
Cross-coupling of aromatic esters and amides†

Abstract
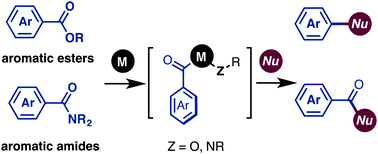
Catalytic cross-coupling reactions of aromatic esters and amides have recently gained considerable attention from synthetic chemists as de novo and efficient synthetic methods to form C–C and C–heteroatom bonds. Esters and amides can be used as diversifiable groups in metal-catalyzed cross-coupling: in a decarbonylative manner, they can be utilized as leaving groups, whereas in a non-decarbonylative manner, they can form ketone derivatives. In this review, recent advances of this research topic are discussed.
 Please wait while we load your content...
Please wait while we load your content...

