
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Plasma technology – a novel solution for CO2 conversion?
Ramses
Snoeckx
 * and
Annemie
Bogaerts
*
* and
Annemie
Bogaerts
*
Research group PLASMANT, Department of Chemistry, University of Antwerp, Universiteitsplein 1, BE-2610 Antwerp, Belgium. E-mail: ramses.snoeckx@uantwerpen.be; annemie.bogaerts@uantwerpen.be
First published on 21st August 2017
Abstract
CO2 conversion into value-added chemicals and fuels is considered as one of the great challenges of the 21st century. Due to the limitations of the traditional thermal approaches, several novel technologies are being developed. One promising approach in this field, which has received little attention to date, is plasma technology. Its advantages include mild operating conditions, easy upscaling, and gas activation by energetic electrons instead of heat. This allows thermodynamically difficult reactions, such as CO2 splitting and the dry reformation of methane, to occur with reasonable energy cost. In this review, after exploring the traditional thermal approaches, we have provided a brief overview of the fierce competition between various novel approaches in a quest to find the most effective and efficient CO2 conversion technology. This is needed to critically assess whether plasma technology can be successful in an already crowded arena. The following questions need to be answered in this regard: are there key advantages to using plasma technology over other novel approaches, and if so, what is the flip side to the use of this technology? Can plasma technology be successful on its own, or can synergies be achieved by combining it with other technologies? To answer these specific questions and to evaluate the potentials and limitations of plasma technology in general, this review presents the current state-of-the-art and a critical assessment of plasma-based CO2 conversion, as well as the future challenges for its practical implementation.
 Ramses Snoeckx | Ramses Snoeckx, born in 1988, obtained his Master's degree in both Environmental Science and Chemistry. Combining these backgrounds, he successfully obtained his PhD in Chemistry (2017) for his research on the plasma-based conversion of greenhouse gases into value-added chemicals and fuels. He actively collaborated with several (inter)national research institutions, for example during research visits at the Instituto Superior Técnico (Portugal), the King Abdullah University of Science and Technology (Saudi Arabia) and the Drexel Plasma Institute (USA). The coming year he will continue his research in this area as post-doctoral fellow at the King Abdullah University of Science and Technology. |
 Annemie Bogaerts | Annemie Bogaerts, born in 1971, obtained her PhD in Chemistry in 1996, from the University of Antwerp in Belgium. She became professor of physical chemistry in 2003, at this university, and has now been full professor since 2012. She is head of the interdisciplinary research group PLASMANT. The research activities of her group include the modelling of plasma chemistry, plasma reactor design and plasma–surface interactions, as well as carrying out plasma experiments for various applications, including environmental and medical applications (mainly cancer treatment), as well as nanotechnology and analytical chemistry. In recent years, special attention has also been given to CO2 conversion by plasma and plasma catalysis. |
1. Introduction
Environmental and energy applications of low temperature plasmas are gaining increasing interest worldwide. The central research question is whether plasma-based solutions can yield a valuable alternative to existing thermal processes and whether they can compete with other novel gas conversion technologies. Nowadays, the conversion of CO2 into chemicals and fuels is a hot topic. The worldwide transition to renewable energy gives plasma processes a clean electricity source, and due to their high operation flexibility, plasmas are very suitable for storing this intermittent renewable energy in a chemical form, i.e. as fuels and chemicals.1.1. CO2 mitigation and valorisation
Throughout history, the use of natural resources has played a major role in the rapid development of the human race. Among these resources, fossil fuels in particular have contributed to a fast and unprecedented development in human society. Still, this comes with a great cost, since burning fossil fuels leads to the emission of large amounts of the greenhouse gas CO2. Because these anthropogenic CO2 emissions outpace the natural carbon cycle, atmospheric CO2 concentrations have been increasing from 280 ppm since the beginning of the industrial revolution to 400 ppm in 2014.1 With high certainty, it can be said that it is this increase that has led to the current adverse global environmental climate changes,1 which have a growing detrimental effect on our climate and environment, and that represent a severe threat to our current society and future generations in general.2–5Therefore, the conversion of this main greenhouse gas into value-added chemicals and liquid fuels is considered as one of the main challenges for the 21st century.1,2 The aim is not only to tackle climate change, but also to provide an answer to our dependence on fossil fuels. As stated by Goeppert et al.,6 “Whether humankind uses up most of the fossil fuel resources (combined with carbon capture) or uses increasingly alternative energies, the need for transportation fuels and materials that we currently obtain from petroleum and natural gas will remain. With increasing population, products based on carbon from plastics to medicines will also be required in increasing quantities. In order to fulfil the demand for carbon-based products, CO2will have to be recycled in an anthropogenic version of nature's own carbon cycle. Carbon capture and recycling (CCR) will capture CO2from any source, and eventually mainly from the atmosphere, and recycle it to new materials and fuels using any alternative energy source.”
Utilization of this waste and converting it into a new feedstock not only complies with the framework of sustainable and green chemistry7,8 but also fits within the ‘cradle-to-cradle’ concept.9 By generating useful products out of CO2 we create the possibility to effectively close the carbon loop. This has already resulted in a booming interest in technologies that can convert CO2 into value-added products,10–12 since they can effectively convert waste into new feedstocks following the cradle-to-cradle principle.9 Besides chemical conversion, which is the principle focus of this review, CO2 also has other applications in the field of carbon dioxide capture, storage and utilization, such as its fixation and technological utilization, for these and other processes, as well as in carbon capture, for which we refer to several existing reviews in the literature.12–19
Besides the traditional thermal CO2 conversion, several alternative technologies are being investigated, such as electrochemical, solar thermochemical, photochemical and biochemical pathways, either with or without catalysts, as well as all their possible combinations. Their advantages and disadvantages are briefly discussed herein, as they form the context for a novel approach considered to have great potential in recent years, which is the approach based on (non-thermal) plasma.20,21 Several options are being investigated in this regard, including both pure CO2 splitting into CO and O2, as well as the reaction with other gases, like CH4 (dry reforming of methane), H2 (hydrogenation of CO2) or H2O (artificial photosynthesis), aiming for the production of syngas and valuable oxygenates, such as methanol, formaldehyde and formic acid.
1.2. Solar fuels
The important difference between the traditional thermal conversion approaches and the emerging technologies is their independence of burning fossil fuels to provide the necessary thermal heat to drive reactions. More precisely, biochemical pathways rely on the natural photosynthesis process to convert light (either natural or artificial) into biomass, which can then be harvested and further processed. Other emerging technologies, such as the solar thermochemical and photochemical processes rely on direct solar energy. The average solar flux striking the earth's surface is 175 W m−2, which represents more energy striking the earth's surface in two hours, i.e. 640 EJ, than the worldwide energy consumption from all sources combined in 2008, i.e. 514 EJ.22 The solar thermochemical process makes use of concentrated solar heat, while the photochemical process relies on the energy of photons. Ultimately though, the electrochemical and plasmachemical processes rely on electricity. Fig. 1 gives an overview of the different novel technologies and their principal use of renewable energy for the conversion of CO2 in a carbon neutral cycle. | ||
| Fig. 1 Overview of the different novel technologies and their principal use of renewable energy for the conversion of CO2 in a carbon neutral cycle. | ||
The reliance of electrochemical and plasmachemical conversions on electricity at first seems to limit their use as a greenhouse gas mitigation technology, since currently, producing electricity generally results in CO2 emissions. This idea though could not be further from the truth due to the worldwide transition to renewable energy sources, such as solar and wind energy. In 2014, the estimated renewable energy share of the global final energy consumption was already 19.2%, while by the end of 2015, the estimated renewable energy share of global electricity production was 23.7%.23 It is even at such a stage that the large-scale adoption of these renewable energy sources poses a challenge for the efficient storage and easy transport of the electricity produced, i.e. not only regarding the need for peak shaving, but more importantly the need for technologies to follow the irregular and at times intermittent supply of renewable electricity in a flexible way. While storage in batteries is possible, it is less efficient than chemical storage in fuels.24 Such fuels, often referred to as carbon neutral fuels or solar fuels, offer a much higher gravimetric and volumetric energy storage capacity, have much higher energy densities than electrical storage techniques and they match the existing worldwide liquid fuel infrastructure.12,24
In the first instance, the reactions in which CO2 is involved can be divided into two categories: the production of chemicals and the production of fuels. The latter is considered as the most suitable target for the conversion of large volumes of CO2 since its market size is 12–14 times larger than the former. One of the most interesting compounds is methanol, which is positioned exactly in the middle of these two categories, being at the same time a raw chemical and a fuel, used in both combustion engines and fuel cells.25
To achieve the transformation of CO2 into value-added chemicals or fuels, the reactions that are of greatest interest involve the conversion of CO2 with a co-reactant that acts as a hydrogen source (like CH4, H2 or H2O). Due to the existing infrastructure, liquid products are preferable to gases, for most applications at least. Two approaches can be considered to achieve this: the indirect oxidative pathway and the direct oxidative pathway. The main product of the former is syngas, a mixture of H2 and CO, which can be converted to almost any commercial bulk chemical or fuel in a second – albeit very energy intensively – step through methanol and/or Fischer–Tropsch synthesis.10 In this case, it is of great importance to have a high degree of control over the H2/CO ratio to be able to steer the synthesis towards the desired products.26 The direct oxidative pathway, on the other hand, tries to eliminate the energy-intensive middle man by converting the reactants immediately into hydrocarbons, short-chain olefins (e.g. ethylene and propylene) and oxygenated products (e.g. methanol, formaldehyde, dimethyl ether and formic acid).
Liquid products are more attractive over gaseous hydrogen, since – while in theory a ‘Hydrogen Economy’27 would be very attractive – the latter has a number of serious drawbacks due to its physico-chemical properties.28 Furthermore, the infrastructure needed to safely transport, store and dispense hydrogen would be very expensive to roll out, while liquid chemicals match the already existing worldwide fuel infrastructure.12,24 Hence, especially in the transportation sector, a transition from liquid–fossil-fuel-derived products (e.g. gasoline, diesel fuel, kerosene) to a renewable and sustainable liquid fuel is highly desirable. Again, methanol is one of the most interesting possible candidates to fulfil these requirements. It is the simplest liquid chemical containing only one carbon. Although to date it is almost exclusively produced from natural gas (and shale gas) for economic reasons, it can easily be obtained from several (future) carbon sources, incl. CO2, biomass, biogas and landfill gas. Therefore, it has been proposed as a key solar fuel for the above-mentioned anthropogenic carbon cycle under the framework of a ‘Methanol Economy’.6
To be economically competitive with the existing structures, the efficient production of these solar fuels is critical during the quest to find an effective CO2 conversion technology with the potential to be commercialized on a large scale.
2. Traditional thermal CO2 conversion approaches
We first briefly discuss the existing traditional (mainly thermocatalytic) approaches used on an industrial scale. As such, this section will act as a comparison for the novel technologies under development. This section is subdivided into: (1) pure CO2 splitting and (2) CO2 conversion in combination with a co-reactant, i.e. CH4, H2 or H2O.2.1. Pure CO2 splitting
Thermal CO2 splitting has not been very effective to date. This is not surprising from a thermodynamic point of view; the carbon–oxygen bonds are relatively strong (783 kJ mol−1)29 and the Gibbs free energy of formation (ΔG° = −394 kJ mol−1)24 clearly shows that CO2 is a highly stable molecule, requiring a substantial energy input, optimized reaction conditions and active catalysts for any chemical conversion to take place. Neither the entropy (TΔS°) nor the enthalpy (ΔH°) term seem favourable for its conversion.2 The overall reaction is written as:| CO2(g) → CO(g) + 1/2O2(g) ΔH° = +283 kJ mol−1 | (R1) |
| CH4(g) + H2O(g) → CO(g) + 3H2(g) ΔH° = +206 kJ mol−1 | (R2) |
It is, however, clear that, without actively removing one of the products (i.e. CO or O2), the equilibrium of this reaction lies strongly to the left. Thus, thermal CO2 splitting is thermodynamically and energetically only favourable at very high temperatures, as can be seen in Fig. 2. At 2000 K for instance, the reaction is not very efficient: we can easily estimate that ca. 92 kJ mol−1 would be needed to heat 1 mole of CO2 from 300 to 2000 K. Furthermore, the reaction enthalpy is equal to 245 kJ mol−1 at 2000 K. Based on a conversion of 1.5% at this temperature, the energy cost for the total conversion is ∼7.9 MJ mol−1, yielding an energy efficiency of only 4.4% with respect to the reaction enthalpy of 283 kJ mol−1 at 300 K. On the other hand, ca. 184 kJ mol−1 would be needed to heat 1 mole of CO2 to 3500 K, and at this temperature the reaction enthalpy is equal to 206 kJ mol−1. Hence, based on a conversion of 80% at this temperature, the energy cost of the total conversion is then only ∼602 kJ mol−1, yielding an energy efficiency of 47% with respect to the reaction enthalpy of 283 kJ mol−1 at 300 K. While the conversion continuing to increase, above 3500 K the energy efficiency starts to decrease. At 5000 K, the conversion is 100% but the energy efficiency is only 35%, as can be deduced from Fig. 2.
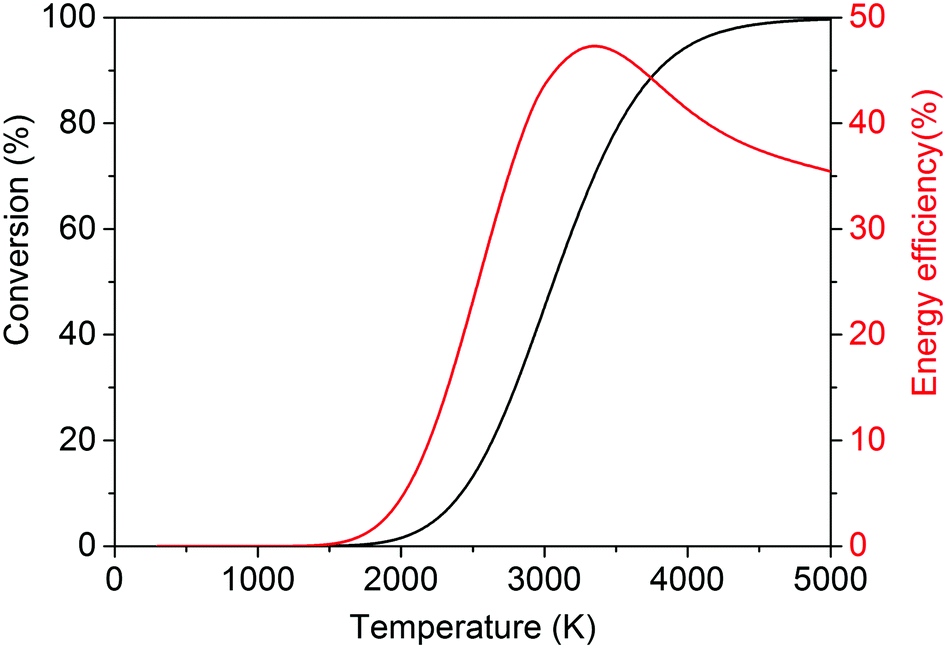 | ||
| Fig. 2 Calculated theoretical thermal conversion (left axis) and corresponding energy efficiency (right axis) as a function of temperature for the pure splitting of CO2 into CO and O2. | ||
Thus, it is clear that the equilibrium production of CO and O2 varies from less than 1% at temperatures below 2000 K up to 45–80% at the temperature range 3000–3500 K.29,30 Therefore, the most pertinent studies regarding thermal CO2 splitting involve membrane reactor systems. Nigara and Cales32 used a calcia-stabilized zirconia membrane and CO as the sweep gas. At a temperature of 1954 K, they were able to reach a conversion of 21.5%, whereas the equilibrium production was a mere 1.2% at the same temperature.32 The overall conversion, however, was much lower due to the permeation of O2 through the membrane, whereby it recombined with the CO sweep gas to form CO2. Itoh et al.33 employed an oxygen permeable yttria-stabilized zirconia membrane and used argon as the sweep gas. Unfortunately, despite the removal of oxygen through the membrane, conversions of only up to 0.5% were obtained for a maximum temperature of 1782 K. Fan et al.34 used a solid oxide (SrCo0.5FeO3) membrane reactor and methane as the sweep gas. Conversions of up to 10% were found at a temperature of 1213 K, which are one to two orders of magnitude higher than what could be expected to be attained conventionally. Nonetheless, the feed gas was diluted with four parts of helium per one part of CO2, and when this is taken into account, an effective conversion of only 2% was actually reached.
To summarize, these studies have demonstrated the possibility of producing CO and O2 by the direct thermal splitting of CO2 – at lower temperatures than the equilibrium predictions – by means of the use of semipermeable membranes to extract oxygen. Nevertheless, the attained overall effective conversions of 0.5–2% are too low to be considered practical for successful application on an industrial scale.
Because to date none of the above-mentioned alternative approaches to split CO2 at lower temperatures have yet realized acceptable conversions and energy efficiencies, thermocatalytic CO2 splitting is currently not applied on an industrial scale. The reason for this is the high energy consumption and, in addition, the lack of effective techniques for separating CO and O2 at high temperatures to avoid ending up with an explosive mixture. Nevertheless, we presented this brief summary of the initial efforts regarding thermal CO2 splitting to support a complete understanding of the matter, and because this work has laid the foundations for one of the novel technologies, i.e. solar thermochemical CO2 splitting, discussed later (see Section 3.2).
2.2. Conversion of CO2 with a co-reactant
Due to the inherent high energy consumption and derived low energy efficiency of thermocatalytic CO2 splitting, the only practical way to reform CO2 consists of using a co-reactant. Thermodynamically speaking, it is significantly easier to convert CO2 when it is paired with a co-reactant that has a higher, i.e. less negative, Gibbs free energy.24 Some suitable candidates are CH4 (ΔG° = −50.7 kJ mol−1) and H2 (ΔG° = 0 kJ mol−1). In essence, these hydrogen-bearing energy carriers give up their intrinsic chemical energy to promote the conversion of CO2.As such, it is no surprise that the most widely investigated traditional processes to convert CO2 involve the reaction with either CH4 or H2. The former is one of the best known traditional processes for reforming CO2 into synthesis gas or syngas, which is a mixture of H2 and CO. The reaction with H2 is known as the Sabatier reaction, which is a well-known process to generate CH4 (and H2O). Additionally, the combination of CO2 and H2 can also be used to produce methanol through the methanol synthesis process. A final process of interest to mention is the combined conversion of CO2 and H2O, a technique for which there is no real traditional approach. Nevertheless, we briefly mention it here because water is an interesting co-reactant to pursue for the growing array of novel techniques. After all, H2O is not only the most ubiquitous and cheapest hydrogen source, compared to CH4 and H2, but converting CO2 in combination with H2O to produce value-added products using renewable energy would successfully mimic natural photosynthesis.35,36 It is interesting to note that besides CH4, H2 and H2O, other possible hydrogen sources could also be considered, such as glycerol. The use of glycerol as a hydrogen donor has already been suggested and successfully implemented for different technologies, such as its use as a hydrogen donor (and green solvent) in catalytic transfer hydrogenation–dehydrogenation reactions of various unsaturated organic compounds;37 its use as a hydrogen donor for the production of CH3OH from CO2 in a hybrid enzymatic/photocatalytic approach38 and its use in the plasma-based reforming of glycerol towards syngas,39 which also led to the suggestion of using glycerol as a hydrogen donor for the possible in situ trapping of oxygen during the plasmachemical splitting of CO2.40 However, in this review, the focus lies on the above-mentioned three most commonly used hydrogen sources CH4, H2 and H2O.
| CH4(g) + CO2(g) → 2CO(g) + 2H2(g) ΔH° = +247 kJ mol−1 | (R3) |
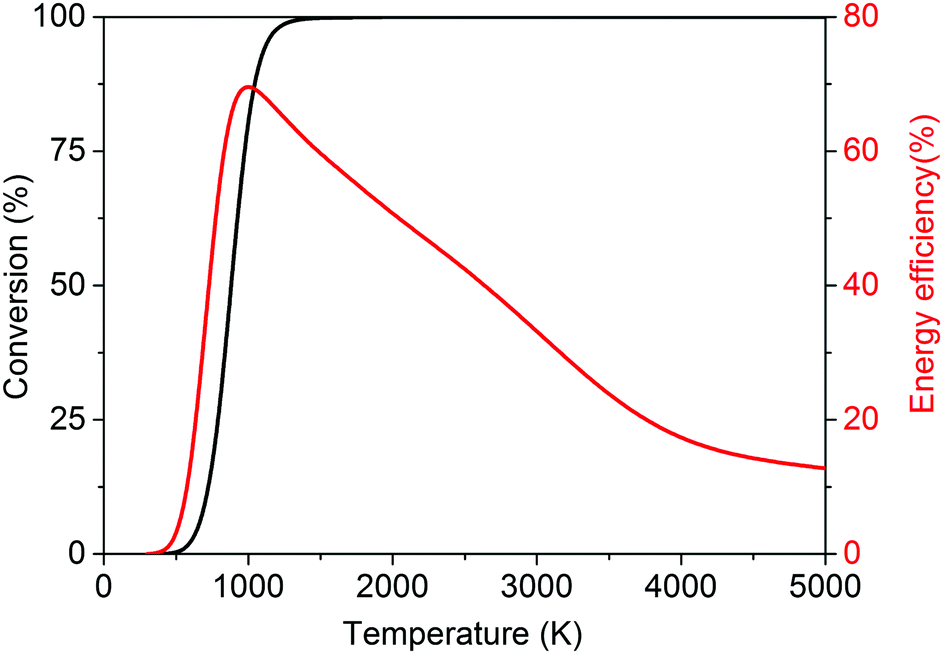 | ||
| Fig. 3 Calculated theoretical thermal conversion (left axis) and corresponding energy efficiency (right axis) as a function of temperature for the dry reforming of methane. | ||
The dry reforming of methane (DRM) has quite a history. It was first studied by Fischer and Tropsch in 1928,42 and has been a challenge for chemical engineering ever since.41 Since that time, the rationale for investigating this process has adapted itself several times to the spirit of the age. In its origin, it arose from a desire for alternative ways to produce fuels and chemicals (in combination with the Fischer–Tropsch synthesis) due to the limited supply of fossil fuels during the Second World War.43 A renewed interest was found in the 1970s in the aftermath of the oil crisis,43 again to circumvent the need for fossil fuels and with the idea of utilizing cheaper and more abundant natural gas. With the beginning of a new millennium and the increasing concern regarding climate change, DRM was seen as a way to convert the major greenhouse gas CO2 into useful products with the aid of natural gas.31,44 To date, a true amalgam of environmental and economical motivations exist, such as the conversion of the greenhouse gas CO2, the capability of using biogas as a feedstock, the search for a convenient way to liquefy CH4 for easier transport, and the availability of cheap CH4 through shale gas.12,18,31,41,42,44–46
Alas, despite all the bright outlooks, there is one major pitfall, namely the process’ inherent susceptibility for soot deposition and the detrimental effect this has on the process through deactivation of the catalyst. Due to this drawback, DRM is to date not yet (widely) used on an industrial scale. Of course, a lot of research is still ongoing towards modified catalysts to circumvent this coking issue, which was originally also a big problem for the currently widely adopted steam reforming of methane. Nevertheless, the inability to transform the alluring promises of DRM into reality through the traditional thermal methods – among other reasons– has sparked and fuelled the growing interest for alternative reforming technologies, as is discussed in Sections 3 and 4.
| CO2(g) + 4H2(g) → CH4(g) + 2H2O(g) ΔH° = −165.3 kJ mol−1 | (R4) |
The selective hydrogenation of CO2 to methanol, on the other hand, is a process that is currently operated on an industrial scale. The annual worldwide production of methanol is estimated to be around 70 M metric tonnes (2015). The most common commercial catalyst is copper supported on high surface area alumina (often promoted with zinc oxide).47 The relevant reactions for the selective hydrogenation of CO2 are:50
| CO2(g) + H2(g) → CO(g) + H2O(g) ΔH° = +40.9 kJ mol−1 | (R5) |
| CO(g) + 2H2(g) → CH3OH(g) ΔH° = −90.8 kJ mol−1 | (R6) |
| CO2(g) + 3H2(g) → CH3OH(g) + H2O(g) ΔH° = −49.9 kJ mol−1 | (R7) |
One of the main drawbacks of the selective hydrogenation of CO2 to methanol in the above case is the production of water as a by-product (see reaction (7)). A third of the H2 is thus converted to water compared to the complete conversion to methanol when starting from syngas (see reaction (6)). Furthermore, the thermodynamics for methanol production from H2 and CO2 are not as favourable as those for the production from syngas (cf.reactions (6) and (7)).51 Therefore, on an industrial scale, methanol production usually relies on syngas in a 3 to 1 ratio from SMR (reaction (2)), while CO2 is added to deal with the excess H2 in the feed (compared to reaction (6)), and finally the produced water (reaction (7)) is recycled via the water–gas shift reaction (the reverse reaction of reaction (5)).
Nevertheless, to conclude, we can state that, currently the selective hydrogenation of CO2 with H2 into methanol is the most – if not to say only – industrially successful traditional process for the direct reforming of CO2 into chemicals and fuels.
| CO2(g) + H2O(g) → CO(g) + H2(g) + 1/2O2(g) ΔH° = +525 kJ mol−1 | (R8) |
| CO2(g) + 2H2O(g) → CH3OH(g) + 3/2O2(g) ΔH° = +676 kJ mol−1 | (R9) |
 | ||
| Fig. 4 Calculated theoretical thermal conversion (left axis) and corresponding energy efficiency (right axis) as a function of temperature for the conversion of CO2 and H2O into H2, CO and O2. | ||
Nevertheless, a process involving the reaction of CO2, H2O, O2 and CH4, called tri-reforming, is gaining quite some interest.24 The concept, proposed by Song et al.,31 involves a synergetic combination of dry reforming, steam reforming and the partial oxidation of methane in a single reactor, which can produce syngas in desired ratios (1.5–2.0), while eliminating carbon formation, as demonstrated for a fixed bed flow reactor at 1123 K over supported nickel catalysts.31
3. Novel CO2 conversion approaches
It stands without doubt that the efficient conversion of CO2 to useful molecules presents an important challenge and a great opportunity for chemists today. Due to the inability of the traditional thermal approaches to address the worldwide CO2 and energy challenge, several promising novel technologies are under development. Plasma technology is one such technology, but before elaborating on this specific technology in more detail, first, a summary of its main ‘frenemies’ in this domain is given. For each technology, first a brief explanation of the working principles and current achievements is given, followed by the major advantages and challenges. From this section, it will become clear that there is, indeed, fierce competition in the quest to find the most effective and efficient CO2 conversion technology with the potential to be used on an industrial scale. It should be noted that only technologies for CO2 conversion are described here. For other (in)direct applications and fixation technologies, we refer to other reviews.12–193.1. Electrochemical conversion
We kick off our discussions with one of the closest competitors to plasma technology, i.e. the electrochemical conversion or reduction of CO2. This closeness derives from the fact that both technologies rely on the use of (renewable) electrical energy, whereas most of the other novel technologies only take direct advantage of renewable energy, i.e. the sun, either based on its focused radiation heat or its emitted photons. Although significant technical and catalytic advances are still required for its large-scale use, electrochemical conversion is becoming a mature technology for H2O splitting. For CO2 reduction, on the other hand, several important challenges remain.7,45,52–55The electrochemical valorization of CO2 is an innovative technology, in which electrical energy is supplied to establish a potential between two electrodes, allowing CO2 to be transformed into value-added chemicals under mild conditions.7,55 This transformation can occur through a wide variety of pathways, which are typically strongly affected by the experimental conditions. The electrochemical reduction of CO2 can proceed through two-, four-, six- and eight-electron reduction pathways in gaseous, aqueous and non-aqueous phases in different cell and electrode configurations.7,53Fig. 5 shows the three main cell types. Fig. 5(a) and (b) illustrate the principle of a solid proton conducting electrolysis cell (SPCEC) for the combined conversion of CO2 and H2O, and of a solid oxide electrolysis cell (SOEC) that could be used for either the pure or combined conversion of CO2 and H2O, respectively, while a typical alkaline electrolysis cell for water splitting is shown in Fig. 5(c). The catalyst and/or electrode materials, the reaction medium, electrolyte solution, buffer strength, pH, CO2 concentration and pressure as well as the reaction temperature all influence and determine the wide variety of products that can be obtained.53 The major reduction products obtained include carbon monoxide (CO), formic acid (HCOOH) or formate (HCOO−) in basic solution, formaldehyde (CH2O), methanol (CH3OH), oxalic acid (H2C2O4) or oxalate (C2O42−) in basic solution, methane (CH4), ethylene (C2H4) and ethanol (C2H5OH).7,45,53
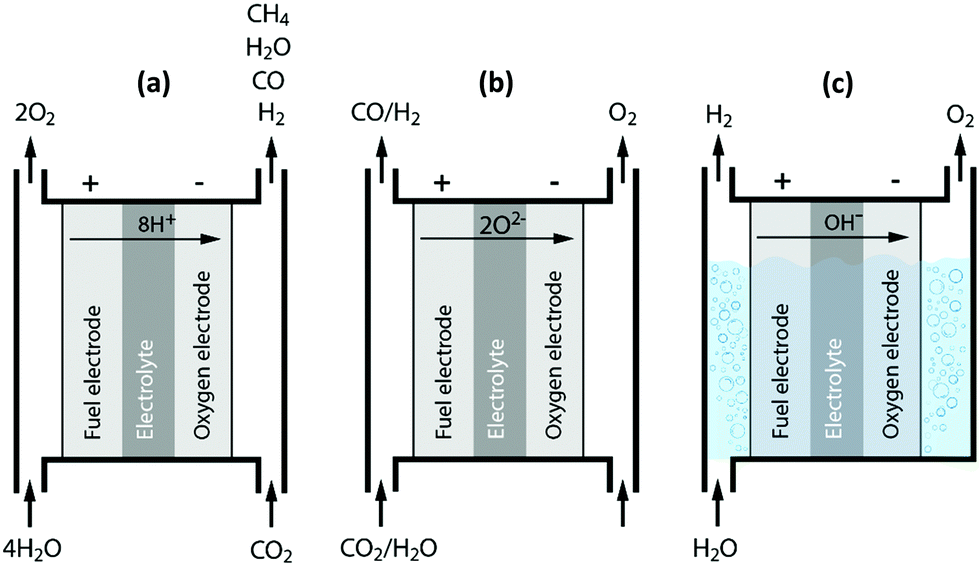 | ||
| Fig. 5 Principles of a solid proton conducting electrolysis cell (SPCEC) (a), solid oxide electrolysis cell (SOEC) (b) and an alkaline electrolysis cell (c) for the conversion of CO2 and/or H2O. | ||
There are a number of reasons why the electrochemical reduction process stands out from the crowd; for instance, the process is controllable by several reaction parameters, including the electrode potential and temperature.7,45,52–54 Furthermore, a wide variety of valuable products can be made, either in mixtures or more importantly in their pure form. For example, besides the direct electrochemical reduction of CO2 to methanol, it is also possible to produce CO and H2 at the cathode in a H2/CO ratio close to 2, while at the anode, a valuable pure oxygen stream is generated (see Fig. 1–5(b)).45 Another advantage is that electrochemical conversion can make use of a wide variety of (intermittent) renewable electricity sources, i.e. more than just solar energy.53 Finally, the electrochemical reaction systems are compact, modular, on-demand and thus easy to utilize for small or large scale-up applications.53
The accomplishments to date in the electrochemical reduction of CO2 have been encouraging, and the potential rewards are enormous.7 Nevertheless, several challenges remain, such as the high overpotential, which is the difference in electrode voltage between the theoretical thermodynamic and actual real-world values to drive a reaction;52,54 the low solubility of CO2 in aqueous solutions;52 the formation of product mixtures, thus requiring expensive separation steps;52 fouling and catalytic deactivation of the electrodes by impurities, reaction intermediates and by-products;7,52,54 the instability of the electrode material;7 the low Faradaic efficiencies, current densities and high energy consumption;52,53 the kinetic barriers leading to low efficiencies;53,54 and the non-optimized electrode/reactor and system design for practical applications.53 In general, it is recognized that the single biggest challenge is the low performance of the electrocatalysts, due to low activity, low selectivity and most importantly insufficient stability. The reported stability tests in the literature are only in the order of, or below, 100 hours, while long-term tests are non-existent to date.53 This makes the development of stable electrocatalyst materials with high activity and selectivity the main priority for this technology.7,53,56
It seems that despite many advances and successful proof-of-concepts being reported, the maturity of electrochemical CO2 reduction technology is still far from reaching the requirements for commercialization, due to the several remaining major technological challenges, as listed above.7,52,53 Particularly for industrial-scale implementation, the low catalyst stability seems to be the major limitation.53 As a result, no electrocatalysts developed to date for the reduction of CO2 would be deemed useful for a large-scale system.54 As stated by Qiao et al.,53 “With continued and extensive efforts focused on developing innovative composite and nanostructured catalyst materials to overcome the challenges of insufficient catalytic activity, product selectivity, and catalytic stability, the technology of CO2electroreduction will become practical in the near future”. Hence, to successfully achieve the transformation of CO2 to liquid fuels and useful chemicals, new methods and approaches for activating the CO2 molecule at lower overpotentials are required.7 In the first instance, novel electrodes enabling operation at current densities close to commercially available H2O electrolyzers have to be developed, for which solid oxide electrodes appear to be suitable candidates.52 Furthermore, a better understanding of the mechanistic role of metal and metal oxides in the reduction process is needed to open the possibility to design electrodes with certain compositions.52 To conclude, efforts to optimize system designs and at the same time to develop durable catalysts still need to be carried out.53 However, the final grand question remains: can all of this be done with inexpensive earth-abundant metals?54
3.2. Solar thermochemical conversion
Another technology, which has recently made several huge leaps forward, is the solar thermochemical conversion of CO2. There are several ways to reduce CO2 with the assistance of renewable solar energy, with those using direct solar light irradiation probably the most effective methods because there is no additional extra energy required and no negative influence on the environment.57 Two forms of direct solar energy conversion can be distinguished: (i) thermal conversion – described here – where work can be extracted after sunlight is absorbed as thermal energy and (ii) quantum conversion – described in the next section – where the work output can be taken directly from the light absorber (e.g. a semiconductor, molecule or organic compound).58 For solar thermochemical conversion, concentrated solar radiation is used – in the form of high-temperature heat – as an energy source to drive the highly endothermic reactions.The single step thermal dissociation of CO2 (or H2O) is impeded by the need to operate at high temperatures (>2500 K), as demonstrated in Fig. 2 above, and the need for effective separation techniques to avoid ending up with an explosive mixture of CO/O2 (or H2/O2).58,59 Multi-step thermochemical cycles using metal oxide redox reactions bypass the separation problem and, in addition, they allow operation at relatively moderate temperatures. More specifically, as shown in Fig. 6, solar processes involving heat at ≥1500 K enable a two-step thermochemical cycle using metal oxide redox reactions for CO2/H2O-splitting.58,59 The first – endothermic – step is the solar thermal reduction of the metal oxide MOox (where M is e.g. Ce, Zn, or Fe)60 to the metal or to the lower-valence metal oxide MOred. The second – nonsolar – exothermic step is the oxidation of the reduced metal oxide with CO2 and/or H2O to form CO and/or H2, allowing the (re)oxidized metal oxide to be reused/recycled for the first step.58,59 In general, two cycle categories can be considered: volatile and non-volatile. Non-volatile cycles utilize metal oxides, which remain in the solid state during reduction, while volatile redox cycles consist of metal oxides, which undergo gas–solid phase transitions. The volatile reactions appear more favourable, but the volatile products must be quenched rapidly to avoid recombination, and to date this issue has not been solved in an energetically efficient fashion.59 For the non-volatile cycles, cerium oxide (ceria, CeO2) has emerged as a highly attractive redox active material choice for two-step thermochemical cycling.60,61 Another promising pathway, which operates at lower temperatures than ceria, is the exploration of doped perovskite oxides.62
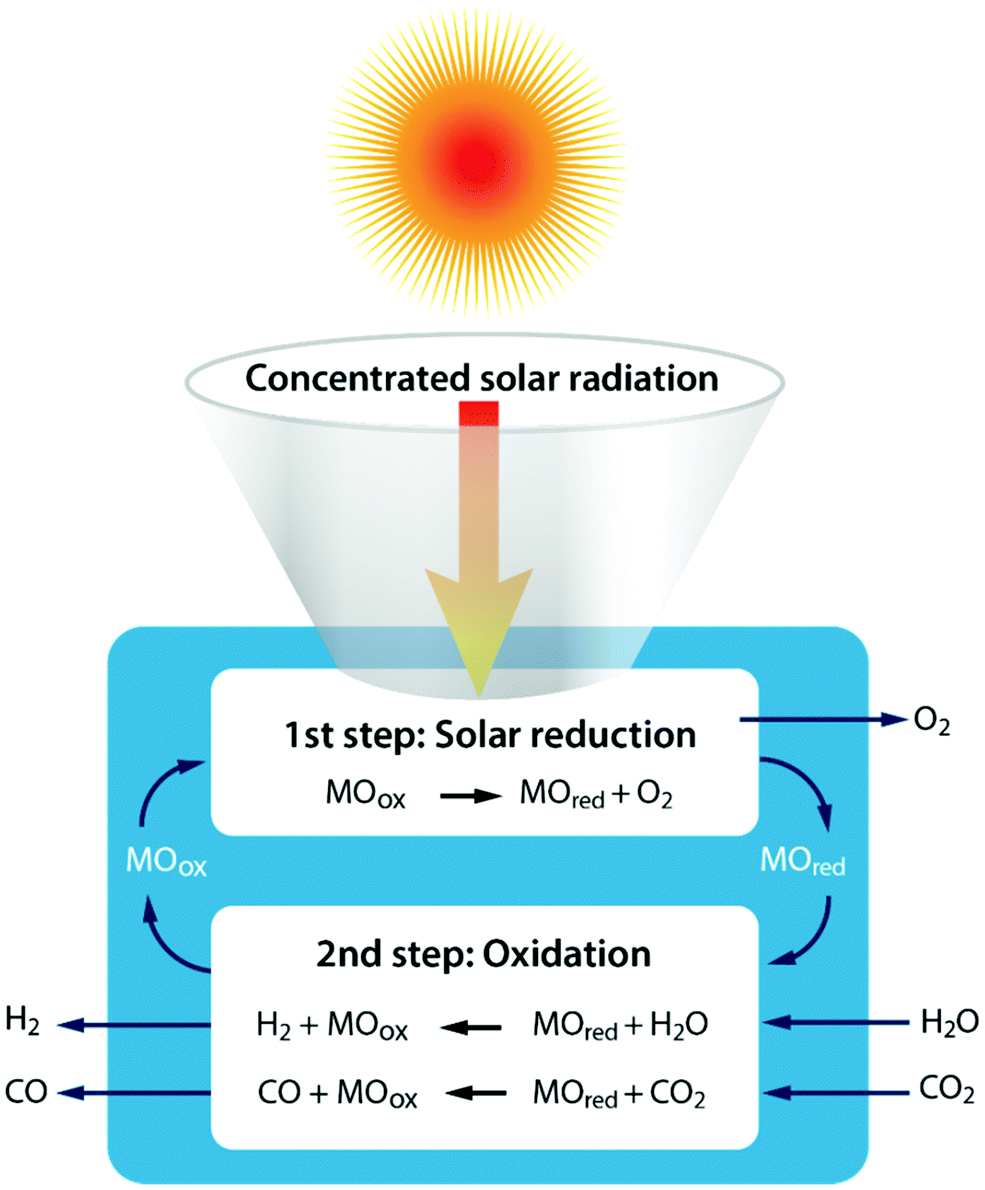 | ||
| Fig. 6 Schematic of the two-step solar thermochemical cycle for CO2 and H2O splitting based on metal oxide redox reactions. | ||
The main advantage of the solar thermochemical conversion of CO2 is obviously the direct use of solar energy. Concentrating solar technologies, which are currently applied commercially for large-scale (megawatt) power generation, can be coupled to high-temperature thermochemical reactors with the potential to achieve high solar-to-fuel energy conversion efficiencies and, consequently, the potential to produce solar fuels on a large scale and at a competitive cost.58 To date, solar flux concentration ratios exceeding 2 MW m−2 are attainable with large-scale solar tower and dish systems. Solar thermochemical applications, although not as far developed as solar thermal electricity generation, employ the same solar concentrating infrastructure, with the solar reactor positioned at the focus of the solar tower (for megawatt centralized applications) or solar dish (for kilowatt decentralized applications).58 A recent comprehensive review of solar concentrating technologies for thermal power and thermochemical fuel production was given by Romero et al.63 Consequently, these cycles inherently have the potential to realize a greater theoretical efficiency than methods using energy vectors or a small part of the solar spectrum and are, in addition, conceptually simpler.62,64 This potential to achieve high solar-to-fuel energy conversion efficiencies is primarily related to the fact that solar thermal processes inherently operate at high temperatures and utilize the entire solar spectrum, and, as such, provide a thermodynamically favourable path to the production of solar fuels.58,59 A thermodynamic analysis based solely on the material properties of e.g. CeO2 indicated that efficiency values in the range of 16–19% could be attainable, even in the absence of sensible heat recovery. These values are close to the 20% efficiency that is likely to be needed for solar fuels to be considered cost competitive,65 as will be discussed in more detail in Section 6.3.
Although significant advances have been made in the field of solar thermochemical CO2 conversion technologies using metal oxides, a lack of fundamental research into the behaviour of the metal oxides under the high-temperature conditions present in these cycles has hampered the development of materials. Basic questions relating to oxygen transport, surface chemistry, structural changes vs. redox reactions, materials’ synthesis methods, the effects of thermochemical cycling on the material and the role of supports still have to be addressed.66 Furthermore, despite its favourable thermodynamics, both the efficiency and the cycling rates in the reactor can be largely limited by thermal losses, resulting from poor conductive and radiative heat transfer across the porous metal oxide structure.58 Finally, the thermochemical conversion rates are higher than, for example, the photocatalytic rates, but although conceptually simple, focusing lenses for sunlight and high-temperature reactors incur high initial investment costs.60
Solar thermochemistry has clearly emerged as a viable path to utilize concentrated solar technology – currently applied commercially for large-scale power generation – for the conversion of CO2 (and H2O) into CO (and H2). Also, solar thermochemical cycles for the conversion of CO2 and H2O via the metal oxide redox reaction have favourable thermodynamics, but the ultimate factor dictating commercial viability is a high solar-to-fuel energy conversion efficiency, and to date efficiencies above 10% are still pending experimental demonstration with robust and scalable solar reactors.58,59,62 The discovery of new materials with large oxygen exchange capabilities at moderate temperatures and their implementation in efficient solar reactors are thus essential. Additionally, rapid chemical kinetics and material stability over thousands of cycles must be demonstrated for each material considered.59,67 This is the second key to achieving market viability, because materials must remain active for many thousands of redox cycles in order to avoid the high costs that would be associated with frequent replacement. As such, commercial success is again predicated upon finding appropriate materials composed of earth-abundant elements that can operate at lower reduction temperatures than current systems, together with sufficient activity to achieve high process efficiency.62
3.3. Photochemical conversion
The photochemistry for the photochemical conversion differs from (solar) thermochemistry in the way the solar energy is used: the former uses the energy of a photon in the chemical reactions,60 while the latter uses the absorbed thermal energy to overcome the activation barriers and to affect the chemical equilibria.58The photoreduction of CO2 to formaldehyde and methanol in purified water was already reported back in 1979, using the semiconductors TiO2, ZnO, CdS, GaP, SiC and WO3.60 Based on a correlation between the conduction band energy potential and the yield of methanol, it was suggested that the photoreduction of CO2 proceeds by the photoexcited electrons in the conduction band moving to CO2. This principle mechanism of selective photocatalysts under light irradiation is shown in Fig. 7. Here, the conduction band energy minimum is higher than that for CO2 photoreduction.60 Again the efficiency of the photocatalytic materials in their use of sunlight for the conversion of CO2 to fuel is of critical importance.68 This efficiency is influenced by several factors, such as catalyst dosage, reactant ratio, reaction temperature, time, system pressure, pH, light intensity and wavelength.57 A wide variety of reduction products can be obtained, just like with the electrochemical technique, including carbon monoxide (CO), formic acid (HCOOH), formaldehyde (CH2O), methanol (CH3OH), methane (CH4), ethylene (C2H4), ethane (C2H6) and ethanol (C2H5OH).68
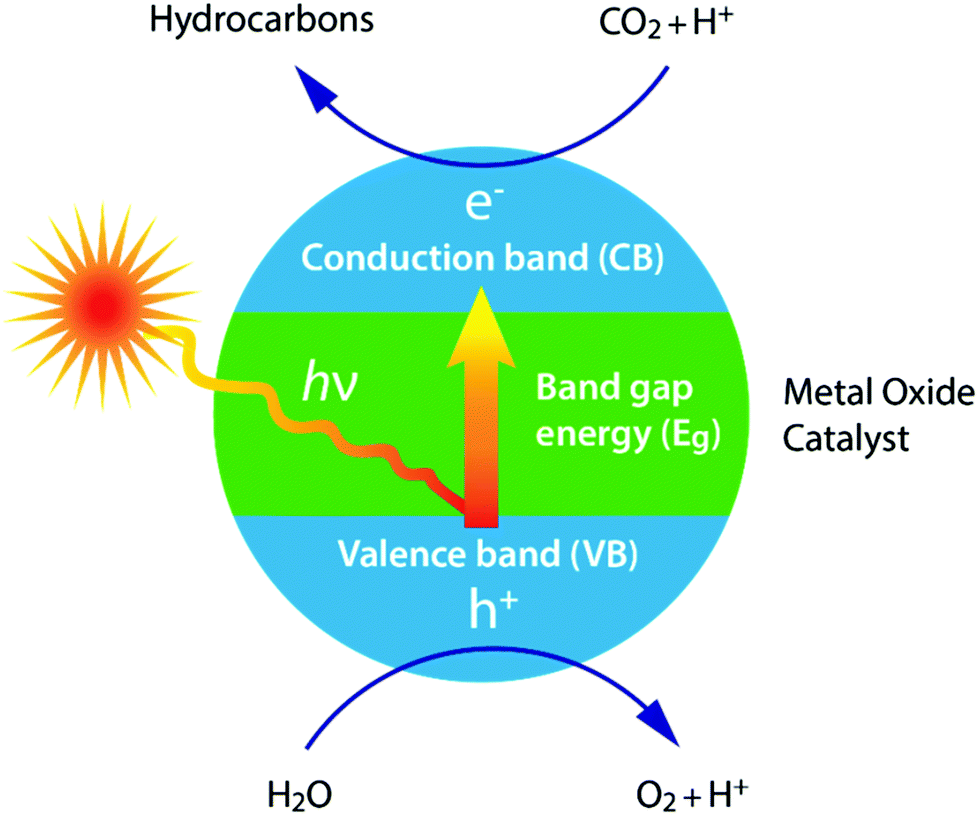 | ||
| Fig. 7 Principle of the photochemical reduction of CO2 by water on a photocatalyst. | ||
From a sustainable point of view, solar light is the ideal energy source. In combination with photocatalytic H2O splitting, the solar-driven reduction of CO2 to fuels is a very attractive approach.52 The advantages of photochemical systems include the assertion that they are composed of only a few parts and are therefore theoretically less likely to fail, providing the remaining parts are reliable.58 The most extensively investigated catalyst for the photoreduction of CO2 is TiO2.60 Several attempts have already been made to enhance the photocatalytic activity of TiO2, including by the addition of a metal, Rh/TiO2 or Rh/WO3–TiO2,60 the use of highly dispersed active Ti ion species,60 through atomically dispersing TiO2 on zeolites or ordered mesoporous SiO2 or by doping with Pt, Cu, N, I, CdSe or PbS.60 The most significant breakthrough was achieved using nitrogen-doped TiO2 nanotube arrays co-catalyzed with Cu and/or Pt nanoparticles, in which water-vapour-saturated CO2 was reduced to methane and other hydrocarbons without the application of an electrical bias.60,68
It should be noted, however, that for many oxide semiconductors (incl. TiO2) this electrical bias is necessary, because the conduction band is located below the acceptor level. This is one factor that limits the efficiency of metal oxide materials.58 Furthermore, most work is performed using artificial (UV) light sources,57,68 because the large band gap of metal oxides results in a poor photo-responsiveness to visible light.52,57,58 Theory dictates that a band gap between 2 and 2.4 eV is optimal, which limits the maximum attainable efficiency to about 17%.68 However, solar energy conversion efficiencies obtained to date are much lower (at present, at <2%),58 mainly as a result of the energy associated with this electrical bias.58 Furthermore, a remaining challenge lies in the separation and collection of the hydrogen and oxygen gas produced (often produced in close proximity).58 Finally, many of the photocatalysts presently being studied are metal complexes employing rare and expensive transition metals, hence efforts must be made using earth-abundant elements that could support large-scale undertakings.54
From the literature, it is clear that encouraging progress has been made towards the photocatalytic conversion of CO2 using sunlight, but nevertheless, the existing techniques are insufficient to date and further efforts are required to increase the solar-to-fuel conversion efficiencies.54,55,57,58,68 This appears to be a general remark for photochemistry: very few examples exist of chemical processes operating effectively on the basis of photocatalysis technology. Not only do the photon efficiency of materials and the resulting achievable rates remain insufficient, also sub-optimal photocatalytic reactors often induce inefficiencies in operation, which limit their practical application.52 The immediate requirement in this technology is to develop visible-light-sensitive photocatalysts, which are prominent in CO2 recycling.57 What we ultimately seek is a means for achieving the high-rate photocatalytic reduction of CO2, using solar radiation as the only input energy source. Since visible light comprises the majority of the solar spectrum energy, it behoves us to consider photocatalysts sensitive to sunlight.68 Immediate research opportunities include uniform co-catalyst sensitization of the entire nanotube array surface for enhanced conversion rates, and the design of co-catalysts to improve and control the product selectivity.68 Although the photocatalytic reduction of CO2 may become an important stepping stone to solar fuel production, much progress remains to be achieved before it could be considered practical as an industrial process.54 Based on the highest reported activities, one can conclude that game-changing rates have not yet been achieved. Reported turnover frequencies are far from those required for an efficient catalytic process, and an efficiency improvement of at least 3 orders of magnitude is needed.52 It is thus clear that photochemical systems have a long way to go to achieve their full potential and to be able to successfully compete with the alternative approaches to producing fuels from sunlight.58
3.4. Biochemical conversion
Another pathway converting solar energy into chemical energy is by ‘natural’ photosynthesis for the production of biofuels.69 The biological conversion of CO2 for producing chemicals or fuels is an attractive route. Nevertheless, the use of first generation biofuels has generated a lot of controversy, mainly due to their competition with agriculture for arable land use for food production, thus impacting global food markets and food security.69,70 The use of microalgae, on the other hand, could meet the conditions for technically and economically viable biofuel production. More specifically, viable biofuel production should be competitive or cost less than petroleum fuels, it should require low to no additional land use, it should enable air quality improvement and should require minimal water use.69,70 Microalgae can typically be used to capture CO2 from three different sources: atmospheric CO2, CO2 emissions from power plants and industrial processes, and CO2 from soluble carbonates.70 The pathways for CO2 fixation have evolved over billions of years and use many diverse mechanisms and enzymes for processing CO2 by forming C–H and C–C bonds and cleaving C–O bonds.47 Furthermore, algae are more photosynthetically efficient than terrestrial plants, making them without doubt very efficient CO2 fixers.71Microalgae are currently considered to be one of the most promising alternative sources for biodiesel; hence, most of the current research and developmental efforts are focused on microalgae, in particular due to their high growth rate and high oil content (up to 77% of the dry cell mass). Algae contain oils, sugars, and functional bioactive compounds that can be used for commercial products. In addition to fuels, the development of appropriate technologies for high-efficiency algal biodiesel production is also applicable to biohydrogen, biogas, bioethanol and biomass-to-liquid (BTL) approaches using fast growing algae.69,70 Furthermore, valuable co-products, such as biopolymers, proteins and animal feed, can be produced during the process.71,72 Other advantages include very short harvesting cycles (∼1–10 days), thus allowing multiple or continuous harvests with significantly increased yields. Furthermore, the cultivation can potentially be carried out on marginal or non-arable land, and even the use of waste water for algal cultivation is a viable option.69,70 To obtain the best performing microalgae strains for biofuel production, one can: (1) screen a wide range of natural isolates, (2) improve them by metabolic (genetic) engineering or (3) obtain them by selection and adaptation. There are algae collections worldwide, which contain thousands of different algal strains that can be accessed.69
It should, however, be emphasized that the significant drawback in all the biochemical techniques is the big share of the cost for cultivation. Among others, the harvesting of algal biomass accounts for the highest proportion of energy input during production, but currently, there are no standard harvesting techniques.69–71 Therefore, currently algal biomass is not suitable to be cultivated solely for bioenergy applications and instead it must be integrated with the production of other value-added products, e.g. pharmaceutics, cosmetics and food. Unfortunately, processes for the recovery of complex molecules from algal biomass are expensive and significant technological progress is still required before commercial deployment.73 Algae can be grown in many ways, such as in freshwater, saltwater or wastewater, in closed photobioreactors or in open ponds.69 One key advantage of algae is that its cultivation does not require cropland, although, on the other hand, other resources are needed.72 Other inorganic nutrients required for algae production include nitrogen and phosphorus,70 resulting in the need for unsustainable inputs of nitrogenous fertilizers, which are produced from fossil fuels and that require huge inputs of energy for production.72 Furthermore, it has been reported that between 3.15 and 3650 litres of freshwater are needed to produce algal biofuel equivalent to 1 litre of gasoline using current technologies. Thus, the integration of the upstream production and downstream processing of microalgae, and the framing of these in the context of water savings and net energy gain is required to build up credibility.72Fig. 8 shows a schematic overview of the different steps for the application of a photobioreactor for CO2 conversion. Finally, optimization of the strain-specific cultivation conditions is complex, with many interrelated factors that can each be limiting factors, including temperature, mixing, fluid dynamics and hydrodynamic stress, gas bubble size and distribution, gas exchange, mass transfer, light cycle and intensity, water quality, pH, salinity, mineral and carbon regulation/bioavailability, cell fragility, cell density and growth inhibition.69
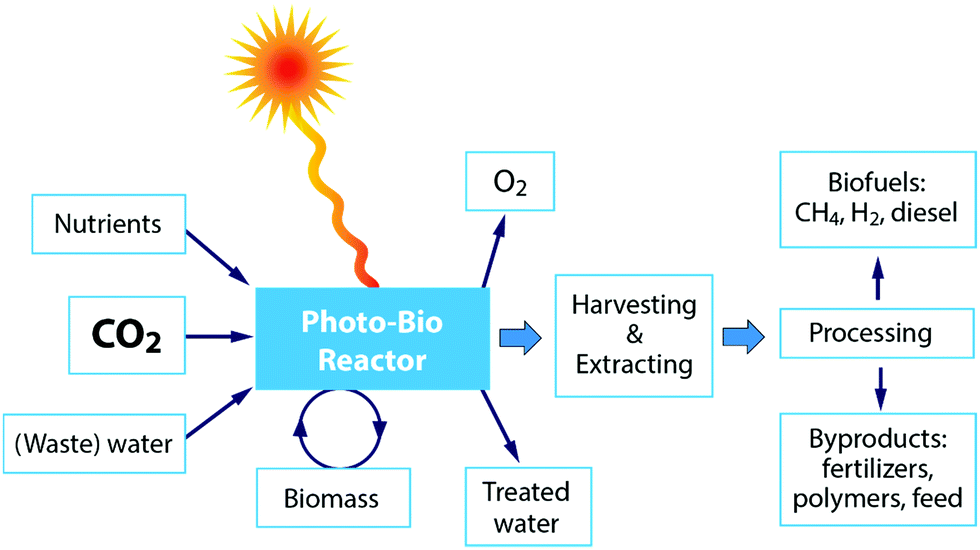 | ||
| Fig. 8 Schematic of photo-bioreactor application for CO2 conversion. | ||
Despite its inherent potential as a biofuel resource, many challenges have impeded the development of algal biofuel technology as a commercially viable solution that could support sustainable production and utilization.70 Consequently, the large-scale cultivation of algae for biofuel production is still in the research and development phase. The long-term potential of this technology can be improved by the following approaches: (1) identifying and developing cost-saving growth technologies of oil-rich algae;69,72 (2) utilizing integrated bio-refineries to produce biodiesel, animal feed, biogas and electrical power, thereby reducing the overall cost of production;71,72 (3) enhancing the algal biology by genetic modification and metabolic engineering to aid the selection and successful outdoor large-scale cultivation of a robust microalgal strain;69,71,72 (4) identifying area-efficient techniques to capture CO2 from industrial power plants;72 (5) recycling nutrients from municipal sewage and industrial wastewaters to reduce the demand for fertilizers to grow the algae;69,70,72 (6) improving the economics of microalgae production by generating additional revenues from wastewater treatment and greenhouse gas emissions abatement;72 (7) most importantly, by developing cost-effective and energy-efficient harvesting methods to make the whole biofuels production process economical;71 in this respect, strain selection is an important consideration since certain species are much easier to harvest than others.69
3.5. Catalytic conversion
In Section 2, we already covered the main traditional thermocatalytic approaches used. Of course, there is still a lot of research ongoing towards finding new improved (thermo-)catalytic pathways for the different processes involved in CO2 conversion. There are two main catalysis types that can be applied for this process: homogeneous and heterogeneous catalysts. The former (e.g. Ru-, Rh- and Ir-based catalysts) are efficient for the formation of formic acid and formates, but are more challenging to be used in commercial applications, while the latter (e.g. Fe-, Cu- and Ni-based catalysts) are more practical for industrial applications, but they frequently suffer from low yields and poor selectivity. As a result, significant improvements in new catalytic systems are necessary to make thermocatalytic CO2 reduction economically feasible.48Nonetheless, the catalytic conversion is briefly discussed here; especially since it is clear from the sections above that catalytic materials can play an important role in the development and further advancement of most of the novel technologies under study.25 These scientific advances give rise to intriguing new combinations, and corresponding names, such as electrocatalytic, photocatalytic, biocatalytic, as well as their even more advanced hybrids forms, e.g. photoelectrocatalytic and bioelectrocatalytic processes.12,14,53 Kumar et al.54 stated quite frankly that virtually every approach under consideration for the conversion of CO2 requires catalysts to facilitate the formation and cleavage of chemical bonds, as illustrated by Fig. 9. In general, these required catalysts fall into three classifications: (1) they already exist and show good performance but are too rare/costly to be scaled up; (2) they already exist but in forms that are not optimal or practical for adaptation to an integrated solar fuels system; (3) they do not exist yet and await discovery.54
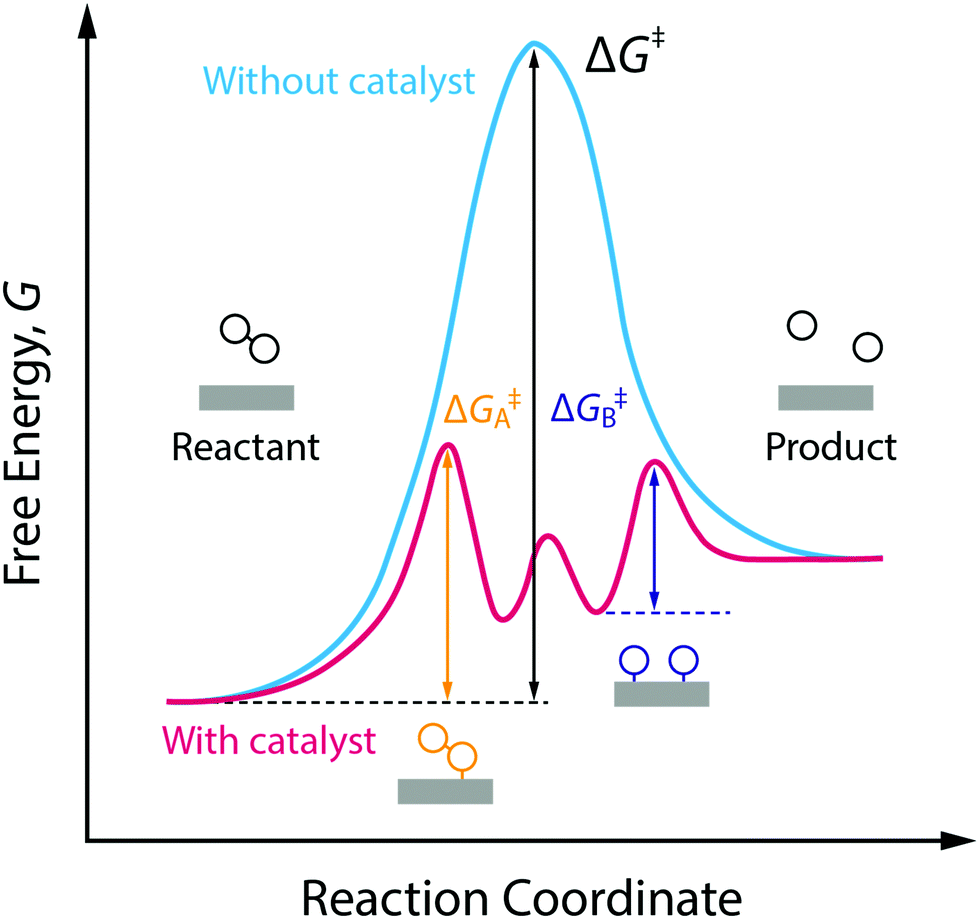 | ||
| Fig. 9 Reaction coordinate diagram showing the working principle of a catalyst. | ||
From a scientific point of view, the development of catalysts with inexpensive metals, such as iron and copper compounds, which can also be active under mild conditions, is a big challenge.48 Still, it is evident that due to their reliance on thermal heat – as currently derived from burning fossil feedstocks – the current pure thermocatalytic routes can only make a limited net contribution to CO2 conversion. Hence, techniques based on the use of (in)direct solar energy and other renewable energy sources are needed to contribute to avoiding large volumes of CO2.25 One exciting thermocatalytic advancement to circumvent this reliance on fossil fuels – besides the solar thermochemical conversion discussed in Section 3.2 above – is the use of microwaves.74 Here, microwave-assisted CO2 conversion over carbon-based catalysts combines the catalytic and dielectric properties of carbonaceous materials with the advantages of microwave heating, which favours catalytic heterogeneous reactions due to, among other reasons, the generation of hot spots.
Microwave radiation has been shown to have beneficial effects on the reaction rate of heterogeneous (catalytic) reactions.75,76 The combination of microwave heating and a carbon material acting as both a catalyst and microwave receptor gives rise to enhanced conversions compared to conventional heating, both in the case of CO2 gasification and in the dry reforming of methane.75,76 Fidalgo et al.76 observed conversions that were a factor of 1.6 to 1.9 higher for MW-heating compared to conventional heating. Unlike in conventional heating, the applied microwave energy is transferred directly to the catalyst without any heat flux. As a result, the temperature inside the material is usually higher than the temperature of the surrounding atmosphere near the surface, and the uniformity of heat distribution is improved with respect to conventional heating, as shown in Fig. 10.76 Furthermore, the formation of hot spots, possibly due to the generation of microplasma within the catalyst bed, which may be at higher temperatures compared to the bulk catalyst, have been reported to be responsible for an enhancement in the reaction rate, higher yields and the improved selectivities of heterogeneous (catalytic) reactions.75,76
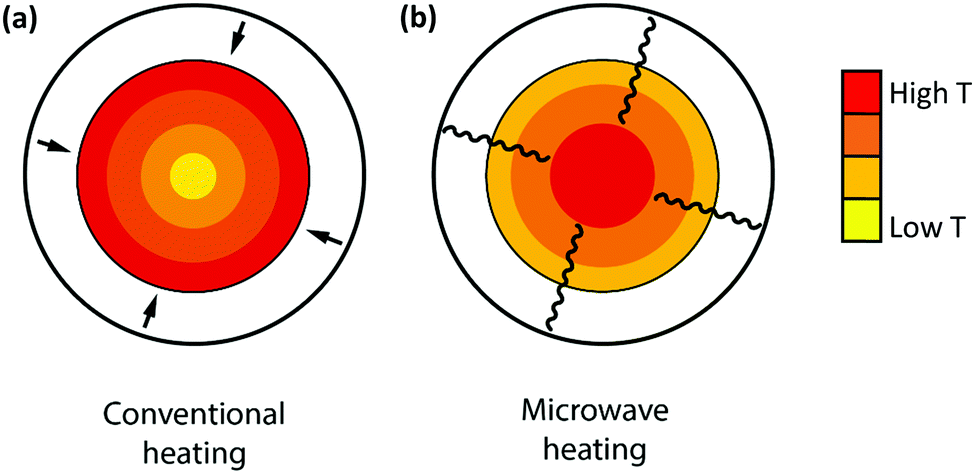 | ||
| Fig. 10 Comparison of the temperature gradients produced by conventional (a) and microwave (b) heating. | ||
3.6. Summary
It should be of no surprise that the field of CO2 conversion is rapidly evolving. As such, it is not the purpose of this section to cover all the (recent) work performed for these novel technologies, such as CO2 mineralization and utilization, which converts the chemical energy of CO2 mineralization into electricity, while producing valuable mineralization products,77 or the hybrid enzymatic/photocatalytic approach for the production of CH3OH from CO2 with the use of water–glycerol, as mentioned in Section 2.38 Clearly, giving a complete comprehensive overview of all the carbon capture, utilization, and storage (CCUS) technologies is outside the scope of this review. Nevertheless, by discussing the main novel technologies for CO2 conversion, we were able to roughly sketch out the current landscape, one in which we want to orient another emerging technology with the potential for CO2 conversion, i.e. plasmachemical conversion. It is evident that, for all technologies discussed above, there are several distinctive up- and downsides. This will not be different for plasma technology – as will become evident from the following section. In the final benchmarking section (Section 6) of this review, we make a visual comparison between all the emerging technologies, based on their versatility, the mentioned distinctive advantages and limitations and – in what appears to be most important from an economical point of view – their solar-to-fuel conversion efficiency.4. Plasma technology for CO2 conversion
From the previous section, it is clear that several alternative (non-conventional) CO2 conversion technologies are, more or less, successfully being investigated. In recent years, another novel technology has started to be considered, but has not spent much time in the spotlight yet: plasma technology. Herein, we first give a brief introduction to plasma technology, and highlight its general advantages and unique features for CO2 conversion. Next, we describe the different kinds of plasma reactors used for CO2 conversion, focusing on their advantages and disadvantages. Subsequently, we discuss the possibly fruitful combination of plasma with catalysts, in so-called plasma-catalysis. Finally, in the next section (Section 5), we present a critical assessment of plasma-based CO2 conversion for these different set-ups, for both pure CO2 splitting and CO2 conversion, together with the specified co-reactants.4.1. Properties of plasma and its unique features for CO2 conversion
The term ‘plasma’ was first introduced by Irving Langmuir (1928). Plasma is an ionized gas, which means that at least one electron is unbound, creating positively charged ions. In practice, the ionization degree in plasma can vary from fully ionized gases (100%) to partially ionized gases (e.g. 10−4–10−6). Besides the various types of ions (both positive and negative), plasma also consists of a large number of neutral species, e.g. different types of atoms, molecules, radicals and excited species. The latter can lead, among other things, to the emission of light. More importantly, all these species can interact with each other, making plasma a highly reactive and complex chemical cocktail, which is of interest to many potential applications.21,78 Indeed, plasmas can already be found in several applications in materials science (e.g. coating deposition, surface modification, nanomaterial fabrication) and in the microelectronics industry (for microchip manufacturing), but also as light sources, lasers and displays (owing to their light emitting characteristics) as well as for many emerging environmental and even medical applications (such as sterilization, wound treatment and even cancer treatment). More details about these applications can be found in the above-cited references, as well as in the references therein. In this review, however, we focus only on their application in CO2 conversion into value-added chemicals and fuels.Plasma is sometimes referred to as the ‘fourth state of matter’, owing to the observation that with increasing temperature matter transforms in the sequence: solid, liquid, (neutral) gas and finally an ionized gas or plasma. Although the concept of plasma is less known than the other states of matter, more than 99% of the visible matter in the universe is in the plasma state, mainly attributed to interstellar matter and the stars. Hence, our own sun is a perfect example of a plasma. Furthermore, basically, many – if not to say all – natural occurring weather phenomena emitting light are in fact plasma, e.g. Saint Elmo's fire, lightning, red sprites, auroras (Borealis and Australis), where we must thank the excited species for the emission of these colourful lightshows. Other natural plasmas close to home are the earth's ionosphere, plasma sphere and the outer magnetosphere.
Beside these natural plasmas, we can distinguish between two main groups of man-made plasmas. The first ones are the high-temperature or fusion plasmas, which are in general completely ionized plasmas. Applications include tokomaks, stellarators, plasma pinches and focuses. The second group comprises the weakly ionized plasmas or so-called gas discharges, which are under study and covered in this review.
A second sub-division can be made based on whether the plasma is in thermal equilibrium or not. The temperature in a plasma is determined by the average energies of the different species (electrons, neutrals, ions) and their relevant degrees of freedom (translational, rotational, vibrational and electronic). Since plasma is a multicomponent system, it can exhibit multiple temperatures. When the temperature of all these species is the same in a localized area, the plasma is said to be in ‘local thermodynamic equilibrium’ (LTE), and these kinds of plasmas are usually called ‘thermal plasmas’. When the plasma is characterized by multiple different temperatures and thus is far from thermodynamic equilibrium, the plasma is said to be in ‘non-local thermodynamic equilibrium’ (non-LTE) and these discharges are usually called ‘non-thermal plasmas’.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 K, depending on the ease of ionization, or at high gas pressure. The latter can be explained as follows. Initially, the electrons receive energy from the electric field during their mean free path in between collisions, and they lose a small portion of this energy during collisions with so-called heavy particles (e.g. gas molecules or atoms). Subsequent collisions of this nature, also known as ‘Joule heating’, can lead to the temperatures changing to reach equilibrium between the electron and heavy particle temperature. At high pressures, the mean free path becomes smaller, so more collisions occur; hence, leading to a more efficient energy exchange between the electrons and the heavy particles. More specifically, it is the square of the ratio of the electric field (E) to the pressure (p), i.e. (E/p)2, which is proportional to the temperature difference in gas discharges.21
000 K, depending on the ease of ionization, or at high gas pressure. The latter can be explained as follows. Initially, the electrons receive energy from the electric field during their mean free path in between collisions, and they lose a small portion of this energy during collisions with so-called heavy particles (e.g. gas molecules or atoms). Subsequent collisions of this nature, also known as ‘Joule heating’, can lead to the temperatures changing to reach equilibrium between the electron and heavy particle temperature. At high pressures, the mean free path becomes smaller, so more collisions occur; hence, leading to a more efficient energy exchange between the electrons and the heavy particles. More specifically, it is the square of the ratio of the electric field (E) to the pressure (p), i.e. (E/p)2, which is proportional to the temperature difference in gas discharges.21
Thermal plasmas have numerous advantages, compared to traditional technologies, due to their interesting characteristics, including high temperature, high intensity non-ionizing radiation and high-energy density. The heat source is also directional, with sharp interfaces and steep thermal gradients that can be controlled independently of the chemistry. Whereas the upper temperature limit when burning fossil fuels is 2300 K, thermal plasmas can reach temperatures of 20000 K or more, as mentioned above. As a result, this type of plasma is already being used for a wide range of applications, such as for coating technology, fine powder synthesis, (extractive) metallurgy (e.g. welding, cutting) and the treatment of hazardous waste materials.79
On the other hand, the inherent nature of thermal plasmas makes them unsuitable for the efficient conversion of CO2. More specifically, the ionization and chemical processes in thermal plasmas are determined by the temperature. As a result, the maximum energy efficiency is limited to the thermodynamic equilibrium efficiency and corresponding conversions of 47% and 80% at 3500 K, respectively (see Fig. 2 above). This is in contrast to non-thermal plasmas, where lab-scale efficiencies of up to 90% have already been reported (see further, Section 5.1).21
Although this way of plasma creation, i.e. by applying a potential difference between two electrodes, is only one possibility to create a plasma, the principle of gas activation by electrons, creating reactive species (ions, excited species, radicals) is equally valid for other types of plasmas as well, and will be discussed in Section 4.2 below. In general, this gas activation by electrons is also the reason for the good energy efficiency of CO2 conversion by non-thermal plasmas, as will be explained below.
In non-thermal plasmas, the electrons are indeed characterized by a much higher temperature than the heavy particles, leading to the non-LTE condition, as mentioned above. As a result of all the different species, the relationship between all their different temperatures can become quite complex, but conventionally the temperature of the electrons (Te) is the highest, followed by the vibrationally excited molecules (Tv), while the lowest temperature is shared by the neutral species (T0, or simply the gas temperature, Tg), the ions (Ti) and the rotational degrees of freedom of the molecules (Tr); hence the temperature order is: Te ≫ Tv > Tr ≈ Ti ≈ T0.21 In most cases, the electron temperature is in the order of 1 eV (∼10000 K), while the gas temperature remains close to room temperature. This high electron temperature is due to the small mass of the electrons, allowing them to be easily accelerated by the applied electromagnetic fields, whereas the heavy particles – even the ions – are not easily accelerated. Furthermore, due to the large mass difference, the electrons lose less energy during elastic collisions with heavy particles, so they can easily keep their high energy gained from the electric field.
As mentioned above, the electrons can be considered as the initiators of the highly reactive chemical mixture. This is obviously one of the key advantages of non-thermal plasma technology: it allows gases – even as unreactive as CO2 – to be ‘activated’ at room temperature by the highly energetic electrons. Accordingly, there is no need to heat the entire reactor or the gas, because the discharge and the associated reactions are easily initiated by applying an electromagnetic field. This results in the second key advantage of non-thermal plasma technology for CO2 conversion, namely that it is a very flexible, or so-called turnkey, process, since it can easily and, more importantly, instantaneously be switched on and off, with conversion and product yield stabilization times generally lower than 30 minutes. Furthermore, its power consumption can easily be scaled and adjusted. As such, it has the inherent ability to be(come) one of the most suitable technologies to utilize excess intermittent renewable energy (e.g. energy originating from wind turbines or solar panels) and for storing it in a chemical form. Indeed, this makes it suitable for both peak shaving and, more importantly, grid stabilization, by being able to adapt to the irregular supply of renewable electricity. Hence, in essence the current transition to renewable energy sources not only gives plasma processes a clean electricity source, but plasma-based CO2 conversion technology can also provide a solution for the imbalance between energy supply and demand by enabling the storage of electrical energy in a desirable chemical form according to the market needs.
Additional advantages compared to the other emerging technologies for CO2 conversion are the low investment cost for the reactors, as they do not rely on rare earth materials, and last but not least, their simple scalability from watt to megawatt applications, as already demonstrated by the successful development of ozone generators.80 Due to their extreme scalability, the applications can vary hugely in both scale and application type, starting from small devices, such as on board vehicles for exhaust treatment or to provide on board fuel cell feeds81 as well as in household-scale devices for indoor air treatment,82,83 to medium on-demand installations, such as modular containerized plant concepts84 or in in situ CO production,85 and finally to large-scale industrial plants. This scalability and flexibility in applications also gives plasma technology a high locational flexibility. As such, both production on demand and remote production become possible, which is a critical point for carbon capture and utilization (CCU) techniques.
On the other hand, for the comparison made here with other novel technologies, the reliance of non-thermal plasma technology on indirect solar energy in the form of electricity is at the same time a limiting factor for the overall solar-to-fuel conversion efficiency, compared to technologies that can harvest solar energy in a direct form. However, when looking at the broader picture, its ability to rely on multiple energy sources again gives plasma technology a huge advantage regarding locational flexibility, not to mention that it can be operated 24/7, even when the sun is not shining.
Although non-thermal plasma is good at creating a non-equilibrium condition by ‘activating’ stable molecules, it is absolutely non-selective in the formation of targeted products. More specifically, the reactive species created by the electrons react according to the laws of chemical kinetics, and as such, they recombine into a (large) number of different products, depending on the reaction conditions, not to mention that the formed products can again be destroyed by new electron collisions. For pure CO2 splitting, this is not much of an issue, since CO and O2 (together with small amounts of O3) are basically the only products that can be formed. However, when combining CO2 with other reactants, such as CH4, H2O or H2, a wide variety of products can be formed, including syngas (CO and H2) and higher hydrocarbons (C2Hx, C3Hy, C4Hz) as well as several oxygenates, e.g. methanol, ethanol, dimethyl ether, formaldehyde, acetaldehyde and carboxylic acids. For that reason, the combination with catalysis is also highly desirable for plasma technology when targeting the selective production of specific compounds (see Section 4.3).
For the application of CO2 conversion, however, it is not the increased temperature that makes these discharges most interesting, but rather their characteristic electron energy distribution, which leads to most of the electron energy going into the vibrational excitation of CO2, as will be discussed below. The latter is indeed known to provide the most energy efficient and hence most important channel for CO2 dissociation.21,87 In fact, in recent modelling studies, it was suggested that the higher gas temperature inherent to these warm discharges is actually an unwanted effect.88–90 As such, the development of ‘cooler’ warm discharges might be beneficial for further increasing the CO2 conversion and energy efficiency.
 | ||
| Fig. 11 Fraction of electron energy transferred to different channels of excitation, as well as to the ionization and dissociation of CO2, as a function of the reduced electric field (E/n), as calculated from the corresponding cross-sections of the electron-impact reactions. The E/n regions characteristic for MW and GA plasma and for DBD plasma are indicated. | ||
From Fig. 11, it is clear that the value of the reduced electric field will have wide implications on the distribution of the electron energy among the different channels. In the region above 200 Td, 70–80% of the electron energy goes into electronic excitation, about 5% is transferred to dissociation, 5% is used for ionization (increasing with E/n), while only 10% goes into vibrational excitation (decreasing with E/n). Around 50 Td, however, only 10% goes into electronic excitation and 90% of the energy goes into vibrational excitations. It is important to keep in mind that the addition of different gases (e.g. Ar, He, N2, H2O, H2, CH4) has an influence on the distribution of these channels.92 Hence, even during the pure decomposition of CO2 into CO and O2, there will be an effect on this distribution.
The distribution of energy into different modes, and especially the fraction going into vibrational excitation, is very important, since, as mentioned above, it is known that the vibrational levels of CO2 can play an important role in the efficient dissociation of CO2. To achieve direct electron-impact dissociation, an electron needs to have enough energy (>7 eV) to excite CO2 into a dissociative (i.e. repulsive) electronic state, which will lead to its dissociation into CO and O (see Fig. 12). As such, the amount of energy spent is much higher than the theoretical value necessary for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond breaking (5.5 eV). Due to the special nature of the CO2 molecule, a more efficient dissociation pathway is based on its vibrational excitation. This pathway starts with electron-impact–vibrational-excitation of the lowest vibrational levels, followed by vibrational–vibrational (VV) collisions. This so-called ladder-climbing gradually populates the higher vibrational levels, which eventually leads to dissociation of the CO2 molecule (see also Fig. 12). In this way, it is possible to dissociate CO2 more efficiently, since only the minimum amount of 5.5 eV for bond breaking is needed, compared to the overshoot in the case of electronic excitation–dissociation.91
O bond breaking (5.5 eV). Due to the special nature of the CO2 molecule, a more efficient dissociation pathway is based on its vibrational excitation. This pathway starts with electron-impact–vibrational-excitation of the lowest vibrational levels, followed by vibrational–vibrational (VV) collisions. This so-called ladder-climbing gradually populates the higher vibrational levels, which eventually leads to dissociation of the CO2 molecule (see also Fig. 12). In this way, it is possible to dissociate CO2 more efficiently, since only the minimum amount of 5.5 eV for bond breaking is needed, compared to the overshoot in the case of electronic excitation–dissociation.91
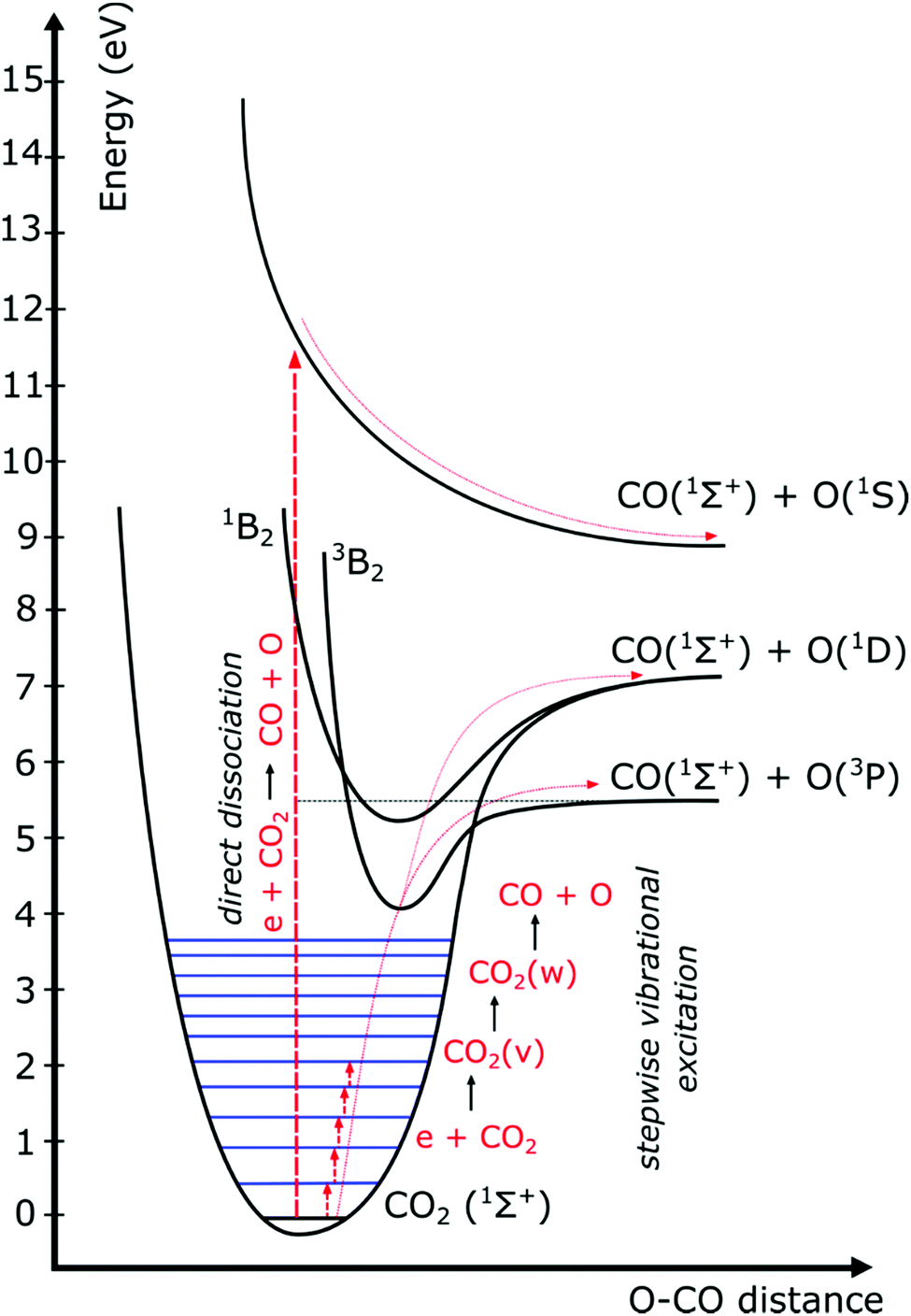 | ||
| Fig. 12 Schematic of some CO2 electronic and vibrational levels, illustrating that much more energy is needed for direct electronic excitation–dissociation than for step-wise vibrational excitation, i.e. the so-called ladder-climbing process (reproduced from ref. 91 with permission from the Royal Society of Chemistry). | ||
4.2. Different types of plasma set-ups used for CO2 conversion
As mentioned above, in recent years, there has been an increasing interest in the use of plasma technology for CO2 conversion. Experiments have been carried out in several types of plasmas. The most common types reported in the literature are dielectric barrier discharges (DBDs), microwave (MW) and gliding arc (GA) discharges, although other types have been used as well (e.g. radiofrequency, corona, glow, spark and nanosecond pulse discharges). A DBD is a typical example of a ‘non-thermal plasma’, where the gas is more or less at room temperature, and the electrons are heated to temperatures of 2–3 eV (∼20000–30000 K) by the strong electric field in the plasma. The MW and GA discharges are examples of ‘warm plasmas’ (see Section 4.1.3 above). The gas can reach temperatures of up to 1000 K and more, and the electron temperature is typically up to a few eV. The operating conditions and characteristic features of the three major plasma types for CO2 conversion are explained here, as well as for some other plasma types that have also been applied for CO2 conversion.
A DBD consists of two plane-parallel or concentric metal electrodes and, as its name suggests, it contains at least one dielectric barrier (e.g. glass, quartz, ceramic material or polymers) in between the electrodes.78,80,86,93 The purpose of the dielectric barrier is to restrict the electric current and thus to prevent the formation of sparks and/or arcs.93 A gas flow is applied between the (discharge) gap, which can typically vary from 0.1 mm (e.g. in plasma displays), to over 1 mm (e.g. for ozone generators) to several cm (e.g. in CO2 lasers).78,86 Some typical planar (top) and cylindrical (bottom) DBD configurations are shown in Fig. 13. In general, DBDs operate at approximately atmospheric pressure (0.1–10 atm, but usually 1 atm), while an alternating voltage with an amplitude of 1–100 kV and a frequency of a few Hz to MHz is applied between both electrodes.
 | ||
| Fig. 13 Basic planar (top) and cylindrical (bottom) dielectric barrier discharge configurations. | ||
To ignite the discharge, or more specifically, to transport current in the discharge gap, an electric field high enough to cause breakdown in the gas needs to be applied. This breakdown voltage (Vb) can be determined according to Paschen's Law, and is a function of the pressure (p) and the gap distance (d):
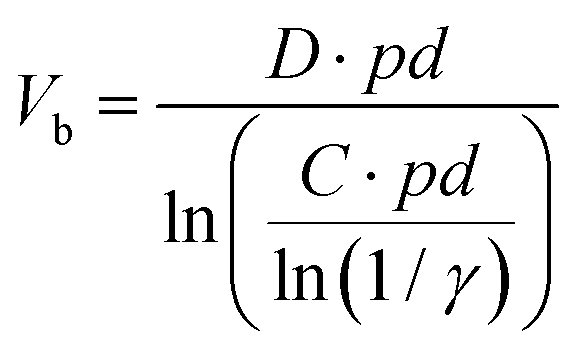
In the so-called surface-wave discharge, which is most commonly used for CO2 conversion research, the gas flows through a quartz tube – which is transparent to microwave radiation – intersecting with a rectangular wave guide, where the discharge is initiated, see Fig. 14. This system, also called ‘guide-surfatron’, involves surface waves. The microwaves propagate along the interface between the quartz tube and the plasma column. The wave energy is absorbed by the plasma. Characteristic features are the wavelength (815 MHz or 2.45 GHz), which is comparable to the length of the apparatus, and the short period of the exciting microwave field. Surface-wave discharges can be created both at reduced and at atmospheric pressure, but in the pressure regime above 0.1 atm they approach a state of LTE.78
 | ||
| Fig. 14 Schematic (left) and image (right; courtesy of Dutch Institute For Fundamental Energy Research (DIFFER)) of a MW discharge. | ||
A classical GA plasma is an auto-oscillating periodic discharge, where the gas flows between two diverging flat electrodes. When applying a potential difference between both electrodes, an arc plasma is formed at the narrowest gap, which is then dragged by the gas flow towards a rising interelectrode distance, until it extinguishes and a new arc is ignited at the shortest interelectrode gap. More specifically, the length of the arc column increases together with the voltage until it exceeds its critical value (lcrit). At this point, heat losses from the plasma column begin to exceed the supplied energy and it is no longer possible to sustain the plasma in its LTE state, resulting in a fast transition into a non-LTE state. The discharge cools rapidly to gas temperature but the plasma conductivity is maintained by a high value of the electron temperature (Te ∼ 1 eV, which is most suitable for efficient vibrational excitation of CO2). After this fast transition, the GA continues its evolution under non-LTE conditions, until the length reaches a new critical value (l ∼ 3·lcrit), leading to decay of the discharge, after which the evolution repeats from the initial breakdown. During this cycle, up to 75–80% of the energy can be dissipated in the non-LTE zone of the gliding arc. It is this effect that permits the stimulation of chemical reactions in regimes quite different from conventional thermal reaction chemistry. When the GA is operated under milder conditions, i.e. lower currents and high frequencies, this transition to the non-LTE phase can occur in the order of nanoseconds. As a result, the GA operates in the non-LTE regime starting almost immediately after its ignition, and a higher fraction of the discharge energy can be consumed by the non-LTE phase.97–99
The classical GA plasma, however, also exhibits some disadvantages. The flat 2D electrode geometry makes it less compatible with industrial systems, and also the gas conversion is non-uniform and quite limited, because a considerable fraction of the gas does not pass through the active plasma region. Moreover, a high gas flow rate is needed to drag the arc, so the gas residence time is limited, thereby further limiting the conversion. Therefore, a 3D cylindrical GA plasma reactor was developed a few years ago, which makes use of vortex flow stabilization.100 The gas flows in the reactor through a tangential inlet. An arc is again formed between both electrodes, and is dragged with the tangential gas flow, thereby expanding until it extinguishes, followed by a new cycle. Basically, the arc is again gliding between the anode and cathode, and it is stabilized in the centre of the reactor. Depending on the diameter of the anode tube, a forward or reverse vortex gas flow is created. If the anode diameter is equal to the cathode diameter, the gas can leave the reactor through the anode outlet, leading only to a forward vortex flow. On the other hand, if the anode diameter is smaller than the cathode diameter, the gas cannot immediately leave the reactor, and will first flow upwards close to the walls, in a forward vortex. As this gas is still cold, it creates an isolating and cooling effect, protecting the reactor walls from the warm plasma arc in the centre. When it reaches the upper end of the reactor, it will have lost some speed due to friction and inertia, and when flowing downwards, it moves in a smaller inner vortex, a so-called reverse vortex, where it mixes with the plasma arc, resulting in a more energy-efficient conversion. The reverse vortex flow GA is also called a ‘gliding arc plasmatron’ (GAP). Fig. 15 shows the difference between these two types of GAs.
 | ||
| Fig. 15 Schematic of the classical GA (top left) and the GAP (top right) configuration, and pictures of the classical GA (bottom left; courtesy of the University of Manchester) and GAP (bottom right; courtesy of the University of Antwerp). | ||
RF discharges usually operate in the 1–100 MHz frequency range, resulting in a corresponding wavelength (300–3 m) much larger than the plasma reactor dimensions. The power coupling can be done through capacitive or inductive coupling, resulting in capacitively coupled plasma (CCP) and inductively coupled plasma (ICP). These kind of discharges are mainly applied to thin-film deposition, plasma etching, the sputtering of materials and as an ion source in mass spectrometry,96 but some research is also being performed for CO2 conversion applications, as is illustrated in Section 5. An RF source operating at low pressure, both with and without the influence of an external magnetic field, has been used. The advantages for CO2 conversion are its capability to allow obtaining high electron densities at low gas temperature.
When a sufficiently large electric field is applied, corona discharges occur near sharp points, edges or thin wires when used as an electrode. When a high negative voltage is applied to the electrode, it acts as a cathode and is considered as a negative corona discharge. When a positive voltage is applied, it is considered as a positive corona discharge. Corona discharges are always non-uniform, with a strong electric field, ionization and luminosity close to the wire/sharp electrode, whereas the charged particles are dragged to the other electrode by weak electric fields. They are often operated in a pulsed mode, mostly to increase the power, while inhibiting the transition of the streamer formation into sparks.21 An advantage of corona discharges compared to DBDs is that they are relatively easy to establish; however, their performance in CO2 conversion processes is quite similar, as will become clear from Section 5.
When a streamer is able to connect two electrodes, without the presence of a pulsed power supply (see corona discharge above) or the presence of a dielectric (see DBD), a spark discharge can develop by further growth of the current.21 However, when the amount of power provided is restricted, the discharge cannot develop into a stationary (thermal) arc and instead the discharge extinguishes within several hundred microseconds. Hence, spark discharges consist of an initiation of streamers that develop into highly energetic spark channels, which then extinguish and reignite periodically. Lightning is a typical example of a spark discharge.
A wide variety of plasma set-ups fall under APGDs, such as miniaturized direct current (dc) GDs, microhollow cathode dc discharges and RF discharges as well as DBDs.78 The main advantage of APGDs is the absence of vacuum conditions compared to with regular glow discharges, and also operating without elevated temperatures. Depending on the gas mixture and electrode configuration, the discharge can operate in a stable homogeneous glow or filamentary glow mode. For example, a DBD can operate in APGD mode, and in this case the discharge benefits from the average power densities of a DBD but operating in a uniform homogeneous glow mode, thus benefiting uniform gas treatment.21 This discharge is interesting for DRM due to its special characteristics of electron density (3 × 1012 cm−3), electron temperature (∼2 eV, suitable for the vibrational excitation of CO2) and plasma gas temperature (900 K), which is lower than that of thermal plasma but higher than that of DBDs and corona discharges.
Finally, nanosecond pulsed discharges are basically discharges that rely on repetitively pulsed excitation, through a nanosecond scale pulse rise time and duration. This leads to a highly non-LTE state with very high plasma densities for a relatively low power consumption due to the short pulse durations. Whereas (sub)microsecond pulsed discharges are initiated by Townsend discharges, nanosecond pulsed discharges ignite instantaneously, i.e. without the involvement of secondary electrons. Furthermore, the discharge remains in a glow-like (rather than filamentary) mode despite the high electric field. The interest in shorter pulses is not because of the duration of the discharge itself, but rather because they offer better control of the electron energy than continuous wave discharges, depending on the pulse length.101 Thus, more energy can be directed towards the desired dissociation channels.
4.3. Plasma-catalysis
As with most of the technologies described in Section 3, plasma set-ups can also be combined with a packing material or catalyst, giving rise to plasma-catalysis. Plasma-catalysis is an emerging branch of plasma processing at the interface of a variety of disciplines, including physical chemistry, material science, nanotechnology, catalysis, plasma physics and plasma chemistry. In short, its objective is to enhance plasma reactions by adding a catalyst to the reaction cycle and vice versa. Theoretically speaking, combining plasma with catalysis offers the best of both worlds. Inert molecules are activated by the plasma under mild conditions, and subsequently the activated species selectively recombine at the catalyst surface to yield the desired products. This will be especially important to further advance and optimize the direct oxidative liquefaction pathway of plasmachemical CO2 conversion, in order to selectively produce the desired liquid products. In this section, we present a brief overview of the different approaches and possible interactions between the plasma and the catalyst. For more extensive details, we refer to several broad reviews on this specific topic that have recently been published.83,102–104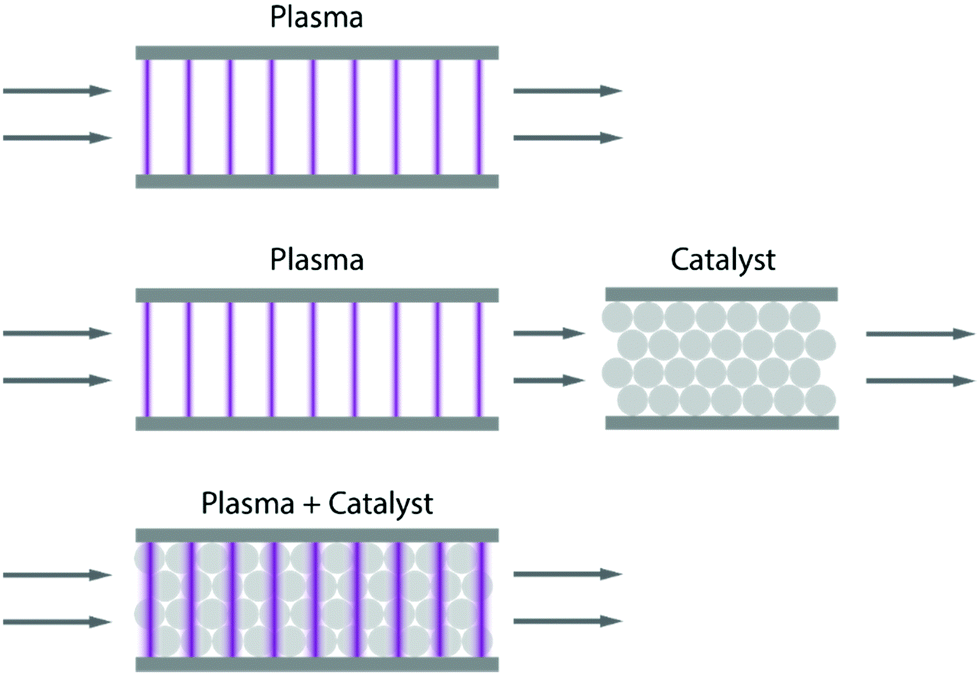 | ||
| Fig. 16 Schematic of the different plasma-catalyst configurations: (a) plasma alone without a catalyst, (b) catalyst downstream of the discharge, and (c) catalyst directly inside the discharge zone. These examples mainly apply to a DBD. For MW and GA discharges, the catalyst is most commonly placed in the downstream region. | ||
The major difference between the one-stage and two-stage configuration is the kind of species to which the catalyst is exposed. In the two-stage configuration, the end-products and the long-lived intermediates will interact with the catalyst, while in the one-stage configuration, the catalyst can also interact with all the short-lived species, including excited species, radicals, photons and electrons. Furthermore, in this configuration the catalyst may also be influenced by the plasma and vice versa (see below). Besides plasma-catalysis, the preparation and modification of catalysts by plasma treatment is gaining increased attention, especially for catalysts with a low thermal stability.103
The most widely investigated and, for this review, most interesting configuration is the one-stage configuration, in which the catalyst is placed inside the discharge region, either completely or only partially occupying the discharge zone. The catalyst itself can be introduced in the discharge as pellets, (fine) powders, foams, honeycomb monoliths, different electrode materials and electrode coatings or as coated quartz wool. The ease of adding a catalyst into the discharge zone greatly varies depending on the type of plasma reactor used. In general, due to their simple geometry and operation close to room temperature, implementation of a catalyst in a DBD reactor is very easy. Although MWs also have a simple geometry, due to the high gas temperature inside the discharge zone (1000–2000 K compared to 300–400 K for DBD) catalysts are often placed downstream, due to their low thermal stability. Finally, GA discharges have rather complex geometries and the same higher gas temperatures, thus catalysts are typically introduced downstream, although the use of a spouted bed has also been reported.105 If these MW and GA discharges can be operated at slightly lower temperatures (≤1000 K), this would yield new possibilities. It would also open up the way for using sufficiently thermally stable catalysts inside the discharge zone, and at the same time it would allow thermal activation of catalysts inside the discharge zone by the plasma.
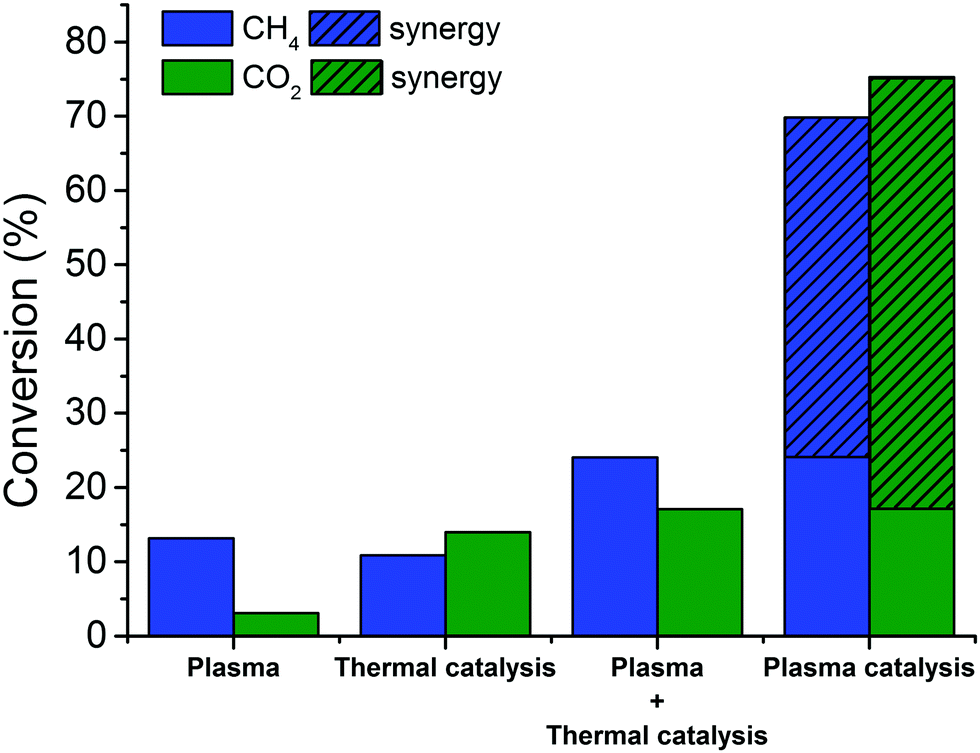 | ||
| Fig. 17 Demonstration of the synergy of plasma-catalysis for the dry reforming of methane, adapted from Zhang et al.106 | ||
The complex interactions can be subdivided into two categories: the effects of the plasma on the catalyst, and vice versa, the effects of the catalyst on the plasma.102,107 Second, it is important to distinguish between the two types of effects: physical and chemical effects. While the physical effects are mainly responsible for gaining a better energy efficiency, the chemical effects can lead to an improved selectivity towards the targeted products. In the case of CO2 splitting, mainly CO and O2 are formed, so the primary added value of the catalyst is to increase the energy efficiency, although the conversion can also be improved by chemical effects, such as enhanced dissociative chemisorption due to the catalyst acid/basic sites. When adding a co-reactant (e.g. CH4, H2O, H2), the catalyst allows modifying the selectivity towards the targeted products.
An overview of the plasma–catalyst interactions is given in Fig. 18. When adding a catalyst, the following effects on the plasma have been reported: (i) an enhancement of the electric field through the geometric distortion and surface roughness; (ii) the formation of micro-discharges inside the pores of the catalyst material, due to the very strong electric field inside the pores, leading to different characteristics compared to the bulk; (iii) a change in the discharge type, because the presence of insulating surfaces promotes the development of surface discharges; (iv) the adsorption of species on the catalyst surface, which affects their concentration and conversion due to longer retention times. All the effects of the catalyst on the plasma can thus generally be considered as physical effects. The reported effects of the plasma on the catalyst, on the other hand, are of both a physical and chemical nature. They include: (i) changes in its physico-chemical properties, such as a higher adsorption probability at the catalyst surface, a higher catalyst surface area, a changed catalyst oxidation state, the reduction of metal oxide catalysts to their metallic form, reduced coke formation on the surface and a change in the catalyst work function; (ii) the formation of hot spots on the surface due to the formation of strong micro-discharges; (iii) catalyst activation by photon irradiation emitted by the excited plasma species (although this effect is probably of only minor importance due to the low photon fluxes); (iv) lowered activation barriers due to there being more reactive vibrationally excited plasma species and consequently a greater possibility for non-adiabatic barrier crossings; (v) changes in the reaction pathways due to the presence of a wide variety of (re)active species.
 | ||
| Fig. 18 An overview of the possible effects of the catalyst on the plasma and vice versa, possibly leading to synergism in plasma-catalysis. | ||
While the physical effects have been more extensively studied, such as the plasma behaviour in packed-bed DBD reactors108 or inside catalyst pores,109 the chemical effects are less understood, mainly because they are often correlated with the physical effects. Indeed, in a DBD, the catalyst is mostly introduced as a packed bed, making it difficult to distinguish between the two effects. Hence, more systematic studies on both the physical and chemical effects of the catalyst material are highly needed.102 Furthermore, as summarized in Fig. 19, the plasma and catalyst both have an influence on the gas composition, and at the same time the plasma influences the catalyst properties and vice versa, as mentioned above. Therefore, both the gas composition and the plasma and catalyst properties continuously influence each other, and at the same time are influenced by the plasma chemistry and surface chemistry. Thus, it is recommended to develop tailored catalysts for plasma-catalysis, rather than simply relying on classical catalysts. This is discussed further in Section 5.
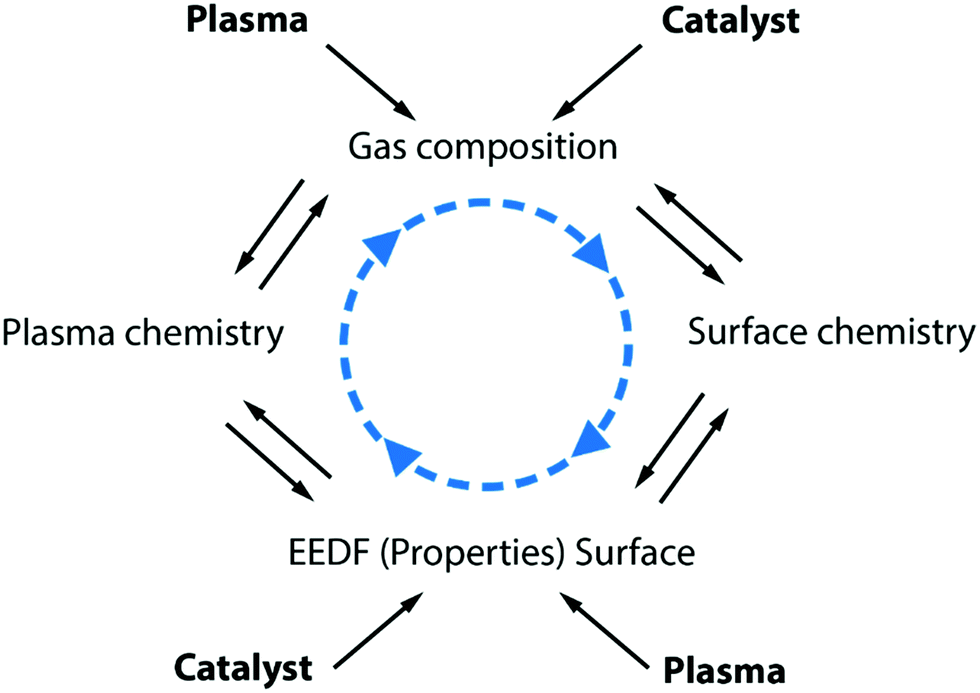 | ||
| Fig. 19 Visualization of the continuous influence of plasma and the catalyst on the gas conversion process (EEDF = electron energy distribution function). | ||
Finally, it is worth mentioning that besides CO2 conversion, the combination of plasma with catalysts is also promising for nanomaterial synthesis, such as for the growth of carbon nanotubes, nanofibres, and graphene. To date, research on this topic has been mainly focused on pure methane plasmas, instead of CO2 plasmas. This is, however, outside the scope of the present review, but we refer to another recent review paper, where these applications are discussed in detail.102
4.4. Definitions
In this section, an overview of the different expressions for the specific energy input (SEI), conversion (χ), energy efficiency (η) and energy cost (EC) are given, which are used for the critical assessment in Section 5.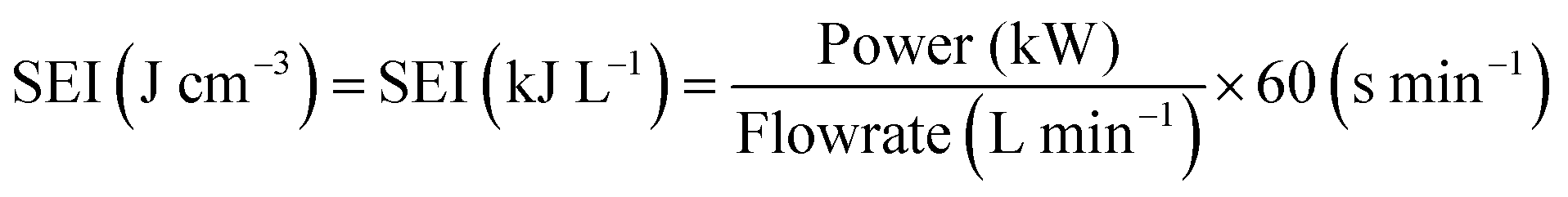 | (1) |
The SEI is also commonly expressed in electron volts per molecule:
 | (2) |
Note that the value of 24.5 L mol−1 is only valid for 298 K and 1 atm.
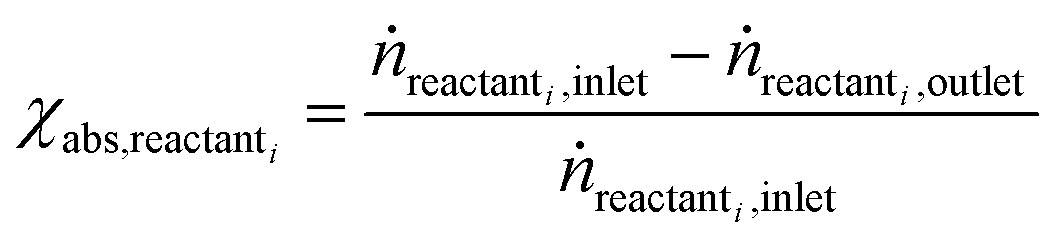 | (3) |
When more than one gas is present in the feed mixture, the effective conversion takes the dilution into account:
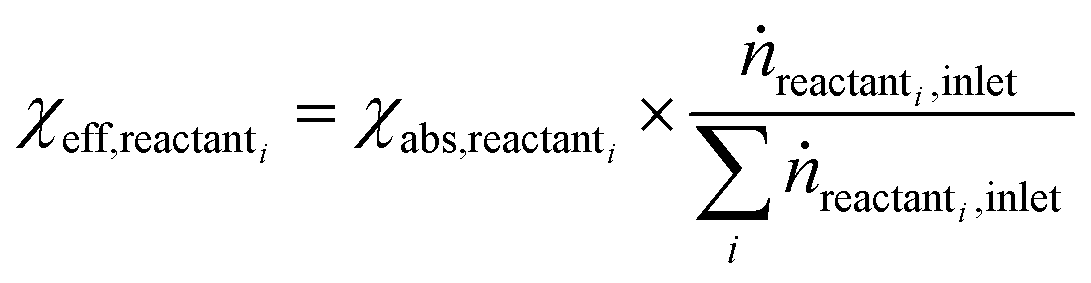 | (4) |
This alternative definition of conversion is important for comparing the conversion of a specific reactant in different mixtures, since it shows how the conversion rate of the reactant is affected, rather than the absolute value of its conversion.
For an easy comparison of DRM mixtures, it is also interesting to determine the total conversion, which is the sum of the effective conversions:
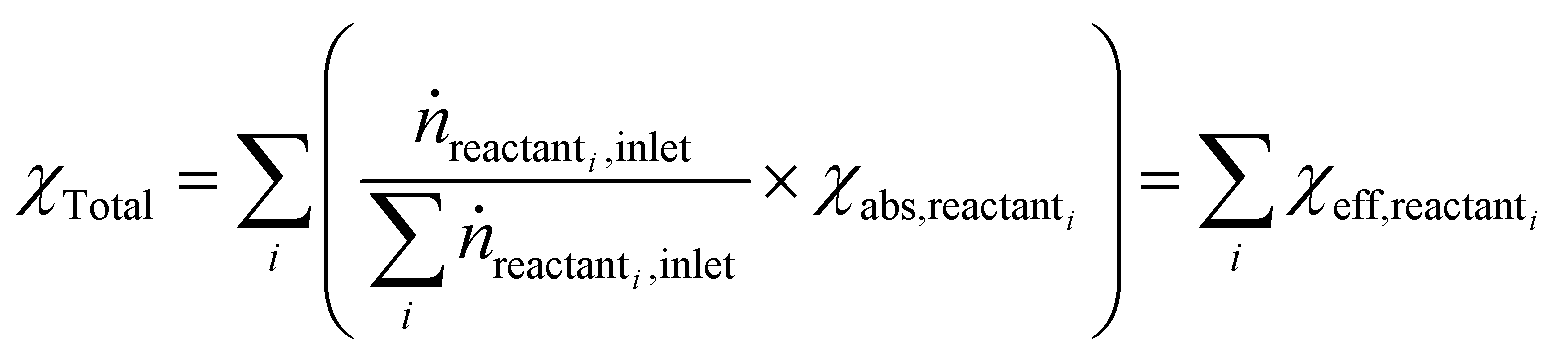 | (5) |
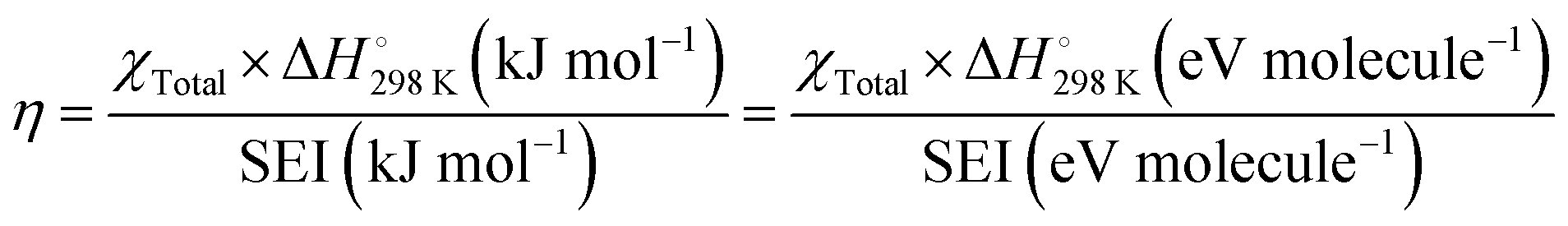 | (6) |
As mentioned in Section 2,  is 283 kJ mol−1 (or 2.93 eV per molecule) for pure CO2 splitting and 247 kJ mol−1 (or 2.56 eV per molecule) for DRM.
is 283 kJ mol−1 (or 2.93 eV per molecule) for pure CO2 splitting and 247 kJ mol−1 (or 2.56 eV per molecule) for DRM.
The energy cost is the amount of energy consumed by the process (generally expressed as kJ per converted mol or eV per converted molecule):
 | (7) |
Note that the value of 24.5 L mol−1 is only valid for 298 K and 1 atm.
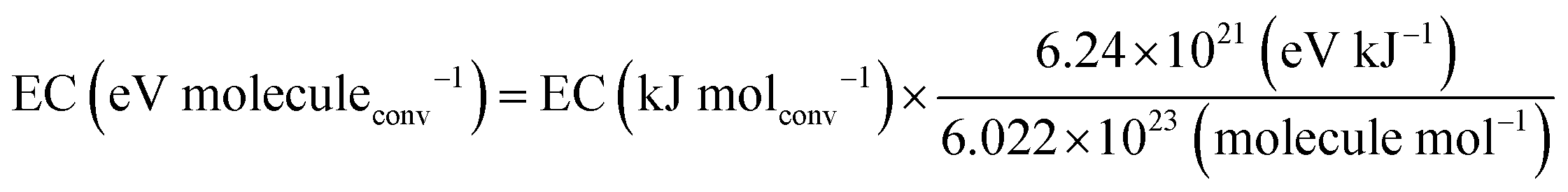 | (8) |
5. Critical assessment of plasma-based CO2 conversion
In this section, we address the present state-of-the-art with respect to plasma-based CO2 conversion, focusing on a critical assessment of the advantages and disadvantages of the various set-ups described in Section 4.2. The aim is to unveil their future challenges, risks and opportunities for successful implementation. Several options are being investigated, including both pure CO2 splitting into CO and O2, as well as the reaction with other gases, such as CH4 (the dry reforming of methane), H2O (artificial photosynthesis) and H2 (hydrogenation). By adding a hydrogen source, the main aim is to produce value-added chemicals and/or fuels, e.g. syngas, hydrocarbons and valuable oxygenates, such as methanol, formaldehyde and formic acid. As mentioned above in Section 4, most research on plasma-based CO2 conversion is performed with DBDs, MW and GA plasmas, with a main focus on improving the energy efficiency of the conversion, as well as the selectivity towards value-added chemicals, in combination with catalysis.In order for plasma technology to be competitive with traditional as well as emerging novel technologies (as described in Sections 2 and 3) we can define two main goals. First, plasma technology will have to be competitive with electrolysis, which is its main competitor and that also has the same advantage as being able to rely on all sorts of renewable electricity. For electrochemical water splitting, commercial energy efficiencies of 65–75% have been obtained; hence, the goal of plasma technology should be at least to aim for an energy efficiency comparable to this, and better than the thermal equilibrium energy efficiency limits of ∼45–50% (see Section 2). Second, when comparing with other novel technologies that can make use of direct solar energy, such as solar thermochemical conversion, we should look at the solar-to-fuel conversion efficiency. As such, for novel technologies, a solar-to-fuel conversion efficiency of approximately 20% is considered industrially competitive, e.g. when looking at the production of syngas,110 as will be elaborated in Section 6.3 below. When relying on solar energy, taking a solar panel efficiency of 25%111 and a plasma-conversion energy efficiency of 60–80% towards the syngas components CO and H2, this would yield a competitive solar-to-fuel efficiency of 15–20%. Hence, in the following critical assessment of plasma-based CO2 conversion technology, we should keep in mind that – from an energy efficiency point of view – to be considered as a competitive and worthy alternative, plasma technology should aspire to an energy efficiency of at least 60%, at least when focusing on the production of syngas.
On the other hand, the conversion of syngas into more suitable fuels and chemicals through the Fischer–Tropsch process or for methanol synthesis (and subsequently for methanol or ethanol to olefin) is a very energy-intensive process. As a result, the energy efficiency requirements for the direct production of these compounds is highly dependent on the formed products. Hence, the target energy cost to be competitive can be significantly lower for a direct one-step process. For example, a solar-to-methanol conversion efficiency of 7.1% is already economically feasible.110 In this case, a plasma-conversion energy efficiency of 30% (instead of the above 60%) would already suffice. This is very important and it shows that, in contrast to the indirect approach through syngas, for the direct oxidative pathway – which aims to synthesize oxygenated liquid products in one step – the energy efficiency target is much lower, depending on the products formed, due to the circumvention of the energy-intensive step of converting syngas into the desired liquids. The possibility to proceed through this direct oxidative pathway is one of the key benefits of plasma technology, and in theory this pathway is the most promising. For now, however, a lot of research is still needed to understand the underlying processes, in order to improve the yield of the desired (oxygenated) liquid products.112 Hence, the analysis in this section is based on the production of the syngas components CO and H2 using plasma technology, based on the energy efficiency target of 60%. Nevertheless, it is important to keep in mind that this 60% energy efficiency target applies to syngas production, but this target would change completely, i.e. it would decrease by a factor two to three, if a direct oxidative pathway towards liquids could be successfully realized using plasma technology.
5.1. CO2 splitting
As outlined in Section 4 above, in the case of pure CO2 splitting, the dominant products are CO and O2 in a 2-to-1 ratio. Hence, this is a simple chemical process and there are no concerns regarding the complexity and the wide variety of products that can be formed – such as in the case with an added H-source – and thus one does not have to contemplate how to steer the different selectivities. As such, the research can solely focus on optimizing the CO2 conversion and the energy efficiencies, and will be judged on those indicators.The most commonly used geometry to study CO2 conversion uses a coaxial DBD reactor,95,113–115 while parallel plate reactors116 (see Section 4.2.1; Fig. 13) are mainly used to study the system with advanced optical diagnostics.117 To achieve higher values of CO2 conversion and energy efficiency, several approaches have already been investigated, including changing the applied frequency, applied power, gas flow rate, discharge length, discharge gap, reactor temperature, dielectric material, electrode material, mixing with gases, i.e. Ar, He, N2, and by introducing (catalytic) packing materials. Furthermore, extensive modelling has also been performed to obtain a more fundamental insight into the plasma chemistry in a DBD and in turn to aid in the improvement of future experiments. Fig. 20 summarizes most of the data available in the literature. From this figure, several main trends become clear. First of all, the conversion increases with increasing specific energy input (SEI), while the energy efficiency generally decreases with increasing SEI, especially above an SEI of 10 eV per molecule. When plotting the energy efficiency as a function of the conversion, it is clear that most of the results are situated below an energy efficiency of 15% and a conversion of 40%, with some exceptions. The highest conversions of 42% were obtained for packed-bed DBDs while the highest energy efficiency of 23% was obtained with a pulsed power DBD. It is important to note that some of the data in this figure has been recalculated to represent coherent values for the conversions and energy efficiencies. For instance, in plasma research, the mixture is sometimes diluted with He, Ar or N2 to obtain an more easily ignitable and more stable discharge, but as will be mentioned below, this influences the results – an effect that was, among others, not always taken into account in the original data. A more detailed influence of the different parameters is discussed below.
 | ||
| Fig. 20 Experimental data collected from the literature for CO2 splitting in a DBD, showing the conversion (a) and energy efficiency (b) as a function of the SEI, as well as the energy efficiency as a function of the conversion (c). The open symbols represent the data with a packed bed (PB). Note, some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
It is well known that the specific energy input (SEI), which is defined as the plasma power divided by the gas flow rate, is the dominant determining factor for the conversion and energy efficiency.95,114,115,118 It has also been observed that the same values of SEI defined by different combinations of plasma power and gas flow rate can result in different conversions. It is the gas flow rate, and hence the residence time, which has the most important effect, while the effect of the power is less significant. As such, a lower power with a lower gas flow rate will result in a higher conversion and energy efficiency than a higher power with a higher gas flow rate for the same SEI.95,115
Some possible geometry modifications are the discharge length and discharge gap. Yu et al.114 found that varying the discharge length for a fixed SEI has no significant effect on the discharge characteristics, the conversion and the energy efficiency. On the other hand, the discharge gap appears to affect the discharge behaviour, and thus also the CO2 conversion. Aerts et al.95 found that above a certain gap width (3.3 mm in their case) less streamer formation occurs, leading to a decrease in the effective plasma volume, and hence lower conversions and efficiencies result.
Furthermore, Aerts et al.95 also reported that the applied frequency seems to have a negligible effect on the conversion and energy efficiency. However, the plasma appears more filamentary at high frequency (75 kHz) compared to at low frequency (6 kHz). Ozkan et al.,115 however, found that both the conversion and energy efficiency decrease slightly upon increasing the frequency from 15 to 30 kHz. This was explained by Paulussen et al.,113 who suggested that the optimum discharge frequency depends on the power input, and as such it cannot be unambiguously stated that higher or lower frequencies give rise to an increased CO2 conversion and energy efficiency.
In thermochemical reactions, the gas temperature is one of the most important parameters governing the reaction rates. For the plasma-based conversion in a DBD, however, the effect of temperature is not so clear. Paulussen et al.113 observed a slight and linear increase in the conversion from 26% to 28.5% when the inlet gas was heated from 303 K to 443 K. Ozkan et al.,115 on the other hand, reported that the wall and gas temperature should remain as low as possible. In addition, Wang et al.118 detected an increase in conversion of 0.5–3% when using an external fan to cool the reactor, and they suggested that higher flow rates are preferred, since the latter remove large amounts of heat from the reactor. Brehmer et al.117 noted a big effect of the wall temperature on the O3 density and suggested that the recombination of CO and O on the wall would also increase with rising wall temperatures.
The effect of the dielectric material used in the DBD reactor is another topic of debate. By studying the formation of a conductive coating, Belov et al.119 observed that the conductivity of the dielectric might be the most crucial parameter affecting the discharge properties. However, Aerts et al.95 reported no significant effect between quartz or alumina under a wide range of conditions, although it was still concluded that alumina has several advantages in terms of reactor stability. Ozkan et al.115 found that alumina and quartz perform better than mullite and Pyrex, and that a thicker dielectric leads to a higher conversion and energy efficiency.115 More sophisticated dielectric materials were created and tested by Li et al.120,121 and Wang et al.122 The former investigated the influence of Ca0.8Sr0.2TiO3 (CST) with 0.5 wt% Li2Si2O5 as the dielectric barrier and achieved an improvement in the conversion by a factor 9 of up to 9, compared to silica glass. Wang et al.122 investigated the performance of CST ceramics with various amounts of CaO–B2O3–SiO2 (CBS) glass addition (0.5–5 wt%), and found that the addition of 5.0 wt% CBS resulted in an increase in the conversion by a factor of 2.6, while the energy efficiency almost tripled compared to that with 0.5 wt% CBS. However, from the calculation of the plasma power in both studies, it appears that the more sophisticated dielectrics mainly increased the efficiency between the input power and plasma power, and not the effective plasma-conversion energy efficiency. Nevertheless, these dielectrics allowed operation under lower voltages, which could be beneficial for certain processes. In summary, different dielectrics may allow for easier igniting and streamer formation, but not necessarily more energy-efficient plasma-conversion chemistry.
Besides the dielectric material, also the electrode material can be varied. Wang et al.118 studied the effect of changing – or coating – the high voltage electrode and obtained an order of activity as Cu > Au > Rh > Fe ∼ Pt ∼ Pd. The Cu and Au electrodes yielded a relative increase in the conversion by a factor 1.5 compared to an Fe electrode. Furthermore, the maximum energy efficiency of the Au electrode was almost three times higher than the energy efficiency of the Rh electrode under the same conditions. However, besides the fact that some of these electrodes or coatings are more expensive, compared to the inert stainless steel electrode, they also are susceptible to chemical erosion (i.e. oxidation) and plasma sputtering (as observed for Au).
As mentioned above, some researchers have also added inert gases, such as N2, Ar and He, to ignite the plasma more easily. This also has several effects on the discharge characteristics, conversion, energy efficiency and even by-product formation in the case of N2. The addition of He and Ar leads to an increase in the CO2 conversion, but the effective conversion decreases, since there is less CO2 present in the mixture and the increased conversion is not sufficient to counteract this drop in the CO2 fraction. As a result, the energy efficiency decreases as well.118,123,124 The addition of N2, on the other hand, shows a completely different behaviour. Snoeckx et al.125 discovered that the presence of N2 in the gas mixture of up to 50% barely influences the effective CO2 conversion and the corresponding energy efficiency. Indeed, N2 enhances the absolute CO2 conversion, due to the dissociation of CO2 upon collision with N2 metastable molecules, and this effect is strong enough to compensate for the lower CO2 content in the mixture. However, the presence of N2 in the mixture leads to the formation of unwanted by-products, i.e. N2O and several NOx compounds, with concentrations in the range of several 100 ppm,125 which can give rise to severe air pollution problems.
Other modifications being investigated to improve the CO2 conversion in a DBD reactor are the use of micro plasma reactors,126 a hybrid DBD reactor on the surface of an SOEC127 and a pulsed power supply.115 A micro plasma reactor provides a stronger electric field and a higher concentration of reactive species, while offering better control of the processing parameters.126 Applying DBD plasma on the surface of an SOEC allows the in situ exclusion of O2 during CO2 splitting, resulting in an increase in the conversion by a factor of 4.127 Finally, the use of a power supply in the pulsed or so-called burst mode, instead of injecting the power in a continuous AC mode, is reported to lead to an increase in conversion and energy efficiency by a factor of 1.5 for a duty cycle of 50%.115
Besides all the experimental work, great advances have also been made in modelling the plasma chemistry for CO2 conversion in a DBD.90,95,124,128–131 The main findings are that the splitting of CO2 is dominated by electron-impact reactions with ground-state molecules and, predominantly, by electron-impact excitation followed by dissociation. Electron-impact ionization is also important, but is compensated by the fact that a large fraction of the formed ions will eventually recombine, resulting in the formation of CO2. Splitting from the vibrationally excited states is found to be of minor importance in a DBD.95,128 A reduced chemistry model consisting of only 9 species and 17 reactions was presented by Aerts et al.,95 which allowed identifying the main dissociation mechanisms. A 1D fluid model, with roughly the same chemistry, was developed by Ponduri et al.130 Furthermore, recently, a thorough examination of the cross-sectional data was performed,94,132 as well as a careful examination of all the rate coefficients and a comparison of the performances of different models.129
Finally, as mentioned in Section 4.3, a packing can be added to the DBD reactor to enhance the conversion and energy efficiency. In the case of pure CO2 splitting, the addition of a packing will not influence the formation of products, since no hydrogen source is available. Hence, most of research work focuses on increasing the conversion and energy efficiency by physical effects. Generally, when tested, the CO2 conversion in packed-bed DBD reactors is always higher than in the corresponding empty reactors, albeit Fig. 20 clearly illustrates that the highest efficiencies found to date are for regular DBD reactors, achieved by varying the different parameters discussed above. Nevertheless, at high conversions, we see that the packed-bed reactors are generally more efficient. As such, in general, adding a packing seems to allow the system to operate at the same energy efficiency, but significantly increases the conversion. Several materials have already been investigated, more specifically, glass wool,133 glass beads,134 silica gel,114 quartz,114 quartz wool,133,135 quartz sand,135 Al2O3,114,133,135,136 CaTiO3,114 ZrO2,133,137 SiO2,133 BaTiO3,133,134,136 MgO135 and CaO.135 The best results have been obtained for ZrO2137 and CaO,135 with conversions in the range of 30–45% and energy efficiencies in the range of 5–10% (see Fig. 20). One of the more recent and interesting works is from Butterworth et al.136 The authors stated that the testing of different materials should be performed according to a more predefined protocol, since the packing particle size affects the discharge phenomena and the chemistry within the packed bed. More specifically, the efficacy of CO2 conversion is strongly affected by the particle size, whereby small particle sizes (180–300 μm) can increase the CO2 conversion by up to 70%. However, they also increase the reactor breakdown voltage and they lead to partial discharging, i.e. a drop in the fraction of the reactor where plasma formation occurs. Comparison with the works of other researchers shows that quite often, insufficient electric field strengths are applied for complete reactor discharging to occur. Hence, packing materials for plasma-catalysis should be tested with equivalent reactor operating conditions. It is therefore important to ensure that either: (a) complete discharging occurs in the reactor, or (b) the partial reactor discharging is quantified.136 A similar message, regarding the need to compare material performances with similar reactor set-ups, was given in the recent work of Michielsen et al.133
From all these data in the literature, we can conclude that a DBD reactor can provide reasonable conversions of up to 40%, but the energy efficiency is still at least a factor of 3–4 away from the necessary 60% mark. DBDs have the advantage of being very scalable and easy to operate, but their current energy efficiency makes it doubtful that they will be the most suitable technology for pure CO2 splitting.
The main set-ups used for CO2 conversion are the (surfaguide) MW discharge (2.45 GHz and 915 GHz) and RF discharges (13.56 MHz). More details about these set-ups can be found in Section 4. To obtain the best values for the conversion and energy efficiency, several approaches have already been proposed, including changing the applied power, gas flow rate, flow type, reactor geometry, gas temperature, and admixture gases, or by introducing (catalytic) packing materials, as well as by carrying out extensive plasma chemistry modelling to gain a better insight into the underlying mechanisms. Fig. 21 summarizes all the data available from the literature. The highest energy efficiencies reported in the 1970–1980s were up to 80% for subsonic flow conditions, and up to 90% for supersonic flows.21,139,140 Recently, several attempts have been made to reproduce these high energy efficiencies.141–144 To date, maximum energy efficiencies of 50% have been successfully achieved, with both MW and RF discharges.141–143 The results obtained by Bongers et al.142 were near the thermal dissociation limit, indicating that the thermal dissociation mechanism is predominant. The authors concluded that the applied values for a reduced electric field, i.e. 70–80 Td, were too high, and future experiments should be directed towards values around 20–50 Td, while keeping the temperature in the discharge as low as possible. It should be noted that all the experiments reporting such high energy efficiencies were performed at reduced pressures, which might not be beneficial for high-throughput industrial implementation, despite the high flow rates of up to 75 SLPM.142 Indeed, increasing the pressure leads to a significant decrease in energy efficiency.140 At atmospheric pressure, Spencer et al.144 obtained a maximum energy efficiency of 20%, which is nevertheless still a factor two better than that obtained for a DBD (see previous section).
 | ||
| Fig. 21 Experimental data collected from the literature for CO2 splitting in MW and RF discharges, showing the conversion (a) and energy efficiency (b) as a function of the SEI, as well as the energy efficiency as a function of the conversion (c). The open symbols represent the data from the 1970–1980s. Note, some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
Fig. 21 also demonstrates that, in general, surfaguide MWs achieve the highest energy efficiencies (up to 90%). Furthermore, just like for the DBD results, the conversion increases with increasing the SEI, while the energy efficiency decreases above an SEI of 0.1–1 eV per molecule. When plotting the energy efficiency as a function of the conversion, it is clear that the more recent results typically have an energy efficiency of 10–50% in the entire range of conversions up to 95%. Again, like for the DBD results above, it is important to note that some of the data have been recalculated to represent coherent values for the conversions and energy efficiencies. A more detailed influence of the different parameters is discussed below.
For the MW discharges, the same trade-off as described for the DBD between energy efficiency and conversion as a function of SEI is present.141,142,144–146 Hence, to successfully increase the energy efficiency, one must be able to increase the conversion degree without increasing the specific energy input of the system, which essentially requires using techniques other than increasing the input power.145 It is also clear that the highest energy efficiencies are obtained for low SEI values (0.1–1 eV per molecule), while the conversion is low at these conditions, hence conflicting requirements are encountered.143
As already mentioned, pressure has one of the most important influences on the MW discharge and on its performance for CO2 conversion, and most studies are carried out at reduced pressure. One of the reasons for this is that at low pressure the three-body recombination reaction CO + O + M → CO2 + M, which is the most important reaction limiting the effective CO2 conversion in the gas phase, becomes negligible.146 An optimum operation pressure seems to exist around 150 mbar (at 5 SLPM), but the latter depends on other operating conditions, such as the gas flow rate.141 Furthermore, the pressure plays an important role in which mode – contracted, diffuse or combined – the plasma is operating.141
Several more complex flow types and geometries have already been studied to optimize the MW discharge performance for CO2 conversion. Supersonic flows in MW discharges have proven to reduce the losses of vibrational levels upon collision with ground-state molecules (i.e. the so-called VT relaxation), which occurs mainly at higher temperature, and has allowed achieving an energy efficiency of up to 90%,140 in agreement with theoretical calculations.139 Bongers et al.142 observed an energy efficiency of only 15% for a supersonic flow, but by additionally quenching the plasma, an energy efficiency of up to 47% was observed, as predicted for plasmas in the thermal regime (see Section 2, Fig. 2). Similarly, the addition of a vortex gas flow, more specifically a reverse vortex, led to a significant improvement in CO2 conversion and energy efficiency compared to a forward vortex flow.142
For MW discharges, the effect of gas temperature is quite complicated. A signature of the desired non-equilibrium conditions in a MW plasma is a low (or moderate) gas temperature (in the order of 1000–2000 K), while vibrational and electron temperatures are higher (i.e. 3000 up to 8000 K and 1–5 eV, respectively).89,141 For low pressures and reduced electric fields (E/n), gas temperatures around 2000 K have been observed. However, when increasing pressure at an SEI of about 0.5 eV per molecule, the discharge undergoes a transition from a diffuse to a contracted regime, and the temperature rises steeply to 14000 K.141 When this transition occurs and the vibrational and rotational gas temperature are in thermal equilibrium, low values of energy efficiency are observed.144 Recent modelling studies suggest that keeping the temperature under control is beneficial for the vibrational excitation, and is a key parameter for more efficient CO2 conversion.88–90 Bongers et al.142 noted that the E/n in their MW plasma set-up was too high to achieve VT non-equilibrium conditions and that the high temperatures in the plasma core could account for the observed high values of E/n. It is thus clear that the gas temperature should be kept as low as possible to reduce vibrational energy losses via VT relaxation.88
Just like for the DBD plasma, some papers have also reported on the addition of other gases to CO2 in the MW plasma, i.e. Ar,144,147,148 He148 and N2.149 For Ar, Spencer et al.147 observed no effect on CO production, suggesting it does not affect the collisional processes benefiting dissociation, but, depending on the set-up, it might be necessary as an electron source to ignite the discharge.144 Tsuji et al.148 observed higher CO2 conversions when diluted in Ar compared to He, and furthermore, the conversion increased but the energy efficiency decreased when adding more Ar or He. In contrast, just like for the DBD, N2 can play a very important role in the CO2 conversion, but the mechanism in the MW plasma is different than in the DBD plasma. Indeed, while in the DBD, the metastable electronically excited N2 molecules give rise to enhanced CO2 conversion (see the previous section),125 while in the MW plasma, the improvement is due to the vibrationally excited N2 molecules.149 The energy difference between the first vibrational level of N2 and CO2 is very small, making the fast resonance transfer of vibrational energy from N2 to CO2 possible.150 As such, N2 can help with the vibrational pumping of the asymmetric mode of CO2 and thus can enhance the CO2 conversion. On the other hand, the vibrationally excited N2 molecules can also react with O atoms, leading to the production of NOx in undesirable concentrations, as was also observed for a DBD (see previous section).149
Just like for DBDs, big leaps forward have been made in the past few years regarding modelling the plasma chemistry to better understand and improve CO2 conversion in MW discharges.87–90,94,131,151,152 The added complexity for MW plasmas compared to the plasma chemistry in DBDs stems from the very effective excitation to vibrational states, which can lead to the so-called ladder-climbing effect and eventually very efficient dissociation of the vibrationally excited CO2 molecule (as discussed in Section 4.1.4). This means that all the vibrational levels up to the dissociation limit need to be taken into account in accurate models for a CO2 MW plasma, as well as all the reactions with these different vibrationally excited levels. The models predict that, besides electron-impact dissociation of the vibrationally excited levels, also collisions with neutrals will become important as dissociation mechanisms, as was also suggested by one optical characterization study,153 and this is the key to achieving maximum energy efficiency of the CO2 splitting process.87 It has also been shown that at low E/n values,151 and in post-discharge conditions,152 the rates from the vibrational-dissociation mechanism overcome the electron-impact dissociation. To be able to perform multidimensional modelling investigations for these type of discharges, i.e. where the vibrational levels play an important role, a reduced chemistry set has been developed in which, among others, the vibrational levels are lumped into a limited number of groups, to avoid the need to solve equations for all the individual CO2 vibrational levels.154
Finally, in contrast to the DBD research, no work has been performed on adding a (catalyst) packing in the MW discharge zone, and only a few papers have reported on adding a post-discharge (catalytic) packing.144,155 Of course, as mentioned in Section 4.3, this is due to the added complexity of adding packing materials to a MW discharge. Spencer et al.144 added a monolith structure with and without a Rh/TiO2 catalytic coating in the post-discharge zone. For both cases, a slight energy efficiency loss was observed and the Rh/TiO2 was deemed inappropriate due to the possible stimulation of the backward reaction of CO + O2 → CO2 + O. On the other hand, Chen et al.155 reported that the use of plasma-pretreated-TiO2-supported NiO catalysts in the post-discharge zone led to an energy efficiency of 17%, which was an increase by a factor 2 compared to the plasma-only case. However, this increase was suggested to arise from the dissociation of CO2 at the catalyst surface with oxygen vacancies through dissociative electron attachment, which is an inherently less efficient dissociation process than the step-wise vibrational excitation (as shown in Section 4.1.4; Fig. 12). We believe that the addition of a catalyst to a MW discharge for pure CO2 splitting will only be beneficial if it is capable of either effectively lowering the E/n value of the discharge due to its physical effects and/or stimulating the dissociation of vibrationally excited CO2 molecules on the surface. Furthermore, the development of MW discharges operating at lower temperatures (≤1000 K) would allow the implementation of thermal catalysts in the discharge zone, rather than use of the current post-discharge packing.
From all these data, it should be clear that MW discharges are more than capable of surpassing the 60% efficiency mark for pure CO2 splitting. However, the best results of this earlier work21,139,140 have not yet been reproduced to date, while the best energy efficiency in the more recent work appears to be around the thermodynamic equilibrium value of 45–50%. This observation, together with the reported gas temperatures, makes it questionable whether vibrational excitation plays the major role here. Nonetheless, from modelling insights,88,89 it is evident that when the set-up can be tailored to achieve the correct strong non-equilibrium conditions,141 vibrational excitations can lead to energy efficiencies of up to 90%. The main disadvantage of MW discharges is, however, their current requirement to operate at low pressures, in order to reach this strong non-equilibrium, and thus these high energy efficiencies.
As explained in Section 4.2.3, there are two main GA reactor geometries used for CO2 conversion. The first one relies on simple two-dimensional (2D) electrode blades. However, this configuration has a few disadvantages: the residence time in the plasma is quite short, flow rates are more limited and, due to its geometry, only a limited fraction of the gas flow is processed by the discharge (e.g. about 20% depending on the actual geometry).97,156,157 As a result, this limits the theoretical maximum possible conversion to ∼20%.97 The GAP configuration, on the other hand, is based on cylindrical electrodes, and the gas follows a vortex flow pattern. The gas in the reverse (inner) vortex flow passes exactly through the arc in the longitudinal direction, which ensures longer residence times in the discharge zone, even at high flow rates. Based on the gas flow calculations, about 40% of the gas flow can be processed by the discharge,158 doubling the theoretical maximum conversion compared to the classical 2D electrode configuration.
Beside these geometry variations, other work has focused on changing the applied power, gas flow rate, flow type, interelectrode gap, admixture gases and plasma chemistry modelling. Again, all the data available in the literature are plotted in Fig. 22. Most experiments report a maximum energy efficiency of around 40–50%,97,100,159 with the highest experimentally reported and also calculated energy efficiency being 65%.97,160 Most of the conversion results, however, remain below 15%, for both the regular GA and the GAP set-ups. The only exception is the work of Indarto et al.161 (35% conversion), where the mixture was diluted with N2, but at the expense of the energy efficiency (<5%). As such, compared to DBDs, which also work at atmospheric pressures, GA plasmas deliver about the same conversion, but the energy efficiency is in general 3–4 times higher. A more detailed discussion on the influence of the different parameters is given below.
 | ||
| Fig. 22 Experimental data collected from the literature for CO2 splitting in a GA, showing the conversion (a) and energy efficiency (b) as a function of the SEI, as well as the energy efficiency as a function of the conversion (c). Note that some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
It should be no surprise that again a trade-off between the energy efficiency and conversion as a function of SEI is observed.97,100,159,161,162 In general, the conversion increases and the energy efficiency decreases with increasing the SEI, as can be seen in Fig. 22, and the same optimal SEI range of 0.1–1 eV per molecule, in terms of energy efficiency, is observed as for the MW discharges. For a regular GA, the conversion clearly increases and the energy efficiency decreases when more power is supplied.97 The GAP geometry, on the other hand, can operate in two regimes depending on the power input, i.e. a low and a high current regime, with the highest energy efficiency but lowest conversion observed for the former and the opposite for the latter.159 Furthermore, it is reported that the lower the gas flow rate is, the higher the conversion is, both in the low and high current regimes, which is a direct result of the longer residence time of the gas in the discharge.159 The same, albeit a slightly less pronounced effect, was observed for the regular GA.161 Kim et al.,160 however, observed an optimum conversion while varying the gas flow rate. This was probably the result of reaching an optimum SEI (0.45 eV per molecule), since this value is in the same order as the SEI range at which Nunnally et al. reported a peak energy efficiency (0.30–0.37 eV per molecule).100
Besides the gas flow rate, an important parameter for the GAP is the vortex flow type, which can be modulated by adjusting the reactor geometry.100,159,163 Operating in a reversed vortex flow (RVF), compared to a forward vortex flow (FVF), provides an increase in residence time and thermal insulation of the discharge from the reactor walls. As a result, the RVF delivers higher energy efficiencies at higher SEI values, leading to an improved conversion.100,159 It is also this vortex flow that allows for the higher gas flow rates to be processed compared to a regular GA, and theoretically also to obtain higher maximum conversions because the gas passes through the arc in the longitudinal direction, thereby yielding a longer residence time.
For the regular GA, the interelectrode gap can be varied to improve the CO2 conversion, and the best result was observed for the smallest interelectrode distance. Indeed, increasing this distance leads to a larger arc volume and a corresponding drop in plasma power and electron density, and consequently also a drop in CO2 conversion.97
Only one research group has reported the use of additive gases, other than CH4, H2O or H2; more specifically, the addition of N2, O2 and air.161 The experiments revealed that adding N2 had a positive effect on the conversion (up to a factor 2 upon the addition of 95% N2), but due to the dilution, a detrimental effect on the energy efficiency was observed (down by a factor of 10–20). Furthermore, the presence of N2 leads to the unwanted production of NOX. The addition of O2 and air showed a decrease in the conversion, indicating the possible strong negative effect of the presence of O2 (impurities), presumably due to Le Chatelier's principle.
Due to the more complex behaviour of the gas flow and of the arc movement, and also the relationship between both in a GA, modelling work is more limited in the literature. For the regular GA, a simple plasma kinetic model has been developed,161 while recently a more detailed 0D chemical kinetic model97 and a 1D quasi-gliding arc mode164 have been presented. The results from these modelling studies show that the electron-impact dissociation of vibrationally excited CO2 is predominant for an arc temperature of 1200 K and the recombination between CO and O atoms is the main conversion limiting reaction.97 Reducing the arc temperature to 1000 K can significantly increase the conversion and energy efficiency, because it limits the recombination reaction rate and enhances the importance of the higher vibrational levels in CO2 dissociation.97 Just like for MW chemistry, lumping of the vibrational levels into a limited number of groups has been successfully performed, opening future perspectives for 2D and 3D GA modelling.164 A successful first attempt at modelling the more complex GAP with a 0D chemical kinetics model supported by 3D gas flow modelling has also been reported, and has confirmed the assumption, in line with the regular GA, that the vibrational levels play an important role for the energy-efficient conversion of CO2.159
In summary, the data in the literature show that GA discharges succeed in exploiting the most energy-efficient CO2 dissociation channel based on vibrational excitation, while operating at atmospheric pressure. Energy efficiencies of 45% are no exception and even results above the target value of 60% have already been reported. On the other hand, model calculations reveal that the non-equilibrium character of the GA – making full potential of the vibrational excitation/dissociation pathway – could be further exploited to improve the energy efficiency. Just like for MW discharges, operating GA plasmas at lower gas temperatures might be the key for achieving this. In addition, the main limiting factor compared to MW discharges appears to be the conversion, due to the limited fraction of the gas flow that is currently processed by the discharge. Smart reactor design, such as enhancing the processed gas fraction, should lead to the necessary advancements to overcome this limitation in the future.
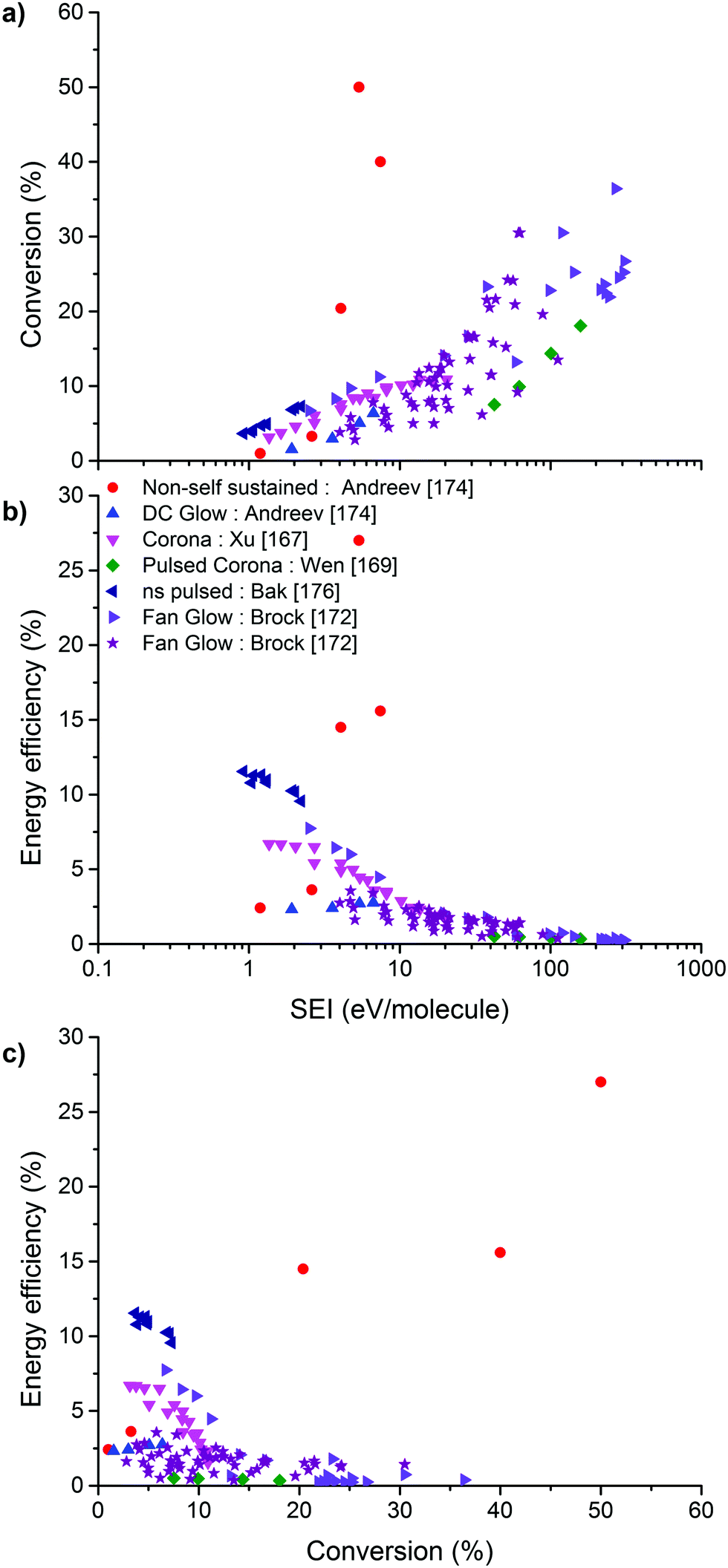 | ||
| Fig. 23 Experimental data collected from the literature for CO2 splitting in other plasmas, besides DBD, MW/RF and GA, showing the conversion (a) and energy efficiency (b) as a function of the SEI, as well as the energy efficiency as a function of the conversion (c). Note that some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
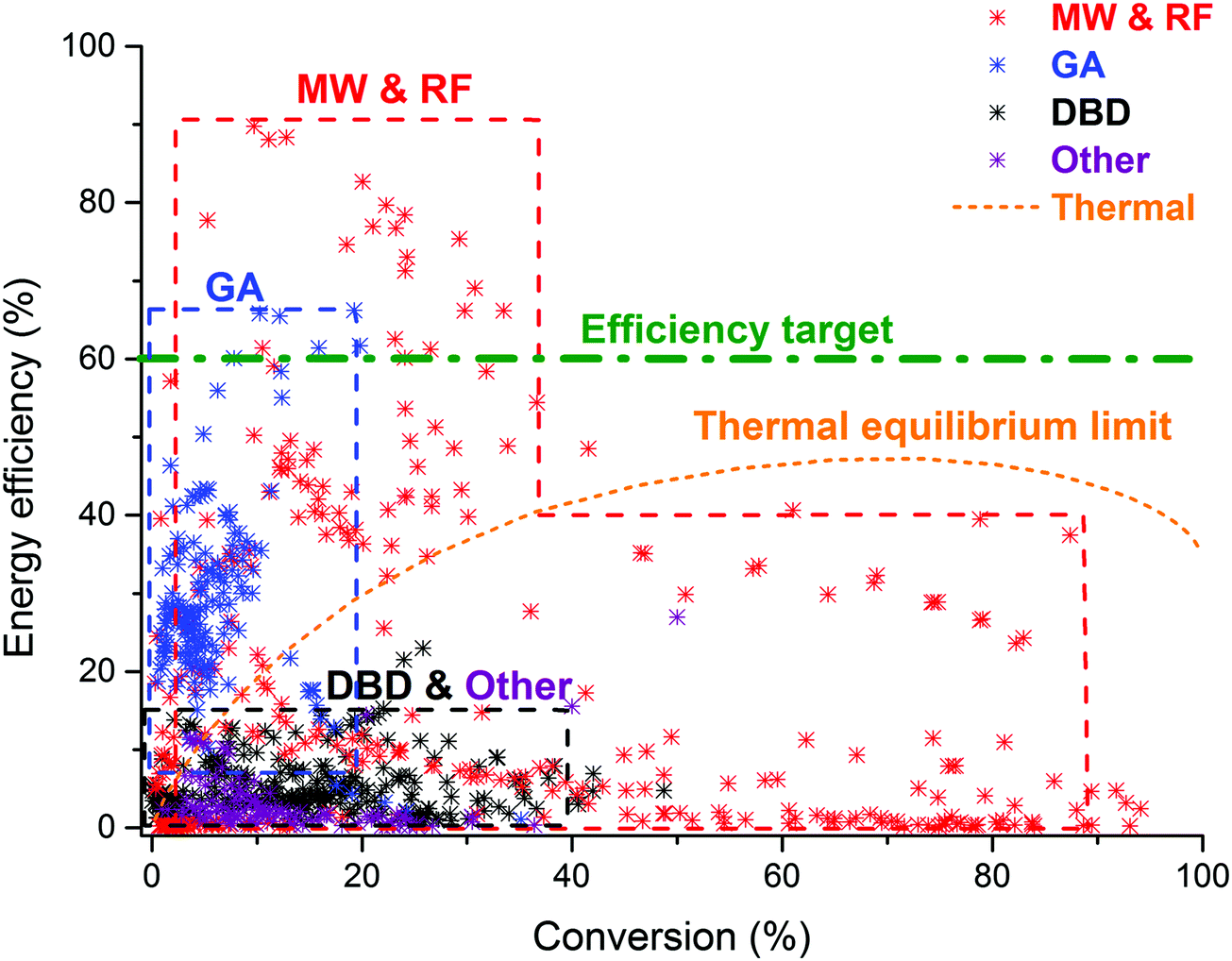 | ||
| Fig. 24 Comparison of all the data collected from the literature for CO2 splitting in the different plasma types, showing the energy efficiency as a function of the conversion. The thermal equilibrium limit and the 60% efficiency target are also indicated. | ||
First of all, it is clear that, although DBDs are among the most extensively studied for CO2 conversion, and indeed are already successfully applied80 for commercial O3 production and VOC removal, they appear to be unsuitable for the efficient conversion of CO2. Their energy efficiency remains a factor of 4 too low, even when combined with a packing, in order to justify them as industrially competitive. The same applies to most of the other plasma types listed in Section 5.1.4 above.
Second, the best results for GA plasmas are capable of reaching the set energy efficiency target, namely 60%. Moreover, almost all of the results obtained are far above the thermal equilibrium limit, which is especially interesting, keeping in mind that the GA plasmas operate at atmospheric pressure. This demonstrates the non-equilibrium character of this type of plasma, even at atmospheric pressure, and the benefits of being able to exploit this behaviour through the energy-efficient dissociation of the CO2 vibrational levels. Moreover, modelling has revealed that this non-equilibrium character could (and should) be further exploited, to further enhance the energy efficiency. To date, the main challenge is the limited conversion, which remains below 20% because only a limited fraction of the gas passes through the active arc plasma.
Finally, if, for now, we ignore the fact that most MW discharges used for CO2 conversion operate at reduced pressure, in contrast to the commercially more interesting atmospheric pressure of GA plasmas, it is clear that MW discharges offer a wide variety of possibilities. Even up to conversions of 40%, the energy efficiency target is easily crossed and they clearly operate in a non-equilibrium regime, thus favouring the step-wise vibrational-dissociation mechanism. Conversions in the range of 40–90% are also possible, albeit with maximum energy efficiencies of only up to 40%. Under these conditions, the MW discharges most probably operate in the thermal regime. Nevertheless, this shows the wide variety of both conversions and energy efficiencies achievable with MW discharges for the conversion of pure CO2.
5.2. CO2 + CH4: dry reforming of methane
Contrary to pure CO2 splitting, DRM can yield a wide variety of products. This has several implications for the evaluation and comparison of the various studies in the literature, mainly because it affects the definition of the energy efficiency. To determine the true energy efficiency of the process, we would need to take all the formed products into account – both gaseous and liquids – to determine the theoretical reaction enthalpy. Another possibility would be to determine the thermal energy efficiency (see Section 6), based on the higher (or lower) heating value of the output, i.e. the products, relative to that of the input, i.e. the reactants and power input. However, typically only the selectivity (or yield) towards the syngas components, CO and H2, and light hydrocarbons is given for most studies in the literature. Thus, at the moment these strategies are impossible to pursue when presenting an overview of the available literature results in the sections below. Therefore, in this section, we use the energy cost, in units of eV per converted molecule, to compare the different discharges, together with their total conversion. As an energy efficiency target for syngas production, we take the same 60% value for the general stoichiometric DRM process, which equals an energy cost of 4.27 eV per molecule converted. Indeed, 100% energy efficiency would yield an energy cost equal to the standard reaction enthalpy of 2.56 eV per molecule (as outlined in Section 2.2.1). In our opinion, this is the best representation to compare the results in the literature, considering the fact that in most of the studies, the higher hydrocarbons and the valuable liquid fraction containing oxygenates (e.g. formaldehyde, methanol, ethanol, formic acid, acetic acid) are neglected. However, as explained at the beginning of Section 5, this 60% energy efficiency target is only valid for the comparison towards syngas production. When liquids (such as methanol) are formed through the direct oxidative pathway, which has already been demonstrated using the plasmachemical conversion of CO2 with a co-reactant, the energy efficiency requirements are drastically lowered by a factor of two to three. This is a direct result of circumventing the energy-intensive step of further processing the syngas into the desired liquid products.Plasma-based DRM has received a lot of attention in recent years. Some overviews can be found in the literature.21,41,177–179 In a recent review on the liquefaction of methane,112 DRM was considered as oxidative plasma-based CH4 liquefaction. It can be performed in two ways: either as a two-step process, yielding mainly syngas, which can then be further processed into Fischer–Tropsch liquids or methanol, or as a one-step process, aiming for the direct formation of value-added oxygenated products, such as formaldehyde, methanol, ethanol, dimethyl ether and formic acid. These two options are therefore called the indirect and direct oxidative plasma liquefaction approaches, respectively. An overview of the progress in the field of plasma-based DRM is given below, starting again with DBD, MW and GA plasmas. However, in contrast to CO2 splitting, a vast amount of research has been performed with some other discharges as well for DRM, so these have been given their separate sections below.
First some general trends applicable for all discharges are summarized. We start off with the SEI, because for all discharges studied, it is clear that for DRM – just like for pure CO2 splitting – the SEI is the major determining factor for the conversion and energy cost, as it combines the effects of power and residence time. Typically, a higher SEI leads to a higher conversion, but also incurs a higher energy cost. The conversions of CH4 and CO2 increase almost linearly when a higher voltage or input power is applied, as observed for DBD,106,180–190 MW,191 GA,192–194 corona,195–198 spark,199–205 APGD206,207 and nanosecond pulsed discharges.208–212 Generally, this is accompanied by an increase in energy cost. Regarding product selectivity, some different trends are reported as a function of power for the different discharge types. In general, in DBDs, higher H2 and CO yields are reported, with the ratio of H2 to CO remaining constant,184,187,188 as well as a sharp drop in the selectivity of the light hydrocarbons.186,189,190 Furthermore, some studies observed no significant changes in the selectivities in a DBD, except for an increase in carbon deposition,184 while other studies report an increase in CO and H2 selectivity,186,187,190 or even the opposite trend.188 For corona discharges, the H2 selectivity seems to decrease, while the CO selectivity exhibits the opposite trend,195,196 and as a result, the H2/CO ratio greatly depends on the discharge power.197 In spark discharges, a more pronounced carbon deposition at higher input power has been reported,202 as well as an increased selectivity for H2 and CO and a drop in C2H2 selectivity, pointing to an enhancement of the reforming reactions over coupling reactions.202,203 APGD discharges exhibit a higher CO selectivity and coke deposition at higher input voltages and these conditions favour the production of unsaturated hydrocarbons, such as C2H2.206 On the other hand, for nanosecond pulsed discharges, upon a higher SEI, the mass balance points to a loss of oxygen and carbon, which are converted into water and carbon powder or are deposited, respectively.208 Finally, for MW and GA plasmas, no specific trends have been reported yet for the effect of power on the product selectivities. Thus we can conclude that, albeit with some exceptions, a higher discharge power generally improves the conversion, but does not change the reaction pathways significantly.
A higher total flow rate has the opposite effect to the power, and leads to lower conversions and energy costs for DBD,106,180,183,185,186,188,189,213 GA,194 corona,196–198,214 spark,199,202 APGD206,207 and nanosecond pulsed discharges.209 No results have been reported yet for MW plasmas. On the other hand, the flow seems to have almost no effect on the selectivity towards the syngas components CO and H2 and hence also not on the syngas ratio itself, for DBD,183,185,189,213 GA194 and APGD.206,207 At higher flow rates, some researchers have also reported a decrease in H2 and CO selectivity in DBDs,186,188 while the selectivity towards hydrocarbons increases.180,183,186,188,213 For corona discharges, a slightly higher H2 and CO selectivity is observed, but the syngas ratio remains almost unchanged.196–198,214 For spark discharges, it is reported that the H2 selectivity rises and the CO selectivity drops, at higher flow rates.199 Finally, for MW and nanosecond pulsed discharges, again no specific data have been reported yet for the effect of flow rate on the product selectivities. Nevertheless, in general we may again conclude that the dissociation and formation mechanisms are, albeit with some exceptions, not significantly influenced by a change in the flow rate, although the selectivities can be altered.
Besides the SEI (or the power and gas flow rate), the CH4/CO2 mixing ratio has a tremendous influence on the CH4 and CO2 conversions, as well as on the product selectivities. In general, when adding more CH4 to the mixture, the effective CH4 conversion rises, while the effective CO2 conversion drops. Note that we talk about the effective conversions (as defined in Section 4.4.2; eqn (4)) and not about the (absolute) conversion itself, in order to take the effect of the change in gas mixture into account. At the same time, the selectivity towards (light) hydrocarbons increases, as well as the selectivity towards H2, while the CO selectivity decreases, when more CH4 is present in the mixture. This inevitably leads to an increase in the syngas ratio. Thus, the H2/CO ratio is highly dependent on the inlet feed, which makes the ratio easily adjustable over a wide range to fit the Fischer–Tropsch or methanol synthesis requirements. This was found for all discharges, i.e. DBD,182,183,186–190 GA,192–194,215 MW,191 corona,197,214,216–218 spark,200,202,204,205 APGD207,217,219,220 and nanosecond pulsed discharges.209,211,221 The formation of carbon black and its deposition on the electrode and reactor walls for mixtures seems to occur with a high CH4/CO2 ratio (>1), and this deposition can highly influence the discharge operation.119 For lower CH4/CO2 ratios, this is not observed, neither in DBD,182,189,190 GA,192–194,215 corona,195–197 spark,200,205 APGD207,217,219,220 nor nanosecond pulsed discharges,209 as CO2 prevents carbon formation. For APGDs, a higher CH4/CO2 ratio is reported to increase the water formation. In contrast, for a DBD187 and spark discharge,202 a higher water production is observed at low CH4/CO2 feed ratios, presumably due to the occurrence of the reverse water–gas shift reaction.
These trends can be explained by the following reactions: increasing the CH4/CO2 ratio leads to a more pronounced H2 formation and at the same time it reduces the amount of O species in the mixture. Hence, the reaction C + O → CO is reduced, leading to a lower CO selectivity, and increased carbon deposition. On the other hand, the reaction H2 + O → H2O becomes more important, giving rise to the higher H2O production, but it is not significant enough to balance the higher H2 production, thus explaining the increased H2 selectivity. This also means that the deposited C is still an active species in the reactions, and it is suggested that the addition of O2207 and/or H2O217 can prevent carbon deposition. Finally, it is reported that the rate of CO production is always higher than the rate of CO2 reduction, proving that CO is formed by both the reduction of CO2 and the oxidation of CH4.214
The effect of the CH4/CO2 mixing ratio on the total conversion and hence on the energy cost is less straightforward. For nanosecond pulsed discharges, depending on the set-up, different optimum values have been reported for this mixing ratio for achieving the lowest energy cost. Ghorbanzadeh et al.209,212 reported a considerable increase in the energy cost when methane becomes predominant in the mixture, while Zhang et al.211 reported the energy cost was cut in half when going from a 1:3 to a 3:1 CH4/CO2 ratio. For a DBD, the effect of the mixing ratio on the total conversion also appears to depend on the specific set-up, since both an increase182,186,222,223 and decrease106,183,188 are widely reported upon the addition of more CH4. Snoeckx et al.,181 Zheng et al.213 and Pinhão et al.224 all reported an initial increase in total conversion upon adding CH4 when the CH4/CO2 ratio was <1, and a decrease when the ratio became >1. These trends suggest that the CH4 conversion is strongly affected by the CO2 conversion, especially when the ratio is <1 and when CO2 is thus the main component in the mixture. For the other plasma types, no specific data have yet been reported in the literature on the effect of the CH4/CO2 mixing ratio on the total conversion. Finally, as the energy cost is inversely proportional to the obtained conversion (cf. Section 4.4.3; eqn (7)), the effect of the CH4/CO2 mixing ratio on the energy cost is opposite to the effects described above for the total conversion.
Besides these general trends for the effect of the SEI and the CH4/CO2 mixing ratio, which are very similar for the various discharge types, some specific trends characteristic for each plasma reactor are reported for DRM. These are summarized in the next sections, along with a summary of all the values for conversion and energy cost reported in the literature.
Fig. 25 summarizes most of the results, in terms of both conversion and energy cost, available in the literature. Due to the large amount of data available for packed-bed reactors, those data are discussed and presented further below in Fig. 26. The conversion and energy cost increase again upon a rising SEI. When plotting the energy cost as a function of the conversion, it is clear that most of the results are situated above an energy cost of 20 eV per molecule and below a conversion of 60%, albeit with some exceptions. The highest experimental conversion reported is 66%,186 and the lowest experimentally observed energy cost is 18 eV per molecule.181 This is more than a factor of 4 higher than the set efficiency target of 4.27 eV per molecule (see the beginning of Section 5.2). Model calculations predict that higher conversion and lower energy costs should be achievable by a careful selection of the operating conditions, as will be outlined below.181 It is important to note that some of the data in this figure have again been recalculated to represent coherent values for the conversions and energy costs. For instance, as also explained for pure CO2 splitting, the mixture is sometimes diluted with He, Ar or N2 to obtain an more easily ignitable and more stable discharge, but this influences the results – an effect that was not always taken into account in the original data. A more detailed influence of the different parameters is discussed below.
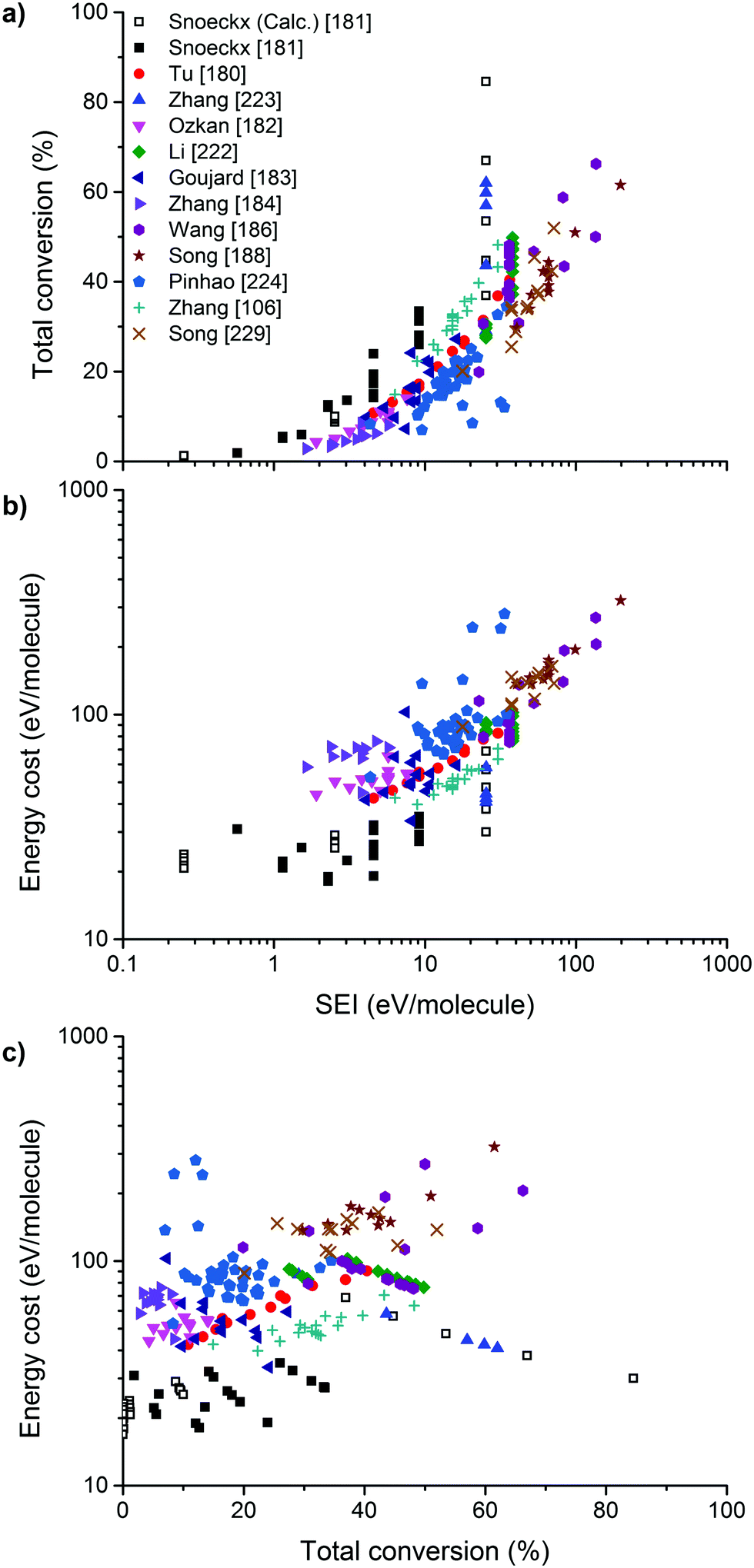 | ||
| Fig. 25 Experimental data collected from the literature for DRM in a DBD, showing the conversion (a) and energy cost (b) as a function of the SEI, as well as the energy cost as a function of the conversion (c). Note that some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
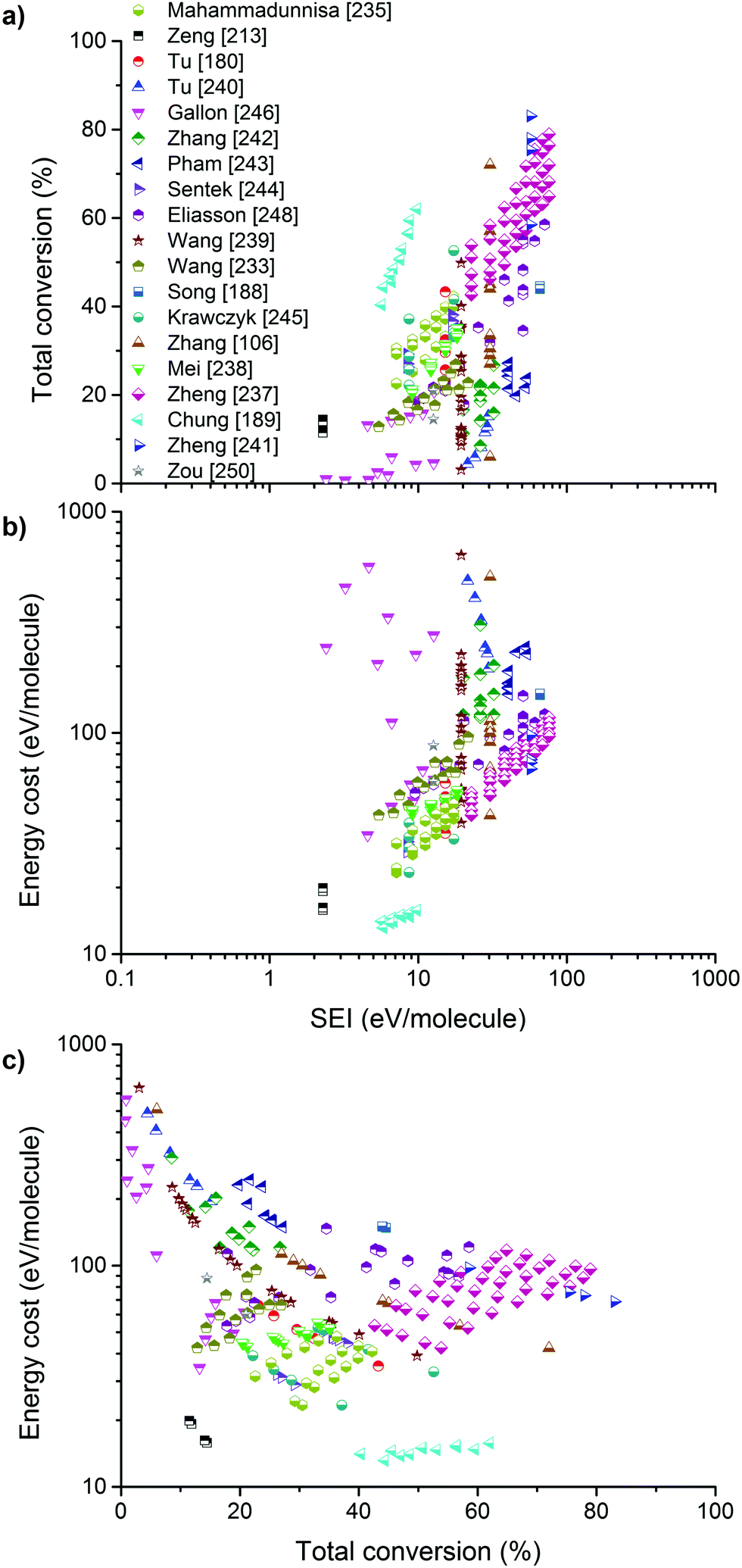 | ||
| Fig. 26 Experimental data collected from the literature for DRM in a packed-bed DBD, showing the conversion (a) and energy cost (b) as a function of the SEI, as well as the energy cost as a function of the conversion (c). Note that some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
The effect of the temperature and pressure on DRM in a DBD has been investigated by several authors. Low pressures seem to favour the reactant conversion, whereas the syngas ratio does not show any pressure dependence.187 On the other hand, the selectivity of both CO and light hydrocarbons is reported to increase with rising pressure.190 For a wall temperature range of 353–523 K, Zhou et al.187 observed slightly higher conversions and product yields upon increasing the temperature, with the syngas ratio being independent of the temperature. Zhang et al.184 and Goujard et al.183 investigated the temperature effect in the range of 297–773 K and 298–873 K, respectively. Both the conversions and the hydrocarbon selectivity were reported to increase with temperature, while the H2 and CO selectivity decreased. However, Zhang et al.184 found an increase in the syngas ratio, whereas Goujard et al.183 observed the opposite, noting also that when the temperature reached 773 K, the CO2 conversion appeared to increase more rapidly than the CH4 conversion, indicating a shift in the dominant reforming processes around 673 K.183 These results indicate that while the electron-impact dissociation reactions govern the reactant conversions, the thermochemistry can be used to control the consecutive product reaction pathways.
Several changes with respect to the electrode configuration and materials have been reported as well. Wang et al.186 found little influence of the discharge gap on the reactions, while Li et al.222 observed higher conversions upon reducing the discharge gap. At the same time, Wang et al.186 reported a significant influence on the conversions using multi-stage ionization (i.e. multiple DBD and ‘afterglow’ zones in one reactor by placing several electrodes in series), while only small effects were observed by Li et al.222 Both groups, however, found a significant positive influence on the syngas product selectivities when applying multi-stage ionization. Rico et al.185 reported better conversions with porous electrodes, while the (catalytic) effect of different inner electrodes only showed very little influence on the conversions,227,228 but a significant increase in the oxygenate synthesis for nickel and copper, suggesting a catalytic role of the metallic surface.228 Finally, applying a pulsed power also seemed to have a beneficial effect on the conversion.229
As mentioned above, some researchers added inert gases, such as N2, Ar and He, to ignite the plasma more easily. However, this also has several effects on the discharge characteristics, conversion and energy cost. Ozkan et al.182 investigated the effect of Ar and He dilution, but did not compare the results with pure CH4/CO2 mixtures. The CH4 conversion was higher for He than for Ar addition, while the opposite was found for CO2 conversion. This could be related to a change from filamentary to glow discharge when switching from Ar to He, which significantly influences the shape of the EEDF and thus the electron-impact reactions. Zhang et al.106 indicated that, although the conversion increases when adding Ar, the latter is not sufficient to counteract the lower CH4/CO2 feed content, since the conversion rates are lower compared to a mixture without Ar. On the other hand, they observed a positive effect towards the syngas component selectivities. Pinhão et al.224 reported a positive effect on the breakdown voltage with increasing He content, resulting in a rise in the conversion rates with increasing the He content up to 80%. However, a still higher He content showed detrimental effects on both the conversion and selectivity, but again no comparison for the mixture without He was made. In a follow-up modelling study by Janeco et al.,92 a shift in the electron velocity distribution function due to the addition of He was observed.
Kolb et al. studied the addition of the molecular gases O2 and H2O on the performance of DRM in a DBD.230,231 They found that O2 aids in the conversion of CH4 but also produces CO2,231 which is not beneficial. H2O, on the other hand, only has a small negative influence on the conversion of CO2, but significantly enhances the conversion of CH4 and, more importantly, the production of valuable oxygenates, such as formaldehyde and methanol.230 This study showed that a tri- or more general multi-reforming process, using a mixture of CO2/CH4/H2O or a combination of different reforming processes in series, could be a very interesting next step to pursue using plasma technology.
Due to the historical interest in thermocatalytic DRM and the ease of implementing a (catalytic) packing in the discharge zone of a DBD, as explained in Section 4.3, a lot of research has already been performed towards plasma-catalytic DRM and hybrid reactors. A complete but brief overview is given below, but for more detailed information on the effects and mechanisms behind plasma-catalysis, we refer to the existing excellent literature on this topic.102,103,232 Most of the work to date has been performed with packed-bed reactors, but some research also exists on fluidized-bed reactors.233,234 Contrary to pure CO2 splitting, for DRM, the addition of packing can greatly influence the formation of products, due to the availability of CH4 as a hydrogen source. Hence, the research work focuses both on increasing the conversion and lowering the energy cost by physical effects, as well as on chemical effects to steer the product distribution towards more value-added chemicals, such as light hydrocarbons and oxygenates. Packed-bed DBD reactors generally yield similar conversions and energy costs as the corresponding empty reactors, as can be seen when comparing Fig. 25 and 26. Nevertheless, the use of a catalytic packing can drastically alter the chemical pathways and thus the selectivity and product distributions, which would be beneficial for exploiting the direct oxidative pathway of DRM, instead of utilizing the indirect process through syngas production.
A broad spectrum of materials have already been investigated for the plasma-catalytic DRM, of which Ni is by far the most commonly used active phase, such as in Ni/γ-Al2O3,106,180,188,213,233,235–240 Ni/SiO2,238,241,242 Ni–Fe/γ-Al2O3,237 Ni–Fe/SiO2,237,242 Ni–Cu/γ-Al2O3,106 Ni0/La2O3,183 Ni/MgO,238 Ni/TiO2,238 NiFe2O4,237 NiFe2O4#SiO2,237 LaNiO3/SiO2,241 LaNiO3241 and LaNiO3@SiO2.241 Furthermore, alumina is the most commonly used support, i.e. in Ni/γ-Al2O3,106,180,188,213,233,235–240 Ni–Fe/γ-Al2O3,237 Mn/γ-Al2O3,213 Cu/γ-Al2O3,106,213,234 Co/γ-Al2O3,213 La2O3/γ-Al2O3,243 Ag/γ-Al2O3,244 Pd/γ-Al2O3,234,244 Fe/γ-Al2O3245 and Cu–Ni/γ-Al2O3,106 or even in its pure form.185,188,244,246 Many other catalytic systems are based on zeolites, e.g. 3A,246 A4,247 NaX,248 NaY245 and Na-ZSM-5.245 Besides, studies have also been conducted using BaTiO3,185,242 a mixture of BaTiO3 and NiSiO2,242 ceramic foams (92% Al2O3, 8% SiO2) coated with Rh, Ni or NiCa,249 quartz wool,246 glass beads,189 a stainless steel mesh,242 starch,250 BZT (BaZr0.75T0.25O3) and BFN (BaFe0.5Nb0.5O3).189 For a regular AC-packed DBD, the best result was obtained for the Zeolite Na-ZSM-5, with a total conversion of 37% and an energy cost of 24 eV per converted molecule (see Fig. 26).245 Just like for pure CO2 splitting, the addition of a catalyst does not seem to make the process more energy efficient, but it does yield higher conversions at the same energy cost. The best overall results in a packed-bed DBD were obtained for a quasi-pulsed DBD packed with BFN and BZN, with total conversions in the range of 45–60% and an energy cost in the range of 13–16 eV per converted molecule (see Fig. 26),189 which is lower than that for a DBD without packing, but this might also be due to the pulsed operation.
Besides the experimental work, major insights have been obtained in recent years based on modelling of the DRM process for a DBD. Different kinds of models and computational techniques have been successfully developed, including semi-empirical kinetic models,187,251,252 zero-dimensional chemical kinetic models with both simplified253 and extensive chemistry sets,131,181,254 a one-dimensional fluid model,255 a so-called 3D Incompressible Navier–Stokes model combined with a convection–diffusion model,256 a hybrid artificial neural network-genetic algorithm,257 a model focusing on a more accurate description of the electron kinetics92 and density functional theory (DFT) studies, to investigate the reaction mechanisms.226,258 Due to the complex chemistry taking place in a DRM, the development of accurate multidimensional models with extensive chemistry is currently restricted by computational limits.
Some key findings of these models are presented here. Snoeckx et al.181 performed an extensive modelling study, with detailed plasma chemistry, spanning a wide range of experimentally accessible conditions. The model predicted that increasing the SEI at a constant gas ratio and frequency results in a higher total conversion. However, the increase in conversion is not entirely proportional to the rise in SEI, resulting in somewhat higher energy costs with increasing the SEI. The lowest energy cost was predicted to be 16.9 eV per molecule, but this corresponds to a very low value for the total conversion, i.e. 0.015%. On the other hand, the highest total conversion predicted by the model, i.e. 84.2%, corresponded to an energy cost of 30.1 eV per molecule. A larger amount of CO2 was observed to lead to a higher total conversion and lower energy cost. This is attributed to the O atoms formed by the electron-impact dissociation of CO2, which reacts very effectively with the H atoms originating from the electron-impact dissociation of CH4. As shown in the kinetics analysis of Snoeckx et al.,254 the conversion of CH4 is normally limited by the fast backward reaction, i.e. CH3 + H → CH4, but when more O atoms are available, this reaction is of minor importance compared to the reaction O + H/OH → OH/H2O. Thus, by limiting the backward reaction, the conversion of CH4 rises dramatically with increasing CO2 content, leading to a higher total conversion.181 Another important effect is the total number of micro-discharge filaments that the gas molecules experience when passing through the DBD reactor. It seems that for most cases a larger number of filaments, but with lower energy, yields higher values for the conversion and lower energy costs, compared to a smaller number of filaments, but with more energy deposited per filament.181 In some other models by Janeco et al.92 and Goujard et al.,253 the effect of adding He to the CH4/CO2 mixture was investigated. Both groups reported that He, as well as all other species that may result from the electron collisions in the gas mixture, can significantly change the electron velocity distribution function, the electron reaction rates and the energy losses, which can ultimately affect the energy cost when taken into account in the model.
Summarizing all the data in the literature, we can conclude that a DBD reactor can provide reasonable conversions of up to 60% and 80% for empty (i.e. non-packed) and packed-bed DBDs, respectively. However, the energy cost lies in the range of 20–100 eV per molecule, which corresponds to an energy efficiency of 12.8–2.6%, assuming that only syngas is formed. This is at least a factor 5 away from the necessary 4.27 eV per molecule benchmark for the energy cost (or the 60% benchmark for energy efficiency), as defined at the beginning of this section, with no positive outlook on the horizon. Indeed, even extensive modelling studies have predicted that the lowest achievable energy cost, obtained after careful selection of the operating conditions, would be 16.9 eV per molecule (see above).181 Hence, despite a simple reactor design for scalability, the ease of implementing a wide variety of packings and its industrial success for ozone generation (and possibly VOC abatement), at this point, the high energy cost – and hence low energy efficiency – makes it doubtful that DBDs will be the most suitable technology for DRM into syngas. Nevertheless, due to its ease of use, research with DBDs still can yield valuable insights and knowledge, which may be transferable to other more efficient discharges. On the other hand, if suitable catalysts could be found for the production of valuable oxygenates with high yields through direct (plasma-catalytic) oxidative liquefaction,112 the energy efficiency target would be significantly lower (by a factor of 2–3 in the case of methanol), so this could change the analysis drastically. However, many challenges for this pathway remain, such as finding suitable catalysts, and advances in energy efficiency are also still required.
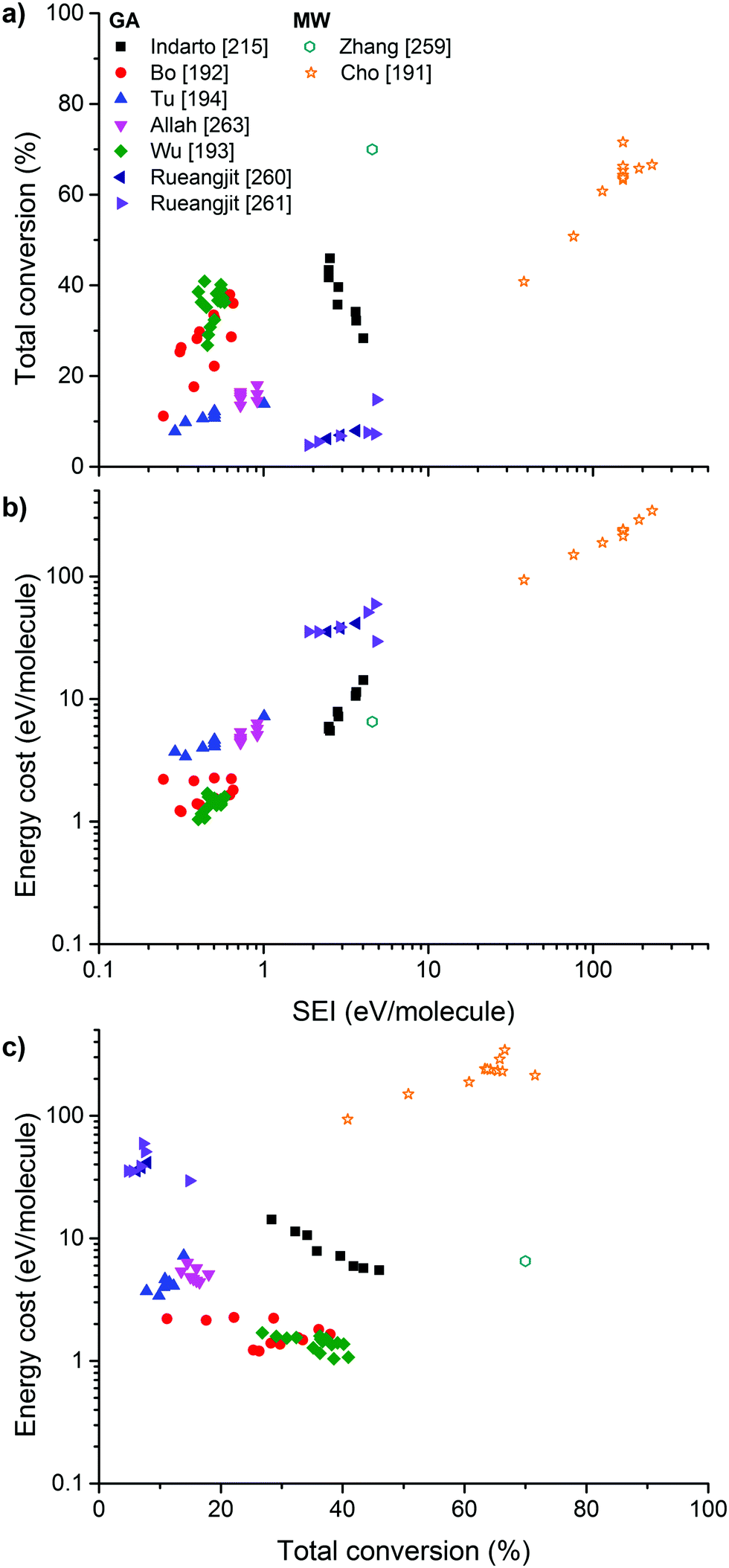 | ||
| Fig. 27 Experimental data collected from the literature for DRM in MW (open symbols) and GA plasma (closed symbols), showing the conversion (a) and energy cost (b) as a function of the SEI, as well as the energy cost as a function of the conversion (c). Note that some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
Cho et al.191 obtained conversions in continuous MW plasma in the same range as in the pulsed MW plasma of Zhang et al.,259 but at much high powers, up to 1.5 kW compared to 120 W, and at low flow rates (100 mL min−1vs. 200 mL min−1), so their energy costs were unacceptably high, i.e. between 93 and 343 eV per molecule. Adding a Ni catalyst to the discharge was reported to lead to a higher conversion by 10% for CH4 and by 15% for CO2. Furthermore, the H2/CO ratio changed drastically from 1.7 for the plasma-only case to 1.2 when combined with the catalyst. As additional products, only C2H4 was detected, with increasing selectivity when more CH4 was added to the feed.191
In Section 3.5, we also discussed catalysis based on MW-heating. Fidalgo et al.76 observed the formation of microplasmas in their so-called microwave receptor/catalyst set-up. However, the MW source was simply used in that case to heat the gas, with the creation of microplasmas at the catalyst surface only a side effect of the strong electric fields at the sharp edges.
In general, the few studies reported so far in literature for DRM by MW plasmas show high conversions, as well as a high selectivity towards CO and H2. Furthermore, based on the pure CO2 splitting results presented in the previous section, we might expect that a large treatment capacity is in principle possible, as well as high energy efficiencies. Nevertheless, experimental evidence to date remains extremely scarce. One of the major unanswered questions is related to the role played by the vibrational levels of CH4 in the CO2 dissociation, and more specifically in the ladder-climbing effect, which was explained in Section 4.1.4 (cf.Fig. 12). In particular, it is not clear whether they will affect (i.e. stimulate or inhibit) this process or not, as well as how the dissociation of CH4 itself will be influenced.
For GA discharges, the addition of inert gases has not yet been studied in the literature for DRM, but some studies have reported mixtures with reactive gases. Rueangjit et al.260,261 investigated the dry reforming of simulated natural gas with a high CO2 content, represented by CH4:C2H6:C3H8:CO2 in a molar ratio of 70:5:5:20. However, only a comparison with mixtures without CO2 was made, and it was then concluded that CO2 considerably enhances the conversion of the hydrocarbons in the feed.260 In addition, very high syngas ratios, i.e. around 10–20, could be obtained by adding these small amounts of hydrocarbons in the feed, due to their lower C–H bond energy.260,261
Furthermore, some research has also been performed on the combination of dry reforming with O2 and steam using GA plasmas, to mimic biogas feeds as an input source.261,262,264–266 However, in this case, CO2 is not only converted but is also being produced from the reaction of the CH4 dissociation products with O atoms,261,264,265 and in the case of H2O addition, even a net CO2 formation can be observed.262,266 It is suggested that when adding O2, the main process becomes the partial oxidation of methane, while the main involvement of CO2 is in the reverse water–gas shift reaction.264 The main positive effect of adding O2 is minimization of the carbon deposit.261 When adding steam, it seems again possible to obtain high syngas ratios, i.e. around 7.262,266
For DRM, the addition of a catalytic bed after the GA discharge zone has been investigated in two studies.262,263 In both studies, a NiO/Al2O3 catalyst was used. It was found that the CO2 and CH4 conversion increased by 24% and 16%, respectively. The selectivity of the syngas components, on the other hand, was found to drop slightly and the unsaturated hydrocarbons, C2H2 and C2H4, were formed with selectivities ranging from 16% to 19%. Lower NiO loading and smaller particle sizes appeared to be beneficial. Finally, it was suggested that there is scope for further development and optimization using a fluidized bed for maximizing the plasma–catalyst interactions.263
It is clear that the obtained energy costs for DRM using a GA plasma are much lower than those obtained for DBDs, while at the same time they allow achieving much higher feed processing capacities. Furthermore, the energy costs achieved already, i.e. around 1 eV per molecule, are already a factor of 4 better than the required target for syngas production. No data is available yet for the DRM of the GAP set-up, but we could expect even further improvements, as already demonstrated for pure CO2 splitting (see Section 5.1.3 above). Finally, the use of a catalytic spouted bed might be able to influence the product distributions, as already reported for pure CH4 reforming.105
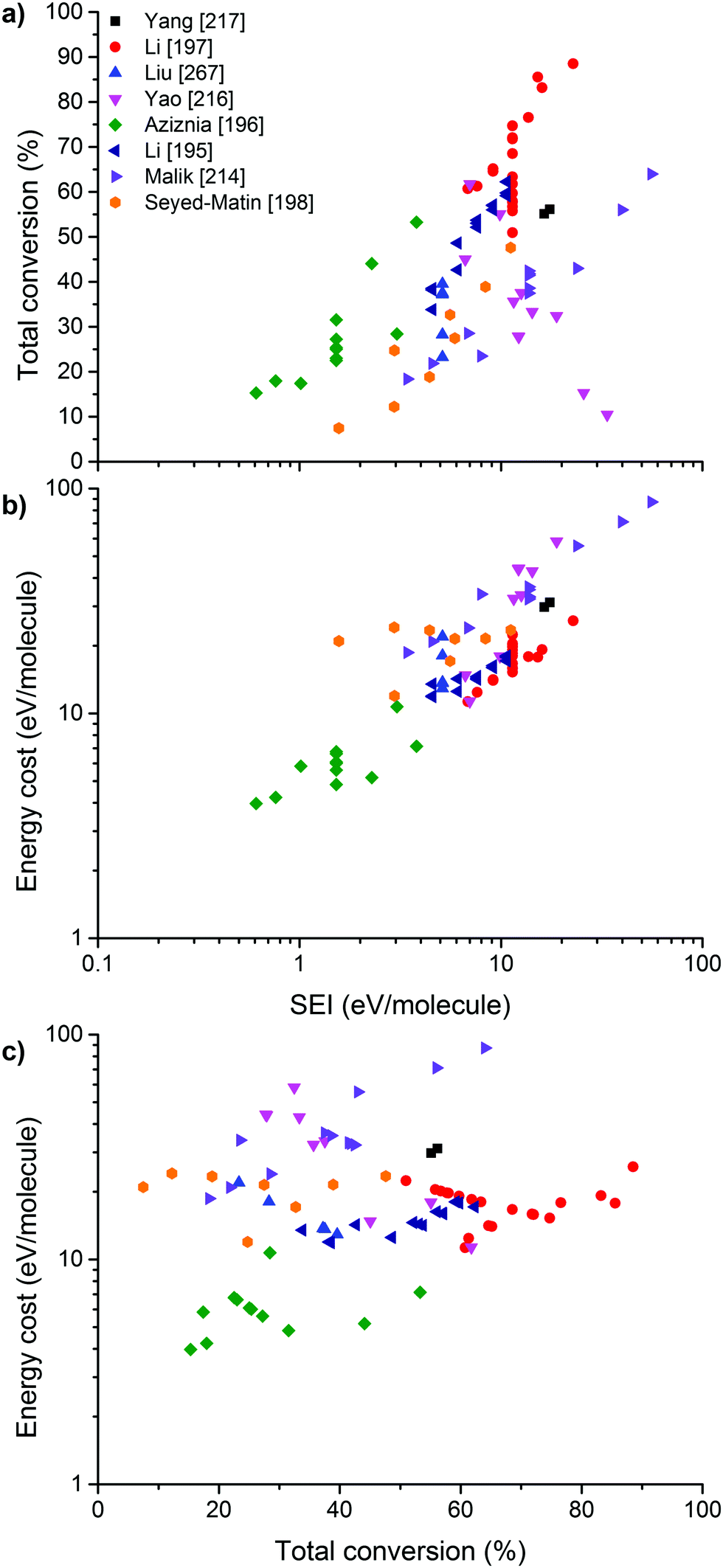 | ||
| Fig. 28 Experimental data collected from the literature for DRM in a corona discharge, showing the conversion (a) and energy cost (b) as a function of the SEI, as well as the energy cost as a function of the conversion (c). Note that some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
A pulsed corona was investigated by Yao et al.,216 who reported that the conversion and CO selectivity increase with rising frequency, while the hydrocarbon selectivities showed the opposite trend.
Some experiments were also performed by placing catalysts in the corona discharge. Liu et al.267 observed a significant conversion of CH4 and CO2 over a NaOH-treated Y zeolite placed in the discharge zone. The selectivity shifted towards higher hydrocarbons, e.g. propane, n-butane, isobutane, 1-butene and n-pentane. Aziznia et al.196 investigated the effect of Ni/Al2O3 in the discharge zone. Upon the addition of Al2O3, the conversions decreased slightly; the CO selectivity also decreased, but the H2 selectively increased remarkably. When adding up to 20 wt% Ni, the CH4 and CO2 conversions increased from 18% and 25% to 23% and 36%, respectively. Furthermore, the selectivity towards CO was favoured over other carbon-containing compounds, and as a result, the H2/CO ratio decreased, i.e. from 0.7 to 0.6 for Ni 20 wt%. It was suggested that CO2 molecules are readily chemisorbed and dissociated into CO on the surface of Ni-based catalysts, and in a high electric field the Ni sites become active and affect the conversion and selectivity. Finally, Li et al.195 used both Ni/Al2O3 and the HZSM-5 zeolite (SiO2/Al2O3 = 38) as catalysts. No significant effect on the conversions was observed, but the catalysts did affect the selectivities, whereby a higher CO and lower H2 selectivity were observed for Ni/Al2O3, while higher H2 and lower CO selectivities were measured for the HZSM-5 zeolite as the catalyst.
It is clear that corona discharges exhibit several similarities with DBDs, with an energy cost in the range of 4–100 eV per molecule and conversions of up to 60% (with exceptions even up to 80%). Thus, much work would still be required to lower the energy cost by a factor 2–3, which would be needed before corona plasmas could become a competitive alternative. In addition, it is difficult to achieve a high treatment capacity due to a corona's localized breakdown.177 This leads us to believe that, at this point, corona discharges have the same negative outlook as DBDs.
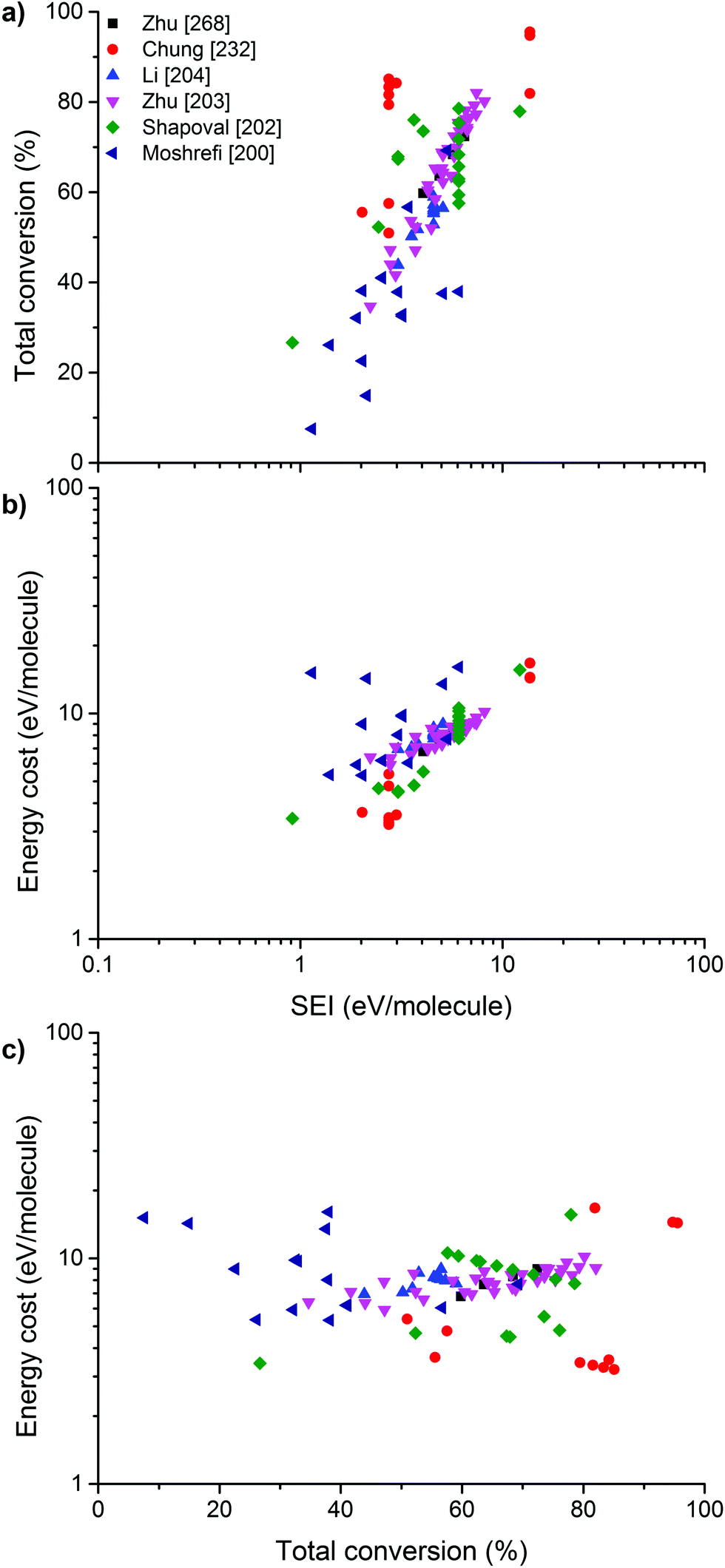 | ||
| Fig. 29 Experimental data collected from the literature for DRM in a spark discharge, showing the conversion (a) and energy cost (b) as a function of the SEI, as well as the energy cost as a function of the conversion (c). Note that some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
A spark discharge with different interelectrode distances and varying pressure and frequency was investigated by Zhu et al.203 With the interelectrode distance rising from 3 to 9 mm, a higher conversion was observed, followed by a slight decrease for even larger gap widths, while the selectivities were only slight affected. At increased pressures, i.e. up to 2.5 bar, higher conversions were reported for the same SEI.203,268 The selectivities again appeared unaffected. When increasing the frequency from 5 to 80 kHz at the same SEI, an increase in the conversions of CH4 and CO2 from 67% and 58% to 74% and 63%, respectively, was observed. This might be explained by the relatively higher density of active species at higher frequencies. The product selectivities, on the other hand, showed only a minor dependence on the frequency.
Just like aforementioned for the GA discharge, for spark discharges, a lot of research has already been performed for the addition of O2 to the mixture to simulate biogas reforming.204,269–272 The same observations were made regarding a shift from DRM to partial oxidation reactions,204,269–271 with the suggestion that the water–gas shift reaction is the major or even the only effective contributor to the CO2 conversion.270,271
Finally, some experiments were also performed for the implementation of catalysts in spark discharges. Chung et al.199 used BaZr0.05Ti0.95O3 (BZT), with a perovskite structure and ferroelectric properties, as a packing material inside the spark discharge zone. Compared to the empty reactor, the CH4 and CO2 conversions increased from 53% and 49% to 84% and 77%, respectively. This indicated that the combination induced synergistic effects to reduce the specific energy cost. Two types of packing were tested: coarse (C-BZT) and fine (F-BZT) particles. C-BZT showed a better performance than C-BZT, possibly because the void space was larger in the former, leading to a higher electron density.
In summary, spark discharges allow a high conversion, i.e. up to 85%, while demonstrating low energy costs, i.e. 3–10 eV per molecule. At the same time, they can achieve high selectivities towards syngas, which is interesting for the indirect oxidative liquefaction. The direct formation of oxygenates has, however, not been observed up to this point.
 | ||
| Fig. 30 Experimental data collected from the literature for DRM in an APGD, showing the conversion (a) and energy cost (b) as a function of the SEI, as well as the energy cost as a function of the conversion (c). Note that some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
Long et al.220 added N2 to the CH4/CO2 mixture to generate a stable discharge, but increasing the N2 flow rate led to a decrease in the CH4 and CO2 conversions from 46% and 34% to 37% and 22%, respectively, while the H2 and CO selectivity only changed slightly.
Furthermore, Long et al.220 combined the APGD plasma jet with a post-discharge catalyst bed. A γ-Al2O3 carrier and 12 wt% Ni/γ-Al2O3 catalyst were used. The performance for the γ-Al2O3 was similar to the plasma-only results, while the Ni/γ-Al2O3 catalyst significantly enhanced the process. The conversion of CH4 and CO2 increased by 14% and 6%, respectively, while the yields of H2 and CO increased by 18% and 11%, respectively. The single pass conversion was, however, too low and N2 was needed to maintain a stable discharge.220
In summary, the APGD seems to be promising for DRM, based on its proper plasma temperature for vibrational excitation and its high electron density, as it has a higher conversion ability compared to DBD and corona discharges, i.e. conversions up to 90% are reported for energy costs as low as 1 eV per molecule. Nevertheless, more research will be needed to fully demonstrate the capabilities. One of the major challenges remains a further enlargement of the process to treat large volumes.
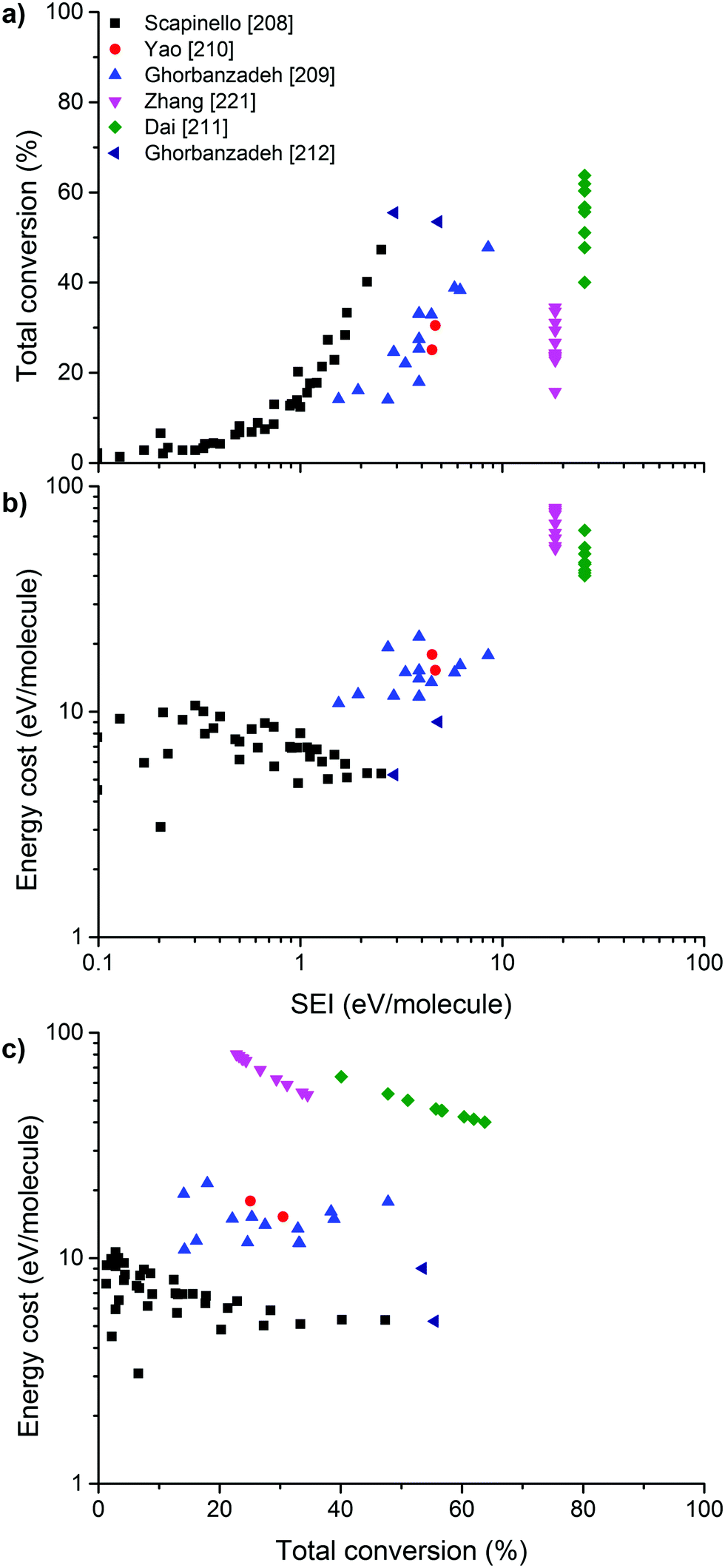 | ||
| Fig. 31 Experimental data collected from the literature for DRM in nanosecond pulsed discharges, showing the conversion (a) and energy cost (b) as a function of the SEI, as well as the energy cost as a function of the conversion (c). Note that some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
In this type of discharge, the amount of power dissipated in the plasma is determined by the pulse voltage and pulse frequency.209–212 A higher pulse voltage or repetition frequency leads to a higher power, which hence increases the SEI and conversions. The influence of the pulse voltage seems to be stronger than that of the pulse frequency.211 Regarding product selectivity, increasing the pulse frequency gives rise to a higher C2H4 selectivity and a decrease in C2H2 selectivity.210,211
Zhang et al.221 investigated the effects of adding various catalysts to the discharge and observed significant effects. Pure γ-Al2O3 significantly increases the CH4 conversion, while both the CO2 conversion and C2 selectivity decrease. La2O3/γ-Al2O3 catalysts, in contrast, give lower CH4 conversions and higher C2 selectivities, i.e. more than 60%. Pd/γ-Al2O3 and Pd-La2O3/γ-Al2O3 catalysts show the same results as pure γ-Al2O3 and La2O3/γ-Al2O3, respectively, but the C2 product distribution shifts from C2H2 to C2H4.221
In summary, these nanosecond pulsed discharges allow a high conversion, i.e. of up to 50–60%, while demonstrating relatively low energy costs, i.e. 3–10 eV per molecule. In previous sections, pulsed power DBD and corona discharges were mentioned, operating in the microsecond pulse regime, but it is clear that the latter did not attain the high conversions and energy efficiencies of nanosecond pulsed discharges. This leads us to believe that the nanosecond timescale is essential to create the necessary strong non-equilibrium,209 as is also the case in the filaments of a DBD.
To summarize, in Fig. 32 we plot the energy cost as a function of the total conversion, grouped per discharge type, for all the data discussed above. Furthermore, both the thermal equilibrium limit (see Section 2.2.1; Fig. 3) and the target energy cost of 4.27 eV per molecule for the production of syngas (see the beginning of Section 5.2; indicated as “Efficiency target”) are displayed. Note that the y-axis is reversed, to allow a better comparison with Fig. 24 for pure CO2 splitting, where we plotted the energy efficiency. This figure allows us to draw the following conclusions.
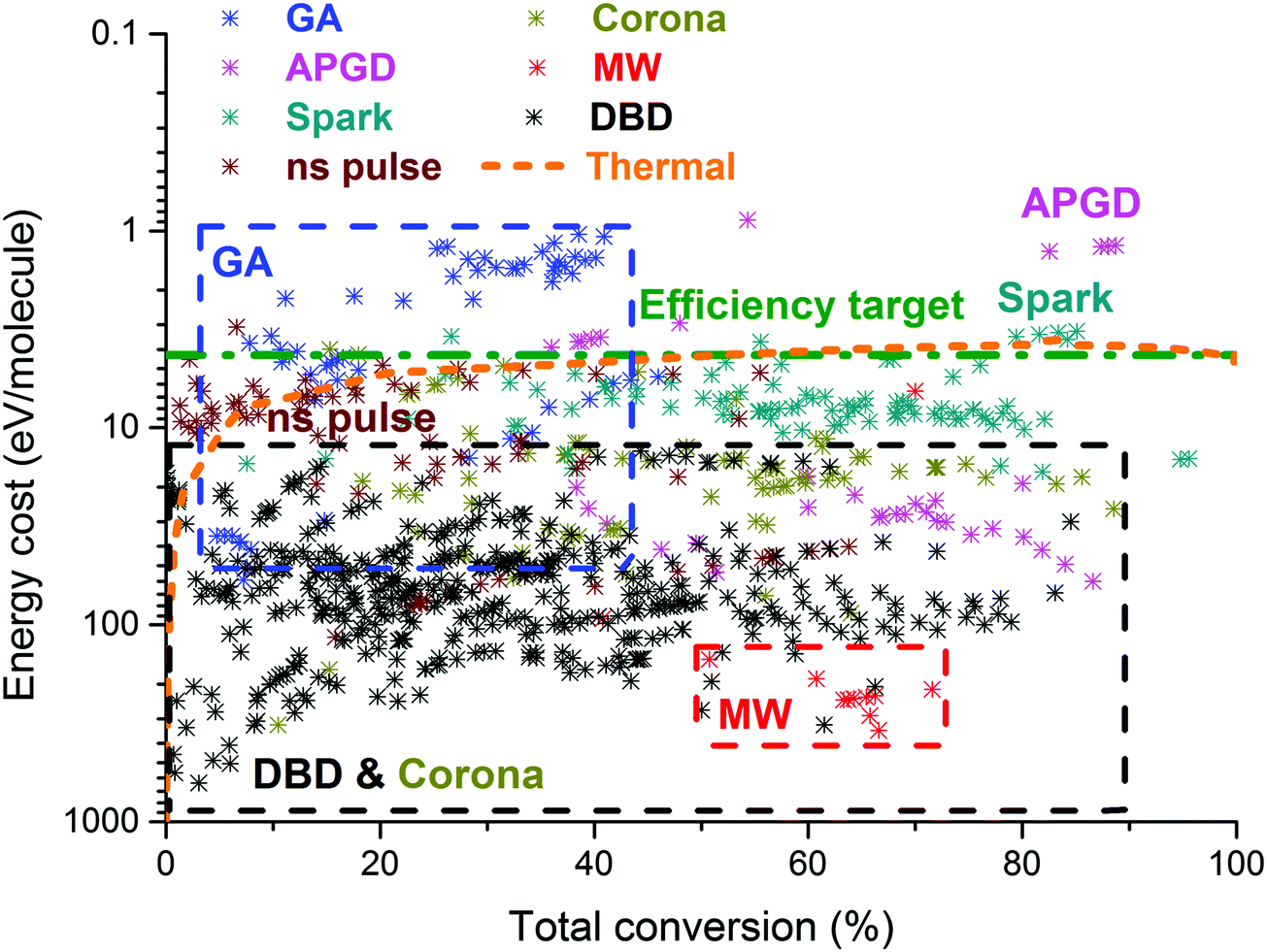 | ||
| Fig. 32 Comparison of all the data collected from the literature for DRM regarding the different plasma types, showing the energy cost as a function of the conversion. The thermal equilibrium limit and the target energy cost of 4.27 eV per molecule for the production of syngas (corresponding to a 60% efficiency target) are also indicated. Note that the y-axis has been reversed for easy comparison with Fig. 24. | ||
First of all, it is clear that for DRM – just like for CO2 splitting – although DBDs are by far the most extensively studied, they do not allow the energy-efficient conversion of CO2 and CH4 into syngas. Their energy cost currently remains a factor of 5 too high, even when combined with a packing, and we do not expect this to be sufficiently improved in the near future for them to become industrially competitive. On the other hand, if suitable catalysts could be found that would allow the direct high yield production of valuable oxygenates, this would change the analysis drastically, due to the more favourable energy requirements for the one-step process, as explained above. The same conclusion applies to most studies on corona discharges and the several studies on APGDs, with some exceptions as mentioned below.
Second, there is a significant lack of data with MW plasmas on DRM compared to pure CO2. Research in this area is highly recommended to evaluate whether the potential of MW discharges for DRM is as high as for pure CO2 splitting.
Third, the best results for GA plasmas are easily capable of surpassing the energy efficiency (or energy cost) target set for DRM. Even for conversions up to 40%, the energy efficiency target is easily crossed and they clearly operate in a strong non-equilibrium regime, as evidenced when compared to the thermal equilibrium limit. Moreover, further improvements can still be expected from this discharge type, as the full capabilities of the novel type of GAP, which has many advantages compared to classical GA discharges (see Sections 4.2.3 and 5.1.3 above), have not yet been explored for DRM.
Fourth, although the use of spark discharges and nanosecond pulsed discharges is relatively new and studies are still scarce, energy costs in the range of 10 eV per molecule are reported, and even as low as 3 eV per molecule, thus already reaching the efficiency target, even with the limited amount of research performed to date. For the nanosecond pulsed discharges, conversions are currently limited to 40–50% at these energy costs, while for spark discharges, conversions up to 85–95% have already been achieved. This clearly shows the potential for this type of discharge, and more research should be performed to further exploit their possibilities.
Finally, as mentioned above, for APGDs, some results obtained in the literature seem to be very promising. Indeed, as appears from Fig. 32, the best overall results were clearly achieved for APGDs, which could reach a high conversion, such as 88% for the spark discharge, in combination with low energy costs, such as 1.2 eV per molecule for the GA discharges. Thus, at this stage, APGDs together with GA discharges seem to be the most powerful, providing significant conversions at high energy efficiencies for DRM.
5.3. CO2 + H2O: artificial photosynthesis
As is clear from the sections above, to date a lot of research is based on both pure CO2 splitting and the dry reforming of methane. There has also been some research on pure H2O splitting with plasmas, i.e. for the production of hydrogen.21,274–278 However, research on the simultaneous conversion of CO2 and H2O – or so-called artificial photosynthesis – into syngas or oxygenated products by plasma is rather limited. The different plasma set-ups used to date for combined CO2/H2O conversion are: a regular DBD,279,280 ferroelectric packed-bed DBD,279 DBD packed with Ni/γAl2O3,281 MW discharge,282–285 GA,100,162 surface discharge286 and a negative DC corona discharge.287Mahammadunnisa et al.281 investigated the effect of a DBD reactor packed with a Ni/γ-Al2O3 catalyst, in both its unreduced and partially reduced forms, on combined CO2/H2O conversion. For the measurements without a catalyst, a CO2 conversion of 12–25% was obtained, depending on the SEI. The higher the power or the longer the residence time, the higher the conversion was. The reported selectivities were 18–14% for H2 and 97–99% for CO, leading to a syngas ratio of 0.55–0.18. When adding the catalysts, the conversion and syngas ratio increased, to 18–28% and 0.95–0.45, respectively, for the unreduced catalyst (NiO/γ-Al2O3), and to 24–36% and 0.66–0.35, respectively, for the partially reduced catalyst (Ni/γ-Al2O3). In this case, the added catalyst leads to a combination of physical and chemical effects, since, beside the enhanced conversion, the NiO catalyst is responsible for a further reduction of the produced CO to CH4 (and CH3OH), as well as the other compounds that were detected, i.e. C2H2, propadiene and carbon nanofibres of the partially reduced catalyst. Furthermore, it was concluded that higher flow rates lead to a higher H2/CO ratio, and thus to more economical syngas production.
Snoeckx et al.280 recently performed a combined experimental and computational study for CO2/H2O conversion in a DBD. CO2 and water vapour were used as the feed gases, with varying the H2O content in the mixture between 0% and 8% for three different SEI values. It was demonstrated that adding a few % of water to a CO2 plasma leads to a steep drop in the CO2 conversion, and then both the CO2 and H2O conversion keep on decreasing slightly when adding more water. The main products formed were CO, H2 and O2, as well as H2O2 – up to 2% for high SEI values and water contents. The combination of the experiments with a computational chemical kinetics study allowed the researchers to analyse the chemical kinetics and to construct and investigate the different chemical pathways to clarify the experimental results. In general, it was concluded that for a DBD, the main reactive species created are OH, CO, O and H, of which the OH radicals will quickly recombine with CO into CO2, thereby limiting the CO2 conversion upon the addition of water. At the same time, the O and H atoms will undergo subsequent reactions to form H2O again, thus explaining why the H2O conversion is also limited. Furthermore, the fast reaction between O/OH and H atoms also explains why no oxygenated products were formed, because it occurs much faster than the possible pathways that might otherwise lead to oxygenates.280
:1 mixture of CO2/H2O was investigated for a MW plasma. They detected low yields of oxalic acid and H2O2 in the cold trap condensate by reversed-phase chromatography using UV and conductivity detectors. The H2O2 production was very dependent on the discharge power, and a maximum yield of 0.024% was obtained. Furthermore, they assumed that H2 and O2 are generated, although these products were not measured. Interestingly, they also found the deposition of a transparent solid crystal film on the reactor walls, corresponding to oxalic acid.
In their follow-up study,283 they used the same MW set-up, but alternative detection techniques, i.e. steam chromatography and gas chromatography–mass spectrometry (GC–MS). The CO2/H2O gas mixture was varied from a 1:4 to a 1:1 ratio, but the full conditions for the presented results were not mentioned. This time, methanol formation, instead of H2O2 and oxalic acid, was reported, albeit in very low concentrations <0.01%, both in the effluent stream and in a transparent solid crystal film that was deposited on the reactor walls. Therefore, the authors concluded that two pathways for methanol formation could be considered: the direct formation from CO2 and H2O in the plasma and the reformation of deposited polymeric material on the walls during the plasma reaction with H2O. Most importantly, they observed that the methanol yield increased by a factor of 3.5 when increasing the pressure from 240 to 400 Pa, and so they stated that the system pressure is one of the most important parameters to consider.
Chen et al.284 applied a surface-wave MW discharge for the simultaneous dissociation of CO2 and H2O. The formation of syngas and O2 was observed, but no hydrocarbons or oxygenates were detected. The influences of the gas mixing ratio, the SEI and the feed flow rate on the H2 and CO production were studied. It was found that syngas with a ratio close to 1 can be produced when the CO2/H2O ratio in the gas mixture is 1:1.
In a follow-up study,285 the authors combined their MW set-up with NiO/TiO2 catalysts. In this work, they reported an increase in CO2 conversion from 23% to 31%, along with a lower energy cost from 30.2 to 22.4 eV per molecule, when adding 10% H2O for the plasma-only conditions. At the same time, a lower gas temperature was observed, which might be due to the higher heat capacity of water and the induced endothermic reactions. In turn, this lower temperature might be responsible for the higher conversion, since a lower gas temperature in MW plasmas is beneficial for energy-efficient CO2 conversion, due to there being lower vibrational losses by VT relaxation (see Section 5.1.2 above) and a reduced backward reaction rate, i.e. the recombination of CO back into CO2.88,89 When adding a NiO/TiO2 catalyst treated with an Ar plasma, the CO2 conversion further increased to 48%, with an energy cost of 14.5 eV per molecule. Still though, no oxygenated products were detected. It was suggested that CO2 is adsorbed at an oxygen vacancy on the catalyst surface, reducing the threshold for the dissociative electron attachment process to 2 eV, creating CO, an adsorbed O atom at the vacancy and an electron. Subsequently, the adsorbed O atoms interact with OH to form O2 and H as well as with gas phase O atoms to recombine to O2. As such, this seems to be some evidence that the reactions and species limiting the CO2 and H2O conversion, as described in the chemical reaction pathways for a pure DBD plasma280 (see previous section), can indeed, as suggested, be hijacked by the implementation of a catalyst.
:1 mixture of CO2/H2 and CO2/H2O in a comparative study by Hayashi et al.286 The CO2 conversions achieved were 15% and 5%, respectively. In both cases, the major products were the same, i.e. CO, CH4, dimethyl ether (DME) and formic acid, and in the case of H2 as co-reactant, also water formation.
Guo et al.287 reported the use of a negative DC corona discharge for the reaction between CO2 and H2O, varying the water vapour content between 10% and 43%, and the pressure between 1 and 4 bar. They observed a drop in CO2 conversion with increasing gas flow rates (hence decreasing SEI) and increasing water content. The main products formed were ethanol and methanol, in roughly a 3:1 ratio, with a total molar yield of up to 4.7% and CO2 conversion reaching a maximum of 16% at 1 bar, 50 mL min−1 and 23% water vapour. Other compounds detected were H2 and CO. Increasing the pressure had a beneficial effect on the methanol and ethanol yields. For example, when going from 1 to 4 bar, the ethanol yield increased from 3.2% to 11.9%. Unfortunately, some of the reported results seem to be contradictory/inconsistent, making it difficult to interpret the obtained results.
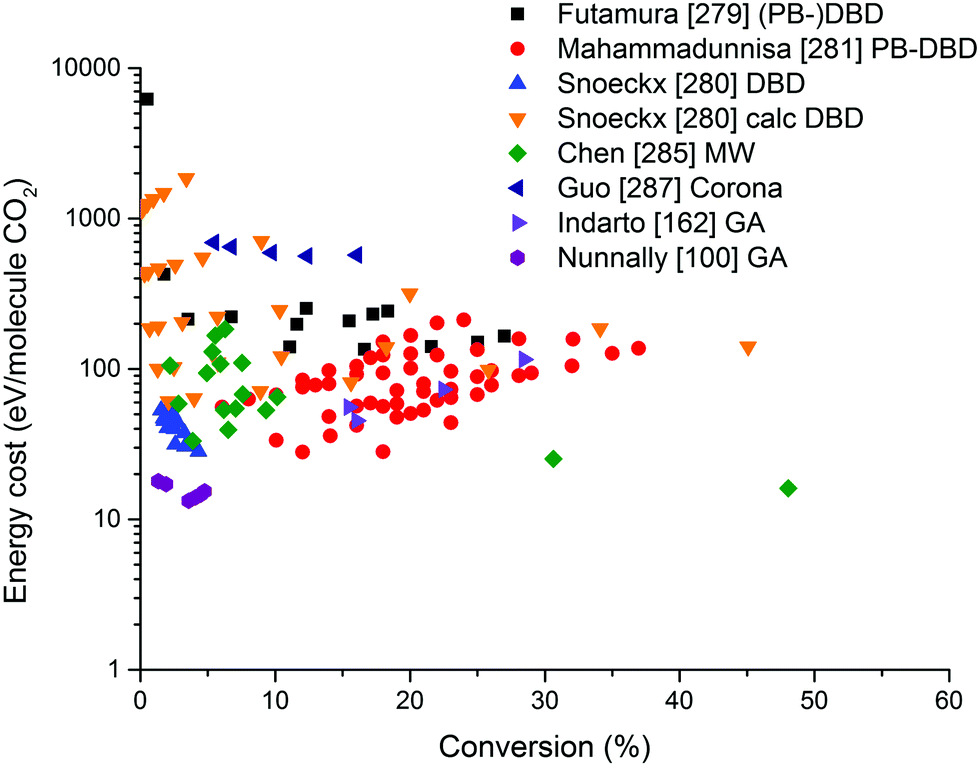 | ||
| Fig. 33 Comparison of all the data collected from the literature for the artificial photosynthesis (CO2 + H2O) in the different plasma types, showing the energy cost per converted CO2 molecule as a function of the conversion. Note, some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
The main products formed are again H2 and CO, like in the case of DRM (see Section 5.2), as well as O2, but some papers also report the production of hydrogen peroxide (H2O2),280,282 oxalic acid (C2H2O4),282 formic acid (CH2O2),286 methane (CH4),281,286 dimethyl ether (C2H6O, DME),286 methanol (CH3OH),281,283,287 ethanol (C2H5OH),287 acetylene (C2H2),281 propadiene (C3H4)281 and even carbon nanofibres (CNF).281 Unfortunately, most data on the formation of these products are only qualitative and mainly incomplete, making it impossible to deduce a general trend on product yields or selectivities. Although research on this process is more limited than for DRM – and hence the road ahead is longer – this clearly indicates that, just like for DRM above, the plasmachemical conversion of CO2 with H2O as a co-reactant allows for the development of the highly desirable direct oxidative pathway.
It is clear from the results obtained in the literature that CO2 and H2O seem to be unsuitable to create oxygenated hydrocarbons in a one-step process by means of a pure non-thermal plasma, i.e. in the absence of a catalyst, with the exception of the negative DC corona discharge. There are too many steps involved in generating these oxygenates, such as CH3OH, in an efficient way, and all of them involve H atoms, which will have the tendency to quickly recombine with OH into H2O or with O2 into HO2, which also reacts further with O into OH, and hence H2O. The problem at hand is thus that the interactions of H atoms with oxygen species (either OH, O3, O2 or O atoms) are too fast and their tendency to form H2O is too strong.
On the other hand, CO2/H2O plasmas can deliver an easily controllable H2/CO ratio with a rich hydrogen content – even up to 8.6 – when sufficient amounts of water can be added to the CO2 plasma, as was demonstrated in the computational study by Snoeckx et al.280 Hence, they might be suitable to create value-added chemicals, such as oxygenates, in a two-step process, which is good news. However, the interaction between H2O and CO2 dissociation products, i.e. the recombination between OH and CO into CO2, and the recombination of H and OH into H2O, limit the CO2 and H2O conversion, and thus the formation of useful products. Besides syngas, the direct production of sufficient amounts of hydrogen peroxide, which can be used as a disinfectant or for biomedical purposes, seems possible. However, the formation rate of H2O2 is also partially limited by the destruction reaction of OH + H2O2 towards H2O and HO2. Therefore, rapid removal of the formed product (i.e. H2O2), e.g. by means of a membrane, would be an important aspect for further improving this process. Based on these findings, and the work by Mahammadunnisa et al.,281 it is evident that a CO2/H2O plasma should be combined with a catalyst in order to produce value-added chemicals.102,107
As such, it appears that, due to the inherent nature of this mixture and the plasma set-ups, the future for the combined CO2 and H2O conversion by pure plasma technology does not look so bright at the moment. Nevertheless, we believe that a possible promising way forward is by its combination with specific tailored catalysts to produce value-added chemicals.
Such catalysts should be able to selectively let the plasma-generated CO and H2 react into oxygenates, such as methanol, and subsequently allow separation of the methanol from the mixture. As mentioned in the next section, Eliasson et al.288 applied a CuO/ZnO/Al2O3 catalyst in a CO2/H2 discharge, which resulted in an increase in methanol yield and selectivity by more than a factor of 10. Several other reported catalysts used already for CO2 conversion with H2 in traditional thermal catalysis might also be interesting to investigate for their suitability in plasma-catalysis in a CO2/H2O mixture, such as Ni-zeolite catalysts, for which methanation is reported,289 a Rh10/Se catalyst, yielding an ethanol selectivity of up to 83%,11 and a Ni–Ga catalyst for the conversion into methanol.290 Moreover, a lot of research into catalytic CO2 hydrogenation is showing promising results for CuO/ZnO/ZrO2 and for Cu/ZnO-based catalysts promoted with Pd and Ga, as well as Pd/ZnO and Pd/SiO2 with the addition of Ga.51 In general, multicomponent systems (Cu/ZnO/ZrO2/Al2O3/SiO2) have reported good performances for the formation of methanol starting from CO/CO2/H2 mixtures,11 making them potentially also very interesting for plasma-catalysis of a CO2/H2O mixture (as well as for DRM), because the CO/CO2/H2 mixture is anyway generated in situ during plasma-based conversion.
Based on the chemical kinetics pathway analysis presented by Snoeckx et al.280 (see Section 5.3.1 above), two pathways might be interesting and realistic to achieve: (i) promoting the recombination of OH radicals to H2O2 or (ii) promoting the reduction of CO to methanol. In both cases, the thermodynamic aspects at the nanoscale will become very important, especially since plasma-catalysis is a far-from-equilibrium process.107 The critical point will be the arrival and binding (e.g. physi- or chemisorption) of the reactants to the catalyst surface. To be successful, this process should be faster than the recombination rate of OH with H into H2O. Of course, these suggestions are only speculations, and further research will be needed to investigate these possibilities in practice.
We need to give a final critical note concerning safety. We need to be cautious about the explosive mixture that can be formed during this process – which could of course also occur for some of the other novel technologies discussed in Section 3, when O2 and H2 are not produced separately – due to the presence of O2, together with CO, H2 and an ignition source in a plasma set-up. At the research level, this will probably never be a problem due to the low volumes and conversions. However, when going to a pilot or industrial scale, i.e. with larger volumes and conversions, the risk will increase significantly. Consequently, both the capital and operating costs will increase drastically to ensure safe operations. One way to circumvent this problem is by diluting this mixture with an inert gas, such as argon or helium. In this case, however, an additional separation (for the products) and recuperation (for the inert gas) step will need to be included, which will also increase the cost. Furthermore, part of the input energy will be lost due to the electron-impact excitation and ionization of these gases. Therefore, this option will reduce the energy efficiency and increase the operating cost, but it will ensure safe operation.
5.4. CO2 + H2: hydrogenation of CO2
Research on the plasma-based hydrogenation of CO2 – using H2 as a co-reactant – is as limited as the combined conversion of CO2 and H2O. Historically, this is in part due to the high cost of hydrogen. Like for multiple other novel technologies discussed in Section 3, the sustainable and economically viable production of H2 is indispensable for pathways relying on the use of H2 as a co-reactant. Although interest into the plasma-based hydrogenation of CO2 emerged around the same time as the use of H2O as a co-reactant, most of the work performed is actually very recent. The different plasma set-ups used to date: are a DBD,288,291 packed-bed DBD,288,292 MW discharge,285,293,294 RF discharge295 and a surface discharge.286:1. For the plasma-only case, the major products found were CO and H2O, which is not surprising keeping the results of CO2/H2O in mind (see the previous section). Other components detected were CH4 and methanol, with a selectivity of only 3–4% and 0.4–0.5%, respectively. By adding the catalyst, the methanol yield increased about 10 times and the selectivity was 10–20 times higher. Furthermore, by optimizing the system to use low power and high pressure, it became possible to further enhance the methanol selectivity over the CH4 selectivity. The results indicate that the discharge shifts the region of maximum catalyst activity from 220 °C to 100 °C. Nevertheless, the electric power used is considered to be prohibitive for methanol production on an industrial scale, due to the low yield, i.e. ∼1%.
The methanation of CO2 in a DBD packed with Ni/zeolite pellets was investigated by Jwa et al.289 Conventional and plasma-assisted catalytic hydrogenation were compared with a varying nickel loading for a temperature range of 180–360 °C for a stoichiometric 4:1 ratio of H2/CO2. For the conventional catalytic hydrogenation case, a conversion of 96% was observed at 360 °C, while for the plasma-assisted hydrogenation, the same conversion was already reached at 260 °C. It was assumed that the reactive species generated in the plasma reactor could speed up the rate determining step of the catalytic hydrogenation. The hydrogenation of CO2 involves the dissociation of CO2 to CO and O on the active site of the Ni/zeolite (CO2,ads → COads + Oads). The rate determining step in the CO2 conversion into CH4 would be the same as for CO conversion (COads → Cads + Oads). Subsequently, the dissociated species react with hydrogen to produce CH4. This increased methanation rate is believed to arise from an increase in the surface concentration of carbon. The plasma can help to dissociate the adsorbed molecules, and hence surface carbon can be produced by both thermal activation and plasma activation, ultimately resulting in a higher methanation rate. Without Ni (bare zeolite), the conversion of CO and CO2 was found to be less than 1%, implying that the zeolite support together with the plasma cannot efficiently convert CO2 into CH4.289
Nizio et al.292 investigated the effect of a packed-bed DBD with Ni-CexZr1−xO2 catalysts on the hydrogenation of CO2 for a stoichiometric H2/CO2 ratio of 4:1. At 90 °C without a catalyst, the CO2 conversion was around 5%, without any methanation taking place. For the same conditions, 78% CO2 conversion and methanation with a selectivity of 99% and an energy cost <3 eV per molecule CH4 was achieved when adding the catalyst. This demonstrated the activity of plasma-catalytic systems at low temperature (Tg < 260 °C) and the possibility of enhancing both the conversion and selectivity of the process under study, by combining plasma with catalysts. Furthermore, the plasma-catalytic system showed almost no difference in activity for the different ceria-zirconia supports, while in the absence of plasma this was the case. The latter clearly indicates that it is indeed the plasma that initiates the methanation process by dissociating adsorbed CO2 molecules – a reaction that does not take place catalytically below 250 °C.
Recently, Zeng et al.291 investigated the performance of CO2 hydrogenation in a DBD at atmospheric pressure and low temperature, both with and without a catalyst, i.e. Cu/γ-Al2O3, Mn/γ-Al2O3 and Cu-Mn/γ-Al2O3. Some plasma-only results were also presented. The CO2 conversion was found to be 7.5%, and the main products were CO, H2O and CH4. The CO and CH4 selectivity were 46% and 8%, respectively. No methanol production was reported, although this might have been due to the inability to detect this compound by the GC set-up used. Adding the catalysts enhanced the process: the CO2 conversion increased to 8–10%, while the CH4 selectivity remained around 6.9–8.6%, but the CO selectivity and yield were enhanced to 76–80% and 6.4–7.9%, respectively.
Finally, de Bie et al.296 performed an extensive computational study on the hydrogenation of CO2 in a DBD, using a one-dimensional (1D) fluid model. The H2/CO2 mixing ratio was varied in the entire range from 1:9 to 9:1. The most abundant products predicted by the model were CO, H2O and CH4, and to a lower extent also formaldehyde, C2H6, O2 and methanol. The CO2 conversion was found to be rather low (2–7% in all the gas mixing ratios), especially when compared to typical values found for DRM (3–20%) at comparable conditions. This was thought to be the result of the lack of high enough concentrations of CH2 and CH3 radicals, which aid in the CO2 conversion. Moreover, a very similar chemical behaviour as described above for the CO2/H2O mixture280 was reported. Indeed, the CO2 conversion was limited due to the formation of CHO (CO + H + M → CHO + M), which reacted back to CO2 (CHO + O → CO2 + H). Furthermore, a lot of subsequent reactions were needed to form the desired hydrocarbons or oxygenates, such as CH4 and methanol, making their overall production rates negligible. Hence, it was concluded that a CO2/H2 mixture in a DBD-only set-up is not suitable for the production of value-added chemicals.
:1, including acetylene, methane, methanol, ethylene, formaldehyde and formic acid.
A so-called RF impulse discharge was used by Kano et al.295 to study the combined H2/CO2 conversion into CH4 and methanol at low gas pressures (1–10 Torr). For a H2/CO2 mixing ratio of 4:1, the main products detected were CO and H2O, as well as CH4 and to a lesser extent, i.e. one order of magnitude lower, methanol. Formaldehyde and formic acid were not observed. When the repetition frequency was lowered from 60 to 10 kHz, the formation of products decreased as well, while the CH4 production almost disappeared. The maximum CO2 conversion and CH4 selectivity obtained were 26% and 21%, respectively. The most efficient production of CH4 took place for a mixing ratio of 6:1 H2/CO2, which was larger than the stoichiometric ratio (4:1).
De la Fuente et al.294 recently used a surface-wave MW plasma reactor without a catalyst to study the effect of the gas flow rate, H2/CO2 mixing ratio and SEI on the performance for CO2 hydrogenation. The main products found were CO and H2O as well as 200 ppm C2H4 and 10–20 ppm methanol, but remarkably no CH4 was detected. The best performance was obtained for a H2/CO2 mixture of 3:1, obtaining a CO2 conversion of 82% and an energy cost of 28 eV per converted CO2 molecule, which are the best values reported in the literature for plasma-only operation. The CO2 conversion was even higher than for pure CO2 splitting, which was 65%, with an energy cost of 35 eV per molecule. It was predicted by means of a zero-dimensional (0D) reactor model that the dominant intermediate species are H and O, which was also found by OES measurements. The higher conversion of CO2 when adding H2 was thought to be the result of the slightly lower ionization energy of H atoms, resulting in higher electron densities. Furthermore, as these atoms cannot be excited vibrationally and/or rotationally, it might lead to higher electron temperatures.294
Chen et al.285 added a Ni/TiO2 catalyst to a surface-wave MW plasma, to not only investigate a CO2/H2O mixture (see Section 5.3.2 above), but also a H2/CO2 mixture in a ratio of 1:9. For the plasma-only case, a reduction in the CO2 conversion from 23% to 14% occurred. When adding the catalyst, the conversion was enhanced by a factor of 2, up to 28%, which was, however, still lower than the value for the pure CO2 case (41%), and also no methanol or CH4 formation was observed.
:1 mixing ratio was used at atmospheric pressure. The products observed were again CO and H2O as the main components and additionally CH4, DME and formic acid. For the highest SEI conditions, the maximum CO2 conversion was approximately 15%. Although higher than for the CO2/H2O case, these conversions and yields are considered to be too low.
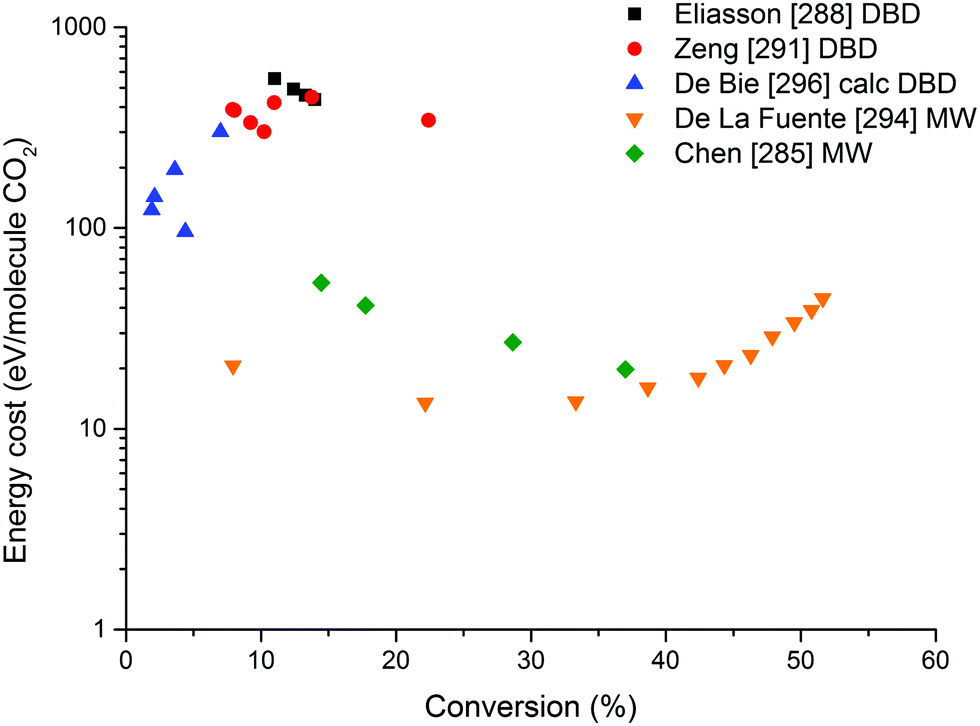 | ||
| Fig. 34 Comparison of all the data collected from the literature for CO2 hydrogenation in the different plasma types, showing the energy cost per converted CO2 molecule as a function of the conversion. Note, some of the data have been recalculated from the original references to take, among others, dilution effects into account. | ||
From all the data presented in the available literature, the main products formed are clearly CO and H2O. Some secondary products are also reported, but always in much smaller, indeed almost negligible, amounts, with the most important ones being CH4286,288,291,293,295,296 and methanol,288,293–296 although some papers also report the production of formaldehyde (CH2O),293,296 formic acid (CH2O2),286,293 dimethyl ether (C2H6O, DME),286 acetylene(C2H2),293 ethylene (C2H4)293,294 and ethane (C2H6).296 In spite of these low amounts, the fact that these products can be formed is an indication that the plasmachemical conversion process has the potential for the development of the highly desirable direct oxidative pathway if suitable catalysts could be found.
Thus, CO2 conversion using H2 as an additive, without a catalytic packing, shows a high resemblance to the use of H2O, which is of course not surprising, since the same reactive intermediate species and hence reactions are responsible for the plasma chemistry taking place. In accordance with that observation, in a pure plasma set-up, the combination of CO2 and H2 also seems to be unsuitable to create methane and/or methanol (or other oxygenated hydrocarbons) in a one-step process. The same number of steps – still too many – are involved in generating these value-added hydrocarbons or oxygenates in an efficient way. All these steps involve H atoms, which will have the same tendency to quickly recombine into OH and subsequently H2O.
Nevertheless, from the limited studies available, it appears that the combination with a catalytic packing is a viable option to produce value-added chemicals in an efficient way. We would like to make the same suggestions as in the previous section, namely that the catalyst should be able to selectively let the plasma-generated C (or CO) and H2 react into methane (or oxygenates). Multicomponent systems (Cu/ZnO/ZrO2/Al2O3/ SiO2) that show good performance for CO/CO2/H2 mixtures11 are potentially very interesting, since this mixture is anyway generated in situ during the plasma-based conversion of CO2/H2. Additionally, based on the work of Jwa et al.289 and Nizio et al.,292 the most effective and efficient pathway seems to be a low-power Ni-based plasma-catalytic set-up, in which the plasma takes care of the rate determining step, namely dissociating the adsorbed CO into C, allowing for the catalyst to reduce the C with H to CH4.
6. Benchmarking of plasma-based CO2 conversion against the other (traditional and novel) technologies
Finally, in this section, we compare plasma technology with the traditional catalytic approach and with the other novel approaches for CO2 conversion, based on all aspects explained in the previous sections. Furthermore, we identify the advantages and limitations of plasma-based CO2 conversion with respect to the other technologies, which will help us to define future priorities for the development of plasma-based CO2 conversion systems.6.1. Process versatility
A first comparison is made based on the versatility of the different CO2 conversion technologies. Table 1 shows the different technologies and the four different CO2 conversion approaches that were discussed. Traditional thermal catalysis, as discussed in Section 2, can be successfully applied for DRM and the hydrogenation of CO2, but seems unsuccessful at this point for pure CO2 splitting, while data for the combined conversion of CO2 and H2O are unavailable. The novel technologies, discussed in Section 3, are all applicable to converting CO2 together with H2O. Furthermore, the electrochemical and solar-thermochemical-based technologies are also capable of pure CO2 splitting. The combinations with CH4 and H2 are not generally reported in the literature. Plasmachemical conversion, on the other hand, currently is the only technology reporting successful application for all four processes, i.e. pure CO2 splitting, DRM, the hydrogenation of CO2 and the combined conversion of CO2 and H2O. As discussed in detail in Section 5, plasma-based pure CO2 splitting and DRM are obviously the two most successful processes today, with some types of plasmas already surpassing the posed 60% energy efficiency target for syngas production and achieving conversions of up to 90–95%, even without the combination with catalysis. Furthermore, the production of liquids in a one-step process, through a direct oxidative pathway (in combination with catalysts), has already proven successful, with further research in full progress. In this case, avoiding the intermediate syngas step, and thus circumventing the need for an additional Fischer–Tropsch or methanol synthesis and subsequently the methanol/ethanol to olefins synthesis, would significantly reduce the energy efficiency target and make it more competitive (i.e. by at least a factor of 2–3 in the case of methanol). Studies regarding the hydrogenation of CO2 using plasmas in combination with catalysis are very promising as well, but more research is still required. Although technically possible, the combined conversion of CO2 and H2O – the so-called artificial photosynthesis – with plasma technology seems the least promising from the data available to date. Nonetheless, here as well the combination with catalysis could prove beneficial, if a suitable catalyst could be found, as discussed in Section 5.3.5 above, but to date, research in this area is very scarce.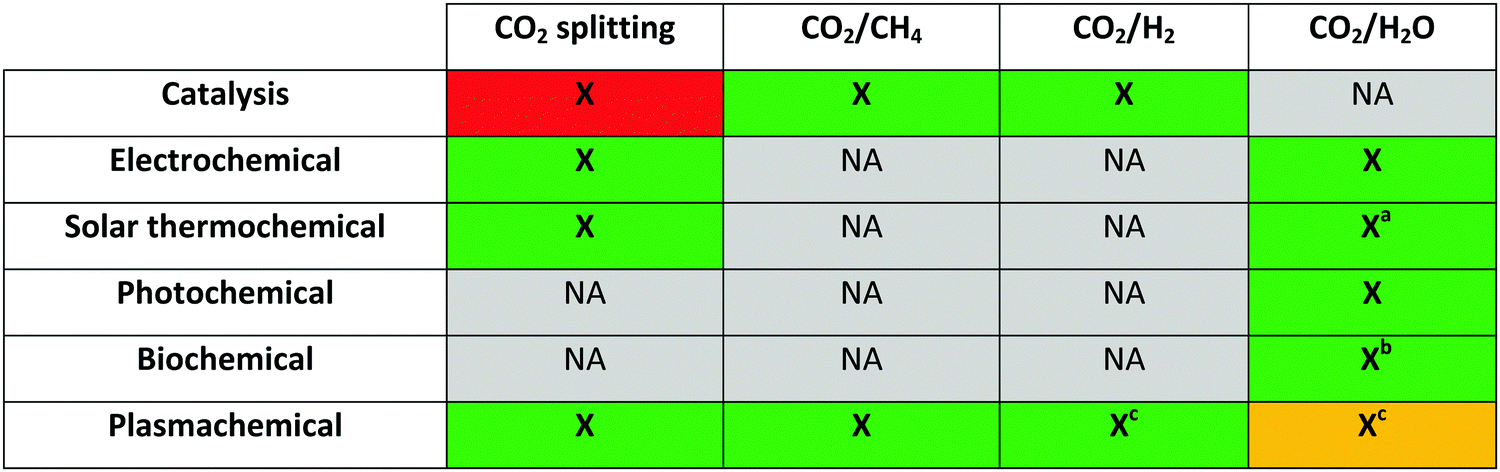
This high process versatility makes the plasmachemical conversion of CO2 very interesting as a CCU technique. It allows the process to be used at a wide variety of locations, independent of the available product feed, and even adjustable to a variable product feed at a specific location by the easily adaptable process. This process versatility gives the plasmachemical conversion a substantial benefit over the other technologies.
6.2. Advantages/disadvantages
It is clear that, although all the technologies, regardless of their maturity, have specific or even unique advantages, there is always a flip side to the coin. Table 2 provides a visual overview of the different technologies discussed in Section 3, as well as for plasmachemical conversion, together with a list of some of their key distinctive advantages and disadvantages.| a Bio- and photochemical processes can also rely on indirect renewable energy when they are coupled with artificial lighting, see Sections 3.3 and 3.4. b Electrochemical cells are turnkey, but generally the cells need to operate at elevated temperatures and the cells are sensitive to on/off fluctuations, see Section 3.1. c The need for post-reaction separation for the electrochemical conversion highly depends on the process and cell type used, see Section 3.1. d Biochemical CO2 conversion requires very energy-intensive post-reaction separation and processing steps, see Section 3.4. e The need for post-reaction separation for plasma technology highly depends on the process, see Section 5. |
|---|
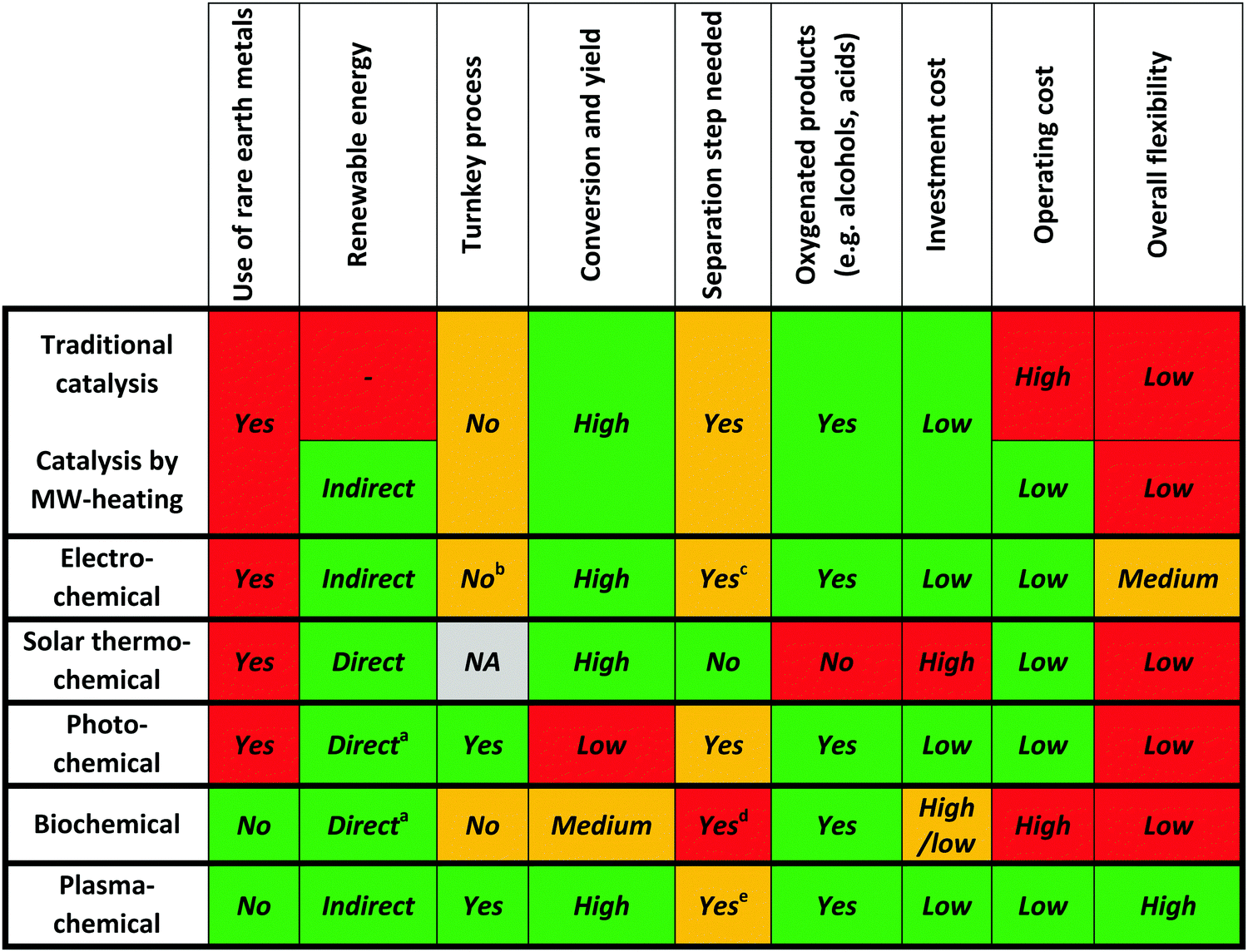
|
From an economical and sustainable point of view, the use of rare earth metals is one of the key disadvantages of most technologies at this point, except for biochemical and plasmachemical conversion. As mentioned before, the grand challenge for the other emerging technologies to be successful (as well as for traditional catalysis) is switching to inexpensive earth-abundant metals. It cannot be stressed enough that this is a critical make or break point, but which is not an issue for the plasmachemical conversion.
The discussion regarding the reliance on renewable energy in a direct or indirect manner (see Sections 1, 3 and 4) is more nuanced. The ability of solar thermochemical, photochemical and biochemical conversion to directly utilize renewable energy, i.e. solar radiation, can be considered to give them an edge over the other novel technologies, i.e., catalysis by MW-heating, electrochemical and plasmachemical conversion, which rely on electricity, and hence indirect renewable energy. The former have the advantage of skipping an energy conversion step, which would otherwise always leads to further energy losses, hence the energy efficiency of the latter is intrinsically determined by the efficiency of the renewable electricity generation. On the other hand, this also means that the MW-heating, electrochemical and plasmachemical processes can rely on other renewable energy sources as well, e.g. wind, hydro, wave and tidal power, increasing their versatility and employability, since they can be installed and operated independent of the availability of solar light. This significantly increases the application flexibility of MW-heating and electrochemical as well as plasmachemical conversion. Note that bio- and photochemical technologies may also rely on indirect renewable energy, when used in combination with artificial lighting.
The wide-scale adoption of these renewable energy sources, and their intermittent character, poses a challenge for the efficient storage and easy transport of the electricity produced. Not only is there a need for peak shaving, but more importantly a need for grid stabilization, which requires technologies to follow the irregular and at times intermittent supply of renewable electricity in a flexible manner. Technologies that are able to harness this energy and convert it into carbon neutral fuels could play an important role for the future energy infrastructure industry. Due to the intermittent character of renewable energies, a flexible storage process, which can easily be switched on and off to follow the supply, i.e. a so-called turnkey process, would be most ideal. Photochemical and plasmachemical conversion are the only two processes that truly meet this condition. They can be turned on with the flick of a switch, since no pre-heating or long stabilization times (<30 minutes) are required, as is the case for traditional catalysis. Likewise, they can be turned off with the flick of a switch, since no sensitive cool-down times are required and there is no risk of damaging the reactors with repeated on–off cycles. The problem with electrochemical cells is that, not only do they operate at elevated temperatures, but they are not designed– indeed, they suffer – for repeatedly being turned on and off, while biochemical processes need to be looked after on a continuous basis, e.g. regarding the nutrients, light, temperature, mixing.
From an industrial point of view, high conversions and yields are required, and most technologies are already capable of delivering on these requirements, with the exception of the photo- and biochemical conversion. A closely related key parameter is the solar-to-fuel conversion efficiency, which is discussed separately in the next section (Section 6.3).
Furthermore, when assuming that all or most of the feed is converted, the question arises as to whether an additional post-reaction separation step is required for the products. Solar thermochemical and electrochemical conversion (depending on the process and cell type) are the only two technologies capable of generating separated product streams, i.e. separated CO and O2 streams in the case of pure CO2 splitting. This is an important advantage, since the separation of CO and O2 is rather energy intensive at this point, although in the future, membranes might bring a convenient solution to this problem. Currently, this is probably the major disadvantage of plasma technology, as the products are all in one feed, i.e. for pure CO2 splitting, a mixture of CO and O2 is produced, and for DRM a mixture of (mainly) CO and H2, with – highly depending on the discharge type – some (minor) side-products, like hydrocarbons and oxygenates. Of course, a mixture of CO and H2 should not be a big problem, if the mixture will be immediately processed further through Fischer–Tropsch or methanol synthesis, since the plasmachemical conversion is able to deliver any desired syngas ratio, mainly depending on the gas mixing ratio in the feed. Nevertheless, research on the in situ trapping of species40 or a combination with other technologies297 is highly valuable, and requires more attention.
At the same time, this problem can also become less significant when focusing on the direct conversion of CO2 into value-added liquid oxygenated products by means of plasma-catalysis,102,103 including alcohols, aldehydes and acids, which are easier to separate. In the long run, as already mentioned before, this direct oxidative pathway to produce more value-added products is highly preferred rather than the current indirect method, which involves first producing CO and H2, which are then further processed. At the moment, except for solar thermochemical conversion, all the technologies are already able to produce more value-added chemicals in a direct fashion, although the yields are still extremely low and most research is still in its early stages.
The investment and operating costs are in general considered to be low for most technologies. The main exceptions are solar thermochemical conversion, which has a high investment cost for concentrating the solar energy, and biochemical conversion, where both investment and operating costs can be high, depending on the bioreactor type. Furthermore, the plasmachemical conversion is a highly modular technology, which is not dependent on an economy of scale and thus allows for local on-demand production capabilities.
Finally, one of the key-features for CCU techniques is their overall flexibility. This is actually a combination of many of the features discussed above. The plasmachemical conversion has a tremendous advantage here, due to its feed flexibility (CO2, CO2/CH4, CO2/H2 and CO2/H2O), its energy source flexibility (solar, wind, hydro, nuclear power), its operation flexibility (instant on/off, power scalability) and its flexibility of scale. None of the other technologies possess this unique combination of features required for successful worldwide implementation as a CCU technique.
In conclusion, plasma technology fares very well in this comparison, with its main disadvantages being: (i) its current need for post-reaction separation processes, (ii) the fact that the energy efficiency is dependent on the reactor type and (iii) the need for combination with catalysis to steer the product yield and selectivity.
6.3. Solar-to-fuel efficiency
In our opinion, the most interesting measure to compare the different technologies is based on how well solar energy is converted into chemical energy, known as the solar-to-fuel conversion efficiency (ηsolar-to-fuel). First, we calculate this value for the various plasma-based technologies, based on the data presented above. Subsequently, we compare these solar-to-fuel efficiencies with the values for the other emerging technologies.Where possible (see the discussion in Section 6.4 below), we calculated the solar-to-fuel conversion efficiency for all the pure CO2 splitting data, based upon:
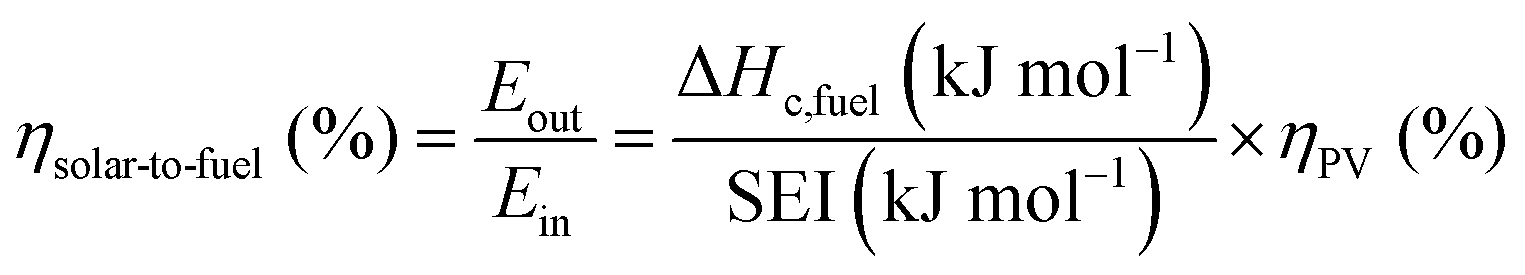 | (9) |
For the simple case of CO2 splitting, this means ΔHc,fuel equals:
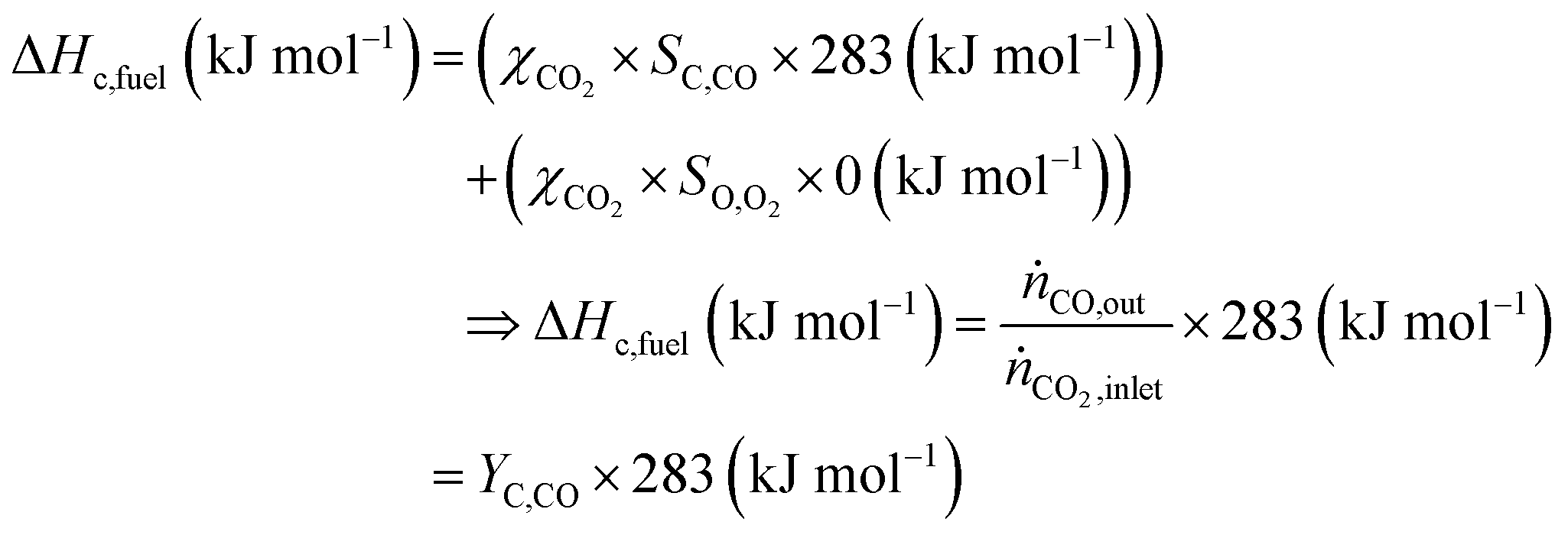 | (10) |
Thus, since the C-based selectivity for CO in the case of pure CO2 splitting in most cases is 100%, the equation for the solar-to-fuel efficiency equals here the definition for energy efficiency (eqn (6); defined in Section 4.4.3) – as well as the thermal energy efficiency (based on the HHV) – multiplied by the photovoltaic efficiency:
 | (11) |
For the DRM data, the equation becomes a bit more delicate. First of all we need to take the HHV of the converted CH4 in the feed into account as part of the denominator, since this counts as an energy input.
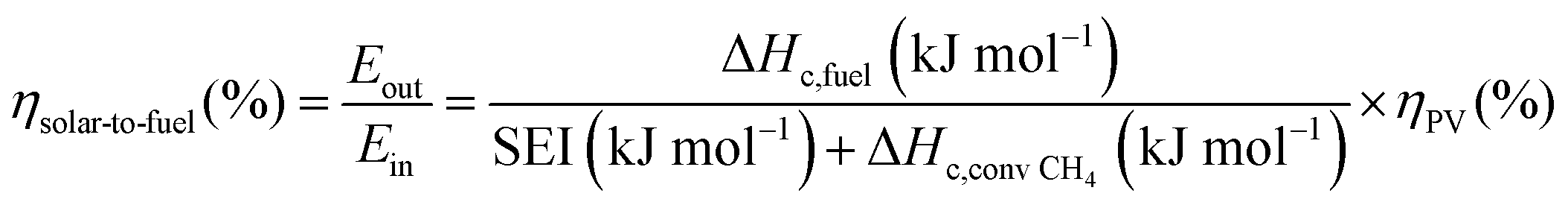 | (12) |
Second, as mentioned in the sections above, a wide variety of products is formed, which all need to be taken into account. Hence, for DRM, the solar-to-fuel efficiency would only equal the definition for energy efficiency (eqn (6)) multiplied by the photovoltaic efficiency, when the stoichiometric reaction as defined in Section 2.2.1 occurs, i.e. CO2 + CH4 → 2CO + 2H2.
Note that since the SEI is based on the plasma power, this efficiency does not take the energy losses into account, which occur at the level of the power supply and power coupling with the plasma. The data regarding these energy losses are generally not reported, since those may vary greatly, depending on the plasma type and power supply used, but they are independent from the plasma process under study. Furthermore, a lot of (successful) research progress is still being made in minimizing these electrical losses when going from outlet power to plasma power.
It should be noted that the solar-to-fuel conversion efficiency could only be calculated based upon the available data in the literature. If certain reaction products were not measured, their contribution to the enthalpy of combustion could not be taken into account. Due to the scarcity of information regarding CO2/H2O and CO2/H2 mixtures, we only consider CO2 splitting and DRM in the following analysis.
For the following discussions, we consider a PV efficiency value of 25% based on the efficiencies of the current commercial PV systems at the cell level.111 It is important to keep in mind that advancements in PV efficiency have a direct positive influence on the solar-to-fuel efficiency of plasma technology. For example, PV efficiencies of up to 45% have already been reported on a lab scale,298 a value which would almost double the solar-to-fuel efficiency of plasma technology as reported below.
It is reported in the literature that – on a purely economic basis – for the various novel technologies to be cost competitive with existing chemical and fuel processes, a solar-to-fuel conversion efficiency of 20% is likely to be needed for the production of syngas.65,110 Nonetheless, as already indicated above, all of these emerging technologies have additional advantages compared to the traditional catalytic technologies currently used. Furthermore, when the intermediate syngas step can be circumvented, i.e. due to the production of liquids through a direct oxidative pathway, this solar-to-fuel conversion efficiency target decreases drastically, e.g. for methanol, a solar-to-methanol efficiency of 7.1% would already be considered feasible.110 This is the greatest advantage of the plasmachemical conversion (in combination with catalysis), namely its already proven capability of producing a wide variety of liquid chemicals in a direct (although currently unselective) manner.
Table 3 presents the best data obtained from the literature on solar-to-fuel efficiencies, together with the obtained conversions, for the different plasma types discussed in Section 5. It should be no surprise that the plasma set-ups that showed the best performance also have the highest solar-to-fuel efficiency. For pure CO2 splitting, MW and GA plasmas are the two most promising discharge types, with maximum achieved solar-to-fuel efficiencies to date of 22.5% and 16.4%, respectively. For DRM, APGDs and GA plasmas have obtained the highest solar-to-fuel efficiencies up to now of 23.0% and 22.1%, respectively. However, most other discharges, except for DBDs, already reach efficiencies between 11% and 15%. This means that the chemicals and fuels produced by the plasmachemical conversion of CO2 into syngas could already be cost competitive, depending on the critical notes made above on product separation costs and power supply efficiencies.
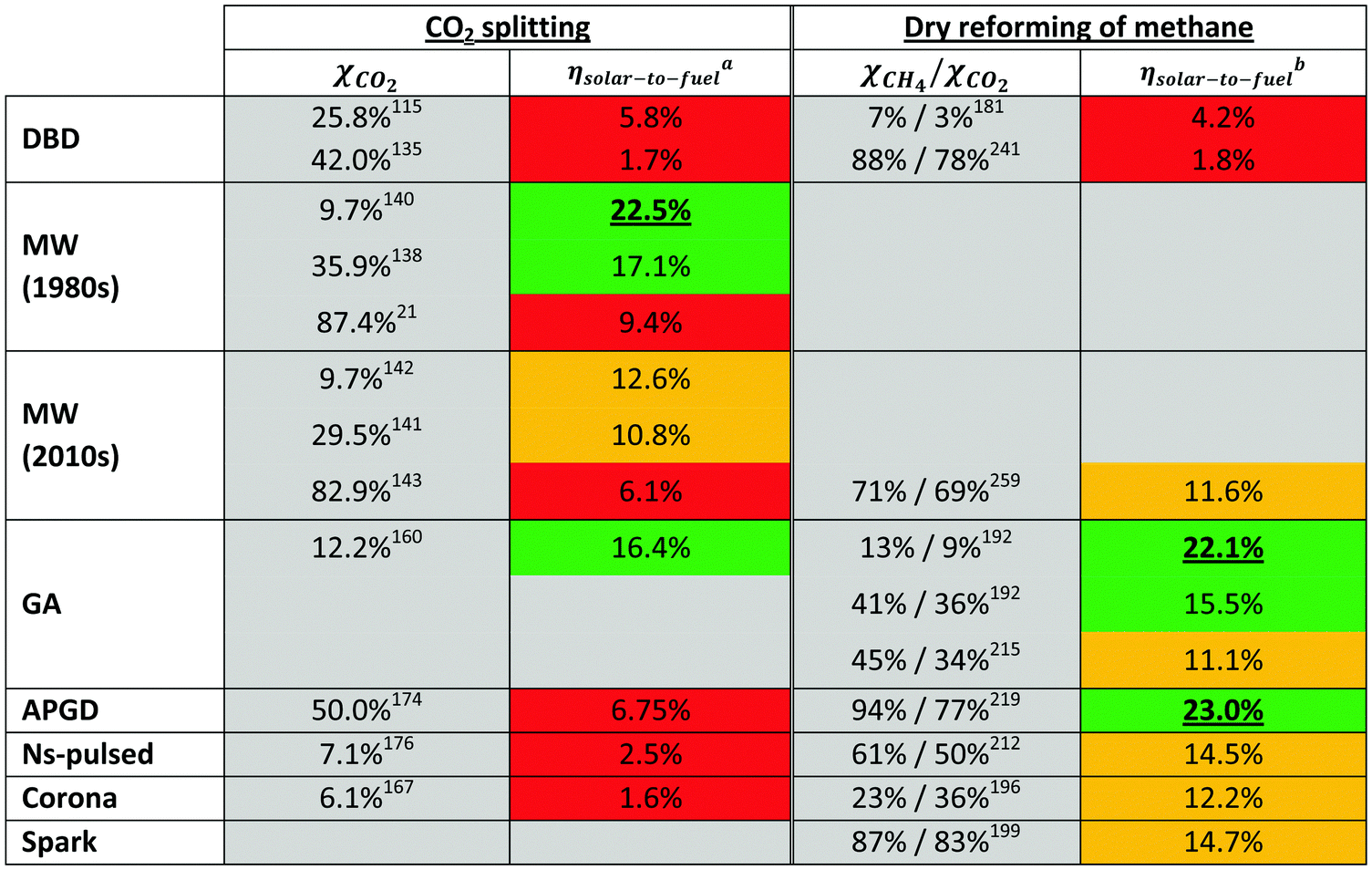
Finally, it is important to realize that this analysis is only a screenshot of the current production of syngas. When a successful shift is made towards the direct oxidative production of liquids, the performance of the different plasma types could change drastically. Indeed, certain plasma types (i.e. DBDs) allow for the easier implementation of catalytic materials, and the latter will play an important role in the selective production of these value-added compounds through the direct oxidative pathway. Furthermore, by circumventing the energy-intensive conversion of syngas to the desired liquids through the Fischer–Tropsch or methanol and subsequent synthesis processes, the required solar-to-fuel conversion efficiency needed to be competitive decreases by a factor of two to three.
Finally, in Table 4, we compare the reported solar-to-fuel conversion efficiencies obtained to date, as well as the theoretical maximum values, of plasmachemical conversion with the other novel technologies. As mentioned in Section 3.1, as water electrolysis powered by renewable energy is already a more mature technology, we have added it here to the comparison.
| Solar-to-fuel efficiency | Currently obtained | Theoretical maximum |
|---|---|---|
| a Based on a PV efficiency of 25%. b Without taking the power supply efficiency into account, due to lack of this data in the literature. | ||
| Water electrolysis | 16–19%a |
65–75%58
(x PV efficiency) |
| Electrochemical conversion | NA | <15%58 |
| Solar thermochemical conversion |
0.4–0.8%61,65
1.7–3.5%59 7.1%110 NA |
16–19%61,65
35–50%58 |
| Photochemical conversion |
<2%58
0.01–10.9%68 |
<5%58
∼17%68 |
| Biochemical conversion | NA |
4.5%35
x harvesting efficiency |
| Plasmachemical conversion | 23%a,b |
90–95%
x power supply efficiency (x PV efficiency) |
Water electrolysis yields efficiencies in the range of 16–19% for a PV efficiency of 25% and an electricity-to-hydrogen efficiency of 65–75%.58 Despite this success, electrochemical CO2 splitting, on the other hand, faces large efficiency losses due to overpotentials, and the theoretical efficiency is expected to be below 15%.58 For the solar thermochemical approach, theoretical efficiencies of 16–19% or 35–50% are assumed,58,61,65 depending on the heat recovery (see details in the cited references). Nevertheless, efficiencies above 10% are still pending experimental demonstration with robust and scalable solar reactors.59,61,65,110 For the photochemical conversion, the efficiency is theoretically limited to a maximum below 5%58 or 17%,68 depending on the band gap energy of the photocatalysts. However, the actual solar-to-fuel conversion efficiencies obtained to date are generally much lower (<2%),58,68 with some exceptions when coupled with an electrolytic cell (10.9%).68 For the biochemical conversion, a maximum efficiency for the conversion of light to stored chemical energy of about 4.5% has been calculated.35 However, this value is rarely achieved. Only in exceptional cases will dry matter yield exceed 1% or 2%.35
Finally, it is clear from Table 4 that the highest value obtained to date on a lab scale for the plasmachemical conversion of CO2, i.e. 23% (see Table 3 above), already appears to be competitive with the current most mature technology (i.e. water electrolysis) to transform renewable energy into chemicals and fuels through the intermediate production of the syngas components CO and H2. Furthermore, the theoretical maximum energy efficiency is 90–95%, when multiplied by the power supply efficiency and the PV efficiency. This is based on the theoretical – and experimentally proven – most energy-efficient CO2 dissociation process through vibrational excitation (see Section 4.1.4). Hence, the construction, use and matching of efficient power supplies for generating the plasma is highly critical. Nonetheless, we should not forget the critical notes made above on product separation. Additional – and indeed substantial – research is needed to provide an answer to these issues, just like in the case with all the other emerging technologies and their specific concerns. Due to its emerging character, interdisciplinary research towards plasma technology, especially in combination with other fields, such as catalysis will, undoubtedly, lead to further advancement and breakthroughs in this field.
6.4. Research recommendations
To conclude, we would like to provide some research recommendations for plasma technology, based on all the collected data from the literature, to further advance the field of plasma-based CO2 conversion. The first recommendation is related to the need for a more standardized framework in reporting data, to allow easier comparison of data within and outside the field of plasma technology. The most important criterion here is to be able to compare the conversions and energy cost/efficiency. Regarding the conversions, this requires a clear reporting on the presence of diluting agents (i.e. N2, He, Ar) when used, since these agents can significantly influence the plasma process, as discussed in Section 5. Furthermore, Pinhão et al.299 elaborately described the effect of gas expansion during plasma processing, which is to date often neglected, but which can have a tremendous influence on data accuracy, for example on the conversions obtained through GC measurements.92,95,125 Concerning the energy cost/efficiency, first of all there should be no room for interpretation about whether the reported power is the applied input power or the measured plasma power and through which electrical techniques, such as Lissajous plots, this power was obtained. Second, one should aim to identify all the important products and their selectivities, to be able to determine the energy efficiency based on the enthalpy of the reaction, as well as the so-called fuel production efficiencies. Third, data on power supply efficiency and its optimization are urgently required to report accurate solar-to-fuel conversion efficiencies. In general, as also discussed by Butterworth et al.136 for the testing of materials for plasma-catalysis, general frameworks on performing experiments and reporting data are becoming essential for the transparent further development of plasma research, both within the plasma community and within the larger CO2 conversion community.Finally, we also have some more personal recommendations. From the plots in the sections above, showing the energy cost/efficiency as a function of the SEI, it becomes more than clear that most applied SEIs are far too high, especially for the combinations with catalysis. An SEI range of 0.1–5 eV per reactant molecule (taking possible dilutions into account) should be the target to achieve energy-efficient conversions, as also recommended by Fridman.21 Furthermore, since plasma processes are very susceptible to the effect of impurities in the feed, more research and insights are needed towards the effect of real-life gas compositions on both the physical side (e.g. discharge stability) and on the chemical side (e.g. product distributions, harmful by-products).
7. Conclusions and outlook
The conversion of CO2 – preferably into value-added products – is considered one of the great challenges of our century. In the past decade, substantial progress with several novel technologies has been made. The last section of this review intended to provide the reader with a state-of-the art and critical assessment of an – up till now – rather underexposed emerging technology: the plasmachemical conversion of CO2. To achieve this, we provided an introduction to the basic concepts of plasma, and we demonstrated its viability as a gas conversion technology, finally putting it in the broader context of its peers.From the advances in the area of CO2 conversion discussed in this review, one outcome stands out without doubt. The question is not ‘if’ one of these novel technologies will be industrially competitive, but rather ‘when’ and ‘which one(s) will play the leading role’. To shine a brighter light on – the final burning question – whether plasma technology could be the answer – or at least be part of the equation – we briefly summarize here the arising opportunities and challenges for plasma technology in the field of CO2 conversion.
Plasmas possess some important advantages over certain other (novel) technologies: (i) they can operate at room temperature using any source of (renewable) electricity, (ii) they have a large flexibility in terms of the feeds that need to be processed, (iii) they provide an extremely flexible ‘turnkey’ process, which allows for the efficient storage of energy, peak shaving and grid stabilization, (iv) the reactors have low investment and operating costs, (v) they have a simple scalability both in size and applicability, and (vi) last but not least, the technology does not rely on rare earth materials – making it rather unique at this point. This unprecedented combination of features gives plasmachemical conversion a very high overall flexibility, making it an extremely useful and valuable technology for CCU.
The flip side of the coin is that the reliance of plasma technology on indirect solar energy in the form of electricity is – at the same time – a limiting factor for the solar-to-fuel conversion efficiency, especially compared to technologies that can directly harvest solar energy. Nonetheless, this also allows plasma technology to rely on other renewable energy sources, and more importantly, it adds to its overall – location and process – flexibility, since it is not dependent on the availability of sunlight. More urgent, however, is the issue of product separation. From all the data available in the literature, it is clear that converting CO2 – with or without an additional hydrogen source – always yields a mixture of products after reaction. This implies the need for an – often expensive – post-processing separation step.
From the benchmarking discussion, it is clear that plasma technology can definitely play an important role in the field of CO2 conversion, and it is not beyond our grasp to think about its eventual commercial implementation, be it on a large or small scale. Nevertheless, as always, only a few candidates seem suitable for the specific task at hand, as we outline here.
Two main CO2 conversion strategies were discussed: (i) pure CO2 splitting and (ii) CO2 conversion in combination with a co-reactant serving as the hydrogen source, i.e. CH4, H2 and H2O, yielding processes named the dry reforming of methane (DRM), the hydrogenation of CO2 and artificial photosynthesis, respectively. At this time, the conversion of CO2 through the latter two processes is – although possible – inefficient in a plasma-only set-up, not to mention industrially undesirable, due to the formation of possibly dangerous mixtures to be handled. In the future, the combination of plasma with other technologies, such as catalysis or electrolysis, could solve these issues. On the contrary, for both pure CO2 splitting and DRM, the proposed energy efficiency target of 60% has already been surpassed by several types of plasma reactors, resulting in a solar-to-fuel conversion efficiency for the production of syngas above – the required – 20% mark.
In the short run, pure CO2 splitting using the most energy-efficient set-ups, i.e. MW and GA discharges, appears to have a high potential. In this case, pure CO and O2 could be produced, after a separation step, or a pure CO stream when in situ trapping of the O2 can be successfully implemented, e.g. by combination with other technologies or through the addition of scavengers.
In the long run, it seems evident that CO2 conversion in combination with a hydrogen containing co-reactant has the highest potential for the efficient production of value-added products, such as alcohols, aldehydes, esters and acids. In particular, the direct oxidative pathway, in which the intermediate syngas step is circumvented, has great potential. For now, however, a lot of research in this area is still needed to increase the selectivity towards these valuable bulk chemicals over the currently produced syngas. This will most probably need to be achieved in combination with catalysts. However, even today, APGD and GA discharges already provide an energy-efficient alternative to produce syngas in any desired ratio through DRM.
By all means, plasma technology is not just an overlooked unicorn – although its combination of features does make it quite unique for CCU – but, as mentioned several times throughout this work, and just like for all other emerging technologies in this field, several important challenges remain. General challenges comprise the need for further fundamental research, concerning: (i) the plasmachemical processes taking place in warm discharges, such as MW and GA, which stimulate vibrational excitation; whereby the latter, representing the most energy-efficient dissociation pathway, should be further exploited by enhancing the non-equilibrium character of these plasmas to further improve the energy efficiency, (ii) the combination of plasma with catalysis and the possibility of taking advantage of the synergetic effects and the selective production of value-added compounds, (iii) in situ O/O2 trapping by using scavengers, or combinations with other technologies, such as catalysis or SOEC, to further enhance the conversions and yields towards the desired value-added compounds, (iv) the demonstration of successful scale-up for other discharges than DBDs and (v) the production and tuning of efficient power supplies for generating and sustaining the plasma.
Moreover, some important discharge-specific research challenges can also be identified. For MW plasmas, the ultimate goal should be to achieve the same energy efficiency at atmospheric pressure as currently obtained at reduced pressure for CO2 splitting, while more research towards DRM is essential. For GA plasmas, the limited amount of gas that passes through the active arc region, and thus that can be converted by the plasma, is currently limiting the overall CO2 conversion, and more research in this direction is needed, e.g. by improving the reactor design. Furthermore, some other plasma reactors show good performance, but the available data and insights are still limited, thus requiring more research to explore their full potential. Of course, inherent to every review, the above analysis represents the current state-of-the-art. Plasma-technology-based CO2 conversion is relatively new and it is clearly a fast advancing field with – just like for any emerging technology – ample room for improvement and new (interdisciplinary) developments.
It appears that the ultimate plasma for CO2 conversion would be a system with a high electron density (Ne) and a low electron temperature (Te) – resulting from a low reduced electric field (E/n) – which are essential to stimulate and achieve a high fraction of vibrational excitation. At the same time, this system should operate at a low gas temperature (Tg) to minimize the VT relaxation losses and backward recombination reactions. Finally, to be industrially attractive, this should be achieved at atmospheric pressure.
Future research towards the direct oxidative production of oxygenates, as well as multi-reforming processes with a combination of gases (e.g. CO2, CH4 and H2O or O2) or several reforming processes in series, using plasma technology appears very promising and should be pursued. Furthermore, more interdisciplinary research into the combination of plasmas with other novel technologies is highly desirable. For catalysis, the combination is already proving alluring, and it is our opinion that combinations with electrolysis, although even more challenging, could also lead to synergetic effects. As is the case for many important issues brought forward in the current age, with a high probability the future will rely on a true amalgam of technologies and solutions, each for its specific tasks, rather than the ‘one solution (or technology) fits all’ mentality of the old age.
From the current analysis, it is evident that pure CO2 splitting could reach industrial implementation at a faster pace. Nevertheless, CO2 conversion using a hydrogen source as a co-reactant should certainly be further pursued, since – when successful – in the long run it offers us the possibility of producing a wide variety of value-added chemicals and fuels, starting from the same building block and allowing the flexibility to tune the output depending on the market's needs. On that account, we believe plasmas could be – at least part of – the bright light that shines on our horizon.
Acknowledgements
We would like to thank W. Wang (University of Antwerp) for providing the data on the thermal equilibrium conversions. Furthermore, we acknowledge financial support from the IAP/7 (Inter-university Attraction Pole) programme ‘PSI-Physical Chemistry of Plasma-Surface Interactions’ by the Belgian Federal Office for Science Policy (BELSPO), the Methusalem financing of the University of Antwerp, the Fund for Scientific Research Flanders (FWO; Grant no. G.0383.16N, G.0254.14N and G.0217.14N), the TOP research project of the Research Fund of the University of Antwerp (grant ID. 32249).References
- R. K. Pachauri and L. A. Meyer, IPCC, 2014: Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Geneva, Switzerland, 2014.
- L. Johnson, J. Grant and P. L. Low, Two degrees of separation: ambition and reality: Low Carbon Economy Index 2014, 2014 Search PubMed.
- C. B. Field, V. R. Barros, D. J. Dokken, K. J. Mach, M. D. Mastrandrea, T. E. Bilir, M. Chatterjee, K. L. Ebi, Y. O. Estrada, R. C. Genova, B. Girma, E. S. Kissel, A. N. Levy, S. MacCracken, P. R. Mastrandrea and L. L. White, IPCC, 2014: Climate Change 2014 Impacts, Adaptation, and Vulnerability. Part A: Global and Sectoral Aspects. Contribution of Working Group II to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 2014.
- C. Lagarde, Int. Monet. Fund, 2013 Search PubMed.
- G. Nichols Roth, G7Climate Change The New Economy, World News -Climate Change The New Economy Ltd, 2015 Search PubMed.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- W. McDonough, M. Braungart, P. Anastas and J. Zimmerman, Environ. Sci. Technol., 2003, 37, 434A–441A CrossRef CAS PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 CAS.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
- M. Aresta, A. Dibenedetto and A. Angelini, Philos. Trans. R. Soc., A, 2013, 371, 20120111 CrossRef PubMed.
- J. A. Martens, A. Bogaerts, N. De Kimpe, P. A. Jacobs, G. B. Marin, K. Rabaey, M. Saeys and S. Verhelst, ChemSusChem, 2017, 10, 1039–1055 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, G. K. Surya Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833 CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M.-C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 CAS.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Müller, Energy Environ. Sci., 2012, 5, 7281 CAS.
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Adv. Inorg. Chem., 2014, pp. 259–288 CrossRef.
- S. Samukawa, M. Hori, S. Rauf, K. Tachibana, P. Bruggeman, G. Kroesen, J. C. Whitehead, A. B. Murphy, A. F. Gutsol, S. Starikovskaia, U. Kortshagen, J.-P. Boeuf, T. J. Sommerer, M. J. Kushner, U. Czarnetzki and N. Mason, J. Phys. D: Appl. Phys., 2012, 45, 253001 CrossRef.
- A. Fridman, Plasma chemistry, Cambridge University Press, New York, 2008 Search PubMed.
- Solar FAQs, ed. J. Tsao, N. Lewis and G. Crabtree, working draft version 20th April 2006, http://www.sandia.gov/˜jytsao/Solar%20FAQs.pdf Search PubMed.
- J. L. Sawin, K. Seyboth and F. Sverrisson, Renewables 2016: Global Status Report, 2016.
- Z. Jiang, T. Xiao, V. L. Kuznetsov and P. P. Edwards, Philos. Trans. R. Soc., A, 2010, 368, 3343–3364 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- P. L. Spath and D. C. Dayton, Natl. Renew. Energy Lab., 2003, 1–160 Search PubMed.
- J. Rifkin, The hydrogen economy: the creation of the worldwide energy web and the redistribution of power on earth, Putnam, New York, 2002 Search PubMed.
- U. Bossel, Proc. IEEE, 2006, 94, 1826–1836 CrossRef CAS.
- S. Rayne, Nat. Preced., 2008, 1–17 Search PubMed.
- D. D. Wagman, J. E. Kilpatrick, W. J. Taylor, K. S. Pitzer and F. D. Rossini, J. Res. Natl. Bur. Stand., 1945, 34, 143–161 CrossRef CAS.
- C. Song, Catal. Today, 2006, 115, 2–32 CrossRef CAS.
- Y. Nigara and B. Cales, Bull. Chem. Soc. Jpn., 1986, 59, 1997–2002 CrossRef CAS.
- N. Itoh, M. A. Sanchez, W.-C. Xu, K. Haraya and M. Hongo, J. Membr. Sci., 1993, 77, 245–253 CrossRef CAS.
- Y. Fan, J. Ren, W. Onstot, J. Pasale, T. T. Tsotsis and F. N. Egolfopoulos, Ind. Eng. Chem. Res., 2003, 42, 2618–2626 CrossRef CAS.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- A. Wolfson, C. Dlugy, Y. Shotland and D. Tavor, Tetrahedron Lett., 2009, 50, 5951–5953 CrossRef CAS.
- M. Aresta, A. Dibenedetto, T. Baran, A. Angelini, P. Łabuz and W. Macyk, Beilstein J. Org. Chem., 2014, 10, 2556–2565 CrossRef CAS PubMed.
- X. Zhu, T. Hoang, L. L. Lobban and R. G. Mallinson, Chem. Commun., 2009, 2908 RSC.
- R. Aerts, R. Snoeckx and A. Bogaerts, Plasma Processes Polym., 2014, 11, 985–992 CrossRef CAS.
- J.-M. Lavoie, Front. Chem., 2014, 2, 1–17 CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- D. Leckel, Energy Fuels, 2009, 23, 2342–2358 CrossRef CAS.
- J. R. H. Ross, Catal. Today, 2005, 100, 151–158 CrossRef CAS.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, J. Org. Chem., 2009, 74, 487–498 CrossRef CAS PubMed.
- J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Today, 2009, 148, 221–231 CrossRef CAS.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. Dubois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- B. Hu, C. Guild and S. L. Suib, J. CO2 Util., 2013, 1, 18–27 CrossRef CAS.
- S. G. Jadhav, P. D. Vaidya, B. M. Bhanage and J. B. Joshi, Chem. Eng. Res. Des., 2014, 92, 2557–2567 CrossRef CAS.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazabal, J. Perez-Ramirez, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- I. Ganesh, Renewable Sustainable Energy Rev., 2014, 31, 221–257 CrossRef CAS.
- S. Verma, B. Kim, H.-R. Molly’ Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- S. Das and W. M. A. Wan Daud, RSC Adv., 2014, 4, 20856–20893 RSC.
- G. P. Smestad and A. Steinfeld, Ind. Eng. Chem. Res., 2012, 51, 11828–11840 CrossRef CAS.
- J. R. Scheffe and A. Steinfeld, Mater. Today, 2014, 17, 341–348 CrossRef CAS.
- Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- A. H. McDaniel, E. C. Miller, D. Arifin, A. Ambrosini, E. N. Coker, R. O’Hayre, W. C. Chueh and J. Tong, Energy Environ. Sci., 2013, 6, 2424–2428 CAS.
- M. Romero and A. Steinfeld, Energy Environ. Sci., 2012, 5, 9234–9245 CAS.
- D. Arifin, V. J. Aston, X. Liang, A. H. McDaniel and A. W. Weimer, Energy Environ. Sci., 2012, 5, 9438–9443 CAS.
- P. Furler, J. R. Scheffe and A. Steinfeld, Energy Environ. Sci., 2012, 5, 6098–6103 CAS.
- E. N. Coker, A. Ambrosini, M. A. Rodriguez and J. E. Miller, J. Mater. Chem., 2011, 21, 10767–10776 RSC.
- B. Meredig and C. Wolverton, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 245119 CrossRef.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- P. M. Schenk, S. R. Thomas-Hall, E. Stephens, U. C. Marx, J. H. Mussgnug, C. Posten, O. Kruse and B. Hankamer, BioEnergy Res., 2008, 1, 20–43 CrossRef.
- L. Brennan and P. Owende, Renewable Sustainable Energy Rev., 2010, 14, 557–577 CrossRef CAS.
- R. Halim, M. K. Danquah and P. A. Webley, Biotechnol. Adv., 2012, 30, 709–732 CrossRef CAS PubMed.
- Y. Shen, RSC Adv., 2014, 4, 49672–49722 RSC.
- W. M. Budzianowski, Renewable Sustainable Energy Rev., 2012, 16, 6507–6521 CrossRef.
- A. Navarrete, G. Centi, A. Bogaerts, Á. Martín, A. York and G. D. Stefanidis, Energy Technol., 2017, 5, 796–811 CrossRef.
- X. Zhang, C. S. Lee, D. M. P. Mingos and D. O. Hayward, Catal. Lett., 2003, 88, 129–139 CrossRef CAS.
- B. Fidalgo, A. Domínguez, J. Pis and J. Menéndez, Int. J. Hydrogen Energy, 2008, 33, 4337–4344 CrossRef CAS.
- H. Xie, W. Jiang, Y. Wang, T. Liu, R. Wang, B. Liang, Y. He, J. Wang, L. Tang and J. Chen, Environ. Earth Sci., 2015, 74, 6481–6488 CrossRef.
- A. Bogaerts, E. Neyts, R. Gijbels and J. Van der Mullen, Spectrochim. Acta, Part B, 2002, 57, 609–658 CrossRef.
- E. Gomez, D. A. Rani, C. R. Cheeseman, D. Deegan, M. Wise and A. R. Boccaccini, J. Hazard. Mater., 2009, 161, 614–626 CrossRef CAS PubMed.
- U. Kogelschatz, Plasma Chem. Plasma Process., 2003, 23, 1–46 CrossRef CAS.
- X. Guofeng and D. Xinwei, Energy, 2012, 47, 333–339 CrossRef.
- O. Koeta, N. Blin-Simiand, W. Faider, S. Pasquiers, A. Bary and F. Jorand, Plasma Chem. Plasma Process., 2012, 32, 991–1023 CrossRef CAS.
- H. L. Chen, H. M. Lee, S. H. Chen, M. B. Chang, S. J. Yu and S. N. Li, Environ. Sci. Technol., 2009, 43, 2216–2227 CrossRef CAS PubMed.
- B. S. Patil, N. Cherkasov, J. Lang, A. O. Ibhadon, V. Hessel and Q. Wang, Appl. Catal., B, 2016, 194, 123–133 CrossRef CAS.
- A. Foote, J. Dedrick, D. O’Connell, M. North and T. Gans, APS Gaseous Electronics Conference, 2016.
- A. Fridman, A. Chirokov and A. Gutsol, J. Phys. D: Appl. Phys., 2005, 38, R1–R24 CrossRef CAS.
- T. Kozák and A. Bogaerts, Plasma Sources Sci. Technol., 2014, 23, 45004 CrossRef.
- T. Kozák and A. Bogaerts, Plasma Sources Sci. Technol., 2015, 24, 15024 CrossRef.
- A. Berthelot and A. Bogaerts, J. Phys. Chem. C, 2017, 121, 8236–8251 CAS.
- A. Bogaerts, A. Berthelot, S. Heijkers, St. Kolev, R. Snoeckx, S. Sun, G. Trenchev, K. Van Laer and W. Wang, Plasma Sources Sci. Technol., 2017, 26, 063001 CrossRef.
- A. Bogaerts, T. Kozák, K. van Laer and R. Snoeckx, Faraday Discuss., 2015, 183, 217–232 RSC.
- A. Janeco, N. R. Pinhao and V. Guerra, J. Phys. Chem. C, 2015, 119, 109–120 CAS.
- C. Tendero, C. Tixier, P. Tristant, J. Desmaison and P. Leprince, Spectrochim. Acta, Part B, 2006, 61, 2–30 CrossRef.
- A. Bogaerts, W. Wang, A. Berthelot and V. Guerra, Plasma Sources Sci. Technol., 2016, 25, 55016 CrossRef.
- R. Aerts, W. Somers and A. Bogaerts, ChemSusChem, 2015, 8, 702–716 CrossRef CAS PubMed.
- H. Conrads and M. Schmidt, Plasma Sources Sci. Technol., 2000, 9, 441–454 CrossRef CAS.
- S. R. Sun, H. X. Wang, D. H. Mei, X. Tu and A. Bogaerts, J. CO2 Util., 2017, 17, 220–234 CrossRef CAS.
- W. Wang, B. Patil, S. Heijkers, V. Hessel and A. Bogaerts, ChemSusChem, 2017, 10, 2145–2157 CrossRef CAS PubMed.
- X. Tu, H. J. Gallon and J. C. Whitehead, Electrical and optical diagnostics of atmospheric pressure argon gliding arc plasma jet, 30th ICPIQ August 28-September 2nd 2011, Belfast, Northern Ireland, UK, C10-081, http://mpserver.pst.qub.ac.uk/sites/icpig2011/081_C10_Tu.pdf Search PubMed.
- T. Nunnally, K. Gutsol, A. Rabinovich, A. Fridman, A. Gutsol and A. Kemoun, J. Phys. D: Appl. Phys., 2011, 44, 274009 CrossRef.
- F. Iza, J. L. Walsh and M. G. Kong, IEEE Trans. Plasma Sci., 2009, 37, 1289–1296 CrossRef.
- E. C. Neyts, K. Ostrikov, M. K. Sunkara and A. Bogaerts, Chem. Rev., 2015, 115, 13408–13446 CrossRef CAS PubMed.
- J. C. Whitehead, J. Phys. D: Appl. Phys., 2016, 49, 243001 CrossRef.
- E. C. Neyts and A. Bogaerts, J. Phys. D: Appl. Phys., 2014, 47, 224010 CrossRef.
- H. Lee and H. Sekiguchi, J. Phys. D: Appl. Phys., 2011, 44, 274008 CrossRef.
- A. Zhang, A. Zhu, J. Guo, Y. Xu and C. Shi, Chem. Eng. J., 2010, 156, 601–606 CrossRef CAS.
- E. C. Neyts and K. Ostrikov, Catal. Today, 2015, 256, 23–28 CrossRef CAS.
- K. Van Laer and A. Bogaerts, Plasma Sources Sci. Technol., 2016, 25, 15002 CrossRef.
- Y. R. Zhang, K. Van Laer, E. C. Neyts and A. Bogaerts, Appl. Catal., B, 2016, 185, 56–67 CrossRef CAS.
- J. Kim, C. A. Henao, T. A. Johnson, D. E. Dedrick, J. E. Miller, E. B. Stechel and C. T. Maravelias, Energy Environ. Sci., 2011, 4, 3122 CAS.
- K. Yoshikawa, H. Kawasaki, W. Yoshida, T. Irie, K. Konishi, K. Nakano, T. Uto, D. Adachi, M. Kanematsu, H. Uzu and K. Yamamoto, Nat, Energy, 2017, 2, 17032 CAS.
- R. Snoeckx, A. Rabinovich, D. Dobrynin, A. Bogaerts and A. Fridman, Plasma Processes Polym., 2017, 14, e1600115 CrossRef.
- S. Paulussen, B. Verheyde, X. Tu, C. De Bie, T. Martens, D. Petrovic, A. Bogaerts and B. Sels, Plasma Sources Sci. Technol., 2010, 19, 34015 CrossRef.
- Q. Yu, M. Kong, T. Liu, J. Fei and X. Zheng, Plasma Chem. Plasma Process., 2012, 32, 153–163 CrossRef CAS.
- A. Ozkan, A. Bogaerts and F. Reniers, J. Phys. D: Appl. Phys., 2017, 50, 84004 CrossRef.
- M. Schiorlin, R. Klink and R. Brandenburg, Eur. Phys. J.: Appl. Phys., 2016, 75, 1–7 CrossRef.
- F. Brehmer, S. Welzel, R. M. C. M. Van De Sanden and R. Engeln, J. Appl. Phys., 2014, 116, 123303 CrossRef.
- J. Wang, G. Xia, A. Huang, S. L. Suib, Y. Hayashi and H. Matsumoto, J. Catal., 1999, 185, 152–159 CrossRef CAS.
- I. Belov, S. Paulussen and A. Bogaerts, Plasma Sources Sci. Technol., 2016, 25, 15023 CrossRef.
- R. Li, Q. Tang, S. Yin and T. Sato, Appl. Phys. Lett., 2007, 90, 3–5 Search PubMed.
- R. Li, Q. Tang, S. Yin and T. Sato, Fuel Process. Technol., 2006, 87, 617–622 CrossRef CAS.
- S. Wang, Y. Zhang, X. Liu and X. Wang, Plasma Chem. Plasma Process., 2012, 32, 979–989 CrossRef CAS.
- M. Ramakers, I. Michielsen, R. Aerts, V. Meynen and A. Bogaerts, Plasma Processes Polym., 2015, 12, 755–763 CrossRef CAS.
- M. A. Lindon and E. E. Scime, Front. Phys., 2014, 2, 1–13 Search PubMed.
- R. Snoeckx, S. Heijkers, K. Van Wesenbeeck, S. Lenaerts and A. Bogaerts, Energy Environ. Sci., 2016, 9, 30–39 Search PubMed.
- X. Duan, Y. Li, W. Ge and B. Wang, Greenhouse Gases: Sci. Technol., 2015, 5, 131–140 CrossRef CAS.
- Y. Tagawa, S. Mori, M. Suzuki, I. Yamanaka, T. Obara, J. Ryu and Y. Kato, Kagaku Kogaku Ronbunshu, 2011, 37, 114–119 CrossRef CAS.
- R. Aerts, T. Martens and A. Bogaerts, J. Phys. Chem. C, 2012, 116, 23257–23273 CAS.
- P. Koelman, S. Heijkers, S. T. Mousavi, W. Graef, D. Mihailova, T. Kozak, A. Bogaerts and J. van Dijk, Plasma Processes Polym., 2017, 14, 1600155 CrossRef.
- S. Ponduri, M. M. Becker, S. Welzel, M. C. M. Van De Sanden, D. Loffhagen and R. Engeln, J. Appl. Phys., 2016, 119, 093301 CrossRef.
- A. Bogaerts, C. De Bie, R. Snoeckx and T. Kozák, Plasma Processes Polym., 2016, 1–21 CAS.
- M. Grofulovic, L. L. Alves and V. Guerra, J. Phys. D: Appl. Phys., 2016, 49, 395207 CrossRef.
- I. Michielsen, Y. Uytdenhouwen, J. Pype, B. Michielsen, J. Mertens, F. Reniers, V. Meynen and A. Bogaerts, Chem. Eng. J., 2017, 326, 477–488 CrossRef CAS.
- D. Mei, X. Zhu, Y.-L. He, J. D. Yan and X. Tu, Plasma Sources Sci. Technol., 2014, 24, 15011 CrossRef.
- X. Duan, Z. Hu, Y. Li and B. Wang, AIChE J., 2015, 61, 898–903 CrossRef CAS.
- T. Butterworth, R. Elder and R. Allen, Chem. Eng. J., 2016, 293, 55–67 CrossRef CAS.
- K. Van Laer and A. Bogaerts, Energy Technol., 2015, 3, 1038–1044 CrossRef CAS.
- V. D. Rusanov, A. A. Fridman and G. V. Sholin, Usp. Fiz. Nauk, 1981, 134, 185–235 CrossRef CAS.
- R. I. Asisov, A. A. Fridman, V. K. Givotov, E. G. Krasheninnikov, B. I. Patrushev, B. V. Potapkin, V. D. Rusanov and M. F. Krotov, 5th International Symposium on Plasma Chemistry, 1983, vol. 1, p. 52 Search PubMed.
- R. I. Asisov, A. K. Vakar, V. K. Jivotov, M. F. Krotov, O. A. Zinoviev, B. V. Potapkin, A. A. Rusanov, V. D. Rusanov and A. A. Fridman, Proc. Acad. Sci. USSR, Chem. Technol. Sect., 1983, 271, 94–98 Search PubMed.
- G. J. van Rooij, D. C. M. van den Bekerom, N. den Harder, T. Minea, G. Berden, W. A. Bongers, R. Engeln, M. F. Graswinckel, E. Zoethout and M. C. M. van de Sanden, Faraday Discuss., 2015, 183, 233–248 RSC.
- W. Bongers, H. Bouwmeester, B. Wolf, F. Peeters, S. Welzel, D. van den Bekerom, N. den Harder, A. Goede, M. Graswinckel, P. W. Groen, J. Kopecki, M. Leins, G. van Rooij, A. Schulz, M. Walker and R. van de Sanden, Plasma Processes Polym., 2017, 14, e1600126 CrossRef.
- A. P. H. Goede, W. A. Bongers, M. F. Graswinckel, R. M. C. M. Van De Sanden, M. Leins, J. Kopecki, A. Schulz and M. Walker, EPJ Web Conf., 2014, 79, 1005 CrossRef.
- L. F. Spencer and A. D. Gallimore, Plasma Sources Sci. Technol., 2013, 22, 15019 CrossRef.
- L. F. Spencer and A. D. Gallimore, Plasma Chem. Plasma Process., 2010, 31, 79–89 CrossRef.
- A. Vesel, M. Mozetic, A. Drenik and M. Balat-Pichelin, Chem. Phys., 2011, 382, 127–131 CrossRef CAS.
- L. F. Spencer, The Study of CO2 Conversion in a Microwave Plasma/Catalyst System, PhD dissertation, The University of Michigan, 2012 Search PubMed.
- M. Tsuji, T. Tanoue, K. Nakano and Y. Nishimura, Chem. Lett., 2001, 22–23 CrossRef CAS.
- S. Heijkers, R. Snoeckx, T. Kozák, T. Silva, T. Godfroid, N. Britun, R. Snyders and A. Bogaerts, J. Phys. Chem. C, 2015, 119, 12815–12828 CAS.
- S. Y. Savinov, H. Lee, H. K. Song and B.-K. Na, Korean J. Chem. Eng., 2002, 19, 564–566 CrossRef CAS.
- L. D. Pietanza, G. Colonna, G. D’Ammando, A. Laricchiuta and M. Capitelli, Plasma Sources Sci. Technol., 2015, 24, 42002 CrossRef.
- L. D. Pietanza, G. Colonna, G. D’Ammando, A. Laricchiuta and M. Capitelli, Chem. Phys., 2016, 468, 44–52 CrossRef CAS.
- T. Silva, N. Britun, T. Godfroid and R. Snyders, Plasma Sources Sci. Technol., 2014, 23, 25009 CrossRef.
- A. Berthelot and A. Bogaerts, Plasma Sources Sci. Technol., 2016, 25, 45022 CrossRef.
- G. Chen, V. Georgieva, T. Godfroid, R. Snyders and M. P. Delplancke-Ogletree, Appl. Catal., B, 2016, 190, 115–124 CrossRef CAS.
- I. Rusu and J. M. Cormier, Chem. Eng. J., 2003, 91, 23–31 CrossRef CAS.
- F. Ouni, A. Khacef and J. M. Cormier, Chem. Eng. Technol., 2006, 29, 604–609 CrossRef CAS.
- S. Heijkers and G. Trenchev, 2017, private communication.
- M. Ramakers, G. Trenchev, S. Heijkers, W. Wang and A. Bogaerts, ChemSusChem, 2017, 1–28 Search PubMed , submitted.
- S. C. Kim, M. S. Lim and Y. N. Chun, Plasma Chem. Plasma Process., 2014, 34, 125–143 CrossRef CAS.
- A. Indarto, J.-W. Choi, H. Lee and H. K. Song, Environ. Eng. Sci., 2006, 23, 1033–1043 CrossRef CAS.
- A. Indarto, D. R. Yang, J.-W. Choi, H. Lee and H. K. Song, J. Hazard. Mater., 2007, 146, 309–315 CrossRef CAS PubMed.
- J. L. Liu, H. W. Park, W. J. Chung and D. W. Park, Plasma Chem. Plasma Process., 2016, 36, 437–449 CrossRef CAS.
- W. Wang, A. Berthelot, S. Kolev, X. Tu and A. Bogaerts, Plasma Sources Sci. Technol., 2016, 25, 65012 CrossRef.
- G. Horváth, J. D. Skalný and N. J. Mason, J. Phys. D: Appl. Phys., 2008, 41, 225207 CrossRef.
- T. Mikoviny, M. Kocan, S. Matejcik, N. J. Mason and J. D. Skalny, J. Phys. D: Appl. Phys., 2004, 37, 64–73 CrossRef CAS.
- W. Xu, M.-W. Li, G.-H. Xu and Y.-L. Tian, Jpn. J. Appl. Phys., 2004, 43, 8310–8311 CrossRef CAS.
- M. Morvová, J. Phys. D: Appl. Phys., 1998, 31, 1865–1874 CrossRef.
- Y. Wen and X. Jiang, Plasma Chem. Plasma Process., 2001, 21, 665–678 CrossRef CAS.
- I. Maezono and J.-S. Chang, IEEE Trans. Ind. Appl., 1990, 26, 651–655 CrossRef CAS.
- S. L. Suib, S. L. Brock, M. Marquez, J. Luo, H. Matsumoto and Y. Hayashi, J. Phys. Chem. B, 1998, 102, 9661–9666 CrossRef CAS.
- S. L. Brock, M. Marquez, S. L. Suib, Y. Hayashi and H. Matsumoto, J. Catal., 1998, 180, 225–233 CrossRef CAS.
- S. L. Brock, T. Shimojo, M. Marquez, C. Marun, S. L. Suib, H. Matsumoto and Y. Hayashi, J. Catal., 1999, 184, 123–133 CrossRef CAS.
- S. N. Andreev, V. V. Zakharov, V. N. Ochkin and S. Y. Savinov, Spectrochim. Acta, Part A, 2004, 60, 3361–3369 CrossRef CAS PubMed.
- S. Mori, A. Yamamoto and M. Suzuki, Plasma Sources Sci. Technol., 2006, 15, 609–613 CrossRef CAS.
- M. S. Bak, S. K. Im and M. Cappelli, IEEE Trans. Plasma Sci., 2015, 43, 1002–1007 CrossRef CAS.
- X. Tao, M. Bai, X. Li, H. Long, S. Shang, Y. Yin and X. Dai, Prog. Energy Combust. Sci., 2011, 37, 113–124 CrossRef CAS.
- A. Lebouvier, S. A. Iwarere, P. D’Argenlieu, D. Ramjugernath and L. Fulcheri, Energy Fuels, 2013, 27, 2712–2722 CrossRef CAS.
- I. Istadi and N. A. S. Amin, Fuel, 2006, 85, 577–592 CrossRef.
- X. Tu and J. C. Whitehead, Appl. Catal., B, 2012, 125, 439–448 CrossRef CAS.
- R. Snoeckx, Y. X. Zeng, X. Tu and A. Bogaerts, RSC Adv., 2015, 5, 29799–29808 RSC.
- A. Ozkan, T. Dufour, G. Arnoult, P. De Keyzer, A. Bogaerts and F. Reniers, J. CO2 Util., 2015, 9, 78–81 CrossRef.
- V. Goujard, J.-M. Tatibouët and C. Batiot-Dupeyrat, Appl. Catal., A, 2009, 353, 228–235 CrossRef CAS.
- X. Zhang and M. S. Cha, J. Phys. D: Appl. Phys., 2013, 46, 415205 CrossRef.
- V. J. Rico, J. L. Hueso, J. Cotrino and A. R. González-Elipe, J. Phys. Chem. A, 2010, 114, 4009–4016 CrossRef CAS PubMed.
- Q. Wang, B.-H. Yan, Y. Jin and Y. Cheng, Plasma Chem. Plasma Process., 2009, 29, 217–228 CrossRef CAS.
- L. M. Zhou, B. Xue, U. Kogelschatz and B. Eliasson, Energy Fuels, 1998, 12, 1191–1199 CrossRef CAS.
- H. K. Song, J.-W. Choi, S. H. Yue, H. Lee and B.-K. Na, Catal. Today, 2004, 89, 27–33 CrossRef CAS.
- W. Chung, K. Pan, H. Lee and M. Chang, Energy Fuels, 2014, 28, 7621–7631 CrossRef CAS.
- C. J. Liu, B. Xue, B. Eliasson, F. He, Y. Li and G. H. Xu, Plasma Chem. Plasma Process., 2001, 21, 301–310 CrossRef CAS.
- W. Cho, W. S. Ju, S. H. Lee, Y. S. Baek and Y. C. Kim, Proceedings of 7th International Conference on Carbon Dioxide Utilization, 2004, pp. 205–208 Search PubMed.
- Z. Bo, J. Yan, X. Li, Y. Chi and K. Cen, Int. J. Hydrogen Energy, 2008, 33, 5545–5553 CrossRef CAS.
- A. Wu, J. Yan, H. Zhang, M. Zhang, C. Du and X. Li, Int. J. Hydrogen Energy, 2014, 39, 17656–17670 CrossRef CAS.
- X. Tu and J. C. Whitehead, Int. J. Hydrogen Energy, 2014, 39, 9658–9669 CrossRef CAS.
- M. Li, C. Liu, Y. Tian, G. Xu, F. Zhang and Y. Wang, Energy Fuels, 2006, 20, 1033–1038 CrossRef CAS.
- A. Aziznia, H. R. Bozorgzadeh, N. Seyed-matin, M. Baghalha and A. Mohamadalizadeh, J. Nat. Gas Chem., 2012, 21, 466–475 CrossRef CAS.
- M. Li, G. Xu, Y. Tian, L. Chen and H. Fu, J. Phys. Chem. A, 2004, 108, 1687–1693 CrossRef CAS.
- N. Seyed-Matin, A. H. Jalili, M. H. Jenab, S. M. Zekordi, A. Afzali, C. Rasouli and A. Zamaniyan, Plasma Chem. Plasma Process., 2010, 30, 333–347 CrossRef CAS.
- W. Chung and M. Chang, Energy Convers. Manage., 2016, 124, 305–314 CrossRef CAS.
- M. M. Moshrefi, F. Rashidi, H. R. Bozorgzadeh and M. Ehtemam Haghighi, Plasma Chem. Plasma Process., 2013, 33, 453–466 CrossRef CAS.
- V. Shapoval, E. Marotta, C. Ceretta, N. Konjevi, M. Ivkovic, M. Schiorlin and C. Paradisi, Plasma Processes Polym., 2014, 11, 787–797 CrossRef CAS.
- V. Shapoval and E. Marotta, Plasma Processes Polym., 2015, 12, 808–816 CrossRef CAS.
- B. Zhu, X. Li, J. Liu, X. Zhu and A. Zhu, Chem. Eng. J., 2015, 264, 445–452 CrossRef CAS.
- X.-S. Li, B. Zhu, C. Shi, Y. Xu and A.-M. Zhu, AIChE J., 2011, 57, 2854–2860 CrossRef CAS.
- S. Kado, K. Urasaki, Y. Sekine and K. Fujimoto, Fuel, 2003, 82, 1377–1385 CrossRef CAS.
- A. Huang, G. Xia, J. Wang, S. L. Suib, Y. Hayashi and H. Matsumoto, J. Catal., 2000, 189, 349–359 CrossRef CAS.
- Q. Chen, W. Dai, X. Tao, H. Yu, X. Dai and Y. Yin, Plasma Sources Sci. Technol., 2006, 8, 181–184 CrossRef CAS.
- M. Scapinello, L. M. Martini, G. Dilecce and P. Tosi, J. Phys. D: Appl. Phys., 2016, 49, 75602 CrossRef.
- A. M. Ghorbanzadeh, S. Norouzi and T. Mohammadi, J. Phys. D: Appl. Phys., 2005, 38, 3804–3811 CrossRef CAS.
- S. L. Yao, F. Ouyang, A. Nakayama, E. Suzuki, M. Okumoto and A. Mizuno, Energy Fuels, 2000, 14, 910–914 CrossRef CAS.
- B. Dai, X. L. Zhang, W. M. Gong and R. He, Plasma Sources Sci. Technol., 2000, 2, 577–580 CrossRef CAS.
- A. M. Ghorbanzadeh, R. Lotfalipour and S. Rezaei, Int. J. Hydrogen Energy, 2009, 34, 293–298 CrossRef CAS.
- Y. Zeng, X. Zhu, D. Mei, B. Ashford and X. Tu, Catal. Today, 2015, 256, 80–87 CrossRef CAS.
- M. a Malik and X. Z. Jiang, Plasma Chem. Plasma Process., 1999, 19, 505–512 CrossRef CAS.
- A. Indarto, J. Choi, H. Lee and H. Song, Energy, 2006, 31, 2986–2995 CrossRef CAS.
- S. L. Yao, M. Okumoto, A. Nakayama and E. Suzuki, Energy Fuels, 2001, 15, 1295–1299 CrossRef CAS.
- Y. Yang, Ind. Eng. Chem. Res., 2002, 41, 5918–5926 CrossRef CAS.
- C. Liu, A. Marafee, R. Mallinson and L. Lobban, Appl. Catal., A, 1997, 164, 21–33 CrossRef CAS.
- D. Li, X. Li, M. Bai, X. Tao, S. Shang, X. Dai and Y. Yin, Int. J. Hydrogen Energy, 2009, 34, 308–313 CrossRef CAS.
- H. Long, S. Shang, X. Tao, Y. Yin and X. Dai, Int. J. Hydrogen Energy, 2008, 33, 5510–5515 CrossRef CAS.
- X. Zhang, B. Dai, A. Zhu, W. Gong and C. Liu, Catal. Today, 2002, 72, 223–227 CrossRef CAS.
- Y. Li, C. J. Liu, B. Eliasson and Y. Wang, Energy Fuels, 2002, 16, 864–870 CrossRef CAS.
- Y. Zhang, Y. Li, Y. Wang, C. Liu and B. Eliasson, Fuel Process. Technol., 2003, 83, 101–109 CrossRef CAS.
- N. R. Pinhão, a Janeco and J. B. Branco, Plasma Chem. Plasma Process., 2011, 31, 427–439 CrossRef.
- G. Scarduelli, G. Guella, D. Ascenzi and P. Tosi, Plasma Processes Polym., 2011, 8, 25–31 CrossRef CAS.
- L. M. Martini, G. Dilecce, G. Guella, A. Maranzana, G. Tonachini and P. Tosi, Chem. Phys. Lett., 2014, 593, 55–60 CrossRef CAS.
- Y. Li, G. Xu, C. Liu, B. Eliasson and B. Xue, Energy Fuels, 2001, 15, 299–302 CrossRef CAS.
- M. Scapinello, L. M. Martini and P. Tosi, Plasma Processes Polym., 2014, 11, 624–628 CrossRef CAS.
- H. Song, H. Lee, J.-W. Choi and B. Na, Plasma Chem. Plasma Process., 2004, 24, 57–72 CrossRef CAS.
- T. Kolb, T. Kroker, J. H. Voigt and K. H. Gericke, Plasma Chem. Plasma Process., 2012, 32, 1139–1155 CrossRef CAS.
- T. Kolb, J. H. Voigt and K.-H. Gericke, Plasma Chem. Plasma Process., 2013, 33, 631–646 CrossRef CAS.
- W. C. Chung and M. B. Chang, Renewable Sustainable Energy Rev., 2016, 62, 13–31 CrossRef CAS.
- Q. Wang, Y. Cheng and Y. Jin, Catal. Today, 2009, 148, 275–282 CrossRef CAS.
- T. Kroker, T. Kolb, A. Schenk, K. Krawczyk, M. Młotek and K.-H. Gericke, Plasma Chem. Plasma Process., 2012, 32, 565–582 CrossRef CAS.
- S. Mahammadunnisa, P. M. K. Reddy, B. Ramaraju and C. Subrahmanyam, 2013.
- S. Kameshima, K. Tamura, Y. Ishibashi and T. Nozaki, Catal. Today, 2015, 256, 67–75 CrossRef CAS.
- X. Zheng, S. Tan, L. Dong, S. Li and H. Chen, J. Power Sources, 2015, 274, 286–294 CrossRef CAS.
- D. Mei, X. Zhu, C. Wu, B. Ashford, P. T. Williams and X. Tu, Appl. Catal., B, 2016, 182, 525–532 CrossRef CAS.
- Q. Wang, B. H. Yan, Y. Jin and Y. Cheng, Energy Fuels, 2009, 23, 4196–4201 CrossRef CAS.
- X. Tu, H. J. Gallon, M. V. Twigg, P. A. Gorry and J. C. Whitehead, J. Phys. D: Appl. Phys., 2011, 44, 274007 CrossRef.
- X. Zheng, S. Tan, L. Dong, S. Li and H. Chen, Int. J. Hydrogen Energy, 2014, 39, 11360–11367 CrossRef CAS.
- K. Zhang, T. Mukhriza, X. Liu, P. P. Greco and E. Chiremba, Appl. Catal., A, 2015, 502, 138–149 CrossRef CAS.
- M. H. Pham, V. Goujard, J. M. Tatibouët and C. Batiot-Dupeyrat, Catal. Today, 2011, 171, 67–71 CrossRef CAS.
- J. Sentek, K. Krawczyk, M. Młotek, M. Kalczewska, T. Kroker, T. Kolb, A. Schenk, K. H. Gericke and K. Schmidt-Szałowski, Appl. Catal., B, 2010, 94, 19–26 CrossRef CAS.
- K. Krawczyk, M. Młotek and B. Ulejczyk, Fuel, 2014, 117, 608–617 CrossRef CAS.
- H. J. Gallon, X. Tu and J. C. Whitehead, Plasma Processes Polym., 2012, 9, 90–97 CrossRef CAS.
- H. H. Nguyen and K. Kim, Catal. Today, 2015, 256, 88–95 CrossRef CAS.
- B. Eliasson, C. Liu and U. Kogelschatz, Ind. Eng. Chem. Res., 2000, 39, 1221–1227 CrossRef CAS.
- M. Kraus, B. Eliasson and A. Wokaun, Phys. Chem. Chem. Phys., 2001, 3, 294–300 RSC.
- J. J. Zou, Y. P. Zhang, C. J. Liu, Y. Li and B. Eliasson, Plasma Chem. Plasma Process., 2003, 23, 69–82 CrossRef CAS.
- M. Kraus, W. Egli, K. Haffner, B. Eliasson, U. Kogelschatz and A. Wokaun, Phys. Chem. Chem. Phys., 2002, 4, 668–675 RSC.
- B. B. Hwang, Y. K. Yeo and B. K. Na, Korean J. Chem. Eng., 2003, 20, 631–634 CrossRef CAS.
- V. Goujard, J.-M. Tatibouët and C. Batiot-Dupeyrat, Plasma Chem. Plasma Process., 2011, 31, 315–325 CrossRef CAS.
- R. Snoeckx, R. Aerts, X. Tu and A. Bogaerts, J. Phys. Chem. C, 2013, 117, 4957–4970 CAS.
- C. De Bie, J. van Dijk and A. Bogaerts, J. Phys. Chem. C, 2015, 119, 22331–22350 CAS.
- H. Machrafi, S. Cavadias and J. Amouroux, J. Phys.: Conf. Ser., 2011, 275, 12016 CrossRef.
- I. Istadi and N. A. S. Amin, Chem. Eng. Sci., 2007, 62, 6568–6581 CrossRef CAS.
- J. Wang, C. Liu and B. Eliassion, Energy, 2004, 153, 148–153 Search PubMed.
- J. Q. Zhang, Y. J. Yang, J. S. Zhang and Q. Liu, Acta Chim. Sin., 2002, 60, 1973–1980 CAS.
- N. Rueangjitt, C. Akarawitoo, T. Sreethawong and S. Chavadej, Plasma Chem. Plasma Process., 2007, 27, 559–576 CrossRef CAS.
- N. Rueangjitt, T. Sreethawong and S. Chavadej, Plasma Chem. Plasma Process., 2008, 28, 49–67 CrossRef CAS.
- Y. N. Chun, Y. C. Yang and K. Yoshikawa, Catal. Today, 2009, 148, 283–289 CrossRef CAS.
- Z. A. Allah and J. C. Whitehead, Catal. Today, 2015, 256, 76–79 CrossRef.
- J. L. Liu, H. W. Park, W. J. Chung, W. S. Ahn and D. W. Park, Chem. Eng. J., 2016, 285, 243–251 CrossRef CAS.
- K. Li, J. Liu, X. Li, X. Zhu and A. Zhu, Chem. Eng. J., 2016, 288, 671–679 CrossRef CAS.
- Y. N. Chun, H. W. Song, S. C. Kim and M. S. Lim, Energy Fuels, 2008, 22, 123–127 CrossRef CAS.
- C.-J. Liu, R. G. Mallinson and L. L. Lobban, Appl. Catal., A, 1999, 178, 17–27 CrossRef CAS.
- B. Zhu, X. S. Li, C. Shi, J. L. Liu, T. L. Zhao and A. M. Zhu, Int. J. Hydrogen Energy, 2012, 37, 4945–4954 CrossRef CAS.
- B. Zhu, X. Li, J. Liu and A. Zhu, Int. J. Hydrogen Energy, 2012, 37, 16916–16924 CrossRef CAS.
- X. Zhu, K. Li, J. Liu and X. Li, Int. J. Hydrogen Energy, 2014, 39, 13902–13908 CrossRef CAS.
- J. Liu, X. Li, X. Zhu, K. Li, C. Shi and A. Zhu, Chem. Eng. J., 2013, 234, 240–246 CrossRef CAS.
- K. Li, J. Liu, X. Li, X. Zhu and A. Zhu, Catal. Today, 2015, 256, 96–101 CrossRef CAS.
- O. Mutaf-yardimci, A. Saveliev, A. A. Fridman and L. A. Kennedy, Int. J. Hydrogen Energy, 1998, 23, 1109–1111 CrossRef CAS.
- P. Bruggeman and C. Leys, J. Phys. D: Appl. Phys., 2009, 42, 53001 CrossRef.
- B. R. Locke and K.-Y. Shih, Plasma Sources Sci. Technol., 2011, 20, 34006 CrossRef.
- M. A. Malik, A. Ghaffar and S. A. Malik, Plasma Sources Sci. Technol., 2001, 10, 82–91 CrossRef CAS.
- P. Sunka, V. Babický, M. Clupek, P. Lukes, M. Simek, J. Schmidt and M. Cernák, Plasma Sources Sci. Technol., 1999, 8, 258–265 CrossRef CAS.
- B. R. Locke and S. M. Thagard, Plasma Chem. Plasma Process., 2012, 32, 875–917 CrossRef CAS.
- S. Futamura and H. Kabashima, Stud. Surf. Sci. Catal., 2004, 153, 119–124 CrossRef CAS.
- R. Snoeckx, A. Ozkan, F. Reniers and A. Bogaerts, ChemSusChem, 2017, 10, 409–424 CrossRef CAS PubMed.
- S. Mahammadunnisa, E. L. Reddy, D. Ray, C. Subrahmanyam and J. C. Whitehead, Int. J. Greenhouse Gas Control, 2013, 16, 361–363 CrossRef CAS.
- T. Ihara, M. Kiboku and Y. Iriyama, Bull. Chem. Soc. Jpn., 1994, 67, 312–314 CrossRef CAS.
- T. Ihara, T. Ouro, T. Ochiai, M. Kiboku and Y. Iriyama, Bull. Chem. Soc. Jpn., 1996, 69, 241–244 CrossRef CAS.
- G. Chen, T. Silva, V. Georgieva, T. Godfroid, N. Britun, R. Snyders and M. P. Delplancke-Ogletree, Int. J. Hydrogen Energy, 2015, 40, 3789–3796 CrossRef CAS.
- G. Chen, N. Britun, T. Godfroid, V. Georgieva, R. Snyders and M.-P. Delplancke-Ogletree, J. Phys. D: Appl. Phys., 2017, 50, 84001 CrossRef.
- N. Hayashi, T. Yamakawa and S. Baba, Vacuum, 2006, 80, 1299–1304 CrossRef CAS.
- L. Guo, X. Ma, Y. Xia, X. Xiang and X. Wu, Fuel, 2015, 158, 843–847 CrossRef CAS.
- B. Eliasson, U. Kogelschatz, B. Xue and L.-M. Zhou, Ind. Eng. Chem. Res., 1998, 37, 3350–3357 CrossRef CAS.
- E. Jwa, S. B. Lee, H. W. Lee and Y. S. Mok, Fuel Process. Technol., 2013, 108, 89–93 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjær, J. S. Hummelshøj, S. Dahl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- Y. Zeng and X. Tu, IEEE Trans. Plasma Sci., 2015, 44, 1–7 Search PubMed.
- M. Nizio, A. Albarazi, S. Cavadias, J. Amouroux, M. E. Galvez and P. Da Costa, Int. J. Hydrogen Energy, 2016, 41, 11584–11592 CrossRef CAS.
- L. Maya, J. Vac. Sci. Technol., A, 2000, 18, 285–287 CAS.
- J. F. de la Fuente, S. H. Moreno, A. I. Stankiewicz and G. D. Stefanidis, Int. J. Hydrogen Energy, 2016, 1–11 Search PubMed.
- M. Kano, G. Satoh and S. Iizuka, Plasma Chem. Plasma Process., 2012, 32, 177–185 CrossRef CAS.
- C. De Bie, J. van Dijk and A. Bogaerts, J. Phys. Chem. C, 2016, 120, 25210–25224 CAS.
- S. Mori, N. Matsuura, L. L. Tun and M. Suzuki, Plasma Chem. Plasma Process., 2016, 36, 231–239 CrossRef CAS.
- F. Dimroth, M. Grave, P. Beutel, U. Fiedeler, C. Karcher, T. N. D. Tibbits, E. Oliva, G. Siefer, M. Schachtner, A. Wekkeli, A. W. Bett, R. Krause, M. Piccin, N. Blanc, C. Drazek, E. Guiot, B. Ghyselen, T. Salvetat, A. Tauzin, T. Signamarcheix, A. Dobrich, T. Hannappel and K. Schwarzburg, Prog. Photovol.: Res. Appl., 2014, 22, 277–282 CrossRef CAS.
- N. Pinhão, A. Moura, J. B. Branco and J. Neves, Int. J. Hydrogen Energy, 2016, 41, 9245–9255 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |