpy)2Cl2, 0.1% Triton X-100 | 7 | 100%269 | |
| Horseradish Peroxidase (HRP) | – Ferrocene modified graphite electrodes | 90 | 96%261 |
| – Au-Nanoparticles with (3-mercaptopropyl)-trimethoxysilane sol–gel on gold electrode | 120 | ∼100%270 | |
| – Synthetic lipid films on glassy carbon electrode | 7 | ∼100%271 | |
| – Layer-by-layer HRP/polyelectrolytes on SAM modified gold electrode | 28 | 65%272 | |
| – Coadsorption with thionine on TiO2 nanotube on titanium foil | 14 | 86%273 | |
| – Au-NPs in chitosan hydrogel | 28 | 85%274 | |
| – MWCNT with methylene blue mediator on glassy carbon | 14 | 80%275 | |
| Cytochrome c | – Graphene nano-flakes in chitosan on glassy carbon electrode | 14 | 97.5%6 |
| – Au-NPs with MWCNTs in room temperature ionic liquid on glassy carbon | 14 | N/A276 | |
| – MWCNT on glassy carbon | 21 | N/A91 | |
| – Porous Au-NP film on ITO | 30 | ∼100%277 | |
| Glucose dehydrogenase | – CNT–chitosan films on glassy carbon | 8 | 20%278 |
| (PQQ–GDH) | – CNT with immobilized ferricyanide mediator on carbon paste electrode | 28 | 87%279 |
| (FAD–GDH) | – Ferricyanide mediator on glassy carbon electrode | 30 | 100%280 |
| Alcohol (ADH) and laccase | – (ADH and LDH) AuNPs on sol–gel network | 8 | 89%281 |
| (LDH) dehydrogenase | – (ADH) Nafion on carbon felt electrode | 5 | 100%282 |
| Hydrogenase | – Tethered nanoconstruct of PSI and H2ase using an alkane dithiol molecular wire to Fe clustersb | 64 | ∼100%33 |
| – 4-Aminophenyl-functionalized carbon electrode | 7 | 90%254 | |
Despite the success of some recent biomimetic and bioinspired interfaces in maintaining long-term protein stability, many devices currently in the literature are not as successful. Kang et al. (2004)283 designed a biofuel cell using a wired blue copper-containing oxidase, bilirubin oxidase (BOD), bioelectrocatalyst to facilitate the reduction of oxygen to water. This system only lasted for a week at ambient conditions, with most of its electrocatalytic activity lost in only a few hours in serum due to the addition of urate, which destabilizes the wired BOD cathode. Similarly, in a study by Mano et al. (2002),284 a biofuel cell consisting of a GOx anode film and a BOD cathode film complexed with an osmium redox polymer remained stable over one week, losing 6% of its power output each day. Although there has been major progress by some groups regarding long-term stability of bioelectrochemical devices, there are still many modifications that need to be made before these devices can be regularly integrated on an industrial scale.
4.4 Necessity of proper orientation in at interfaces
Protein orientation can be crucial to the performance of electron transport proteins assembled at an electrode interface. In protein–protein interactions in nature, complementary electrostatic fields in the active sites of two redox proteins can orient them appropriately to minimize distance between proteins.292 This effect can be translated to bioelectrochemical devices, where electrostatic forces between oppositely charged electrode surfaces and enzyme active sites can immobilize proteins in specific orientations for maximum electron transfer.292 Many experimental examples show the necessity of protein orientation with respect to the interface.130,132,293 For example, in SAMs, the protein's redox center must be oriented specifically with respect to the interface to allow for any observable redox activity. Studies using both LHI–RC complexes and PSI proteins show that maximum electron transport only occurs when the proteins are in a certain orientation.130,133,293 There are several methods employed to control the orientation of proteins within engineered devices; including His-tagging the redox protein133 and co-immobilization of cofactors and proteins.294 Protein orientation is especially important in DET systems to minimize distance between a site and the electrode.Another example of protein orientation that affects the electron transport properties of an interface comes from the wiring of two redox enzymes to produce directional electron flow to an electrode surface. These systems take advantage of the complementary oxidation and reduction properties of two enzymes, as well as the electron transfer capabilities of a mediator polymer that wires the two active sites together. In a recent example, Efrati et al. (2016)294 set out to mimic the communication between PSI and PSII during electron transfer by electrically wiring PSI and GOx using an osmium redox polymer (Fig. 24).294 The cross-linked system was connected to an electrode surface by linking PSI to a monolayer of pyrroloquinoline quinone (PQQ) on an indium tin oxide (ITO) glass electrode. In the electron transfer pathway, photoexcited PSI transfers electrons to PQQ, which are then transferred to the glass electrode. Upon the introduction of glucose, glucose is oxidized to produce a reduced FADH2 cofactor in GOx, which is then oxidized by the osmium polymer to regenerate the biocatalyst, GOx. Finally, osmium mediated electron transfer back to the oxidized PSI regenerates PSI to repeat the process. This process generates increased photocurrent as glucose concentration increases, resulting in a light-to-electrical power conversion efficiency of 0.12%. This system is extremely interdependent, requiring all components to effectively produce photocurrent. It serves as a prime example of a fully integrated bioelectrochemical device that operates by the electronic wiring of two redox enzymes in their proper orientations by a redox polymer mediator.
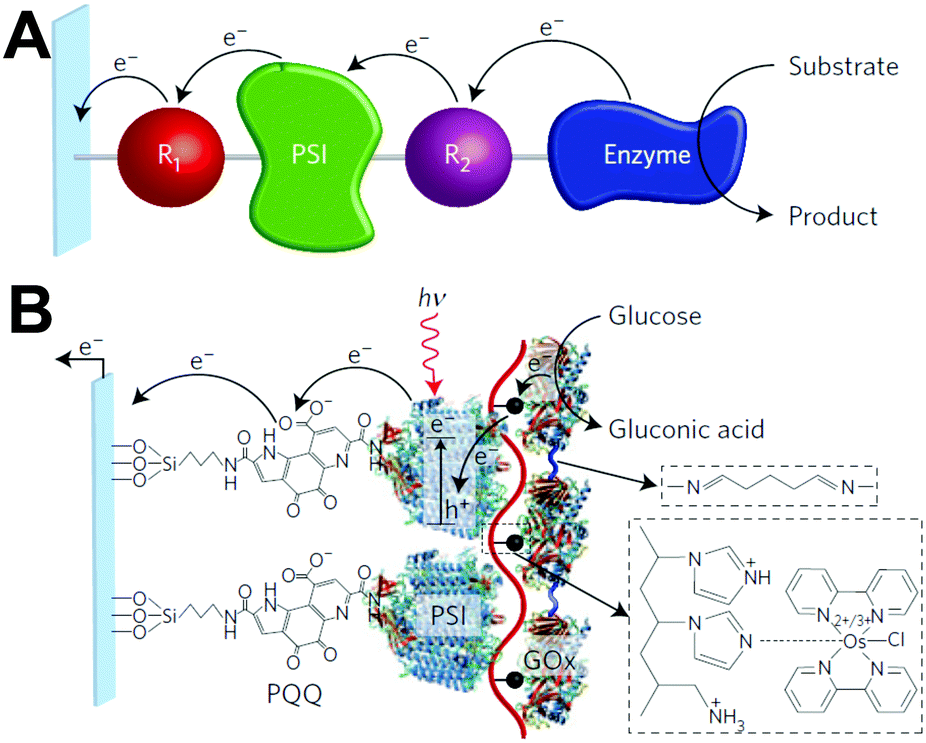 | ||
| Fig. 24 (A) Scheme from Efrati et al. (2016) of electron transfer through co-immobilized enzymes. A photoexcited PSI protein transfers electrons through R1 units to the electrode. The hole in PSI is filled when it oxidizes the active site of the enzyme through mediator groups R2, resulting in the oxidation of the substrate and generation of photocurrent. (B) PSI and glucose are wired by an osmium redox mediator. PSI is linked to the electrode surface by pyrroloquinoline quinone (PQQ) attached to the electrode surface. Copyright (2016) Nature Publishing Group. | ||
5.0 Conclusions and outlook
The biomimetic and bioinspired focus of this review reveals several overall directions and fundamental challenges in interfacing redox enzymes to electrodes. This unique focus also provides insight into opportunities in this rapidly expanding area of research and the practical challenges to the scale up of enzyme-based bioelectronics. These directions, opportunities and challenges are summarized in the following paragraphs.Only a handful of enzymes have been studied extensively in conjunction with an electrode with a major focus on stable enzymes such as photosystem I,33,122,236,256,257,294 glucose oxidase,156,171,176,196,247,258,259,261,295 and laccases.14,36,253,263–265 While the body of literature is large in this area, the lack of variety and diversity underscores the difficulty of wiring proteins, many of which are easily denatured,296 to electrodes. We propose that the recent approach of using the biomimetic perspective of providing a hydrated conductive environment to maximize the stability and function of these enzymes is an excellent general direction that may allow the successful wiring of most redox proteins to electrodes. With more attention to creating interfaces and protein environments that mimic their native environments, the longevity and range of proteins that are active on electrodes can be improved.
The study of longevity of protein electrode devices has been limited but shows promise with bioinspired approaches. Table 4 summarizes longevity of bioelectronics devices reported in literature. Even though surprising longevity of up to a year has been reported in some cases,256 for actual applications, challenges with stability still remain for most enzymes.262,267,268,283,284 In addition to providing a more cell like environment to enhance longevity,134,150 an interesting bioinspired idea in this regard could be the renewability of enzymes such as that seen in biological systems through regeneration or replacement of enzymes. Inactive enzymes could be washed away from the device and replaced by newly purified proteins. We propose that detergents be used to solubilized membrane proteins from the interface as an initial step in recycling the interface. Soluble proteins could be engineered to have a membrane bound domain for insertion into a membrane modified electrode and for detergent extraction.
Membrane protein based bioelectronic devices (beyond PSI) are limited and may benefit from utilizing a membrane environment on electrodes for stabilization. Membrane proteins form a large class of proteins that conduct electron transfer across cell membranes, especially in photosynthetic and electrogenic organisms.23,241–243 They are challenging to stabilize due to the need for a hydrophobic environment around the hydrophobic band around the proteins.227 The use of membrane forming lipids and block copolymers235 could provide an excellent alternative to detergents currently used, as illustrated in a few studies with tethered lipid bilayers.195,228 Additional studies using such bilayer-based systems with a range of membrane proteins could enhance the range of enzyme-based bioelectronics.
Direct electron transfer to multiple layers of proteins on an electrode is challenging but an excellent direction for application in sensors and in some energy-relevant applications. Most devices with high density of proteins on electrodes rely on multiple layers of protein on the electrode184,297 and mediated electron transfer to provide an electrical connection to the electrode. Diffusional limitation and low protein utilization are challenges that have been identified for this approach of random packing and diffusion mediated electron transfer.70,198,294 Novel approaches to engineering thin conductivity polymer electrode interfaces298 and electrodes could increase the efficiency and protein utilization of devices. Directly wiring proteins to electrodes (using short conductive tethers) and the use of membrane spanning conjugated oligoelectrolytes are possible initial approaches.
Actual large-scale applications of bioelectrochemical systems based on enzymes are limited and several challenges need to be overcome to make this possible. The principal obstacles are cost and device lifetime. The cost of protein production has reduced rapidly in recent years, however such scale-up is still lagging for membrane proteins.299,300 The need for aqueous environments while assembling electrodes with proteins is a particular challenge for scale up as well as most industrial applications cast films and materials using organic solvents.301 In some applications, the solvent used to create a desired interface, such as an organic solvent, may not be as compatible with the proteins that are meant to be incorporated.184 Materials for electrode interfaces that provide a stabilizing conductive environment to redox proteins at low cost also need to be further developed and both block copolymers with intercalated COEs236 and conductive polymers302–304 seem promising in this regard. Further, a process scheme for manufacturing of enzyme enhanced electrodes needs to be developed with particular competitive applications such as biosensing, in situ diagnostics, effective energy or chemical production in mind.
The field of enzyme-based bioelectronics is poised to have an excellent future in the upcoming decades due to the ever-tighter integration of biological and electrical systems in recent innovations such as microbial fuel cells305–308 and environmental and biomedical biosensing.52,162,171,250,259,269,271,275,279,281,309 Materials and process innovation inspired by biological systems could provide several ideas for improving the efficiency and scalability of bioelectronic devices and expand their larger scale applications to a wide range of areas relevant to improving human life and the environment.
Acknowledgements
The review has been funded at Penn State in part by the United States Environmental Protection Agency (EPA) under the Science to Achieve Results (STAR) Graduate Fellowship Program. Emelia Conte was funded at Penn State through the Summer Research Fellowship in Biomolecular Engineering. We acknowledge Dr Bryan Ferlez for his critical reading of the manuscript and advice on electron transfer within photosystem I.References
- B. E. Logan, Nat. Rev. Microbiol., 2009, 7, 375–381 CrossRef CAS PubMed.
- J. Du, C. Catania and G. C. Bazan, Chem. Mater., 2013, 26, 686–697 CrossRef.
- A. J. McCormick, P. Bombelli, R. W. Bradley, R. Thorne, T. Wenzel and C. J. Howe, Energy Environ. Sci., 2015, 8, 1092–1109 CAS.
- I. Willner and E. Katz, Angew. Chem., Int. Ed., 2000, 39, 1180–1218 CrossRef.
- E.-H. Yoo and S.-Y. Lee, Sensors, 2010, 10, 4558–4576 CrossRef PubMed.
- J.-F. Wu, M.-Q. Xu and G.-C. Zhao, Electrochem. Commun., 2010, 12, 175–177 CrossRef CAS.
- A. Badura, D. Guschin, B. Esper, T. Kothe, S. Neugebauer, W. Schuhmann and M. Rögner, Electroanalysis, 2008, 20, 1043–1047 CrossRef CAS.
- M. Alkasrawi, I. Popescu, B. Mattiasson, E. Csöregi and V. Laurinavicius, Anal. Commun., 1999, 36, 395–398 RSC.
- S. Cosnier, Biosens. Bioelectron., 1999, 14, 443–456 CrossRef CAS PubMed.
- T. Kothe, S. Pöller, F. Zhao, P. Fortgang, M. Rögner, W. Schuhmann and N. Plumeré, Chem. – Eur. J., 2014, 20, 11029–11034 CrossRef CAS PubMed.
- J. Baur, A. Le Goff, S. Dementin, M. Holzinger, M. Rousset and S. Cosnier, Int. J. Hydrogen Energy, 2011, 36, 12096–12101 CrossRef CAS.
- Y. Liu, X. Dong and P. Chen, Chem. Soc. Rev., 2012, 41, 2283–2307 RSC.
- A. Walcarius, S. D. Minteer, J. Wang, Y. Lin and A. Merkoçi, J. Mater. Chem. B, 2013, 1, 4878–4908 RSC.
- J. W. Gallaway and S. A. Calabrese Barton, J. Am. Chem. Soc., 2008, 130, 8527–8536 CrossRef CAS PubMed.
- B. Unnikrishnan, S. Palanisamy and S.-M. Chen, Biosens. Bioelectron., 2013, 39, 70–75 CrossRef CAS PubMed.
- A. J. Scardino and R. de Nys, Biofouling, 2011, 27, 73–86 CrossRef CAS PubMed.
- J. Wang, Electroanalysis, 2005, 17, 7–14 CrossRef CAS.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- F. Wang, X. Liu and I. Willner, Adv. Mater., 2013, 25, 349–377 CrossRef CAS PubMed.
- A. Badura, T. Kothe, W. Schuhmann and M. Rögner, Energy Environ. Sci., 2011, 4, 3263–3274 CAS.
- C. Léger, S. J. Elliott, K. R. Hoke, L. J. Jeuken, A. K. Jones and F. A. Armstrong, Biochemistry, 2003, 42, 8653–8662 CrossRef PubMed.
- C. C. Moser, T. A. Farid, S. E. Chobot and P. L. Dutton, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 1096–1109 CrossRef CAS PubMed.
- P. Joliot and A. Joliot, Biochim. Biophys. Acta, Bioenerg., 1984, 765, 210–218 CrossRef CAS.
- M. Y. El-Naggar, G. Wanger, K. M. Leung, T. D. Yuzvinsky, G. Southam, J. Yang, W. M. Lau, K. H. Nealson and Y. A. Gorby, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18127–18131 CrossRef CAS PubMed.
- O. Morozova, G. Shumakovich, S. Shleev and Y. I. Yaropolov, Appl. Biochem. Microbiol., 2007, 43, 523–535 CrossRef CAS.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
- C. C. Page, C. C. Moser and P. L. Dutton, Curr. Opin. Chem. Biol., 2003, 7, 551–556 CrossRef CAS PubMed.
- M. Sjödin, S. Styring, H. Wolpher, Y. Xu, L. Sun and L. Hammarström, J. Am. Chem. Soc., 2005, 127, 3855–3863 CrossRef PubMed.
- C. Shih, A. K. Museth, M. Abrahamsson, A. M. Blanco-Rodriguez, A. J. Di Bilio, J. Sudhamsu, B. R. Crane, K. L. Ronayne, M. Towrie and A. Vlček, Science, 2008, 320, 1760–1762 CrossRef CAS PubMed.
- S. Prabhulkar, H. Tian, X. Wang, J.-J. Zhu and C.-Z. Li, Antioxid. Redox Signaling, 2012, 17, 1796–1822 CrossRef CAS PubMed.
- Y. Zhang and R. Holm, Inorg. Chem., 2004, 43, 674–682 CrossRef CAS PubMed.
- J. M. Kuchenreuther, C. S. Grady-Smith, A. S. Bingham, S. J. George, S. P. Cramer and J. R. Swartz, PLoS One, 2010, 5, e15491 Search PubMed.
- C. E. Lubner, P. Knörzer, P. J. Silva, K. A. Vincent, T. Happe, D. A. Bryant and J. H. Golbeck, Biochemistry, 2010, 49, 10264–10266 CrossRef CAS PubMed.
- I. Ivanov, T. Vidaković-Koch and K. Sundmacher, Energies, 2010, 3, 803–846 CrossRef CAS.
- J. Kim, H. Jia and P. Wang, Biotechnol. Adv., 2006, 24, 296–308 CrossRef CAS PubMed.
- A. Zebda, C. Gondran, A. Le Goff, M. Holzinger, P. Cinquin and S. Cosnier, Nat. Commun., 2011, 2, 370 CrossRef PubMed.
- K. MacVittie, J. Halámek, L. Halámková, M. Southcott, W. D. Jemison, R. Lobel and E. Katz, Energy Environ. Sci., 2013, 6, 81–86 CAS.
- H. Sakai, T. Nakagawa, Y. Tokita, T. Hatazawa, T. Ikeda, S. Tsujimura and K. Kano, Energy Environ. Sci., 2009, 2, 133–138 CAS.
- M. Falk, Z. Blum and S. Shleev, Electrochim. Acta, 2012, 82, 191–202 CrossRef CAS.
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, John Wiley & Sons, 2013 Search PubMed.
- P. Loach and G. Fasman, Handbook of Biochemistry and Molecular Biology, 1976, vol. I, pp. 122–130 Search PubMed.
- S. Vogt, M. Schneider, H. Schäfer-Eberwein and G. Nöll, Anal. Chem., 2014, 86, 7530–7535 CrossRef CAS PubMed.
- M. Jormakka, S. Törnroth, B. Byrne and S. Iwata, Science, 2002, 295, 1863–1868 CrossRef PubMed.
- C. Fichtner, C. Laurich, E. Bothe and W. Lubitz, Biochemistry, 2006, 45, 9706–9716 CrossRef CAS PubMed.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed.
- S. Calabrese Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867–4886 CrossRef.
- Y. Wu and S. Hu, Microchim. Acta, 2007, 159, 1–17 CrossRef CAS.
- S. Chen, R. Yuan, Y. Chai, L. Zhang, N. Wang and X. Li, Biosens. Bioelectron., 2007, 22, 1268–1274 CrossRef CAS PubMed.
- P. Cinquin, C. Gondran, F. Giroud, S. Mazabrard, A. Pellissier, F. Boucher, J.-P. Alcaraz, K. Gorgy, F. Lenouvel and S. Mathé, PLoS One, 2010, 5, e10476 Search PubMed.
- J. Zhu, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS PubMed.
- J.-M. Pernaut and J. R. Reynolds, J. Phys. Chem. B, 2000, 104, 4080–4090 CrossRef CAS.
- C. Shan, H. Yang, J. Song, D. Han, A. Ivaska and L. Niu, Anal. Chem., 2009, 81, 2378–2382 CrossRef CAS PubMed.
- M. Rasmussen and S. D. Minteer, Anal. Methods, 2013, 5, 1140–1144 RSC.
- M. Auriol, Y. Filali-Meknassi, R. D. Tyagi and C. D. Adams, Water Res., 2007, 41, 3281–3288 CrossRef CAS PubMed.
- H. Garcia-Arellano, M. Alcalde and A. Ballesteros, Microb. Cell Fact., 2004, 3, 1 CrossRef PubMed.
- M. Husain and Q. Husain, Crit. Rev. Environ. Sci. Technol., 2007, 38, 1–42 CrossRef.
- M. Trovaslet, E. Enaud, Y. Guiavarc'h, A.-M. Corbisier and S. Vanhulle, Enzyme Microb. Technol., 2007, 41, 368–376 CrossRef CAS.
- J. Bollag, K. Shuttleworth and D. Anderson, Appl. Environ. Microbiol., 1988, 54, 3086–3091 CAS.
- C. Michel, M. Brugna, C. Aubert, A. Bernadac and M. Bruschi, Appl. Microbiol. Biotechnol., 2001, 55, 95–100 CrossRef CAS PubMed.
- L. Gianfreda and M. A. Rao, Enzyme Microb. Technol., 2004, 35, 339–354 CrossRef CAS.
- X. Dominguez-Benetton, S. Srikanth, Y. Satyawali, K. Vanbroekhoven and D. Pant, J. Microb. Biochem. Technol., 2013, 2013, 1–20 Search PubMed.
- C. Kohlmann, W. Märkle and S. Lütz, J. Mol. Catal. B: Enzym., 2008, 51, 57–72 CrossRef CAS.
- P. K. Addo, R. L. Arechederra, A. Waheed, J. D. Shoemaker, W. S. Sly and S. D. Minteer, Electrochem. Solid-State Lett., 2011, 14, E9–E13 CrossRef CAS.
- S. Srikanth, M. Maesen, X. Dominguez-Benetton, K. Vanbroekhoven and D. Pant, Bioresour. Technol., 2014, 165, 350–354 CrossRef CAS PubMed.
- A. L. Ghindilis, P. Atanasov and E. Wilkins, Electroanalysis, 1997, 9, 661–674 CrossRef CAS.
- A. Chaubey and B. Malhotra, Biosens. Bioelectron., 2002, 17, 441–456 CrossRef CAS PubMed.
- K. Habermüller, M. Mosbach and W. Schuhmann, Fresenius' J. Anal. Chem., 2000, 366, 560–568 CrossRef.
- S. Shleev, J. Tkac, A. Christenson, T. Ruzgas, A. I. Yaropolov, J. W. Whittaker and L. Gorton, Biosens. Bioelectron., 2005, 20, 2517–2554 CrossRef CAS PubMed.
- A. Guiseppi-Elie, C. Lei and R. H. Baughman, Nanotechnology, 2002, 13, 559 CrossRef CAS.
- F. Barrière, P. Kavanagh and D. Leech, Electrochim. Acta, 2006, 51, 5187–5192 CrossRef.
- H. Kang, R. Liu, H. Sun, J. Zhen, Q. Li and Y. Huang, J. Phys. Chem. B, 2011, 116, 55–62 CrossRef PubMed.
- R. D. Milton, S. Abdellaoui, N. Khadka, D. R. Dean, D. Leech, L. C. Seefeldt and S. D. Minteer, Energy Environ. Sci., 2016, 9, 2550–2554 CAS.
- T. Ohara, Platinum Met. Rev., 1995, 39, 54–62 CAS.
- S. C. Barton, H.-H. Kim, G. Binyamin, Y. Zhang and A. Heller, J. Am. Chem. Soc., 2001, 123, 5802–5803 CrossRef CAS.
- L. P. Christopher, B. Yao and Y. Ji, Front. Environ. Res., 2014, 2, 12 Search PubMed.
- B. Branchi, C. Galli and P. Gentili, Org. Biomol. Chem., 2005, 3, 2604–2614 CAS.
- A. I. Cañas and S. Camarero, Biotechnol. Adv., 2010, 28, 694–705 CrossRef PubMed.
- A. E. Cass, G. Davis, G. D. Francis, H. A. O. Hill, W. J. Aston, I. J. Higgins, E. V. Plotkin, L. D. Scott and A. P. Turner, Anal. Chem., 1984, 56, 667–671 CrossRef CAS PubMed.
- B. Brunetti, P. Ugo, L. M. Moretto and C. R. Martin, J. Electroanal. Chem., 2000, 491, 166–174 CrossRef CAS.
- I. J. Higgins, H. A. Hill and E. V. Plotkin, 1987, 1–18.
- J. Hodak, R. Etchenique, E. J. Calvo, K. Singhal and P. N. Bartlett, Langmuir, 1997, 13, 2708–2716 CrossRef CAS.
- M. Kesik, H. Akbulut, S. Söylemez, Ş. C. Cevher, G. Hızalan, Y. A. Udum, T. Endo, S. Yamada, A. Çırpan and Y. Yağcı, Polym. Chem., 2014, 5, 6295–6306 RSC.
- W. Schuhmann, T. J. Ohara, H. L. Schmidt and A. Heller, J. Am. Chem. Soc., 1991, 113, 1394–1397 CrossRef CAS.
- F. Barrière, Y. Ferry, D. Rochefort and D. Leech, Electrochem. Commun., 2004, 6, 237–241 CrossRef.
- A. Heller, Curr. Opin. Chem. Biol., 2006, 10, 664–672 CrossRef CAS PubMed.
- M. Holzinger, A. Le Goff and S. Cosnier, Electrochim. Acta, 2012, 82, 179–190 CrossRef CAS.
- F.-F. Zhang, Q. Wan, X.-L. Wang, Z.-D. Sun, Z.-Q. Zhu, Y.-Z. Xian, L.-T. Jin and K. Yamamoto, J. Electroanal. Chem., 2004, 571, 133–138 CrossRef CAS.
- T. Tamaki, T. Ito and T. Yamaguchi, Fuel Cells, 2009, 9, 37–43 CrossRef CAS.
- P. Asuri, S. S. Bale, R. C. Pangule, D. A. Shah, R. S. Kane and J. S. Dordick, Langmuir, 2007, 23, 12318–12321 CrossRef CAS PubMed.
- J. J. Gooding, R. Wibowo, J. Liu, W. Yang, D. Losic, S. Orbons, F. J. Mearns, J. G. Shapter and D. B. Hibbert, J. Am. Chem. Soc., 2003, 125, 9006–9007 CrossRef CAS PubMed.
- G.-C. Zhao, Z.-Z. Yin, L. Zhang and X.-W. Wei, Electrochem. Commun., 2005, 7, 256–260 CrossRef CAS.
- N. S. Malvankar, M. Vargas, K. P. Nevin, A. E. Franks, C. Leang, B.-C. Kim, K. Inoue, T. Mester, S. F. Covalla and J. P. Johnson, Nat. Nanotechnol., 2011, 6, 573–579 CrossRef PubMed.
- H. Yan, C. Chuang, A. Zhugayevych, S. Tretiak, F. W. Dahlquist and G. C. Bazan, Adv. Mater., 2015, 27, 1908–1911 CrossRef CAS PubMed.
- S. Pirbadian, S. E. Barchinger, K. M. Leung, H. S. Byun, Y. Jangir, R. A. Bouhenni, S. B. Reed, M. F. Romine, D. A. Saffarini and L. Shi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12883–12888 CrossRef CAS PubMed.
- G. Reguera, K. D. McCarthy, T. Mehta, J. S. Nicoll, M. T. Tuominen and D. R. Lovley, Nature, 2005, 435, 1098–1101 CrossRef CAS PubMed.
- K. Xiao, N. S. Malvankar, C. Shu, E. Martz, D. R. Lovley and X. Sun, Sci. Rep., 2016, 6, 1–9 CrossRef.
- D. E. Holmes, Y. Dang, D. J. Walker and D. R. Lovley, Microb. Genomics, 2016, 1–20 Search PubMed.
- Y. Tan, R. Y. Adhikari, N. S. Malvankar, S. Pi, J. E. Ward, T. L. Woodard, K. P. Nevin, Q. Xia, M. T. Tuominen and D. R. Lovley, Small, 2016, 12, 4481–4485 CrossRef CAS PubMed.
- C. R. Hess, G. A. Juda, D. M. Dooley, R. N. Amii, M. G. Hill, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2003, 125, 7156–7157 CrossRef CAS PubMed.
- R. Ludwig, W. Harreither, F. Tasca and L. Gorton, ChemPhysChem, 2010, 11, 2674–2697 CrossRef CAS PubMed.
- M. T. Meredith and S. D. Minteer, Annu. Rev. Anal. Chem., 2012, 5, 157–179 CrossRef CAS PubMed.
- L. Stoica, T. Ruzgas, R. Ludwig, D. Haltrich and L. Gorton, Langmuir, 2006, 22, 10801–10806 CrossRef CAS PubMed.
- G. Kovacs, R. Ortiz, V. Coman, W. Harreither, I. C. Popescu, R. Ludwig and L. Gorton, Bioelectrochemistry, 2012, 88, 84–91 CrossRef CAS PubMed.
- F. Tasca, L. Gorton, W. Harreither, D. Haltrich, R. Ludwig and G. Nöll, Anal. Chem., 2009, 81, 2791–2798 CrossRef CAS PubMed.
- H. Von Canstein, J. Ogawa, S. Shimizu and J. R. Lloyd, Appl. Environ. Microbiol., 2008, 74, 615–623 CrossRef CAS PubMed.
- P. Bartlett and K. Pratt, J. Electroanal. Chem., 1995, 397, 61–78 CrossRef.
- V. Flexer, E. Calvo and P. Bartlett, J. Electroanal. Chem., 2010, 646, 24–32 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- C. C. Moser, J. M. Keske, K. Warncke, R. S. Farid and P. L. Dutton, Nature, 1992, 355, 796–802 CrossRef CAS PubMed.
- J. Golbeck and A. van der Est, The Biophysics of Photosynthesis, Springer, New York, 2014 Search PubMed.
- G. Grampp, Angew. Chem., Int. Ed. Engl., 1993, 32, 691–693 CrossRef.
- P. F. Barbara, T. J. Meyer and M. A. Ratner, J. Phys. Chem., 1996, 100, 13148–13168 CrossRef CAS.
- D. Leech, P. Kavanagh and W. Schuhmann, Electrochim. Acta, 2012, 84, 223–234 CrossRef CAS.
- A. G. Mauk, Essays Biochem., 1998, 34, 101–124 CrossRef.
- G. Jensen, A. Warshel and P. Stephens, Biochemistry, 1994, 33, 10911–10924 CrossRef CAS PubMed.
- A. Sancar, Chem. Rev., 2003, 103, 2203–2238 CrossRef CAS PubMed.
- I. A. Solov'yov, T. Domratcheva and K. Schulten, Sci. Rep., 2014, 4, 3845 Search PubMed.
- I. Willner and E. Katz, Bioelectronics: From Theory to Applications, John Wiley & Sons, 2006 Search PubMed.
- Y. Tokita, S. Yamada, W. Luo, Y. Goto, N. Bouley-Ford, H. Nakajima and Y. Watanabe, Angew. Chem., Int. Ed., 2011, 50, 11663–11666 CrossRef CAS PubMed.
- M. Curri, A. Agostiano, G. Leo, A. Mallardi, P. Cosma and M. Della Monica, Mater. Sci. Eng., C, 2002, 22, 449–452 CrossRef.
- W. Vastarella and R. Nicastri, Talanta, 2005, 66, 627–633 CrossRef CAS PubMed.
- A. Mershin, K. Matsumoto, L. Kaiser, D. Yu, M. Vaughn, M. K. Nazeeruddin, B. D. Bruce, M. Graetzel and S. Zhang, Sci. Rep., 2012, 2, 1–7 Search PubMed.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS PubMed.
- P. V. Kamat, Chem. Rev., 1993, 93, 267–300 CrossRef CAS.
- V. P. Shinkarev, B. Zybailov, I. R. Vassiliev and J. H. Golbeck, Biophys. J., 2002, 83, 2885–2897 CrossRef CAS PubMed.
- M. Hippler, F. Drepper, W. Haehnel and J.-D. Rochaix, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7339–7344 CrossRef CAS.
- P. Fromme, Photosynthetic Protein Complexes: A Structural Approach, Wiley, 2008 Search PubMed.
- K. Brettel and W. Leibl, Biochim. Biophys. Acta, Bioenerg., 2001, 1507, 100–114 CrossRef CAS.
- P. Jordan, P. Fromme, H. T. Witt, O. Klukas, W. Saenger and N. Krauß, Nature, 2001, 411, 909–917 CrossRef CAS PubMed.
- M. Ciobanu, H. A. Kincaid, G. K. Jennings and D. E. Cliffel, Langmuir, 2005, 21, 692–698 CrossRef CAS PubMed.
- B. S. Ko, B. Babcock, G. K. Jennings, S. G. Tilden, R. R. Peterson, D. Cliffel and E. Greenbaum, Langmuir, 2004, 20, 4033–4038 CrossRef CAS PubMed.
- M. Kondo, Y. Nakamura, K. Fujii, M. Nagata, Y. Suemori, T. Dewa, K. Iida, A. T. Gardiner, R. J. Cogdell and M. Nango, Biomacromolecules, 2007, 8, 2457–2463 CrossRef CAS PubMed.
- M. Kondo, K. Iida, T. Dewa, H. Tanaka, T. Ogawa, S. Nagashima, K. V. Nagashima, K. Shimada, H. Hashimoto and A. T. Gardiner, Biomacromolecules, 2012, 13, 432–438 CrossRef CAS PubMed.
- H. A. Kincaid, T. Niedringhaus, M. Ciobanu, D. E. Cliffel and G. K. Jennings, Langmuir, 2006, 22, 8114–8120 CrossRef CAS PubMed.
- S. A. Trammell, A. Spano, R. Price and N. Lebedev, Biosens. Bioelectron., 2006, 21, 1023–1028 CrossRef CAS PubMed.
- A. Aoki and A. Heller, J. Phys. Chem., 1993, 97, 11014–11019 CrossRef CAS.
- D. H. Kim, M. Abidian and D. C. Martin, J. Biomed. Mater. Res., Part A, 2004, 71, 577–585 CrossRef PubMed.
- R. A. Green, N. H. Lovell, G. G. Wallace and L. A. Poole-Warren, Biomaterials, 2008, 29, 3393–3399 CrossRef CAS PubMed.
- M. Gerard, A. Chaubey and B. Malhotra, Biosens. Bioelectron., 2002, 17, 345–359 CrossRef CAS PubMed.
- Y. Lee, I. Yang, J. E. Lee, S. Hwang, J. W. Lee, S.-S. Um, T. L. Nguyen, P. J. Yoo, H. Y. Woo and J. Park, J. Phys. Chem. C, 2013, 117, 3298–3307 CAS.
- R. Green and M. R. Abidian, Adv. Mater., 2015, 27, 7620–7637 CrossRef CAS PubMed.
- G. Kaur, R. Adhikari, P. Cass, M. Bown and P. Gunatillake, RSC Adv., 2015, 5, 37553–37567 RSC.
- R. A. Green, R. T. Hassarati, J. A. Goding, S. Baek, N. H. Lovell, P. J. Martens and L. A. Poole-Warren, Macromol. Biosci., 2012, 12, 494–501 CrossRef CAS PubMed.
- H. Zhao, W. E. Price and G. G. Wallace, J. Membr. Sci., 1994, 87, 47–56 CrossRef CAS.
- P. Humpolicek, V. Kasparkova, P. Saha and J. Stejskal, Synth. Met., 2012, 162, 722–727 CrossRef CAS.
- A. Vaitkuviene, V. Kaseta, J. Voronovic, G. Ramanauskaite, G. Biziuleviciene, A. Ramanaviciene and A. Ramanavicius, J. Hazard. Mater., 2013, 250, 167–174 CrossRef PubMed.
- H. Yamato, M. Ohwa and W. Wernet, J. Electroanal. Chem., 1995, 397, 163–170 CrossRef.
- B. Cornell, V. Braach-Maksvytis, L. King, P. Osman, B. Raguse, L. Wieczorek and R. Pace, Nature, 1997, 580–582 CrossRef CAS PubMed.
- B. Raguse, V. Braach-Maksvytis, B. A. Cornell, L. G. King, P. D. Osman, R. J. Pace and L. Wieczorek, Langmuir, 1998, 14, 648–659 CrossRef CAS.
- M. Tanaka and E. Sackmann, Nature, 2005, 437, 656–663 CrossRef CAS PubMed.
- G. Valincius, G. Niaura, B. Kazakeviciene, Z. Talaikyte, M. Kazemekaite, E. Butkus and V. Razumas, Langmuir, 2004, 20, 6631–6638 CrossRef CAS PubMed.
- S. M. Schiller, R. Naumann, K. Lovejoy, H. Kunz and W. Knoll, Angew. Chem., Int. Ed., 2003, 42, 208–211 CrossRef CAS PubMed.
- R. Naumann, E. Schmidt, A. Jonczyk, K. Fendler, B. Kadenbach, T. Liebermann, A. Offenhäusser and W. Knoll, Biosens. Bioelectron., 1999, 14, 651–662 CrossRef CAS.
- F. Giess, M. G. Friedrich, J. Heberle, R. L. Naumann and W. Knoll, Biophys. J., 2004, 87, 3213–3220 CrossRef CAS PubMed.
- J. A. Jackman, W. Knoll and N.-J. Cho, Materials, 2012, 5, 2637–2657 CrossRef CAS.
- V. B. Wang, J. Du, X. Chen, A. W. Thomas, N. D. Kirchhofer, L. E. Garner, M. T. Maw, W. H. Poh, J. Hinks and S. Wuertz, Phys. Chem. Chem. Phys., 2013, 15, 5867–5872 RSC.
- J. Du, A. W. Thomas, X. Chen, L. E. Garner, C. A. Vandenberg and G. C. Bazan, Chem. Commun., 2013, 49, 9624–9626 RSC.
- L. E. Garner, J. Park, S. M. Dyar, A. Chworos, J. J. Sumner and G. C. Bazan, J. Am. Chem. Soc., 2010, 132, 10042–10052 CrossRef CAS PubMed.
- H. Hou, X. Chen, A. W. Thomas, C. Catania, N. D. Kirchhofer, L. E. Garner, A. Han and G. C. Bazan, Adv. Mater., 2013, 25, 1593–1597 CrossRef CAS PubMed.
- R. A. Green, S. Baek, L. A. Poole-Warren and P. J. Martens, Sci. Technol. Adv. Mater., 2016, 1–13 Search PubMed.
- L. Zhang, Z. Wang, C. Xu, Y. Li, J. Gao, W. Wang and Y. Liu, J. Mater. Chem., 2011, 21, 10399–10406 RSC.
- Y. Xiang, Z. Peng and D. Chen, Eur. Polym. J., 2006, 42, 2125–2132 CrossRef CAS.
- D. Zhai, B. Liu, Y. Shi, L. Pan, Y. Wang, W. Li, R. Zhang and G. Yu, ACS Nano, 2013, 7, 3540–3546 CrossRef CAS PubMed.
- P. Martens and K. Anseth, Polymer, 2000, 41, 7715–7722 CrossRef CAS.
- J. A. Stammen, S. Williams, D. N. Ku and R. E. Guldberg, Biomaterials, 2001, 22, 799–806 CrossRef CAS PubMed.
- Y. Wang, Z. Li, J. Wang, J. Li and Y. Lin, Trends Biotechnol., 2011, 29, 205–212 CrossRef CAS PubMed.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- X. Wang, S. M. Tabakman and H. Dai, J. Am. Chem. Soc., 2008, 130, 8152–8153 CrossRef CAS PubMed.
- S. Niyogi, E. Bekyarova, M. E. Itkis, H. Zhang, K. Shepperd, J. Hicks, M. Sprinkle, C. Berger, C. N. Lau and W. A. Deheer, Nano Lett., 2010, 10, 4061–4066 CrossRef CAS PubMed.
- H. Fan, L. Wang, K. Zhao, N. Li, Z. Shi, Z. Ge and Z. Jin, Biomacromolecules, 2010, 11, 2345–2351 CrossRef CAS PubMed.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- T. Alava, J. A. Mann, C. c. Théodore, J. J. Benitez, W. R. Dichtel, J. M. Parpia and H. G. Craighead, Anal. Chem., 2013, 85, 2754–2759 CrossRef CAS PubMed.
- J. Liu, H. Bai, Y. Wang, Z. Liu, X. Zhang and D. D. Sun, Adv. Funct. Mater., 2010, 20, 4175–4181 CrossRef CAS.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed.
- A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18–52 CrossRef CAS PubMed.
- E. Katz, I. Willner and J. Wang, Electroanalysis, 2004, 16, 19–44 CrossRef CAS.
- A. Böker, J. He, T. Emrick and T. P. Russell, Soft Matter, 2007, 3, 1231–1248 RSC.
- S. Smart, A. Cassady, G. Lu and D. Martin, Carbon, 2006, 44, 1034–1047 CrossRef CAS.
- A. Magrez, S. Kasas, V. Salicio, N. Pasquier, J. W. Seo, M. Celio, S. Catsicas, B. Schwaller and L. Forró, Nano Lett., 2006, 6, 1121–1125 CrossRef CAS PubMed.
- M. Holzinger, A. Le Goff and S. Cosnier, New J. Chem., 2014, 38, 5173–5180 RSC.
- S. Löfås and B. Johnsson, J. Chem. Soc., Chem. Commun., 1990, 1526–1528 RSC.
- J. J. Gooding and D. B. Hibbert, TrAC, Trends Anal. Chem., 1999, 18, 525–533 CrossRef CAS.
- A. Narváez, G. Suárez, I. C. Popescu, I. Katakis and E. Domınguez, Biosens. Bioelectron., 2000, 15, 43–52 CrossRef.
- E. J. Calvo, F. Battaglini, C. Danilowicz, A. Wolosiuk and M. Otero, Faraday Discuss., 2000, 116, 47–65 RSC.
- Y. Wang, P. P. Joshi, K. L. Hobbs, M. B. Johnson and D. W. Schmidtke, Langmuir, 2006, 22, 9776–9783 CrossRef CAS PubMed.
- C.-A. Lee and Y.-C. Tsai, Sens. Actuators, B, 2009, 138, 518–523 CrossRef CAS.
- H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3534–3539 CrossRef CAS PubMed.
- P. B. Crowley and M. A. Carrondo, Proteins: Struct., Funct., Bioinf., 2004, 55, 603–612 CrossRef CAS PubMed.
- P. C. Dos Santos, D. R. Dean, Y. Hu and M. W. Ribbe, Chem. Rev., 2004, 104, 1159–1174 CrossRef CAS PubMed.
- D. Leys, J. Basran, F. Talfournier, M. J. Sutcliffe and N. S. Scrutton, Nat. Struct. Mol. Biol., 2003, 10, 219–225 CAS.
- J. Lin, I. A. Balabin and D. N. Beratan, Science, 2005, 310, 1311–1313 CrossRef CAS PubMed.
- O. Miyashita, M. Y. Okamura and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3558–3563 CrossRef CAS PubMed.
- E. E. Ross, B. Bondurant, T. Spratt, J. C. Conboy, D. F. O'Brien and S. S. Saavedra, Langmuir, 2001, 17, 2305–2307 CrossRef CAS.
- N. Mano, J. E. Yoo, J. Tarver, Y.-L. Loo and A. Heller, J. Am. Chem. Soc., 2007, 129, 7006–7007 CrossRef CAS PubMed.
- N. Vasylieva, B. Barnych, A. Meiller, C. Maucler, L. Pollegioni, J.-S. Lin, D. Barbier and S. Marinesco, Biosens. Bioelectron., 2011, 26, 3993–4000 CrossRef CAS PubMed.
- D. Chakraborty, E. McClellan, R. Hasselbeck and S. C. Barton, J. Electrochem. Soc., 2014, 161, H3076–H3082 CrossRef.
- M. Breuer, K. M. Rosso, J. Blumberger and J. N. Butt, J. R. Soc., Interface, 2015, 12, 20141117 CrossRef PubMed.
- M. Y. a. F. El-Nagger, E. Steven, Live Wires, http://www.the-scientist.com/?articles.view/articleNo/35299/title/Live-Wires/, accessed 7/5, 2016.
- H. H. Weetall, Trends Biotechnol., 1985, 3, 276–280 CrossRef CAS.
- F. Perreault, A. F. de Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC.
- Q. Zhu, Y. Chai, R. Yuan, Y. Zhuo, J. Han, Y. Li and N. Liao, Biosens. Bioelectron., 2013, 43, 440–445 CrossRef CAS PubMed.
- K. S. Novoselov, V. Fal, L. Colombo, P. Gellert, M. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- X. Huang, Z. Zeng, Z. Fan, J. Liu and H. Zhang, Adv. Mater., 2012, 24, 5979–6004 CrossRef CAS PubMed.
- T. Premkumar and K. E. Geckeler, Prog. Polym. Sci., 2012, 37, 515–529 CrossRef CAS.
- S. K. Min, W. Y. Kim, Y. Cho and K. S. Kim, Nat. Nanotechnol., 2011, 6, 162–165 CrossRef CAS PubMed.
- Y. He, R. H. Scheicher, A. Grigoriev, R. Ahuja, S. Long, Z. Huo and M. Liu, Adv. Funct. Mater., 2011, 21, 2674–2679 CrossRef CAS.
- J. Carrero-Sanchez, A. Elias, R. Mancilla, G. Arrellin, H. Terrones, J. Laclette and M. Terrones, Nano Lett., 2006, 6, 1609–1616 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- K. Parvez, S. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. Feng and K. Müllen, ACS Nano, 2012, 6, 9541–9550 CrossRef CAS PubMed.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- Z. Wang, X. Zhou, J. Zhang, F. Boey and H. Zhang, J. Phys. Chem. C, 2009, 113, 14071–14075 CAS.
- A. D. Ostrowski, E. M. Chan, D. J. Gargas, E. M. Katz, G. Han, P. J. Schuck, D. J. Milliron and B. E. Cohen, ACS Nano, 2012, 6, 2686–2692 CrossRef CAS PubMed.
- S. Kalathil and D. Pant, RSC Adv., 2016, 6, 30582–30597 RSC.
- Y. B. S. Côme, H. Lalo, Z. Wang, G. W. Kohring, R. Hempelmann, M. Etienne, A. Walcarius and A. Kuhn, Electroanalysis, 2013, 25, 621–629 CrossRef.
- B. Wang, J. Zhang, G. Cheng and S. Dong, Anal. Chim. Acta, 2000, 407, 111–118 CrossRef CAS.
- C. Renault, C. P. Andrieux, R. T. Tucker, M. J. Brett, V. r. Balland and B. Limoges, J. Am. Chem. Soc., 2012, 134, 6834–6845 CrossRef CAS PubMed.
- A. De Poulpiquet, A. Ciaccafava and E. Lojou, Electrochim. Acta, 2014, 126, 104–114 CrossRef CAS.
- K. Van Nguyen and S. D. Minteer, Chem. Commun., 2015, 51, 13071–13073 RSC.
- Y. H. Kim, E. Campbell, J. Yu, S. D. Minteer and S. Banta, Angew. Chem., Int. Ed., 2013, 52, 1437–1440 CrossRef CAS PubMed.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS PubMed.
- X. Wei, J. Cruz and W. Gorski, Anal. Chem., 2002, 74, 5039–5046 CrossRef CAS PubMed.
- P. Åsberg and O. Inganäs, Biosens. Bioelectron., 2003, 19, 199–207 CrossRef.
- N. Plumeré, O. Rüdiger, A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- G. B. Sigal, C. Bamdad, A. Barberis, J. Strominger and G. M. Whitesides, Anal. Chem., 1996, 68, 490–497 CrossRef CAS PubMed.
- A. M. Seddon, P. Curnow and P. J. Booth, Biochim. Biophys. Acta, Biomembr., 2004, 1666, 105–117 CrossRef CAS PubMed.
- L. J. Jeuken, S. D. Connell, P. J. Henderson, R. B. Gennis, S. D. Evans and R. J. Bushby, J. Am. Chem. Soc., 2006, 128, 1711–1716 CrossRef CAS PubMed.
- S. Paula, A. Volkov, A. Van Hoek, T. Haines and D. W. Deamer, Biophys. J., 1996, 70, 339 CrossRef CAS PubMed.
- S. Tanizaki and M. Feig, J. Chem. Phys., 2005, 122, 124706 CrossRef PubMed.
- T. Gonen, Y. Cheng, P. Sliz, Y. Hiroaki, Y. Fujiyoshi, S. C. Harrison and T. Walz, Nature, 2005, 438, 633–638 CrossRef CAS PubMed.
- S. Scheuring and J. N. Sturgis, Photosynth. Res., 2009, 102, 197–211 CrossRef CAS PubMed.
- Y.-Y. Won, K. Paso, H. T. Davis and F. S. Bates, J. Phys. Chem. B, 2001, 105, 8302–8311 CrossRef CAS.
- P. O. Saboe, E. Conte, S. Chan, H. M. Feroz, B. H. Ferlez, M. E. Farell, M. F. Poyton, I. T. Sines, H. Yan and G. C. Bazan, J. Mater. Chem. A, 2016, 4, 15457–15463 CAS.
- S. Jia, J. Fei, J. Deng, Y. Cai and J. Li, Sens. Actuators, B, 2009, 138, 244–250 CrossRef CAS.
- P. O. Saboe, C. E. Lubner, N. S. McCool, N. M. Vargas-Barbosa, H. Yan, S. Chan, B. Ferlez, G. C. Bazan, J. H. Golbeck and M. Kumar, Adv. Mater., 2014, 26, 7064–7069 CrossRef CAS PubMed.
- N. D. Kirchhofer, M. A. Rasmussen, F. W. Dahlquist, S. D. Minteer and G. C. Bazan, Energy Environ. Sci., 2015, 8, 2698–2706 CAS.
- A. W. Thomas, L. E. Garner, K. P. Nevin, T. L. Woodard, A. E. Franks, D. R. Lovley, J. J. Sumner, C. J. Sund and G. C. Bazan, Energy Environ. Sci., 2013, 6, 1761–1765 CAS.
- Y. Q. Gao, W. Yang and M. Karplus, Cell, 2005, 123, 195–205 CrossRef CAS PubMed.
- D. Acuña-Castroviejo, M. Martín, M. Macías, G. Escames, J. León, H. Khaldy and R. J. Reiter, J. Pineal Res., 2001, 30, 65–74 Search PubMed.
- L. Ernster and G. Dallner, Biochim. Biophys. Acta, Mol. Basis Dis., 1995, 1271, 195–204 CrossRef.
- M. Cordes and B. Giese, Chem. Soc. Rev., 2009, 38, 892–901 RSC.
- J. Simon, R. J. van Spanning and D. J. Richardson, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 1480–1490 CrossRef CAS PubMed.
- P. V. Iyer and L. Ananthanarayan, Process Biochem., 2008, 43, 1019–1032 CrossRef CAS.
- A. S. Rathore, P. Latham, H. Levine, J. Curling and O. Kaltenbrunner, BioPharm Int., 2004, 17, 46 Search PubMed.
- A. Heller, J. Phys. Chem., 1992, 96, 3579–3587 CrossRef CAS.
- X. Kang, J. Wang, H. Wu, I. A. Aksay, J. Liu and Y. Lin, Biosens. Bioelectron., 2009, 25, 901–905 CrossRef CAS PubMed.
- E. M. Ekanayake, D. M. Preethichandra and K. Kaneto, Biosens. Bioelectron., 2007, 23, 107–113 CrossRef CAS PubMed.
- J. Zhang, F. Zhang, H. Yang, X. Huang, H. Liu, J. Zhang and S. Guo, Langmuir, 2010, 26, 6083–6085 CrossRef CAS PubMed.
- S. Brahim, D. Narinesingh and A. Guiseppi-Elie, Biosens. Bioelectron., 2002, 17, 53–59 CrossRef CAS PubMed.
- J. F. Allen and J. Forsberg, Trends Plant Sci., 2001, 6, 317–326 CrossRef CAS PubMed.
- H. Ruan, X. D. Tang, M.-L. Chen, M. Joiner, G. Sun, N. Brot, H. Weissbach, S. H. Heinemann, L. Iverson and C.-F. Wu, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2748–2753 CrossRef CAS PubMed.
- C. F. Blanford, R. S. Heath and F. A. Armstrong, Chem. Commun., 2007, 1710–1712 RSC.
- O. Rüdiger, J. M. Abad, E. C. Hatchikian, V. M. Fernandez and A. L. De Lacey, J. Am. Chem. Soc., 2005, 127, 16008–16009 CrossRef PubMed.
- A. K. Manocchi, D. R. Baker, S. S. Pendley, K. Nguyen, M. M. Hurley, B. D. Bruce, J. J. Sumner and C. A. Lundgren, Langmuir, 2013, 29, 2412–2419 CrossRef CAS PubMed.
- N. Terasaki, N. Yamamoto, K. Tamada, M. Hattori, T. Hiraga, A. Tohri, I. Sato, M. Iwai, M. Iwai and S. Taguchi, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 653–659 CrossRef CAS PubMed.
- P. N. Ciesielski, F. M. Hijazi, A. M. Scott, C. J. Faulkner, L. Beard, K. Emmett, S. J. Rosenthal, D. Cliffel and G. K. Jennings, Bioresour. Technol., 2010, 101, 3047–3053 CrossRef CAS PubMed.
- Y. Liu, M. Wang, F. Zhao, Z. Xu and S. Dong, Biosens. Bioelectron., 2005, 21, 984–988 CrossRef CAS PubMed.
- B. A. Gregg and A. Heller, Anal. Chem., 1990, 62, 258–263 CrossRef CAS PubMed.
- M. B. Fischback, J. K. Youn, X. Zhao, P. Wang, H. G. Park, H. N. Chang, J. Kim and S. Ha, Electroanalysis, 2006, 18, 2016–2022 CrossRef CAS.
- A. Pizzariello, M. Stred'ansky and S. Miertuš, Bioelectrochemistry, 2002, 56, 99–105 CrossRef CAS PubMed.
- H.-H. Kim, N. Mano, Y. Zhang and A. Heller, J. Electrochem. Soc., 2003, 150, A209–A213 CrossRef CAS.
- Y. Liu, X. Qu, H. Guo, H. Chen, B. Liu and S. Dong, Biosens. Bioelectron., 2006, 21, 2195–2201 CrossRef CAS PubMed.
- D. Quan, Y. Kim and W. Shin, J. Electroanal. Chem., 2004, 561, 181–189 CrossRef CAS.
- M. A. Rahman and H.-B. Noh, Anal. Chem., 2008, 80, 8020–8027 CrossRef CAS PubMed.
- X. Wu, F. Zhao, J. R. Varcoe, A. E. Thumser, C. Avignone-Rossa and R. C. Slade, Biosens. Bioelectron., 2009, 25, 326–331 CrossRef CAS PubMed.
- K. Murata, K. Kajiya, N. Nakamura and H. Ohno, Energy Environ. Sci., 2009, 2, 1280–1285 CAS.
- N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2003, 125, 6588–6594 CrossRef CAS PubMed.
- P. A. Paredes, J. Parellada, V. M. Fernández, I. Katakis and E. Domínguez, Biosens. Bioelectron., 1997, 12, 1233–1243 CrossRef CAS.
- J. Jia, B. Wang, A. Wu, G. Cheng, Z. Li and S. Dong, Anal. Chem., 2002, 74, 2217–2223 CrossRef CAS PubMed.
- J. Tang, B. Wang, Z. Wu, X. Han, S. Dong and E. Wang, Biosens. Bioelectron., 2003, 18, 867–872 CrossRef CAS PubMed.
- S. Yang, Y. Li, X. Jiang, Z. Chen and X. Lin, Sens. Actuators, B, 2006, 114, 774–780 CrossRef CAS.
- S. Liu and A. Chen, Langmuir, 2005, 21, 8409–8413 CrossRef CAS PubMed.
- X.-L. Luo, J.-J. Xu, Q. Zhang, G.-J. Yang and H.-Y. Chen, Biosens. Bioelectron., 2005, 21, 190–196 CrossRef CAS PubMed.
- J. Z. Xu, J. J. Zhu, Q. Wu, Z. Hu and H. Y. Chen, Electroanalysis, 2003, 15, 219–224 CrossRef CAS.
- C. Xiang, Y. Zou, L.-X. Sun and F. Xu, Electrochem. Commun., 2008, 10, 38–41 CrossRef CAS.
- A. Zhu, Y. Tian, H. Liu and Y. Luo, Biomaterials, 2009, 30, 3183–3188 CrossRef CAS PubMed.
- M. Zhang, A. Smith and W. Gorski, Anal. Chem., 2004, 76, 5045–5050 CrossRef CAS PubMed.
- G. Li, H. Xu, W. Huang, Y. Wang, Y. Wu and R. Parajuli, Meas. Sci. Technol., 2008, 19, 065203 CrossRef.
- S. Tsujimura, S. Kojima, K. Kano, T. Ikeda, M. Sato, H. Sanada and H. Omura, Biosci., Biotechnol., Biochem., 2006, 70, 654–659 CrossRef CAS PubMed.
- B. K. Jena and C. R. Raj, Anal. Chem., 2006, 78, 6332–6339 CrossRef CAS PubMed.
- N. L. Akers, C. M. Moore and S. D. Minteer, Electrochim. Acta, 2005, 50, 2521–2525 CrossRef CAS.
- C. Kang, H. Shin, Y. Zhang and A. Heller, Bioelectrochemistry, 2004, 65, 83–88 CrossRef CAS PubMed.
- N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2002, 124, 12962–12963 CrossRef CAS PubMed.
- B. R. Glick, J. J. Pasternak and C. L. Patten, Molecular Biotechnology: Principles and Applications of Recombinant DNA, ASM Press, Washington DC, 4th edn, 2009, p. 683 Search PubMed.
- K. A. Henzler-Wildman, M. Lei, V. Thai, S. J. Kerns, M. Karplus and D. Kern, Nature, 2007, 450, 913–916 CrossRef CAS PubMed.
- L. Foit, G. J. Morgan, M. J. Kern, L. R. Steimer, A. A. von Hacht, J. Titchmarsh, S. L. Warriner, S. E. Radford and J. C. Bardwell, Mol. Cell, 2009, 36, 861–871 CrossRef CAS PubMed.
- A. Bachmair, D. Finley and A. Varshavsky, Science, 1986, 234, 179–186 CAS.
- S. Bassnett and D. C. Beebe, Dev. Dyn., 1992, 194, 85–93 CrossRef CAS PubMed.
- I. Willner and B. Willner, Trends Biotechnol., 2001, 19, 222–230 CrossRef CAS PubMed.
- R. Blonder, S. Levi, G. Tao, I. Ben-Dov and I. Willner, J. Am. Chem. Soc., 1997, 119, 10467–10478 CrossRef CAS.
- I. Matthew, P. Weber, F. Salemme and F. Richard, Nature, 1983, 301, 169–171 CrossRef PubMed.
- I. Lee, J. W. Lee and E. Greenbaum, Phys. Rev. Lett., 1997, 79, 3294 CrossRef CAS.
- A. Efrati, C.-H. Lu, D. Michaeli, R. Nechushtai, S. Alsoub, W. Schuhmann and I. Willner, Nat. Energy, 2016, 15021 CrossRef.
- N. C. Foulds and C. R. Lowe, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 1259–1264 RSC.
- W. Schuhmann, H. Zimmermann, K. Habermüller and V. Laurinavicius, Faraday Discuss., 2000, 116, 245–255 RSC.
- B. Munge, G. Liu, G. Collins and J. Wang, Anal. Chem., 2005, 77, 4662–4666 CrossRef CAS PubMed.
- E. Suraniti, S. Vivès, S. Tsujimura and N. Mano, J. Electrochem. Soc., 2013, 160, G79–G82 CrossRef CAS.
- M. R. Banki, T. U. Gerngross and D. W. Wood, Protein Sci., 2005, 14, 1387–1395 CrossRef CAS PubMed.
- H. Feroz, C. Vandervelden, B. Ikwuagwu, B. Ferlez, C. S. Baker, D. J. Lugar, M. Grzelakowski, J. H. Golbeck, A. L. Zydney and M. Kumar, Biotechnol. Bioeng., 2016, 113 CrossRef CAS PubMed.
- U. Siemann, Scattering Methods and the Properties of Polymer Materials, Springer, 2005, pp. 1–14 Search PubMed.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- A. Rudge, I. Raistrick, S. Gottesfeld and J. P. Ferraris, Electrochim. Acta, 1994, 39, 273–287 CrossRef CAS.
- R. Friend, R. Gymer, A. Holmes, J. Burroughes, R. Marks, C. Taliani, D. Bradley, D. Dos Santos, J. Bredas and M. Lögdlund, Nature, 1999, 397, 121–128 CrossRef CAS.
- S. K. Chaudhuri and D. R. Lovley, Nat. Biotechnol., 2003, 21, 1229–1232 CrossRef CAS PubMed.
- K. Rabaey, G. Lissens, S. D. Siciliano and W. Verstraete, Biotechnol. Lett., 2003, 25, 1531–1535 CrossRef CAS PubMed.
- Y. Qiao, S.-J. Bao, C. M. Li, X.-Q. Cui, Z.-S. Lu and J. Guo, ACS Nano, 2007, 2, 113–119 CrossRef PubMed.
- M.-H. Lee, J. L. Thomas, W.-J. Chen, M.-H. Li, C.-P. Shih and H.-Y. Lin, ACS Sustainable Chem. Eng., 2015, 3, 1190–1196 CrossRef CAS.
- E. D'costa, I. Higgins and A. Turner, Biosensors, 1986, 2, 71–87 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |