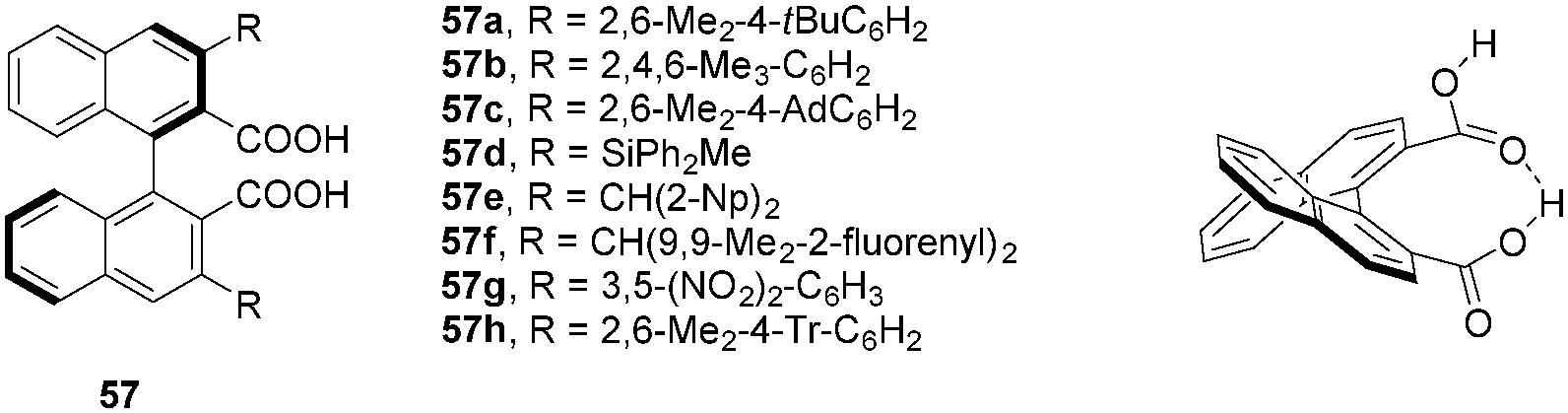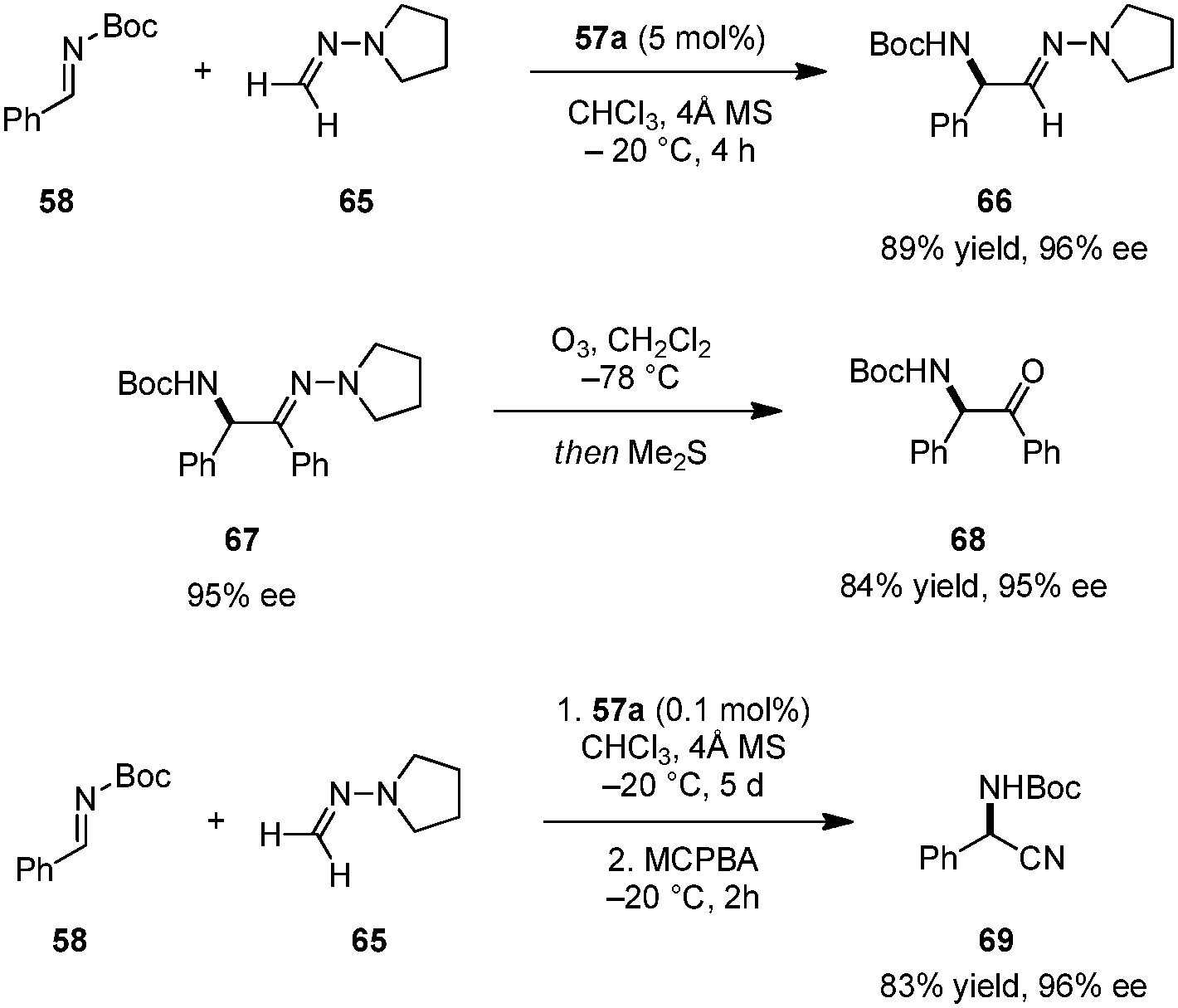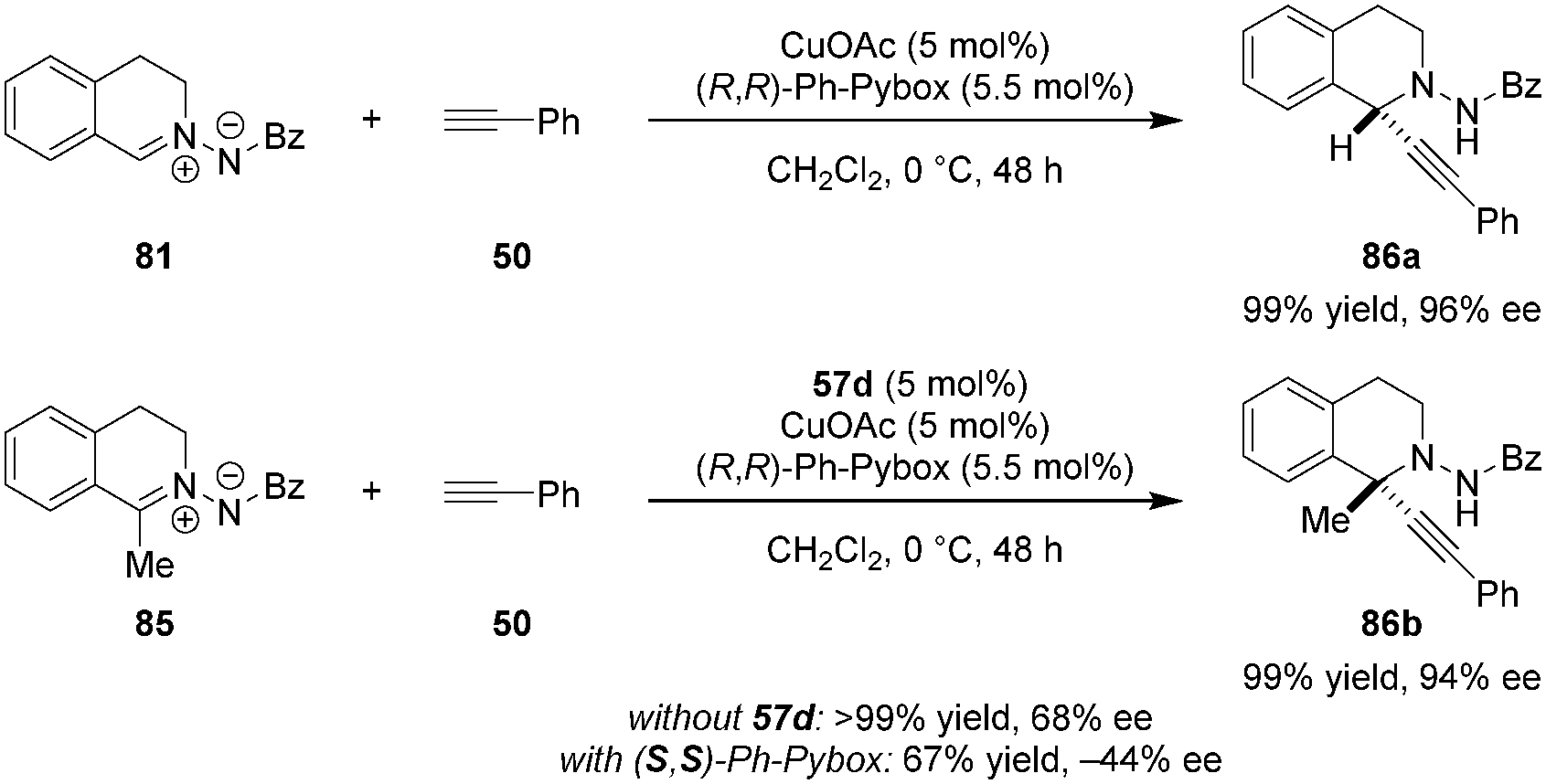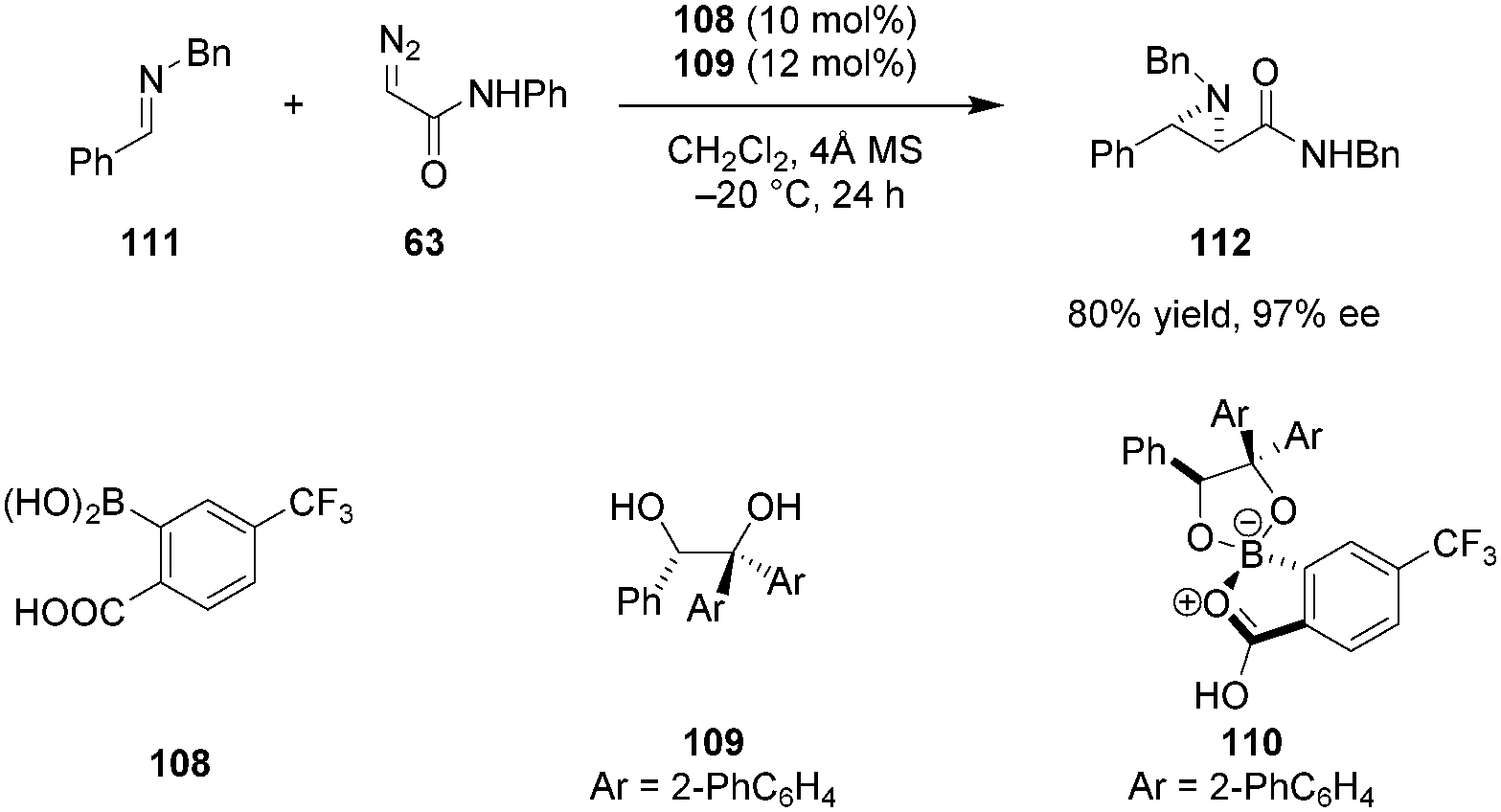DOI:
10.1039/C6CS00239K
(Review Article)
Chem. Soc. Rev., 2017,
46, 5889-5902
Asymmetric Brønsted acid catalysis with chiral carboxylic acids
Received
21st March 2016
First published on 21st July 2017
Abstract
Chiral Brønsted acids have found widespread applications as organocatalysts. Weakly acidic species such as diols and (thio)ureas, typically classified as hydrogen bonding catalysts, as well as stronger phosphoric acids, have proven to be highly effective in catalyzing a wide range of asymmetric transformations. In contrast, the use of chiral carboxylic acids as Brønsted acid catalysts is much less developed but has recently garnered increased attention. Given that their pKa's generally lie between those of typical hydrogen bond donors and chiral phosphoric acids, chiral carboxylic acids provide the opportunity to activate a different set of substrates requiring intermediate catalyst acidity. In addition, by taking advantage of judicious catalyst design, the acidity of carboxylic acids can be modulated, for instance by stabilizing the corresponding carboxylate counterion. This review provides an overview of this field, with the aim of highlighting both catalyst design and synthetic applications.

Chang Min
| Chang Min was born and raised in Wuhan, China. He earned his BS and MS degrees in the Department of Pharmacy at Wuhan University working with Prof. Chune Dong and Haibing Zhou. In 2011, he moved to Rutgers, The State University of New Jersey for his PhD study, working with Prof. Daniel Seidel mainly on organocatalysis. In November 2016, Chang joined Prof. Noah Burns' group at Stanford University as a postdoctoral scholar. |

Daniel Seidel
| Daniel Seidel studied chemistry at the Friedrich-Schiller-Universität Jena and at the University of Texas at Austin (Diplom 1998). He performed his graduate studies in the lab of Prof. Jonathan L. Sessler, obtaining his PhD in 2002. From 2002 to 2005, he was an Ernst Schering Postdoctoral Fellow in the group of Prof. David A. Evans at Harvard University. He started his independent career at Rutgers University in 2005 and was promoted to Associate Professor in 2011 and Full Professor in 2014. In the summer of 2017, he moved to the University of Florida. Research in his group is focused on new concepts for asymmetric catalysis and synthetic methodology. |
1. Introduction
1.1 Overview of chiral Brønsted acid catalysts
Acids are prevailing catalysts in innumerable chemical processes ranging from biological to industrial settings. The last decade has witnessed the rapid development of chiral Brønsted acids for various enantioselective C–C and C–X bond formations. Weakly acidic species such as (thio)ureas,1–7 squaramides,8–12 BINOLs,13,14 TADDOLs,15,16 and others,17–20 which typically activate substrates via hydrogen bonding,21–27 have become some of the most popular types of organocatalysts (Fig. 1). A range of these compounds have also been utilized in anion binding catalysis,28–34 and many other innovative applications have been established.35 Since the first introduction of phosphoric acids as asymmetric Brønsted acid catalysts independently by Akiyama36 and Terada37 in 2004, enantioselective Brønsted acid catalysis has attracted considerable attention as a powerful tool for asymmetric synthesis.38–44 Numerous variations of mostly BINOL-derived phosphoric acids have been developed and shown to enable an ever increasing number of asymmetric transformations. In a continuing trend, derivatives with higher acidity, including the corresponding nitrogen,45–49 thiol,50 and carbon acids,51,52 have been synthesized for the purpose of activating substrates with attenuated basicities. These exciting advances notwithstanding, there is still a large number of known or conceivable acid-promoted reactions that have yet to be realized in catalytic enantioselective fashion. With proper choice of chiral Brønsted acid catalysts, classic transformations mediated by achiral acids,53 whether as catalysts, promoters or solvents, have the potential to be conducted in asymmetric fashion. Thus, the development of novel efficient catalytic systems remains a worthwhile endeavor.
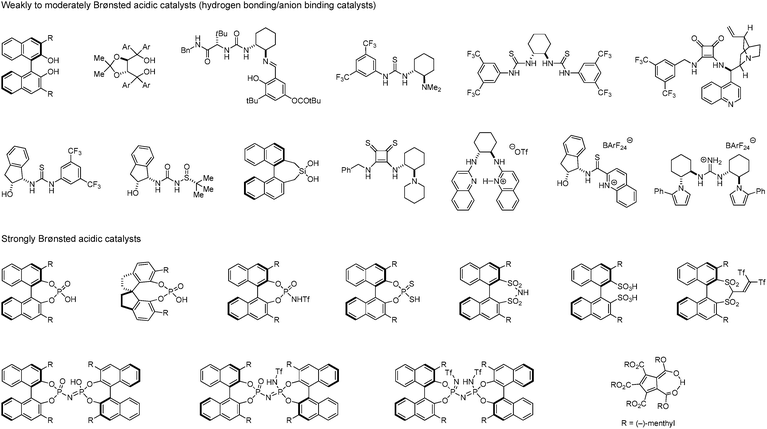 |
| | Fig. 1 Examples of chiral organocatalysts not covered in this review, spanning a broad range of acidities. | |
Carboxylic acids are arguably the most common acids in organic chemistry and this structural motif is present in a myriad of compounds. Chiral carboxylic acids exist widely in nature in the form of amino acids and compounds such as tartaric acid, malic acid, cholic acid, biotin, etc. Despite their abundance, the utilization of carboxylic acids to provide critical substrate activation is largely underdeveloped, especially when compared to phosphoric acids. One major reason is likely related to the relatively weaker Brønsted acidity of carboxylic acids, limiting the scope of substrates that can be activated. However, choosing an acid catalyst with appropriate acidity can be decisive for the successful activation of particular types of substrates. Therefore, chiral carboxylic acids were initially introduced to bridge the gap between weakly acidic hydrogen bond donors and the stronger phosphoric acids. Remarkably, enzymes can easily realize extremely challenging acid–base chemistry in the absence of highly acidic functionality.54 To modulate the pKa of a specific site in a given enzyme, nature has evolved various ingenious ways to create finely tuned microenvironments. When an environment is generated in favor of stabilizing the conjugate base of the acid, the acidity can be significantly increased. In enzymatic reactions, it is common for the acid strength to change by four to five orders of magnitude by taking advantage of electrostatic interactions, hydrogen bonding, desolvation, metal coordination, etc.55 By applying nature's lessons, it may be feasible to create highly reactive catalysts via the combination of components that stabilize the conjugate base in the absence of highly acidic moieties.
1.2 Scope of this review
This review focuses on catalytic enantioselective reactions wherein a catalytic amount of a chiral carboxylic acid is used to activate one or more substrates. While activation can occur via hydrogen bonding or protonation, it is often challenging to differentiate between these modes of activation which really represent different ends of a spectrum of possible interactions. While there are a significant number of plausible interactions between the catalyst and substrate(s), the nature of these interactions may also change during different stages of the reaction. In addition, a range of other non-covalent interactions between the catalyst and substrates are often present simultaneously and may play decisive roles with regard to reactivity and enantioselectivity.56,57 Reports wherein chiral carboxylic acids are used as additives in aminocatalysis to facilitate the formation of enamines or iminium ions,58–60 or in combination with chiral phosphoric acid catalysts to reduce product inhibition and facilitate catalyst turnover,61,62 are beyond the scope of this review. Additionally, there are also cases in which the absolute configuration of the chiral carboxylic acid additive has negligible effects on the reaction enantioselectivity.63 In those reports, carboxylic acids only play supplemental roles and additive effects of this type have recently been reviewed by Wang et al.64 However, in some cases, the chiral carboxylic acid is used synergistically in association with a metal catalyst activating different substrates, or as a precatalyst which is converted to another species during the reaction. Examples of such reactions are included. Because a detailed mechanistic understanding is lacking in many cases, this review may inadvertently include transformations in which chiral carboxylic acids facilitate enantioselective reactions by mechanisms other than strict Brønsted acid/hydrogen bonding catalysis. Lastly, this review is organized according to the structural features of the catalysts, and based on the key design elements or backbones best representing the type of catalyst.
2. Tartaric acid and derivatives
Tartaric acid is one of the most abundant naturally occurring chiral acids. Due to the fact that both of its enantiomers are inexpensive and readily available, tartaric acid and its derivatives have been widely applied as resolving agents,65 auxiliaries and ligands.66 However, even though tartaric acid is one of the strongest chiral carboxylic acids present in the chiral pool (pKa1 = 2.90), examples in which tartaric acid and its derivatives are used directly as chiral Brønsted acid catalysts remain rather limited.
2.1 Historical use of tartaric acid and derivatives as chiral proton sources
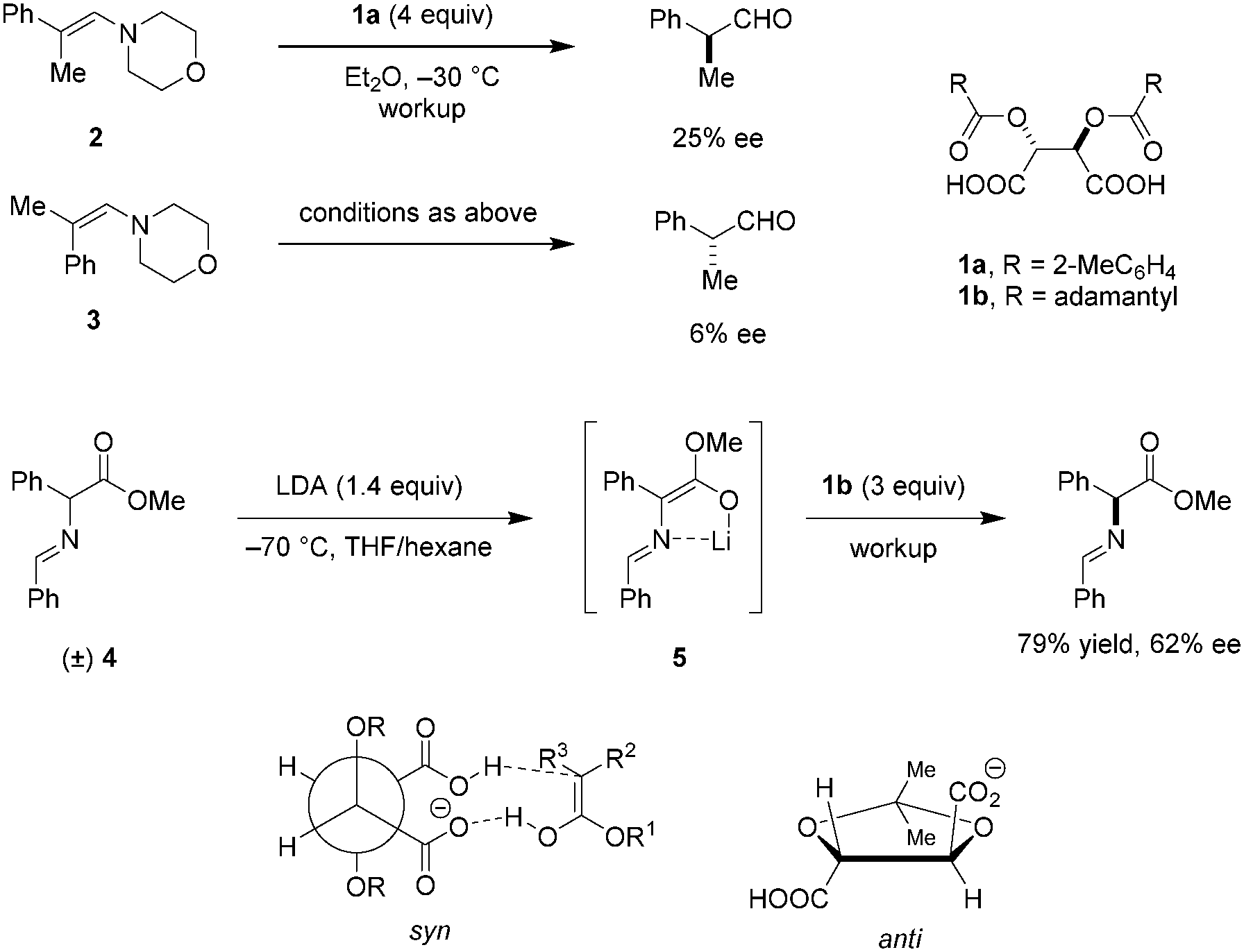
The possibly first use of tartaric acid derivatives as chiral proton sources in enantioselective protonations of preformed enamines was reported by Duhamel and Plaquevent in the late 1970's.67 It was found that not only the nature of the acyl groups of the O,O′-diacyl tartaric acid but also the enamine geometry have a dramatic impact on the absolute configuration and ee of the protonation products (Scheme 1). When E-enamine 2 was exposed to four equivalents of 1a, (S)-2-phenylpropanal was obtained in 25% ee. Under otherwise identical conditions, the Z-enamine 3 gave (R)-2-phenylpropanal in only 6% ee. These results seem to exclude the possibility of equilibration between the two diastereomeric iminium ion intermediates. Thus, the protonation step is most likely under kinetic control. The same research group applied O,O′-diacyl tartaric acid 1b to the deracemization of α-substituted carbonyl compounds.68 The racemic Schiff base methyl ester 4 was converted to the corresponding achiral lithium enolate 5 by deprotonation with LDA, followed by enantioselective protonation with three equivalents of 1b to provide optically active Schiff base ester in 79% yield and 62% ee. Intramolecular coordination between lithium and the imine is thought to result in a rigid conformation that enhances facial selectivity in the protonation step. Hydrolysis of the Schiff base ester product provides the corresponding α-amino acid. While superstoichiometric amounts of O,O′-diacyl tartaric acids are required in the process, the acids can be easily recycled without loss of enantiomeric purity.
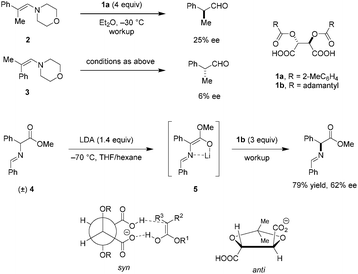 |
| | Scheme 1 Enantioselective protonation with diacyl tartaric acids. | |
To gain a better understanding of the relationship between the structures of O,O′-diacyl tartaric acids and the ee of the protonation products, a thorough study of the effect of the acyl groups was carried out.69 Not surprisingly, the size and shape of the substituents on the carboxyl groups significantly affect the enantioselectivities. It was proposed that the two bulky R-groups on the O,O′-diacyl tartaric acids force the two carboxylic acid groups into a syn-conformation, with one serving as a proton donor and the other as a hydrogen bond acceptor. The syn-conformation appears to be a main reason for ensuring high enantioselectivities, since locking the two carboxylic acid groups of tartaric acid in an anti-conformation results in a dramatic loss of ee. Although early work on enantioselective protonation is limited to the use of (super)stoichiometric amounts of chiral acids,70–74 the systematic study and consideration of structure–enantioselectivity relationships provide a valuable platform for the understanding and future design of chiral Brønsted acid catalysts.
2.2 Tartaric acid and derivatives as chiral Brønsted acid catalysts
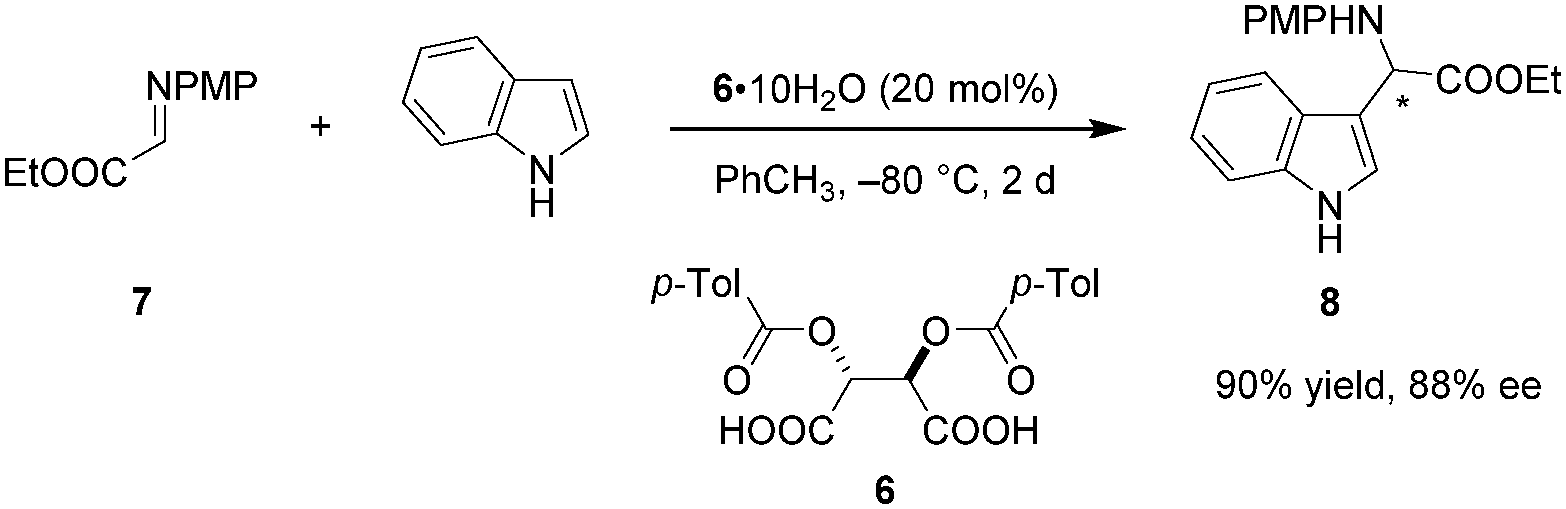
Despite the long history of tartaric acid and its derivatives as stoichiometric chiral proton sources, their utilization as Brønsted acid catalysts began only recently. In 2010, Terada et al. reported the use of tartaric acid derived acid 6 as a catalyst in an enantioselective aza-Friedel–Crafts reaction of indole with α-imino ester 7 (Scheme 2).75 Interestingly, the presence of water is essential for achieving good enantioselectivities. The exact ratio of water and 6 significantly influences the enantioselectivity. In fact, rigorous exclusion of water in the presence of 4 Å molecular sieves results in racemic product. Raising the amount of water from to 0.3 to 1 equivalent dramatically improves the ee, while further increase to 10 equivalents leads to a relatively smaller boost. In a control experiment, the analogous aza-Friedel–Crafts reaction of N-methylindole afforded racemic product (not shown), implicating the indole N–H proton as a crucial element in the enantiodetermining step, most likely as a hydrogen-bond donor that enables secondary interactions. At the same time, such an interaction would increase the nucleophilicity of the indole nucleus. While the scope of this work is somewhat limited, it highlights the important finding that simple tartaric acid derivatives can be utilized as chiral Brønsted acid catalysts.
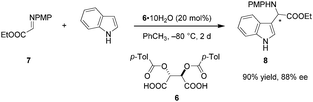 |
| | Scheme 2 Enantioselective aza-Friedel–Crafts reaction with a diacyltartaric acid catalyst. | |
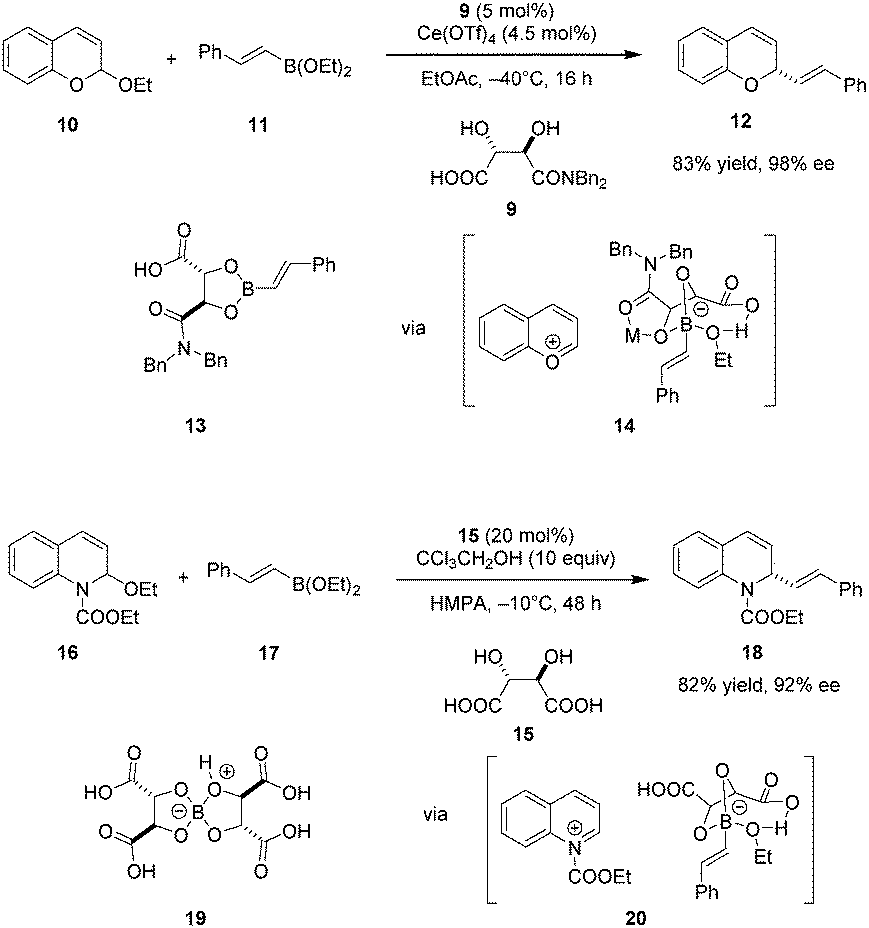
Schaus and coworkers identified tartaric acid derived mono-amide 9 as an efficient carboxylic acid catalyst for enantioselective additions of vinyl or electron-rich aryl boronates to chromene acetals (Scheme 3).76 A dual catalytic system consisting of 9 and an achiral Lewis acid, specifically Ce(OTf)4, was found to be optimal. Only moderate yields and ee's were obtained without added Lewis acid, and the Lewis acid catalyst alone caused substantial decomposition of the chromene starting material and no formation of desired product 12. Based in part on these observations, the authors proposed that the primary mode of enantioselective catalysis is Brønsted acid catalysis, promoting the formation of pyrylium ion 14. Mechanistic studies showed that mixing boronate and catalyst 9 results in the facile formation of isolable dioxaborolane 13. This presumed intermediate was shown to react with chromene acetal 10 under otherwise identical conditions to give nearly the same results as the standard procedure. Supported by mass spectrometry and in situ FT-IR studies, Ce(OTf)4 is proposed to bind to the amide carbonyl group of the dioxaborolane to enhance the acidity of the boronate, thus facilitating the formation of chiral ion pair 14 which ultimately undergoes transformation into enantioenriched product 12.
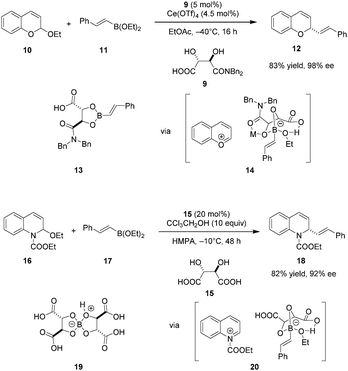 |
| | Scheme 3 Asymmetric addition of boronates to chromene acetals and quinoline-derived N,O-acetals. | |
A modified catalytic system was applied in an enantioselective boronate addition to N-acyl quinolinium ions.77 Interestingly, unmodified tartaric acid (15) was identified as the ideal asymmetric catalyst. In contrast to previous work, the enantioselectivity increased in the absence of Lewis acid cocatalysts. Instead, protic additives such as CCl3CH2OH were employed to facilitate catalyst turnover with concomitant increase in yield. The authors proposed a similar catalytic cycle wherein the N-acyl quinolinium ion 20 is the key ion pair intermediate. The catalytically viable dimeric tartaric acid adduct 19 is thought to be the resting state of the catalyst.
In an intriguing departure from their earlier work, the Schaus group reported the tartaric acid catalyzed enantioselective [4+2] cycloaddition of isochromene acetals (e.g., 21) with vinylboronates (e.g., 22) to generate dihydronaphthalene and dihydrobenzofluorene products such as 23 (Scheme 4).78 Oxocarbenium ions 24 and 25 are thought to be key intermediates along the path to final product 23. In analogy to the 1,2-addition of boronates to chromene acetals (Scheme 3), the tartaric acid–Lewis acid combination proved to be pivotal. No reaction was observed in the absence of any rare-earth triflates. Interestingly, although the substrates and the catalytic system are similar to those shown in Scheme 3, 1,2-addition products were never observed in reactions of 21 and 22.
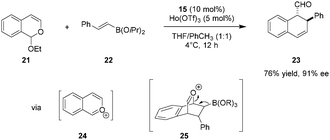 |
| | Scheme 4 Enantioselective [4+2] cycloaddition of isochromene acetals and vinylboronates. | |
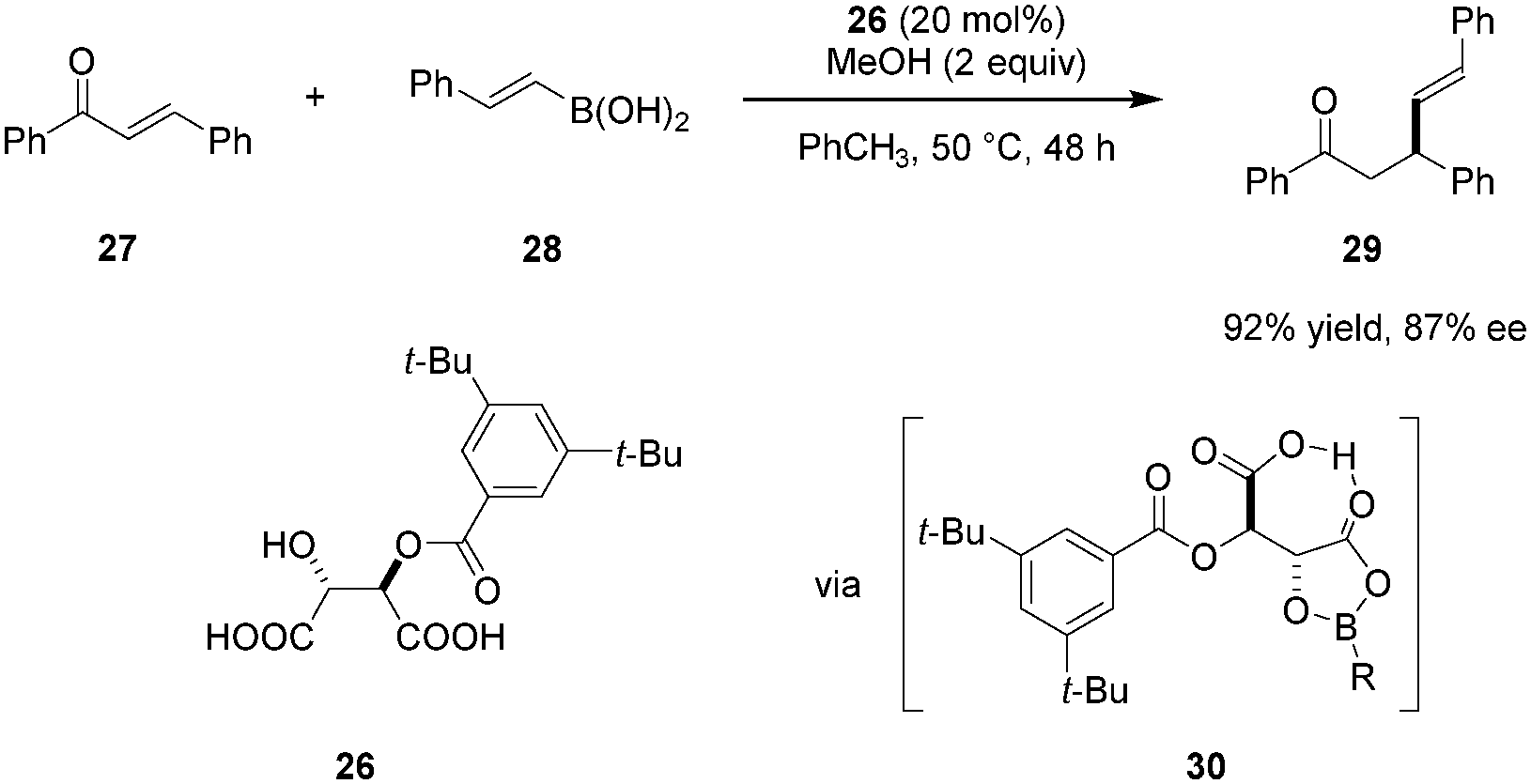
Sugiura and coworkers reported on the use of mono O-acyl tartaric acids as catalysts in enantioselective conjugate additions of boronic acids to enones (Scheme 5).79 Electronic and steric properties of the acyl group significantly impact both catalyst reactivity and product enantioselectivity. Modification of either of the two carboxylic acid groups results in a dramatic drop in reactivity while the addition of methanol was found to suppress the background reaction and improve catalytic efficiency. Tartaric acid derivative 26 emerged as the most effective catalyst. As for the reaction mechanism, the authors proposed that catalyst 26 and boronic acid 28 initially undergo formation of intermediate 30, a key feature of which is the presence of an intramolecular hydrogen bond.80 Due to the electron-withdrawing acyloxy group on boron, intermediate 30 is thought to be relatively Lewis acidic. Enone 27 is then activated toward nucleophilic attack by coordination of its carbonyl oxygen to the boron center in 30. The above mentioned intramolecular hydrogen bond involving the free carboxyl group likely stabilizes the transition state while rigidifying the overall structure. This system is comparable to Yamamoto's chiral acyloxyborane (CAB) catalyst81–86 and is consistent with his definition of a “Brønsted acid assisted Lewis acid catalyst” (BLA).38 Compound 26 was also found to catalyze the conjugate addition of boronic acids to dienones.87
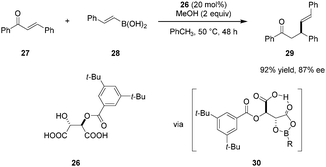 |
| | Scheme 5 Enantioselective conjugate addition of boronic acids to enones. | |
Catalytic enantioselective conjugate additions of bis(neopentyl glycolato)diboron (32) to enones such as 27 was reported by Sugiura et al., utilizing mono O-acyl tartaric acid 31 as a catalyst (Scheme 6).88 Methanol, shown to be an effective additive previously (cf.Scheme 5), completely inhibited the reaction. Instead, the addition of benzoic acid improved the yield, presumably by facilitating catalyst turnover. Adduct 33 can be transformed into β-hydroxy ketone 34 without loss of ee via treatment with sodium perborate. In contrast to previous studies, no significant ligand exchange between diboron species 32 and catalyst 31 was observed in this system. Thus, the mechanism of this reaction remains unclear at present.
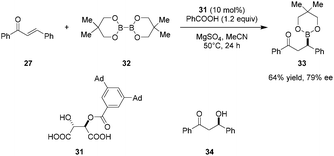 |
| | Scheme 6 Catalytic enantioselective addition of diboron compounds to enones. | |
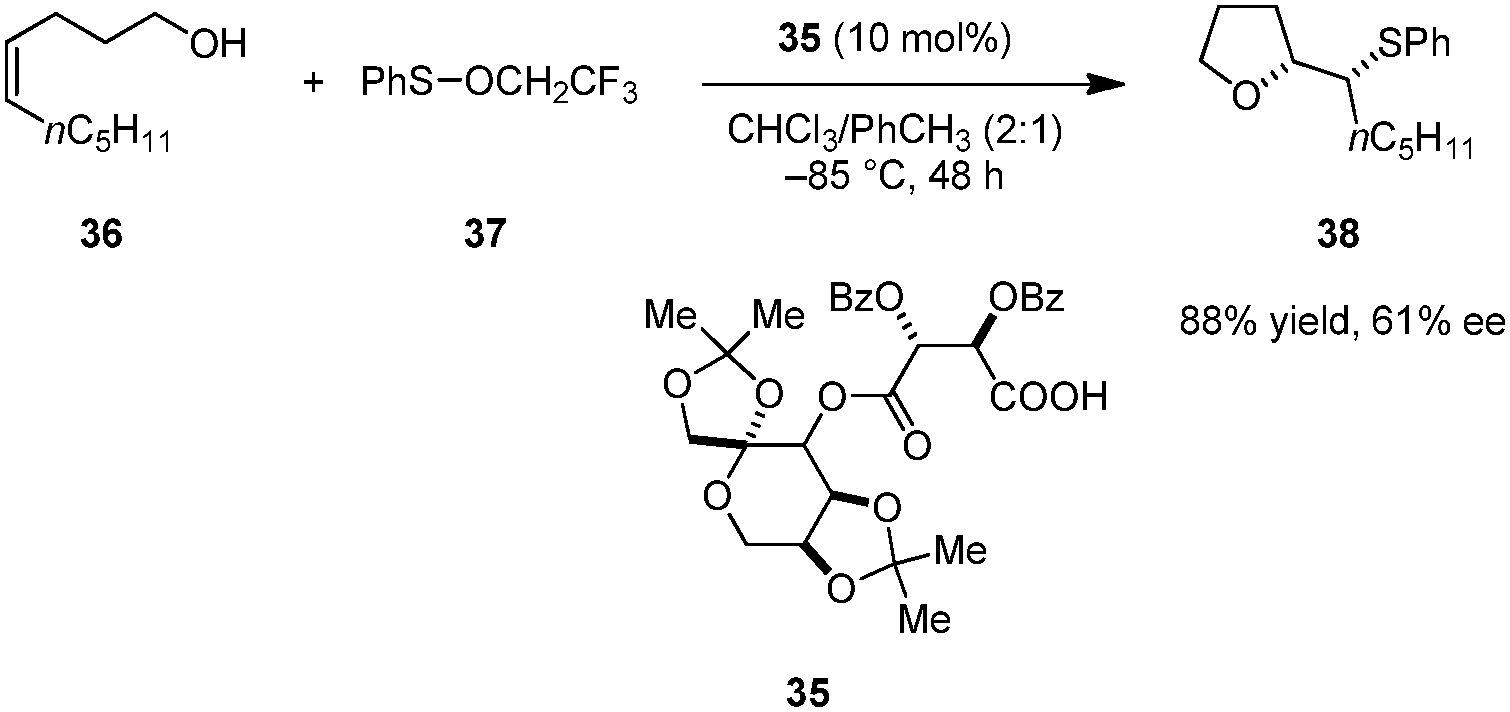
Shi and coworkers reported enantioselective oxysulfenylations of alkenols (e.g., 36) to provide enantioenriched tetrahydrofurans such as 38 (Scheme 7).89 Tartaric acid derivative 35 outperformed several commonly used chiral phosphoric acids and N-triflyl phosphoramides in this transformation. Trifluoroethyl benzenesulfenate 37 displayed significantly higher reactivity than more common electrophilic sulfur reagents such as N-(phenylthio)succinimide. This reaction is only applicable to cis-alkenols; complex mixtures were obtained for the corresponding trans-alkenols. While the mechanism of this transformation has not been investigated in detail, it likely involves an ion pair in which the carboxylate serves as a chiral counteranion to an episulfonium intermediate. Two diastereomeric ion pairs may exist in equilibrium, one of which undergoes ring closure more rapidly.
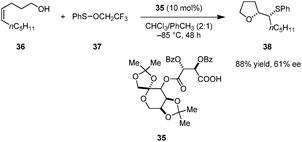 |
| | Scheme 7 Enantioselective oxysulfenylation of olefins. | |
3.
N-Protected amino acids
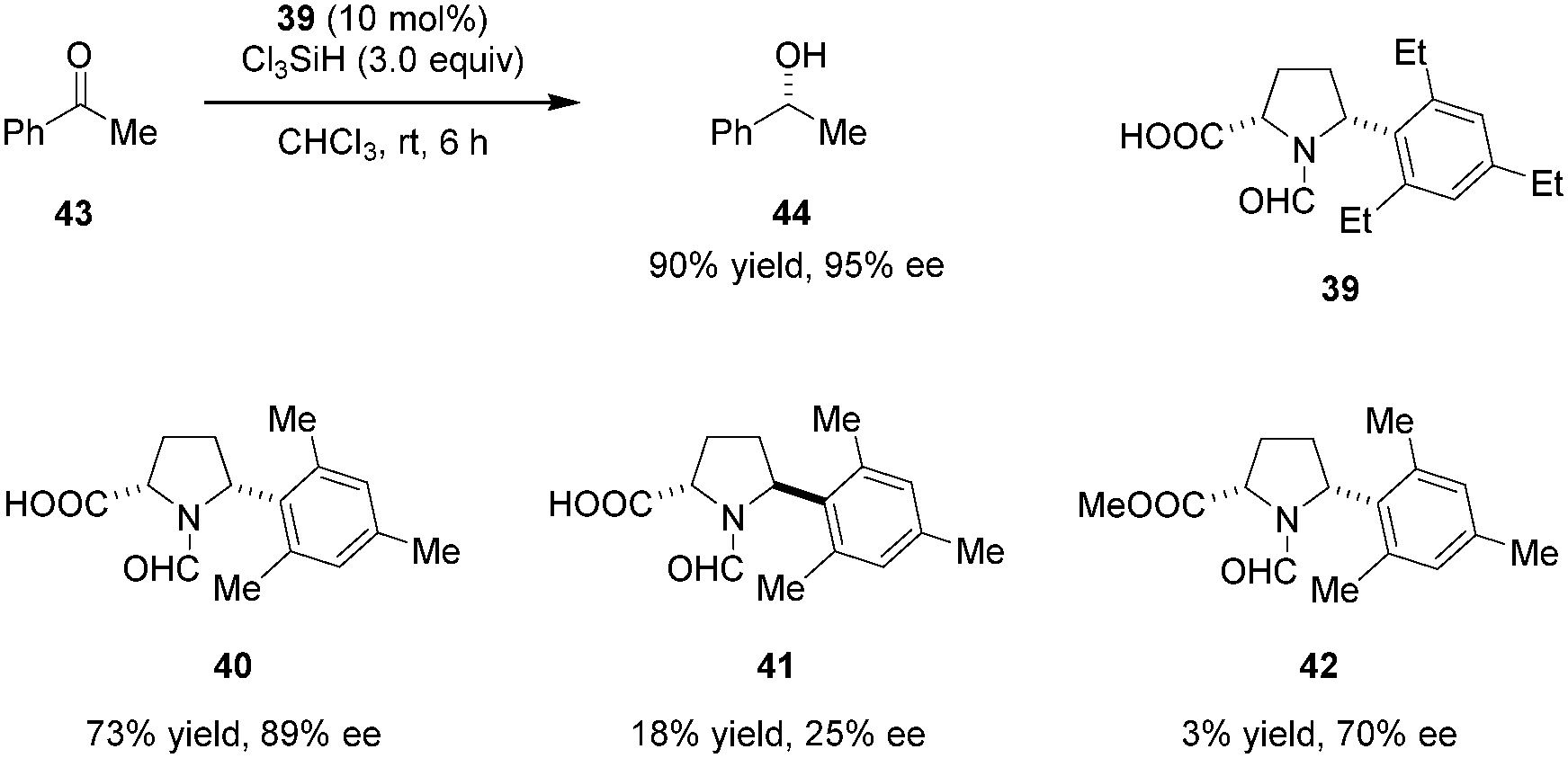
Highly enantioselective reductions of aryl alkyl ketones including acetophenone (43) with trichlorosilane were achieved by the Matsumura group (Scheme 8).90 Reactions are catalyzed by bifunctional N-formyl-α′-aryl-L-proline derivative 39. The Lewis basic formamide site of 39 most likely serves to activate trichlorosilane while the free carboxylic acid group is thought to activate the ketone substrate via hydrogen bonding. Both the α-carboxylic acid and a bulky α′-aryl group are essential to achieve high reactivity and enantioselectivity. In addition, these two substituents are required to be cis-configured (cf. catalysts 40 and 41). The corresponding methyl ester 42, while capable of instilling enantioselectivity, was found to be nearly inactive.
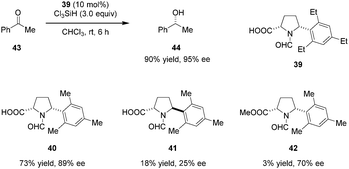 |
| | Scheme 8 Enantioselective reduction of ketones by trichlorosilane. | |
In related work, Benaglia and coworkers reported the enantioselective reduction of ketimines (e.g., 46) by trichlorosilane, catalyzed by simple N-acyl-L-proline derivative 45 (Scheme 9).91 While relatively low yields and moderate ee's were observed in most cases, imine activation by the carboxylic acid group was found to be crucial. The corresponding methyl ester of 45 gave rise to significantly lower yield and ee of the benzylic amine product 47. DFT studies indicate that the proton and hydride transfers are concerted but asynchronous, with the hydride transfer slightly preceding protonation. On this basis, the authors proposed that an asymmetric counteranion-directed catalytic pathway is involved in enantiocontrol.
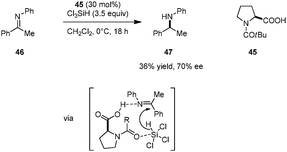 |
| | Scheme 9 Enantioselective reduction of ketimines by trichlorosilane. | |
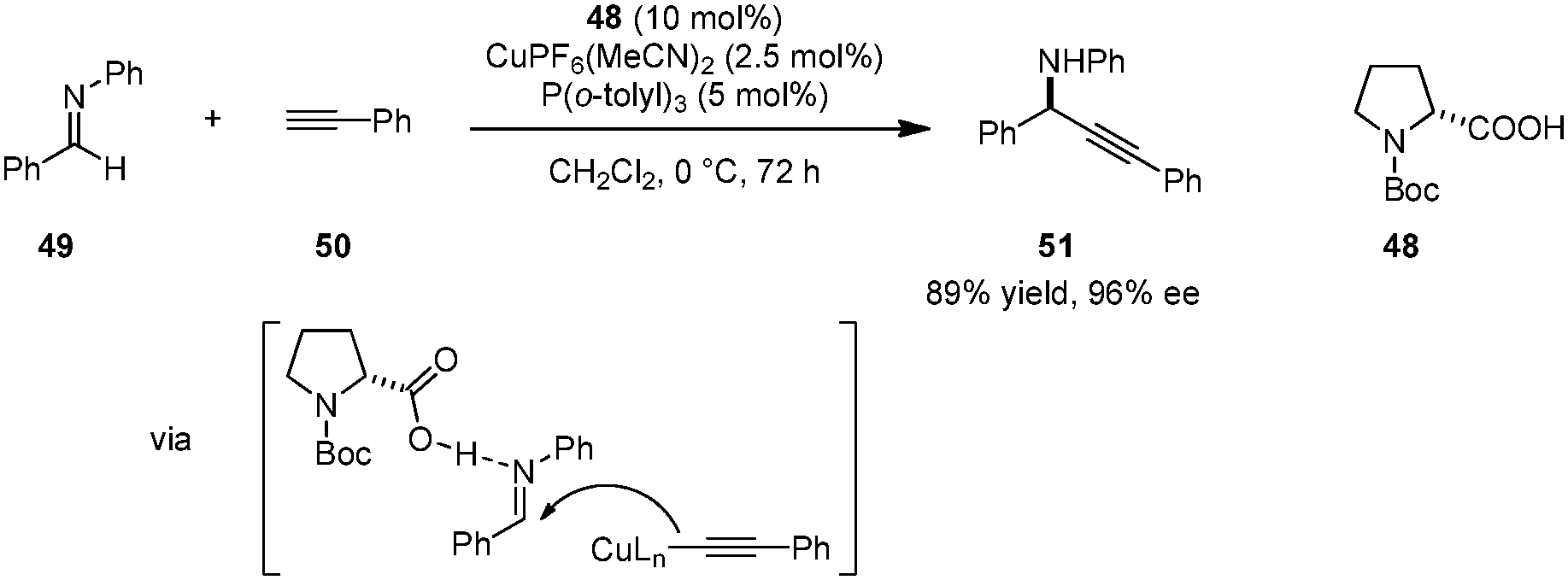
A unique strategy for highly enantioselective alkynylations of imines (e.g., 49 → 51) was disclosed by Arndtsen et al. (Scheme 10).92 The combination of N-Boc proline (48), a Cu(I) salt, and a simple phosphine ligand was found to result in a powerful catalytic system. Hydrogen bonding between the carboxylic acid group and the imine was found to be crucial; poor yields were obtained in the absence of a carboxylic acid additive. The presence of a phosphine ligand is not essential with regard to reactivity but proper choice of this additive leads to higher enantioselectivities. Interestingly, while a first-order dependence on the N-Boc-proline concentration was observed, the reaction was found to be zeroth-order in Cu(I) salt, suggesting different roles for the two components. A key advantage of this system is its easy tunability. The reliance on inexpensive amino acids as the source of chirality obviates the need for chiral phosphine ligands. More challenging substrates such as aliphatic imines, aliphatic alkynes or N-alkyl imines can be accommodated by simply changing the achiral phosphine ligand.
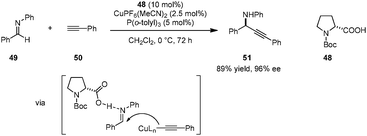 |
| | Scheme 10 Cu(I)/amino acid catalyzed enantioselective imine alkynylation. | |
4. α-Hydroxy carboxylic acids
In 2005, Yamamoto reported regio- and enantioselective nitroso aldol reactions between simple enamines (e.g., 53) and nitrosobenzene (54) (Scheme 11).93 This important achievement is often cited as the first example of utilizing chiral carboxylic acids as asymmetric Brønsted acid catalysts. When mandelic acid analog 52 was applied as the catalyst in ether solution, the highly enantioenriched O-nitroso aldol product 55 was formed exclusively over the corresponding N-nitroso aldol product 56. In contrast, under TADDOL catalysis in toluene, the predominant products were the N-nitroso aldol adducts. The authors noted that a possible intramolecular hydrogen bond between the hydroxyl group and the carbonyl oxygen of the carboxylic acid might contribute to a more rigid structure while increasing the acidity of the catalyst.94 This fits within Yamamoto's classification of a “Brønsted acid assisted Brønsted acid” (BBA) system.38
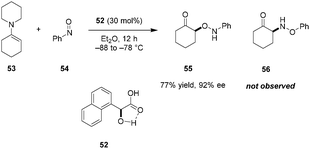 |
| | Scheme 11 Regio- and enantioselective nitroso-aldol reactions. | |
5. Axially chiral dicarboxylic acids
Maruoka and coworkers developed a series of axially chiral dicarboxylic acids possessing a binaphthyl backbone (Fig. 2).95 As is the case for the corresponding phosphoric acids, the presence of substituents96–98 in the 3,3′-positions is essential for reactivity and enantioselectivity. Thus, a number of derivatives with varied steric and electronic characteristics have been prepared. Maruoka's axially chiral dicarboxylic acids catalysts exhibit a number of unique features. For example, an intramolecular hydrogen bonding interaction between the two carboxylic acid groups is responsible for increased acidity and a rigidified catalyst structure. The dihedral angle is larger than that of the corresponding BINOL-derived chiral phosphoric acids, providing a unique chiral pocket. Maruoka's dicarboxylic acids have become the most widely used chiral carboxylic acid catalysts to date. They have effectively been exploited in a wide range of enantioselective transformations.
 |
| | Fig. 2 Maruoka's axially chiral dicarboxylic acids. | |
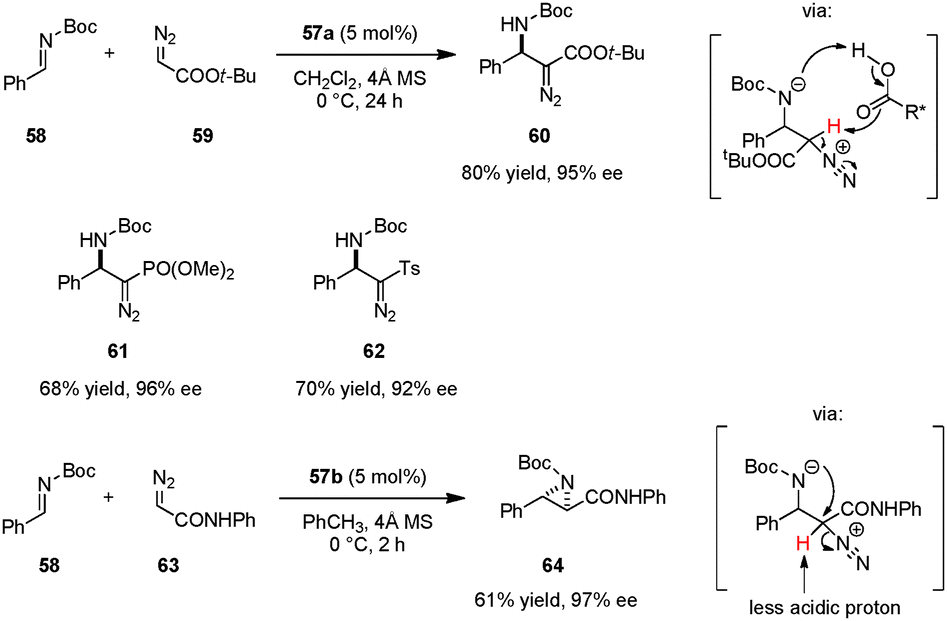
The first example of a catalytic enantioselective reaction utilizing an axially chiral dicarboxylic acid catalyst is the asymmetric Mannich addition of α-diazo acetates to aromatic N-Boc imines (Scheme 12).99 This reaction is complementary to a prior chiral phosphoric acid catalyzed diazo Mannich reaction that required the use of p-dimethylaminobenzoyl aldimines in order to achieve high enantioselectivities.100 The weaker dicarboxylic acid 57a enabled the use of more conventional N-Boc imines such as 58. Furthermore, identical reaction conditions could be applied to the addition of diazomethyl phosphonate and diazomethyl sulfone to 58, providing enantiomerically enriched β-aminophosphonate 61 and β-amino sulfone 62, respectively.101 The authors proposed that deprotonation of the α-proton from the potential intermediate by the catalyst's basic oxygen moiety might facilitate direct alkylation. Interestingly, when diazoacetamides (e.g., 63) were employed as nucleophiles, the reaction outcome was changed in favor of asymmetric aziridination.102 This divergent behavior was attributed to the reduced acidity of the proton in α-position of the diazo group. The aza-Darzens reaction catalyzed by 57b selectively provided aziridines such as 64 as their corresponding trans-diastereomers. Prior to this work, chiral Lewis acid103–105 and achiral Brønsted acid106 catalyzed aza-Darzens reactions were well documented to selectively give rise to cis-aziridine products. Maruoka et al. speculated that the unique trans-selectivity results from the anti-orientation of the carboxamide and aryl groups which avoids steric repulsion. Moreover, a favorable intramolecular hydrogen bond between the carbonyl group of Boc and the amide N–H may contribute to the observed trans-selectivity. Subsequent to Maruoka's work, different trans-107,108 and cis-selective109 asymmetric aza-Darzens reactions catalyzed by chiral phosphoric acids and N-triflyl phosphoramides were also realized.
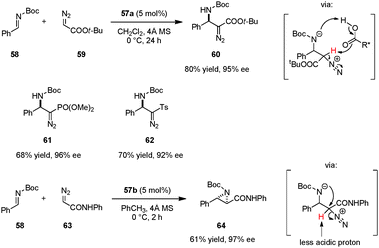 |
| | Scheme 12 Enantioselective Mannich reaction and aza-Darzens reaction of N-Boc imines with α-diazo compounds. | |
Catalyst 57a was successfully employed in imino aza-eneamine reactions of N-Boc imines, in which N,N-dialkyl hydrazones serve as acyl anion equivalents (Scheme 13).110 Earlier reports on this transformation utilizing BINOL derivatives111 or phosphoric acid112 catalysts were only moderately enantioselective and limited to N,N-dialkylhydrazones derived from formaldehyde. In contrast, carboxylic acid catalyst 57a was not only suitable for the addition of unsubstituted N,N-dialkylhydrazone 65, but also accommodated less nucleophilic aromatic-aldehyde-derived N,N-dialkylhydrazones. α-Amino hydrazone products such as 67 can be readily transformed into α-amino ketones (e.g.68) without loss of ee. In a subsequent publication, the authors developed a one-pot synthesis of α-amino nitriles.113 Following completion of the imino aza-enamine reaction of 58 and 65, the reaction mixture was treated with m-chloroperbenzoic acid (MCPBA) to furnish 69. In this process, catalyst loadings can be as low as 0.1 mol% while retaining high yields and ee's. This one-pot imino aza-enamine reaction/oxidation sequence provides a practical alternative to classical Strecker reactions as enantiopure amino acids can be accessed without the need for handling potentially hazardous cyanide sources.
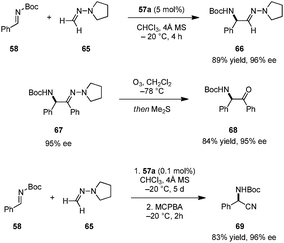 |
| | Scheme 13 Enantioselective imino aza-eneamine reaction. | |
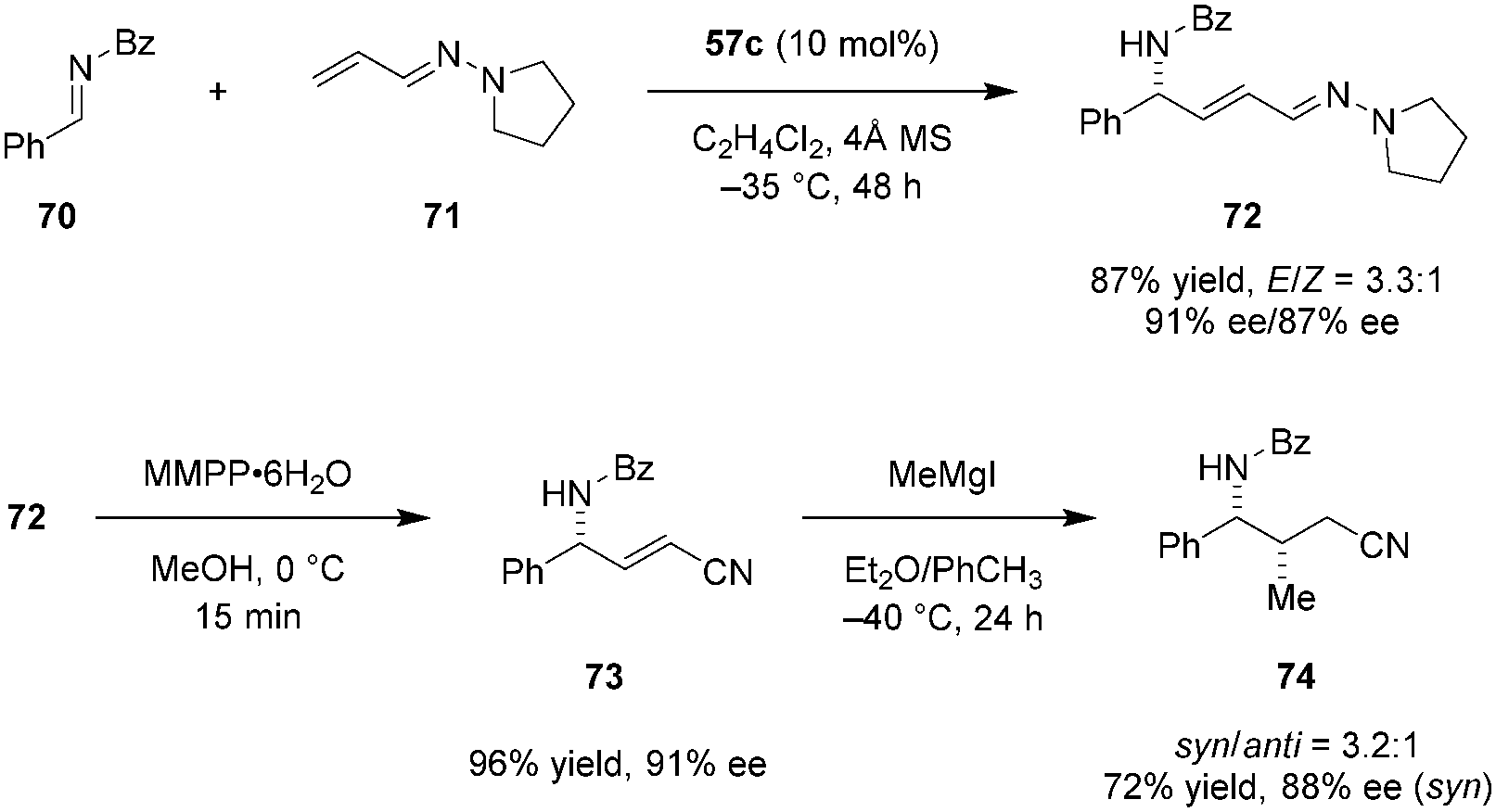
Maruoka et al. later applied a similar catalytic system to the formal alkenylation of imines such as 70 (Scheme 14).114 Vinylogous aza-enamines (e.g., 71) were used as nucleophiles, allowing for the facile construction of highly enantioenriched allylic amines such as 72. Conditions optimized for the corresponding imino aza-eneamine reactions (Scheme 13), only provided the desired alkenynation product in less than 20% yield as a mixture of E/Z isomers (not shown). However, replacement of the N-Boc imine 58 with N-benzoyl imine 70 significantly improved the reactivity and enantioselectivity. The reaction exhibited essentially perfect regioselectivity. No C1 addition of the vinylogous aza-enamine 71 was observed. With regard to synthetic applications, the aza-enamine adduct 72 could be readily transformed into α,β-unsaturated γ-amino nitrile 73 by treatment with magnesium monoperoxyphthalate (MMPP). Product 73 was obtained in nearly quantitative yield without any deterioration of ee. Compound 73 was functionalized further via conjugate addition of methyl magnesium iodide, introducing an additional stereogenic center and providing product 74 in moderate diastereoselectivity.
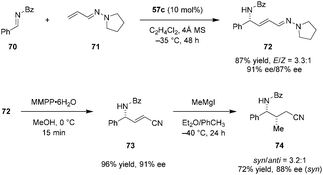 |
| | Scheme 14 Enantioselective vinylogous aza-enamine reaction. | |
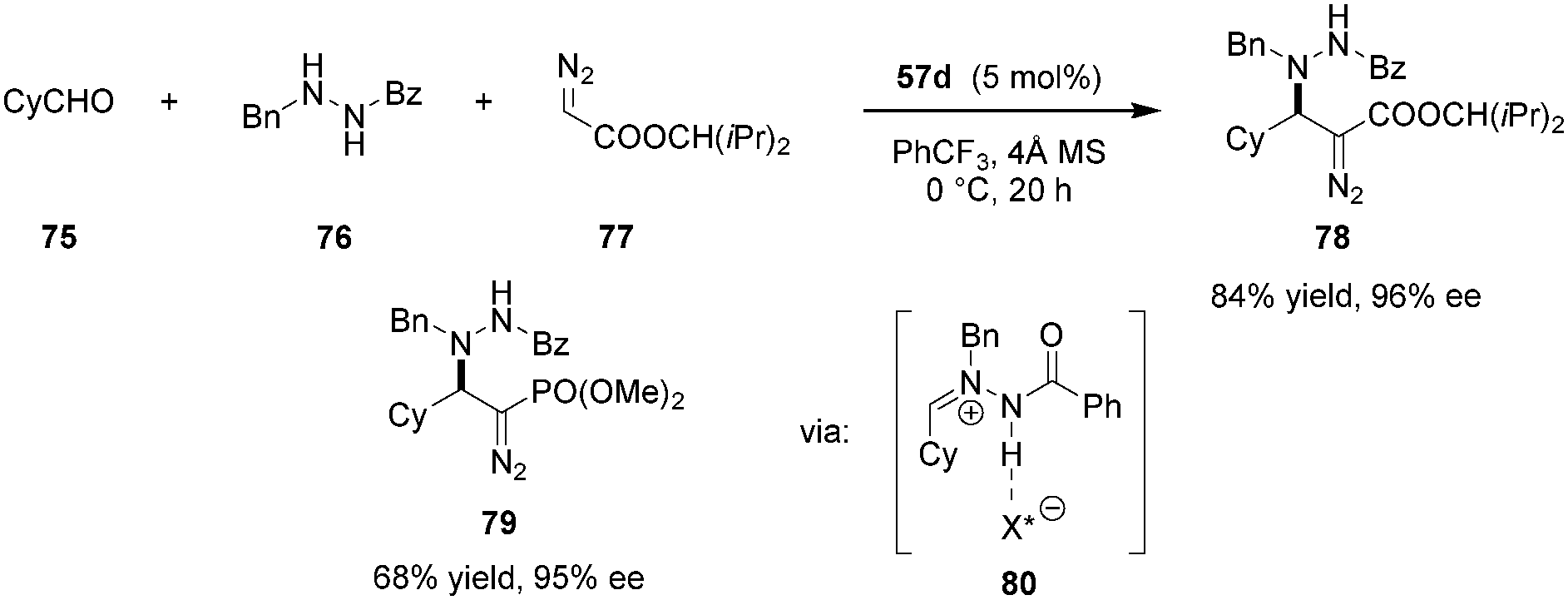
Azomethine imines were also found to be viable electrophiles for activation by Maruoka's carboxylic acid catalysts (Scheme 15).115 In the presence of 57d, transient 80 could be generated by condensation of aldehyde 75 and N′-benzylbenzoylhydrazide 76. Intermediate 80 was trapped via 1,2-addition of alkyl diazoacetates or (diazomethyl)phosphonates, providing a broad range of chiral α-diazo-β-hydrazino esters (e.g., 78) and phosphonates (e.g., 79), analogs of β-amino acids.
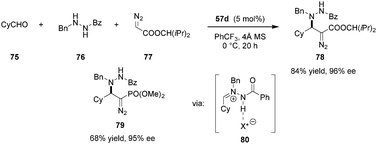 |
| | Scheme 15 Enantioselective addition alkyl diazoacetate to in situ-generated azomethine imines. | |
Applying a similar strategy in which a carboxylic acid catalyst serves to lower the LUMO of an azomethine imine, Maruoka and coworkers reported inverse-electron-demand (IED) 1,3-dipolar cycloadditions of azomethine imines (e.g., 81) with electron-rich olefins such as enol ether 82 (Scheme 16).116exo-Addition products (e.g., 83) were obtained exclusively in excellent yields and ee's. The enal-derived vinylogous aza-enamine 71 was also found to serve as an electron-rich dipolarophile to provide product 84.
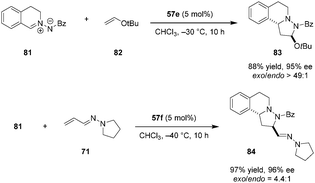 |
| | Scheme 16 Asymmetric inverse-electron-demand (3+2) cycloaddition. | |
N,N′-Cyclic azomethine imines were previously shown by Fu and coworkers to undergo asymmetric (3+2) cycloadditions with alkynes (not shown).117 In contrast, Maruoka et al. found that C,N-cyclic azomethine imines such as 81 exhibit divergent reactivity favoring direct alkynylation, resulting in the formation of C1-substituted tetrahydroisoquinolines (Scheme 17).118 A Cu(I)/Pybox catalyst was found to be suitable for the alkynylation of azomethine imines lacking a substituent at the C1-position. However, only moderate ee's were observed for C1-substituted azomethine imine substrates. It was found that the product ee could be improved significantly by adding the dicarboxylic acid 57d as a cocatalyst, presumably to activate the azomethine imine 85 towards the addition of a chiral Cu-acetylide. In the mismatched case wherein the (S,S)-Ph-Pybox was employed in combination with 57d, the opposite product enantiomer was obtained in significantly diminished yield and ee. This reaction represents the first example of a highly enantioselective catalytic alkynylation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond that furnishes a stereogenic tetrasubstituted carbon center.
N double bond that furnishes a stereogenic tetrasubstituted carbon center.
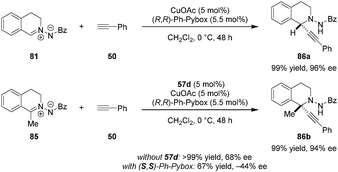 |
| | Scheme 17 Asymmetric alkynylation of C1-substituted C,N-cyclic azomethine imines. | |
The CuOAc/Pybox/axially chiral dicarboxylic acid catalyst system was subsequently applied to the enantioselective 1,3-dipolar cycloaddition of aldehydes, hydrazides and alkynes (Scheme 18).119 In the matched case, the addition of chiral dicarboxylic acid cocatalyst 57d not only improved the enantioselectivity but also suppressed the formation of undesired and competing alkynylation which was observed with the Cu(I)/Pybox catalyst alone. Under the optimized conditions, the in situ-generated acyclic azomethine imines underwent (3+2) cycloaddition with copper acetylides almost exclusively to give a variety of 3,4-disubstituted pyrazolines (e.g., 87a). The substrate scope was found to be broad and encompasses both aromatic and aliphatic aldehydes.
 |
| | Scheme 18 Enantioselective three-component 1,3-dipolar cycloaddition. | |
The desymmetrization of 4-substituted cyclohexanones via enantioselective ring expansion with diazoacetates to give cycloheptanones was achieved by Maruoka and coworkers, utilizing a chiral aluminum-based Lewis acid catalyst (not shown).120 The same research group later showed that the proposed intermediate 91 can also be accessed by protonation of β-diazo alcohols (e.g.88) (Scheme 19). An asymmetric semipinacol rearrangement was achieved by using 57g as a chiral Brønsted acid catalyst.121 For ease of purification and ee determination, the initial rearrangement product 89 was converted to cycloheptanone 90via Krapcho dealkoxycarbonylation. While only moderate yields and ee's were obtained in most cases, this method provides an interesting organocatalytic alternative to access enantioenriched γ-substituted cycloheptanones.
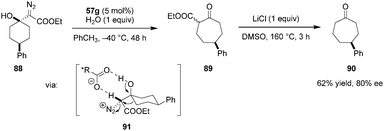 |
| | Scheme 19 Desymmetrization of 4-substituted cyclohexanones via asymmetric semipinacol rearrangement. | |
The development of asymmetric Ugi reactions has been a challenging problem for Brønsted acid catalysis.122 This is particularly true for chiral carboxylic acid catalysts. As a major challenge, the conjugate base of the acid catalyst can attack the highly electrophilic nitrilium intermediate, leading to incorporation of the catalyst into the product. One indirect solution to this problem is the installation of an additional nucleophilic site on the substrate, allowing for intramolecular trapping of the nitrilium intermediate, thus avoiding catalyst deactivation.123,124 Maruoka et al. effectively realized this strategy with dicarboxylic acid catalyst 57g (Scheme 20).125 Following addition of isocyanide 93 to the intermediate chiral ion pair 95, the benzoyl oxygen attacks the nitrilium to provide heterocyclic product 94 in excellent yield and ee. Compound 94 was readily hydrolyzed under acidic conditions to give α-hydrazine amide 97 without loss of ee. The authors also developed a one-pot procedure in which the Ugi reaction was followed by the cleavage of the benzoyl moiety under moderately basic conditions. Utilizing this sequence, the unique benzoxazole product 98 was generated in excellent yield and ee.
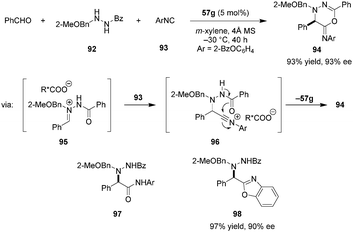 |
| | Scheme 20 Enantioselective Ugi-type reaction with azomethine imines. | |
In another application of their axially chiral dicarboxylic acids, the Maruoka group successfully utilized quinone imine ketals (e.g., 99) as electrophiles (Scheme 21).126 Conjugate addition of enecarbamate 100 to the dicarboxylic-acid-activated quinone imine ketal 99 leads to intermediate 102 which undergoes rearomatization via the elimination of methanol. The resulting iminium ion 103 then reacts with methanol to provide α-amino-β-aryl ether product 101. The latter was obtained in excellent yield, dr, and ee. Quinone imine ketals function as substituted aromatic ring surrogates in this process. Products could be readily transformed into highly enantioenriched β-aryl amines and α-aryl esters. This report represents the first example of using quinone imine ketals in asymmetric Brønsted acid catalysis.
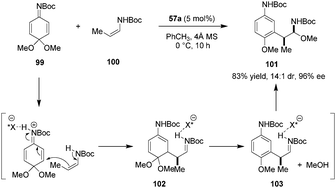 |
| | Scheme 21 Enantioselective arylation of enecarbamates. | |
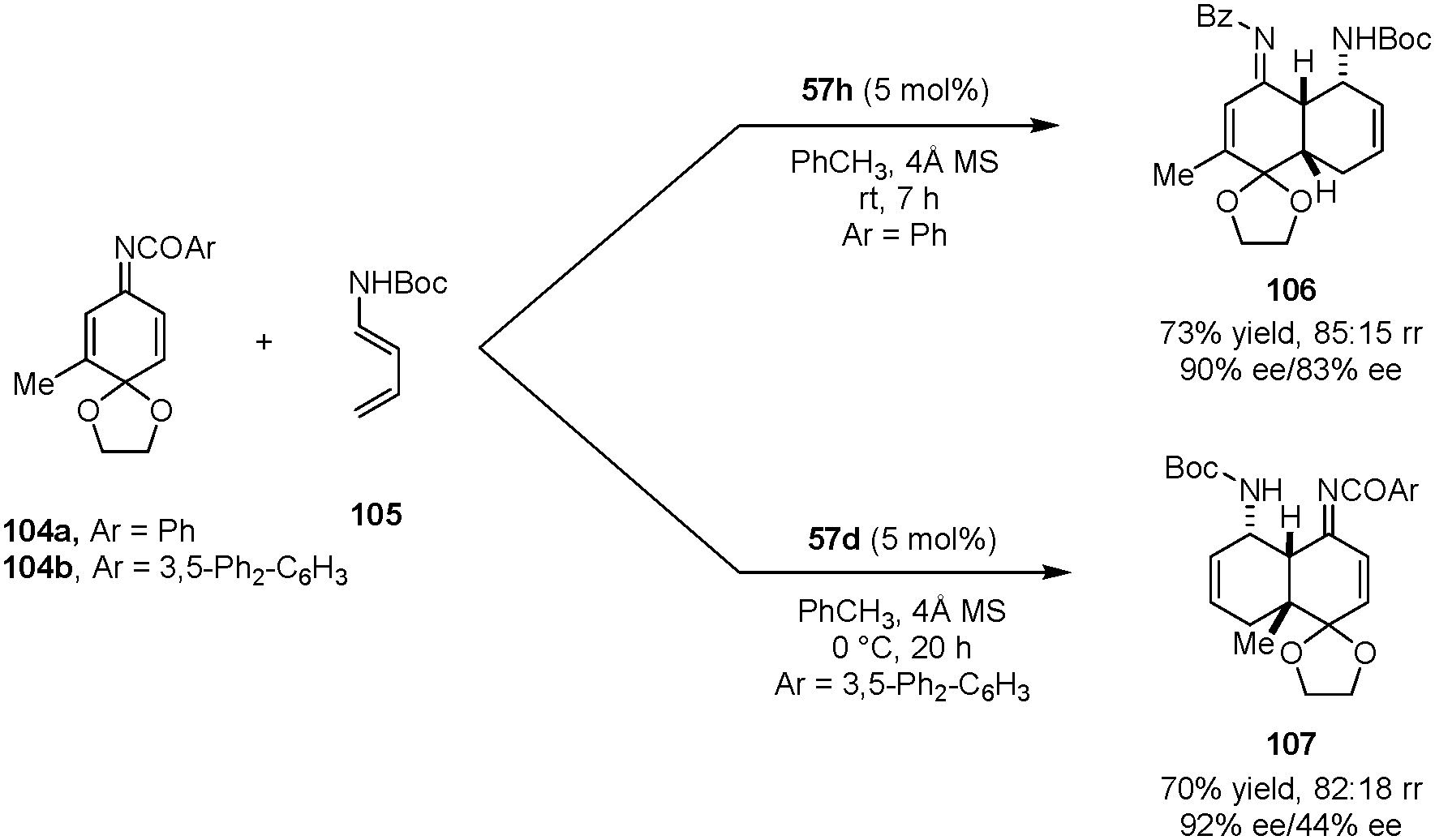
Quinone imine ketals were also employed as dienophiles in asymmetric aza-Diels–Alder reactions with diene carbamates to provide enantioenriched cis-decalin derivatives (Scheme 22).127 In the presence of dicarboxylic acid catalyst 57h, N-benzoyl 3-methyl quinone imine ketal 104a reacts with diene carbamate 105 predominantly on the less hindered CC bond, exclusively providing the endo addition product 106. With the bulkier catalyst 57d and 3,5-diphenylbenzoyl quinone imine ketal 104b, the cycloaddition preferentially occurred on the more substituted alkene site to form product 107. The authors proposed that the regioselectivity originates from the rapid interconversion of the E and Z isomers of the imine. With a bulkier catalyst and protecting group, the Z isomer is preferred in the transition state, directing the diene to add to the sterically more hindered site.
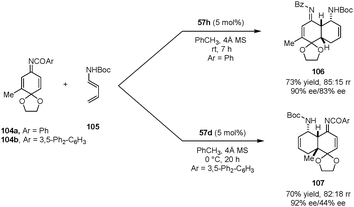 |
| | Scheme 22 Enantioselective and regioselective aza-Diels–Alder reactions of quinone imine ketals. | |
6. Boron-assisted chiral carboxylic acids
In line with Yamamoto's concept of “Lewis acid assisted Brønsted acid catalysis” (LBA), it is well established that the acidity of a Brønsted acid can be increased dramatically upon coordination to a Lewis acid.38 Furthermore, such an interaction can result in more organized catalyst structures and provide an effective chiral environment for asymmetric induction. Taking advantage of this principle, Mattson et al. designed achiral 2-borylbenzoic acid derivatives in which the boron center can internally coordinate to the carboxylic acid.128ortho-Bpin benzoic acid displayed superior activity over simple benzoic acid or silicon-substituted analogs in the addition of indoles to nitroalkenes. Maruoka et al. reported the first successful application of chiral boron-assisted carboxylic acids in asymmetric catalysis. The active catalyst 110 was assembled in situ from 108 and 109 and utilized in trans-selective aziridinations of both N-Boc imines and less electrophilic N-Bn imines (e.g., 111) with diazoacetamides such as 63 (Scheme 23).129 Given the modular nature of catalyst assembly, a diverse range of catalysts could be accessed quickly without the need to isolate individual catalysts. The presence of a stereogenic boron center was found to contribute to the excellent selectivity of the catalyst. Based on 1H-NMR experiments, the authors concluded that only one of the two possible diastereomers of 110 is formed under the reaction conditions. Catalysts based on C2-symmetrical diols provided products in poor enantioselectivities.
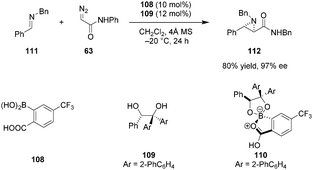 |
| | Scheme 23 Chiral borylbenzoic acid catalyzed enantioselective aza-Darzens reaction. | |
7. Conjugate-base-stabilized carboxylic acids
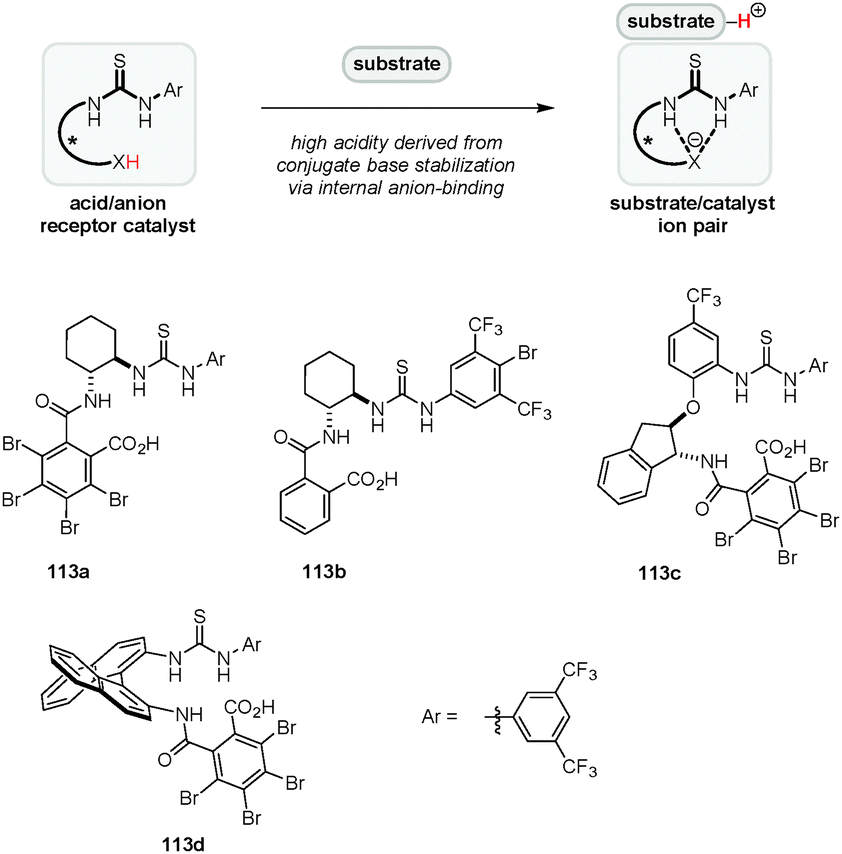
Yamamoto's original concept of “Brønsted acid-assisted Brønsted acid catalysis” (BBA)38 was expanded by Jacobsen and coworkers who utilized a combination of achiral Brønsted acids and chiral (thio)urea catalysts, a highly successful strategy that was utilized in a variety of enantioselective transformations.31,130–134 In these systems, the conjugate base of the Brønsted acid associates with the (thio)urea catalyst by hydrogen bonding/anion-binding. A particular strength of this approach is that the acidity and chiral environment can be readily tuned by altering either catalyst component. A complementary strategy for asymmetric Brønsted acid catalysis was introduced by our group (Fig. 3). We envisioned a new type of chiral Brønsted acid in which the acidic site (e.g., a carboxylic acid) is covalently connected via an appropriate linker to an anion recognition site such as a thiourea moiety. Upon substrate protonation, the conjugate acid binds to the anion recognition site, resulting in the formation of a substrate–catalyst ion pair (Fig. 3).135 The stabilization of the conjugate base by anion-binding is expected to increase the acidity of the catalyst while simultaneously serving to rigidify its structure. Furthermore, anion-binding may reduce the strength of hydrogen-bonding between the carboxylate and the cationic substrate, thus increasing the electrophilicity of the latter. A variety of catalysts possessing different chiral backbones were synthesized and evaluated in a number of reactions.
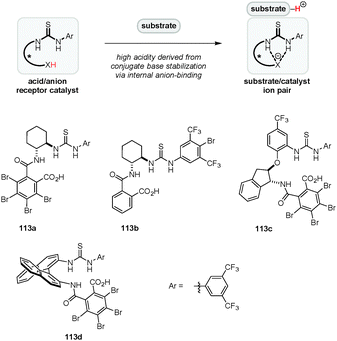 |
| | Fig. 3 Conjugate-base-stabilized chiral carboxylic acids. | |
This concept was applied successfully in the first catalytic enantioselective Povarov reaction of secondary aromatic amines (Scheme 24).135 Out of a series of benzoic acid derivatives, acid-thiourea 113a, a compound that is easily synthesized in two steps from inexpensive starting materials, was identified as the best catalyst for the three-component Povarov reaction involving secondary aromatic amines including indoline (114) and tetrahydroquinoline. Various polycyclic compounds such as 117 were obtained in excellent yields, diastereoselectivities and enantioselectivities. Aliphatic aldehydes, which may be problematic because of the potential for enamine formation, proved to be more reactive than aromatic aldehydes in this system. It should be noted that acid-thioureas, derived from amino acids, were reported prior to our studies.136,137 Thus far, there appear to be no examples in which these compounds are used as chiral Brønsted acid catalysts, although they have been utilized as additives in enamine catalysis.58
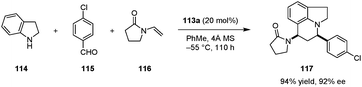 |
| | Scheme 24 Enantioselective Povarov reactions with secondary aromatic amines. | |
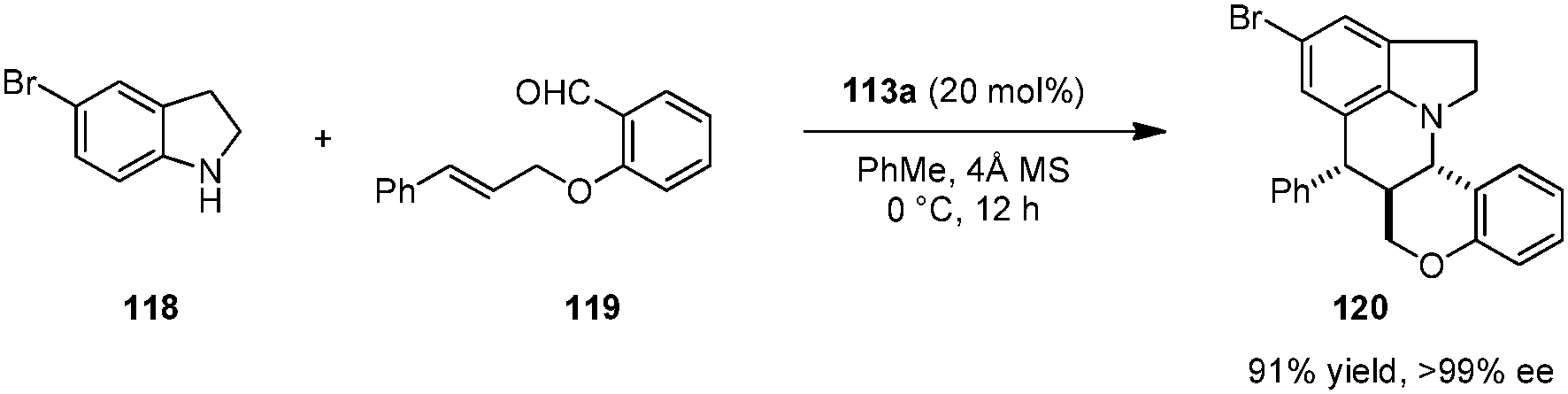
Brønsted acid 113a was also identified as an efficient catalyst for asymmetric intramolecular Povarov reactions.138,139 Tetra- and pentacyclic heterocycles with three contiguous stereogenic centers were obtained with excellent levels of stereoselectivity from secondary aromatic amines and aldehydes possessing a pendent dienophile (Scheme 25).138 The condensation/cycloaddition of 118 and 119 represents the first example of a catalytic enantioselective intramolecular aza-Diels–Alder reaction. Interestingly, the corresponding racemic catalyst (rac-113a) was nearly ineffective in accelerating the formation of racemic products. A non-linear effects study showed that the ee value of the product remained at a virtually identical level down to a catalyst ee value of only 10%. However, the reaction rate and diastereoselectivity dropped dramatically upon reducing catalyst ee. These results point to catalyst aggregation likely playing an important role in the overall catalytic process.
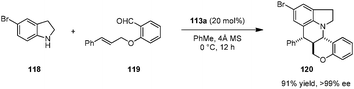 |
| | Scheme 25 Catalytic enantioselective intramolecular aza-Diels–Alder reaction. | |
Catalyst 113a was further applied in asymmetric Pictet–Spengler reactions of unmodified tryptamine (121) with a range of aromatic aldehydes such as 115 to provide β-carboline products (e.g., 122) in excellent yields and enantioselectivities (Scheme 26).140 Catalyst turnover was facilitated via in situ Boc-protection or by using malonic acid as a cheap achiral additive (not shown).
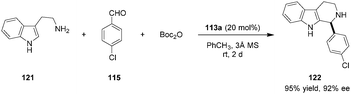 |
| | Scheme 26 Catalytic enantioselective Pictet–Spengler reaction. | |
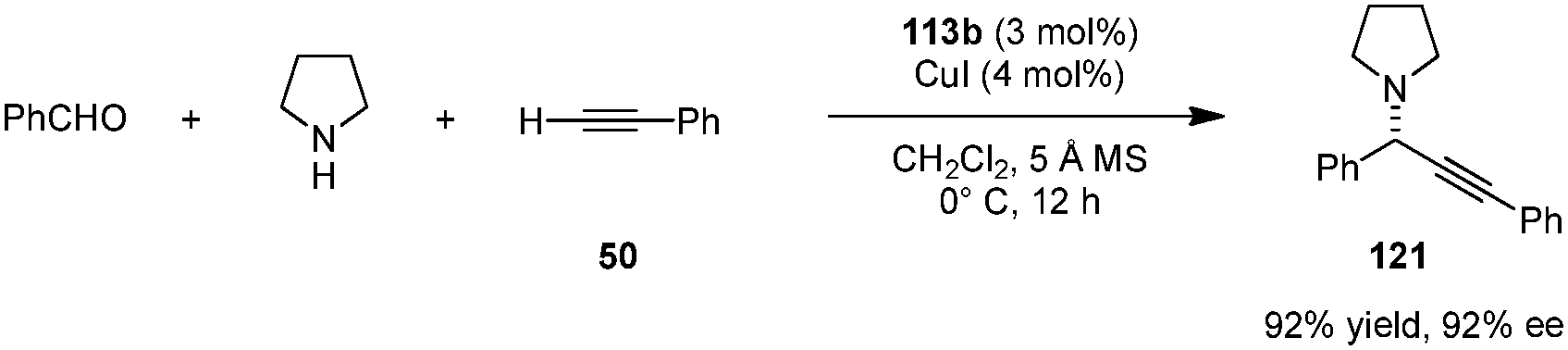
A combination of acid-thiourea catalyst 113b and copper iodide was identified as an efficient catalyst system for enantioselective A3 reactions of secondary amines, in particular pyrrolidine (Scheme 27).141 Addition of terminal alkynes to in situ generated iminium ions provided propargylamines (e.g., 121) in excellent yields and ee's. Pyrrolidine-based propargylamines could be converted to allenes without loss of ee (not shown). The exact roles of each catalyst component in the enantiodetermining step remain unclear.
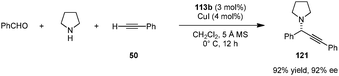 |
| | Scheme 27 Cu(I)/acid-thiourea catalyzed enantioselective A3 reaction. | |
8. Miscellaneous carboxylic acids
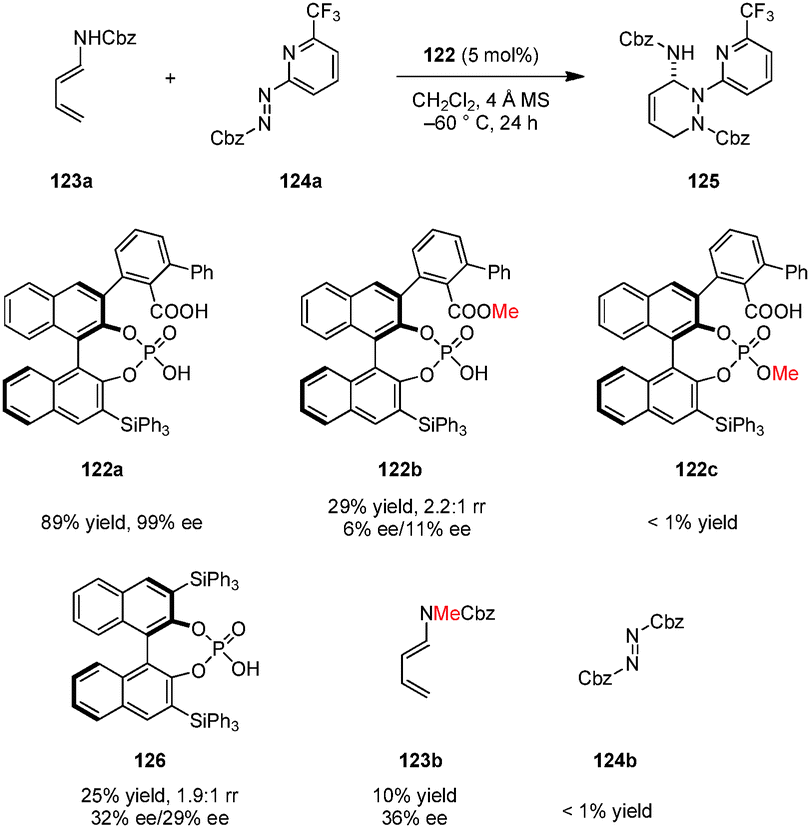
Terada and coworkers devised a novel type of chiral Brønsted acid catalyst possessing both carboxylic acid and phosphoric acid moieties (Scheme 28).142 Catalyst 122a was successfully applied in a highly regio-, diastereo-, and enantioselective aza-Diels–Alder reaction of dienamines such as 123a with azopyridinecarboxylates (e.g., 124a) to provide cycloaddition products (e.g., 125) in excellent yields and ee's. Based on a computational study, the authors proposed that multiple hydrogen bonds between the carboxylic acid, monophosphoric acid and both of the substrates are essential for high selectivity and catalytic efficiency. The carboxylic acid group is thought to act as an intramolecular hydrogen bond donor for a phosphate oxygen atom, an interaction that increases catalyst acidity and results in a more defined chiral environment. Both methyl carboxylate 122b and methyl phosphate 122c were found to be inefficient catalysts. Also, the related chiral phosphoric acid catalyst 126 provided product 125 with significantly decreased yield and ee. Consistent with the proposed hydrogen bonding network between the catalyst and the substrates, both dienamine substrate 123b, lacking a free N–H and 124b, lacking a basic pyridine substituent, gave poor results.
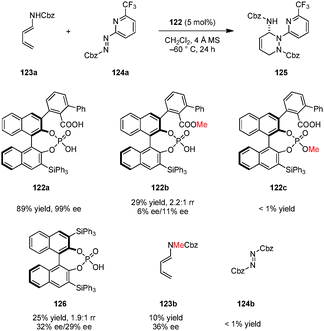 |
| | Scheme 28 Chiral carboxylic acid–monophosphoric acid catalyzed hetero-Diels–Alder reaction. | |
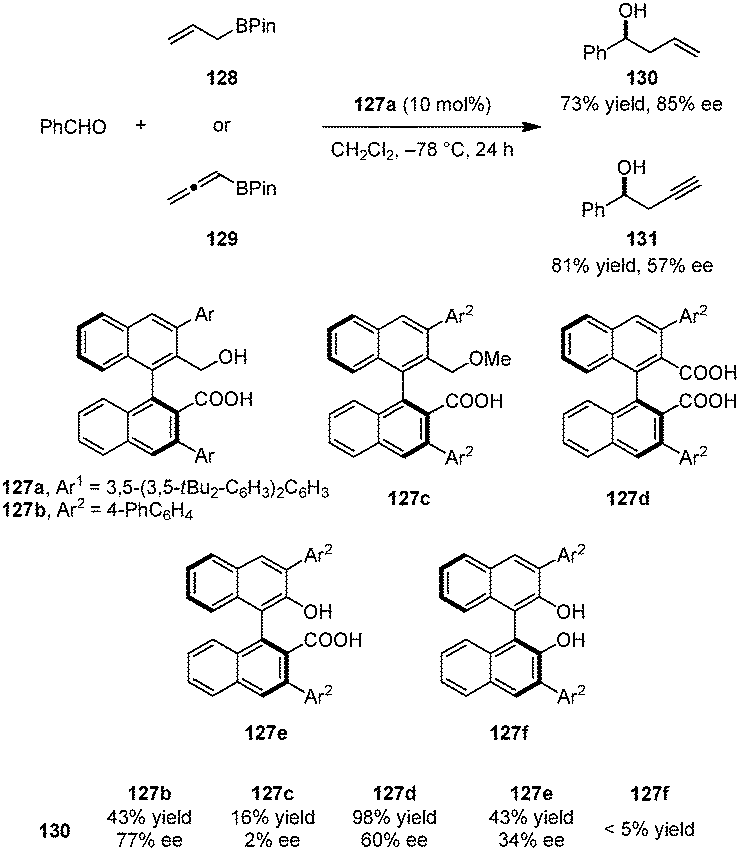
Very recently, the Hamashima group introduced a series of axially chiral hydroxyl carboxylic acid catalysts for the asymmetric allylation and propargylation of aldehydes (Scheme 29).143 Catalyst 127a, whose carboxylate salt was previously employed as an anionic phase-transfer catalyst,144,145 provided the best results in the addition of allyl-boronates (e.g., 128) and allenyl-boronates (e.g., 129) to benzaldehyde to provide the corresponding products 130 and 131. Other catalysts such as the corresponding methyl ether 127c, dicarboxylic acid 127d, naphthol carboxylic acid 127e, and BINOL 127f all gave inferior results, illustrating the importance of both a free carboxyl and a hydroxyl group in suitable positions. In agreement with Antilla's work on chiral phosphoric acid catalyzed allylations of aldehydes,146 the authors proposed that the carboxylic acid serves to protonate the boronate oxygen. In addition, intramolecular hydrogen bonding between the hydroxyl and carboxyl groups is likely to play a significant role in the overall process.
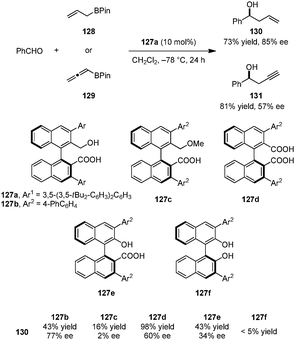 |
| | Scheme 29 Axially chiral hydroxyl carboxylic acids catalyzed asymmetric allylation and propargylation of aldehydes. | |
9. Conclusions
“When the acids compared are neutral (HA) and create negative conjugate bases (A−), it is most convenient to predict the relative acidities by examining the relative stabilities of the anionic conjugate bases. The acid with the most stable conjugate base A− will be the strongest acid.” As elegantly summarized by Anslyn and Dougherty,147 the guiding principle behind increasing the acidity of a neutral acid catalyst is to introduce elements that stabilize its conjugate base. As we hope this review illustrates, the acidity and activity of chiral carboxylic acids can be modulated to such an extent that they become viable candidates for asymmetric catalysis. In addition to classic approaches that focus mostly on introducing strongly electron-withdrawing groups to stabilize the conjugate base by inductive and resonance effects, many other factors can contribute to the stabilization of an anionic conjugate base. These factors include the introduction of additional acidic sites, intramolecular hydrogen bonding/anion-binding, and cooperative action with a Lewis acid. Other factors such as aromaticity,52 electrostatic attraction, and π-effects56,148,149 can also dramatically stabilize an acid's conjugate base and still hold significant potential. Given that multiple design elements can act cooperatively in a single catalyst, as is the case in enzymes, the opportunities for developing highly efficient Brønsted acid catalysts appear to be limitless.
Acknowledgements
This material is based upon work supported by the National Science Foundation under CHE – 1565599.
Notes and references
- M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 4901–4902 CrossRef CAS.
- M. S. Sigman, P. Vachal and E. N. Jacobsen, Angew. Chem., Int. Ed., 2000, 39, 1279–1280 CrossRef CAS PubMed.
- P. R. Schreiner and A. Wittkopp, Org. Lett., 2002, 4, 217–220 CrossRef CAS.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS PubMed.
- Y. Sohtome, A. Tanatani, Y. Hashimoto and K. Nagasawa, Tetrahedron Lett., 2004, 45, 5589–5592 CrossRef CAS.
- R. P. Herrera, V. Sgarzani, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 6576–6579 CrossRef CAS PubMed.
- M. T. Robak, M. Trincado and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 15110–15111 CrossRef CAS PubMed.
- J. P. Malerich, K. Hagihara and V. H. Rawal, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS PubMed.
- R. Ian Storer, C. Aciro and L. H. Jones, Chem. Soc. Rev., 2011, 40, 2330–2346 RSC.
- J. Aleman, A. Parra, H. Jiang and K. A. Jorgensen, Chem. – Eur. J., 2011, 17, 6890–6899 CrossRef CAS PubMed.
- P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Adv. Synth. Catal., 2015, 357, 253–281 CrossRef CAS.
- M. Rombola, C. S. Sumaria, T. D. Montgomery and V. H. Rawal, J. Am. Chem. Soc., 2017, 139, 5297–5300 CrossRef CAS PubMed.
- N. T. McDougal and S. E. Schaus, J. Am. Chem. Soc., 2003, 125, 12094–12095 CrossRef CAS PubMed.
-
M. Shibasaki and S. Matsunaga, Privileged Chiral Ligands and Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 295–332 Search PubMed.
- Y. Huang, A. K. Unni, A. N. Thadani and V. H. Rawal, Nature, 2003, 424, 146 CrossRef CAS PubMed.
- H. Pellissier, Tetrahedron, 2008, 64, 10279–10317 CrossRef CAS.
- B. M. Nugent, R. A. Yoder and J. N. Johnston, J. Am. Chem. Soc., 2004, 126, 3418–3419 CrossRef CAS PubMed.
- A. G. Schafer, J. M. Wieting, T. J. Fisher and A. E. Mattson, Angew. Chem., Int. Ed., 2013, 52, 11321–11324 CrossRef CAS PubMed.
- M. Ganesh and D. Seidel, J. Am. Chem. Soc., 2008, 130, 16464–16465 CrossRef CAS PubMed.
- C. Uyeda and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 9228–9229 CrossRef CAS PubMed.
- P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289–296 RSC.
- M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS PubMed.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed.
-
P. M. Pihko, Hydrogen bonding in organic synthesis, Wiley-VCH, Weinheim, 2009 Search PubMed.
- X. Yu and W. Wang, Chem. – Asian J., 2008, 3, 516–532 CrossRef CAS PubMed.
- Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299–4306 CAS.
- T. J. Auvil, A. G. Schafer and A. E. Mattson, Eur. J. Org. Chem., 2014, 2633–2646 CrossRef CAS.
- J. Lacour and V. Hebbe-Viton, Chem. Soc. Rev., 2003, 32, 373–382 RSC.
- Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC.
- R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed.
- K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed.
- M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed.
- D. Seidel, Synlett, 2014, 783–794 CrossRef CAS.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
-
P. I. Dalko, Comprehensive enantioselective organocatalysis catalysts, reactions, and applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2013 Search PubMed.
- T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed.
- D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
- H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924–1942 CrossRef CAS PubMed.
- T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed.
- M. Terada, Synthesis, 2010, 1929–1982 CrossRef CAS.
- M. Rueping, A. Kuenkel and I. Atodiresei, Chem. Soc. Rev., 2011, 40, 4539–4549 RSC.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed.
-
M. Rueping, D. Parmar and E. Sugiono, Asymmetric Brønsted Acid Catalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2015, pp. 5–86 Search PubMed.
- D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef CAS PubMed.
- P. Garcia-Garcia, F. Lay, P. Garcia-Garcia, C. Rabalakos and B. List, Angew. Chem., Int. Ed., 2009, 48, 4363–4366 CrossRef CAS PubMed.
- S. Vellalath, I. Coric and B. List, Angew. Chem., Int. Ed., 2010, 49, 9749–9752 CrossRef CAS PubMed.
- L. Liu, P. S. J. Kaib, A. Tap and B. List, J. Am. Chem. Soc., 2016, 138, 10822–10825 CrossRef CAS PubMed.
- S. Lee, P. S. Kaib and B. List, J. Am. Chem. Soc., 2017, 139, 2156–2159 CrossRef CAS PubMed.
- N. D. Shapiro, V. Rauniyar, G. L. Hamilton, J. Wu and F. D. Toste, Nature, 2011, 470, 245–249 CrossRef CAS PubMed.
- T. Gatzenmeier, M. van Gemmeren, Y. Xie, D. Hofler, M. Leutzsch and B. List, Science, 2016, 351, 949–952 CrossRef CAS PubMed.
- C. D. Gheewala, B. E. Collins and T. H. Lambert, Science, 2016, 351, 961–965 CrossRef CAS PubMed.
-
H. Yamamoto and K. Ishihara, Acid catalysis in modern organic synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
-
A. J. Kirby, eLS, John Wiley & Sons, Ltd, 2001 Search PubMed.
- T. K. Harris and G. J. Turner, IUBMB Life, 2002, 53, 85–98 CrossRef CAS PubMed.
- C. R. Kennedy, S. Lin and E. N. Jacobsen, Angew. Chem., Int. Ed., 2016, 55, 12596–12624 CrossRef CAS PubMed.
- R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed.
- A. Erkkila, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS PubMed.
- J. Jiang, H. Xu, J. Xi, B. Ren, F. Lv, X. Guo, L. Jiang, Z. Zhang and W. Hu, J. Am. Chem. Soc., 2011, 133, 8428–8431 CrossRef CAS PubMed.
- D. Zhang, H. Qiu, L. Jiang, F. Lv, C. Ma and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 13356–13360 CrossRef CAS PubMed.
- X. Y. Zhou, M. Bao and Y. G. Zhou, Adv. Synth. Catal., 2011, 353, 84–88 CrossRef CAS.
- L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 4006–4123 CrossRef CAS PubMed.
- L. Synoradzki, U. Bernaś and P. Ruśkowski, Org. Prep. Proced. Int., 2008, 40, 163–200 CrossRef CAS.
-
E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive asymmetric catalysis, Springer, Berlin, New York, 1999 Search PubMed.
- L. Duhamel and J.-C. Plaquevent, Tetrahedron Lett., 1977, 18, 2285–2288 CrossRef.
- L. Duhamel and J. C. Plaquevent, J. Am. Chem. Soc., 1978, 100, 7415–7416 CrossRef CAS.
- L. Duhamel and J. C. Plaquevent, Bull. Soc. Chim. Fr., 1982, 75–83 CAS.
- L. Duhamel, P. Duhamel and J. C. Plaquevent, Tetrahedron: Asymmetry, 2004, 15, 3653–3691 CrossRef CAS.
- J. T. Mohr, A. Y. Hong and B. M. Stoltz, Nat. Chem., 2009, 1, 359–369 CrossRef CAS PubMed.
- A. Claraz, S. Oudeyer and V. Levacher, Curr. Org. Chem., 2012, 16, 2192–2205 CrossRef CAS.
- D. Leow, J. Shen, Y. Su and G. Peh, Mini-Rev. Org. Chem., 2014, 11, 410–423 CrossRef CAS.
- S. Oudeyer, J.-F. Briere and V. Levacher, Eur. J. Org. Chem., 2014, 6103–6119 CrossRef CAS.
- H. Ube, S. Fukuchi and M. Terada, Tetrahedron: Asymmetry, 2010, 21, 1203–1205 CrossRef CAS.
- P. N. Moquist, T. Kodama and S. E. Schaus, Angew. Chem., Int. Ed., 2010, 49, 7096–7100 CrossRef CAS PubMed.
- T. Kodama, P. N. Moquist and S. E. Schaus, Org. Lett., 2011, 13, 6316–6319 CrossRef CAS PubMed.
- Y. Luan, K. S. Barbato, P. N. Moquist, T. Kodama and S. E. Schaus, J. Am. Chem. Soc., 2015, 137, 3233–3236 CrossRef CAS PubMed.
- M. Sugiura, M. Tokudomi and M. Nakajima, Chem. Commun., 2010, 46, 7799–7800 RSC.
- N. Grimblat, M. Sugiura and S. C. Pellegrinet, J. Org. Chem., 2014, 79, 6754–6758 CrossRef CAS PubMed.
- K. Furuta, Y. Miwa, K. Iwanaga and H. Yamamoto, J. Am. Chem. Soc., 1988, 110, 6254–6255 CrossRef CAS PubMed.
- K. Furuta, S. Shimizu, Y. Miwa and H. Yamamoto, J. Org. Chem., 1989, 54, 1481–1483 CrossRef CAS.
- K. Furuta, T. Maruyama and H. Yamamoto, J. Am. Chem. Soc., 1991, 113, 1041–1042 CrossRef CAS.
- K. Ishihara, Q. Z. Gao and H. Yamamoto, J. Org. Chem., 1993, 58, 6917–6919 CrossRef CAS.
- K. Ishihara, Q. Z. Gao and H. Yamamoto, J. Am. Chem. Soc., 1993, 115, 10412–10413 CrossRef CAS.
- K. Ishihara, M. Mouri, Q. Z. Gao, T. Maruyama, K. Furuta and H. Yamamoto, J. Am. Chem. Soc., 1993, 115, 11490–11495 CrossRef CAS.
- M. Sugiura, R. Kinoshita and M. Nakajima, Org. Lett., 2014, 16, 5172–5175 CrossRef CAS PubMed.
- M. Sugiura, W. Ishikawa, Y. Kuboyama and M. Nakajima, Synthesis, 2015, 2265–2269 CrossRef CAS.
- H. Guan, H. Wang, D. Huang and Y. Shi, Tetrahedron, 2012, 68, 2728–2735 CrossRef CAS.
- Y. Matsumura, K. Ogura, Y. Kouchi, F. Iwasaki and O. Onomura, Org. Lett., 2006, 8, 3789–3792 CrossRef CAS PubMed.
- M. Bonsignore, M. Benaglia, L. Raimondi, M. Orlandi and G. Celentano, Beilstein J. Org. Chem., 2013, 9, 633–640 CrossRef CAS PubMed.
- Y. Lu, T. C. Johnstone and B. A. Arndtsen, J. Am. Chem. Soc., 2009, 131, 11284–11285 CrossRef CAS PubMed.
- N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 1080–1081 CrossRef CAS PubMed.
- S. Hirashima and H. Yamamoto, J. Synth. Org. Chem., Jpn., 2013, 71, 1116–1125 CrossRef CAS.
- T. Hashimoto and K. Maruoka, J. Synth. Org. Chem., Jpn., 2013, 71, 472–479 CrossRef CAS.
- T. Hashimoto and K. Maruoka, Synthesis, 2008, 3703–3706 CAS.
- T. Hashimoto, T. Takagaki, H. Kimura and K. Maruoka, Chem. – Asian J., 2011, 6, 1936–1938 CrossRef CAS PubMed.
- H. Egami, K. Sato, J. Asada, Y. Kawato and Y. Hamashima, Tetrahedron, 2015, 71, 6384–6388 CrossRef CAS.
- T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2007, 129, 10054–10055 CrossRef CAS PubMed.
- D. Uraguchi, K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2005, 127, 9360–9361 CrossRef CAS PubMed.
- T. Hashimoto, H. Kimura, H. Nakatsu and K. Maruoka, J. Org. Chem., 2011, 76, 6030–6037 CrossRef CAS PubMed.
- T. Hashimoto, N. Uchiyama and K. Maruoka, J. Am. Chem. Soc., 2008, 130, 14380–14381 CrossRef CAS PubMed.
- J. C. Antilla and W. D. Wulff, J. Am. Chem. Soc., 1999, 121, 5099–5100 CrossRef CAS.
- J. C. Antilla and W. D. Wulff, Angew. Chem., Int. Ed., 2000, 39, 4518–4519 CrossRef CAS PubMed.
- Z. Lu, Y. Zhang and W. D. Wulff, J. Am. Chem. Soc., 2007, 129, 7185–7194 CrossRef CAS PubMed.
- A. L. Williams and J. N. Johnston, J. Am. Chem. Soc., 2004, 126, 1612–1613 CrossRef CAS PubMed.
- X. Zeng, X. Zeng, Z. Xu, M. Lu and G. Zhong, Org. Lett., 2009, 11, 3036–3039 CrossRef CAS PubMed.
- T. Hashimoto, H. Nakatsu, K. Yamamoto and K. Maruoka, J. Am. Chem. Soc., 2011, 133, 9730–9733 CrossRef CAS PubMed.
- T. Akiyama, T. Suzuki and K. Mori, Org. Lett., 2009, 11, 2445–2447 CrossRef CAS PubMed.
- T. Hashimoto, M. Hirose and K. Maruoka, J. Am. Chem. Soc., 2008, 130, 7556–7557 CrossRef CAS PubMed.
- D. J. Dixon and A. L. Tillman, Synlett, 2005, 2635–2638 CrossRef CAS.
- M. Rueping, E. Sugiono, T. Theissmann, A. Kuenkel, A. Kockritz, A. Pews-Davtyan, N. Nemati and M. Beller, Org. Lett., 2007, 9, 1065–1068 CrossRef CAS PubMed.
- T. Hashimoto, H. Kimura and K. Maruoka, Tetrahedron: Asymmetry, 2010, 21, 1187–1188 CrossRef CAS.
- T. Hashimoto, H. Kimura and K. Maruoka, Angew. Chem., Int. Ed., 2010, 49, 6844–6847 CrossRef CAS PubMed.
- T. Hashimoto, H. Kimura, Y. Kawamata and K. Maruoka, Nat. Chem., 2011, 3, 642–646 CrossRef CAS PubMed.
- T. Hashimoto, M. Omote and K. Maruoka, Angew. Chem., Int. Ed., 2011, 50, 3489–3492 CrossRef CAS PubMed.
- R. Shintani and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 10778–10779 CrossRef CAS PubMed.
- T. Hashimoto, M. Omote and K. Maruoka, Angew. Chem., Int. Ed., 2011, 50, 8952–8955 CrossRef CAS PubMed.
- T. Hashimoto, Y. Takiguchi and K. Maruoka, J. Am. Chem. Soc., 2013, 135, 11473–11476 CrossRef CAS PubMed.
- T. Hashimoto, Y. Naganawa and K. Maruoka, J. Am. Chem. Soc., 2011, 133, 8834–8837 CrossRef CAS PubMed.
- T. Hashimoto, S. Isobe, C. K. A. Callens and K. Maruoka, Tetrahedron, 2012, 68, 7630–7635 CrossRef CAS.
- S. S. van Berkel, B. G. M. Bogels, M. A. Wijdeven, B. Westermann and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2012, 3543–3559 CrossRef CAS.
- T. Yue, M. Wang, D. Wang, G. Masson and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 6717–6721 CrossRef CAS PubMed.
- Y. Su, M. J. Bouma, L. Alcaraz, M. Stocks, M. Furber, G. Masson and J. Zhu, Chem. – Eur. J., 2012, 18, 12624–12627 CrossRef CAS PubMed.
- T. Hashimoto, H. Kimura, Y. Kawamata and K. Maruoka, Angew. Chem., Int. Ed., 2012, 51, 7279–7281 CrossRef CAS PubMed.
- T. Hashimoto, H. Nakatsu, Y. Takiguchi and K. Maruoka, J. Am. Chem. Soc., 2013, 135, 16010–16013 CrossRef CAS PubMed.
- T. Hashimoto, H. Nakatsu and K. Maruoka, Angew. Chem., Int. Ed., 2015, 54, 4617–4621 CrossRef CAS PubMed.
- T. J. Auvil and A. E. Mattson, Synthesis, 2012, 2173–2180 CAS.
- T. Hashimoto, A. O. Galvez and K. Maruoka, J. Am. Chem. Soc., 2013, 135, 17667–17670 CrossRef CAS PubMed.
- R. S. Klausen and E. N. Jacobsen, Org. Lett., 2009, 11, 887–890 CrossRef CAS PubMed.
- R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030–5031 CrossRef CAS PubMed.
- H. Xu, S. J. Zuend, M. G. Woll, Y. Tao and E. N. Jacobsen, Science, 2010, 327, 986–990 CrossRef CAS PubMed.
- Y. Lee, R. S. Klausen and E. N. Jacobsen, Org. Lett., 2011, 13, 5564–5567 CrossRef CAS PubMed.
- S. Lin and E. N. Jacobsen, Nat. Chem., 2012, 4, 817–824 CrossRef CAS PubMed.
- C. Min, N. Mittal, D. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 14084–14088 CrossRef CAS PubMed.
- D.-Q. Xu, H.-D. Yue, S.-P. Luo, A.-B. Xia, S. Zhang and Z.-Y. Xu, Org. Biomol. Chem., 2008, 6, 2054–2057 CAS.
- Z.-B. Li, S.-P. Luo, Y. Guo, A.-B. Xia and D.-Q. Xu, Org. Biomol. Chem., 2010, 8, 2505–2508 CAS.
- C. Min, C. T. Lin and D. Seidel, Angew. Chem., Int. Ed., 2015, 54, 6608–6612 CrossRef CAS PubMed.
- C. Min and D. Seidel, Chem. – Eur. J., 2016, 22, 10817–10820 CrossRef CAS PubMed.
- N. Mittal, D. X. Sun and D. Seidel, Org. Lett., 2014, 16, 1012–1015 CrossRef CAS PubMed.
- C. Zhao and D. Seidel, J. Am. Chem. Soc., 2015, 137, 4650–4653 CrossRef CAS PubMed.
- N. Momiyama, H. Tabuse, H. Noda, M. Yamanaka, T. Fujinami, K. Yamanishi, A. Izumiseki, K. Funayama, F. Egawa, S. Okada, H. Adachi and M. Terada, J. Am. Chem. Soc., 2016, 138, 11353–11359 CrossRef CAS PubMed.
- Y. Hamashima, Y. Ota, Y. Kawato and H. Egami, Synlett, 2017, 976–980 CrossRef.
- H. Egami, J. Asada, K. Sato, D. Hashizume, Y. Kawato and Y. Hamashima, J. Am. Chem. Soc., 2015, 137, 10132–10135 CrossRef CAS PubMed.
- S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed.
- P. Jain and J. C. Antilla, J. Am. Chem. Soc., 2010, 132, 11884–11886 CrossRef CAS PubMed.
-
E. V. Anslyn and D. A. Dougherty, Modern physical organic chemistry, University Science, Sausalito, 2006 Search PubMed.
- C.-T. Chen and J. S. Siegel, J. Am. Chem. Soc., 1994, 116, 5959–5960 CrossRef CAS.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed.
Footnote |
| † Present address: Department of Chemistry, University of Florida, Gainesville, Florida 32611, USA. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*