
Photoelectron spectroscopy and density functional theory studies of (FeS)mH− (m = 2–4) cluster anions: effects of the single hydrogen†
Shi
Yin
 and
Elliot R.
Bernstein
*
and
Elliot R.
Bernstein
*
Department of Chemistry, NSF ERC for Extreme Ultraviolet Science and Technology, Colorado State University, Fort Collins, CO 80523, USA. E-mail: erb@colostate.edu
First published on 21st November 2017
Abstract
Single hydrogen containing iron hydrosulfide cluster anions (FeS)mH− (m = 2–4) are studied by photoelectron spectroscopy (PES) at 3.492 eV (355 nm) and 4.661 eV (266 nm) photon energies, and by Density Functional Theory (DFT) calculations. The structural properties, relative energies of different spin states and isomers, and the first calculated vertical detachment energies (VDEs) of different spin states for these (FeS)mH− (m = 2–4) cluster anions are investigated at various reasonable theory levels. Two types of structural isomers are found for these (FeS)mH− (m = 2–4) clusters: (1) the single hydrogen atom bonds to a sulfur site (SH-type); and (2) the single hydrogen atom bonds to an iron site (FeH-type). Experimental and theoretical results suggest such available different SH- and FeH-type structural isomers should be considered when evaluating the properties and behavior of these single hydrogen containing iron sulfide clusters in real chemical and biological systems. Compared to their related, respective pure iron sulfur (FeS)m− clusters, the first VDE trend of the diverse type (FeS)mH0,1− (m = 1–4) clusters can be understood through (1) the different electron distribution properties of their highest singly occupied molecular orbital employing natural bond orbital analysis (NBO/HSOMO), and (2) the partial charge distribution on the NBO/HSOMO localized sites of each cluster anion. Generally, the properties of the NBO/HSOMOs play the principal role with regard to the physical and chemical properties of all the anions. The change of cluster VDE from low to high is associated with the change in nature of their NBO/HSOMO from a dipole bound and valence electron mixed character, to a valence p orbital on S, to a valence d orbital on Fe, and to a valence p orbital on Fe or an Fe–Fe delocalized valence bonding orbital. For clusters having the same properties for NBO/HSOMOs, the partial charge distributions at the NBO/HSOMO localized sites additionally affect their VDEs: a more negative or less positive localized charge distribution is correlated with a lower first VDE. The single hydrogen in these (FeS)mH− (m = 2–4) cluster anions is suggested to affect their first VDEs through the different structure types (SH- or FeH-), the nature of the NBO/HSOMOs at the local site, and the value of partial charge number at the local site of the NBO/HSOMO.
Introduction
Iron sulfur clusters are ubiquitous and evolutionarily ancient prosthetic groups that are required to sustain fundamental life processes. Interest in iron sulfur clusters is partially driven by their role as active centers of proteins,1 and their great significance in both industrial and biochemical catalysis.2–5 As has been recognized for many decades, iron sulfur clusters are employed for many purposes in biological systems.6 Owing to their remarkable structural plasticity and versatile chemical/electronic features, iron sulfur clusters participate in electron transfer, substrate binding/activation, iron/sulfur storage, regulation of gene expression, and enzyme activity. Understanding of the iron sulfur system grows with continuing and expanding investigations. Iron sulfur clusters play critical roles in the active sites of a wide variety of metalloproteins and metalloenzymes, and are involved in biological electron transfer processes,7 small molecule activation,8–10 radical based catalytic transformations,11 DNA repair,12 and signal transduction.13Additionally, hydrogen is the most widely distributed element in the world, and it is also the most abundant element in biological systems. Among various aspects of metalloprotein structure, reorganization of metal ligand(s) and/or hydrogen bonding networks around redox-active sites are often utilized in modulating natural redox potentials and functionalities.14–17 Action of sulfhydryl (SH) reagents on enzymes is both varied and complex: most of these reactions produce conformational alterations in the enzyme maintaining the iron–sulfur centers.18 The presence of a hydrogen containing moiety, such as –SH groups, which is essential for catalytic activity in dehydrogenase, has been discovered in recent decades, and has since been widely studied.19–22 Direct evidence for the hydrogen containing groups (–SH) activating enzymes and the important role they play for enzyme function, has been reported recently.23–25
Investigations of iron–sulfur systems, ranging from bare Fe–S clusters to analogous complexes and proteins, are common throughout bioinorganic chemistry.26 Iron sulfur clusters and complexes have been synthesized and characterized, forming a large class of organometallic chemistry.27 Many strategies have been developed to understand the relationship between the remarkable structures of iron sulfur clusters (i.e., Fe2S2, Fe3S4, and Fe4S4) and their associated reactivity in biological systems.28–30 Although a number of studies have been performed on gas phase cationic,31,32 neutral,33,34 and anionic35–42 iron sulfur clusters for investigation of their composition, stability, structure, and reactivity, experimental investigation of single hydrogen containing iron hydrosulfide cluster anions is not been reported to the best of our knowledge.
In this paper, we present a photoelectron spectroscopy (PES) study of a series of single hydrogen containing iron sulfide (FeS)mH− (m = 2–4) cluster anions, employing a magnetic-bottle time-of-flight (MBTOF) PES apparatus. The PES spectra of these cluster anions at 532 nm and 355 nm photon energies are reported, and the structural and bonding properties of these cluster anions not directly detected by the spectroscopic technique employed are investigated at different theoretical levels by Density Functional Theory (DFT). The most probable structures and ground state spin multiplicities of the (FeS)mH− (m = 2–4) anionic cluster series are tentatively assigned by comparing the theoretical VDEs with their experiment values: theory is pursued based on a comparison of the two sets of VDEs (experimental and theoretical) and consequently the appropriate algorithm is employed to determine cluster properties not directly addressed by PES. Values for the electron affinities of their neutral counterparts are presented and analyzed as well. The properties of these single hydrogen containing iron sulfide clusters are compared with their related pure iron sulfur cluster (for example, Fe2S2−/Fe2S2H− and Fe4S4−/Fe4S4H−), which are essential cluster sites in biological systems, to understand the effect of a single hydrogen atom perturbation on the putative (FeS)m− cluster properties.
Methods
A. Experimental
The MBTOF-PES experimental setup, consisting of a laser vaporization cluster/molecular source, an orthogonal acceleration/extraction reflectron time of flight (oaRETOF) mass spectrometer (MS), a mass gate, a momentum decelerator, and a MBTOF electron analyzer, employed in this work has been described previously in detail.43,44 Only a brief outline of the apparatus is given below. In this work, (FeS)mH− clusters are generated by laser ablation of a mixed iron/sulfur target [made by pressing a mixture of iron (99.9%, Sigma Aldrich) and sulfur (99.98%, Sigma Aldrich) powders with a ratio of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S = 20:1] in the presence of a 1% CH4 in helium carrier gas. A 10 Hz, focused, 532 nm Nd3+:YAG laser (Nd3+:yttrium aluminum garnet) with ∼3 mJ pulse−1 energy is used for the laser ablation. The expansion gas is pulsed into the vacuum by a supersonic nozzle (R. M. Jordan, Co.) with a backing pressure of typically 100 psi.
:S = 20:1] in the presence of a 1% CH4 in helium carrier gas. A 10 Hz, focused, 532 nm Nd3+:YAG laser (Nd3+:yttrium aluminum garnet) with ∼3 mJ pulse−1 energy is used for the laser ablation. The expansion gas is pulsed into the vacuum by a supersonic nozzle (R. M. Jordan, Co.) with a backing pressure of typically 100 psi.
Generated cluster anions enter the extraction region of the TOFMS/PES spectrometer through a 6 mm skimmer. Anions present in the expansion are extracted perpendicularly from the beam by pulsed voltage applied to the first extraction plate. The voltages on the extraction plates are −250 V (pulsed), 0 V, and +750 V, respectively. A liner for both anion and electron flight tube regions has the same voltage (+750 V) as the last extraction plate. Two sets of ion deflectors and one ion einzel lens are positioned downstream of the extraction plates. The anions are then analyzed by the oaRETOFMS. The photoelectron technique has the following energy conserving relationship: hν = EKE + EBE, in which hν is the photon energy, EKE is the measured electron kinetic energy, EBE is the electron binding energy. In order to obtain a photoelectron spectrum of the anions of interest, a three-grid mass gate is used for cluster and molecule anion mass selection. Following the mass gate, the mass selected ion beam enters a momentum decelerator. Both the pulse width and the pulse amplitude of the momentum decelerator can be optimized to achieve the best deceleration effect.
The mass selected and decelerated anions are exposed to different laser wavelengths (355 nm, 266 nm) at the photo-detachment region. The photo-detached electrons are energy analyzed by MBTOF-PES spectrometer. A cone shape permanent magnet is used for the high magnetic field (∼700 G) generation at the anion beam/photo detachment laser interaction region. The permanent magnet is mounted on a vacuum motor controlled, linear translation stage (Physik Instrumente LPS-24), so that the position of the permanent magnet can be optimized for the best photoelectron spectrometer resolution. The 1 m electron flight tube is surrounded by a solenoid, which is covered with two layers of GIRON magnetic shielding metal. The electric current for the solenoid is about 0.8 A, which produces a magnetic field of ∼10 gauss at the center of the flight tube. The photo-detached electrons pass through the flight tube and are detected by a microchannel plate (MCP) detector. A resolution of ∼4% (i.e., 40 meV/1.00 eV electron kinetic energy) for the overall MBTOF PES apparatus can be achieved. Under the above operating conditions, PES resolution is no longer limited by Doppler broadening associated with the perpendicular motion of the ion beam with respect to the collection and flight path of the photodetached electrons: in other words, momentum deceleration for the ion cluster beam is efficacious. PES spectra are collected and calibrated at this resolution with known spectra of Cu−.45
B. Theoretical
All calculations are performed using the Gaussian 09 program package.46 The structures of (FeS)mH− (m = 2–4) clusters are optimized for different isomers and spin multiplicities using DFT without constraints. For each cluster, different initial structures are employed as input in the optimization procedure. For each structure, spin multiplicities are scanned from low to high. All relative energies are zero point energy corrected. Vibrational frequency calculations are further performed to confirm global minima, which have zero imaginary frequency. The Perdew–Wang47 correlation functional (BPW91) and the B3LYP functional,48,49 combined with the triple-ζ valence plus polarization (TZVP),50 6-311+G(d),51–53 and aug-cc-PV5Z54 basis sets, which are proved to have good performance in previous studies of iron sulfur and iron hydrosulfide clusters,19,42 are employed to explore description of these (FeS)mH− (m = 2–4) clusters. All calculations are treated in a spin-unrestricted manner.In this approach, for each spin state of the (FeS)mH− anions, the first vertical detachment energy (VDE = Eneutralatoptimizedaniongeometry − Eoptimizedanion) is calculated as the lowest transition from the spin state (M) of the anion into the final lowest spin state (M + 1 or M − 1, M = 2S + 1) of the neutral (FeS)mH species at the geometry optimized for the anion. The optimized anion geometries are used for the further calculations of the photoelectron spectra using time-dependent density functional theory (TDDFT).55 Vertical excitation energies of the neutral species are added to the first VDE to obtain the second and higher VDEs of these (FeS)mH− anion clusters. The outer valence Green function method (OVGF/TZVP)56 is also used to calculate the second and higher VDEs. Calculated VDEs for each spin state of each (FeS)mH− (m = 2–4) cluster are compared with experimental results to determine the appropriate theoretical algorithm for determination of cluster structures, charge distributions, wave functions, and other anion and neutral cluster properties.
An NBO analysis is an often employed orbital (wave function) localization and population analysis method to help understand the electron distribution in a molecule or cluster around particular sites or moieties of interest. Within this method, natural atomic orbitals (NAOs), determined for the particular species under consideration, are evaluated and employed: NAOs are the effective orbitals of an atom in the particular molecular environment (rather than for isolated atoms). NAOs are also the maximum occupancy orbitals. Information obtained from an NBO analysis, such as partial charges and HOMO–LUMO orbitals, is reported to explain, for example, a number of experimental phenomena of gas phase 1-butyl-3-methylimidazolium chloride ion pairs.57 The NBO calculations in this work are performed using the NBO 3.1 program as implemented in the Gaussian 09 package.
Partial charge distributions of cluster anions studied in this work are calculated using Mulliken population analysis, Breneman's CHELPG algorithm,58 and an NBO analysis as implemented in Gaussian 09. CHELPG employs a grid method to select a point at which the electrostatic potential of the molecule is sampled. Fe is assigned a van der Waals radius of 1.95 Å59 and default atomic radii are used for all other atoms for the CHELPG method.
Results and discussion
A. General results for the (FeS)2–4H− clusters
The obtained PE spectra for the (FeS)2–4H− cluster anions at different photon energies are shown in Fig. 1–3, respectively. The first VDE, proving important in establishing a cluster's electronic and geometric structure, is derived from the energy of the first peak maxima in the photoelectron spectra. The VDEs are the energies of maximum overlap between the nuclear wave functions of the ground states of the anions and their respective neutrals. Broad peaks containing no reproducible fine structure are observed in the spectra of (FeS)2–4H− clusters probably due to hot band related features, multiple populated spin states, effects of vibrational anharmonicity, and geometry differences between anions and neutrals. The spin multiplicity for the ground state of a cluster anion (FeS)mH− (m = 2–4) is assigned mainly based on agreement of the first calculated VDEs for the different spin multiplicities compared to the experimental values. The relative energy differences (ΔE) between the spin states under consideration are also evaluated and employed to generate the assignment. The details of assignments of (FeS)2–4H− cluster spectra will be described and discussed in following sections, respectively. | ||
| Fig. 1 Photoelectron spectra of (FeS)2H− cluster anions at 355 nm and 266 nm. X and X′ label the ground state transition peaks of isomer I and II, respectively (presented in Fig. 4). The identifier A in (b) labels the first low-lying excited electronic state transition peak associated with isomer I (X). The downward pointing arrow (↓) in the figure indicates the value of the assigned EA (the slope of the first onset is linearly extrapolated to the base line of signal to assign the experimental EA). | ||
 | ||
| Fig. 2 Photoelectron spectra of (FeS)3H− cluster anions at 355 nm and 266 nm. X, X′′ and X′ label the ground state transition peaks of isomer I, II and III, respectively, presented in Fig. 4. The downward pointing arrow (↓) in the figure indicates the value of the assigned EA (the slope of the first onset is linearly extrapolated to the base line of signal to assign the experimental EA). | ||
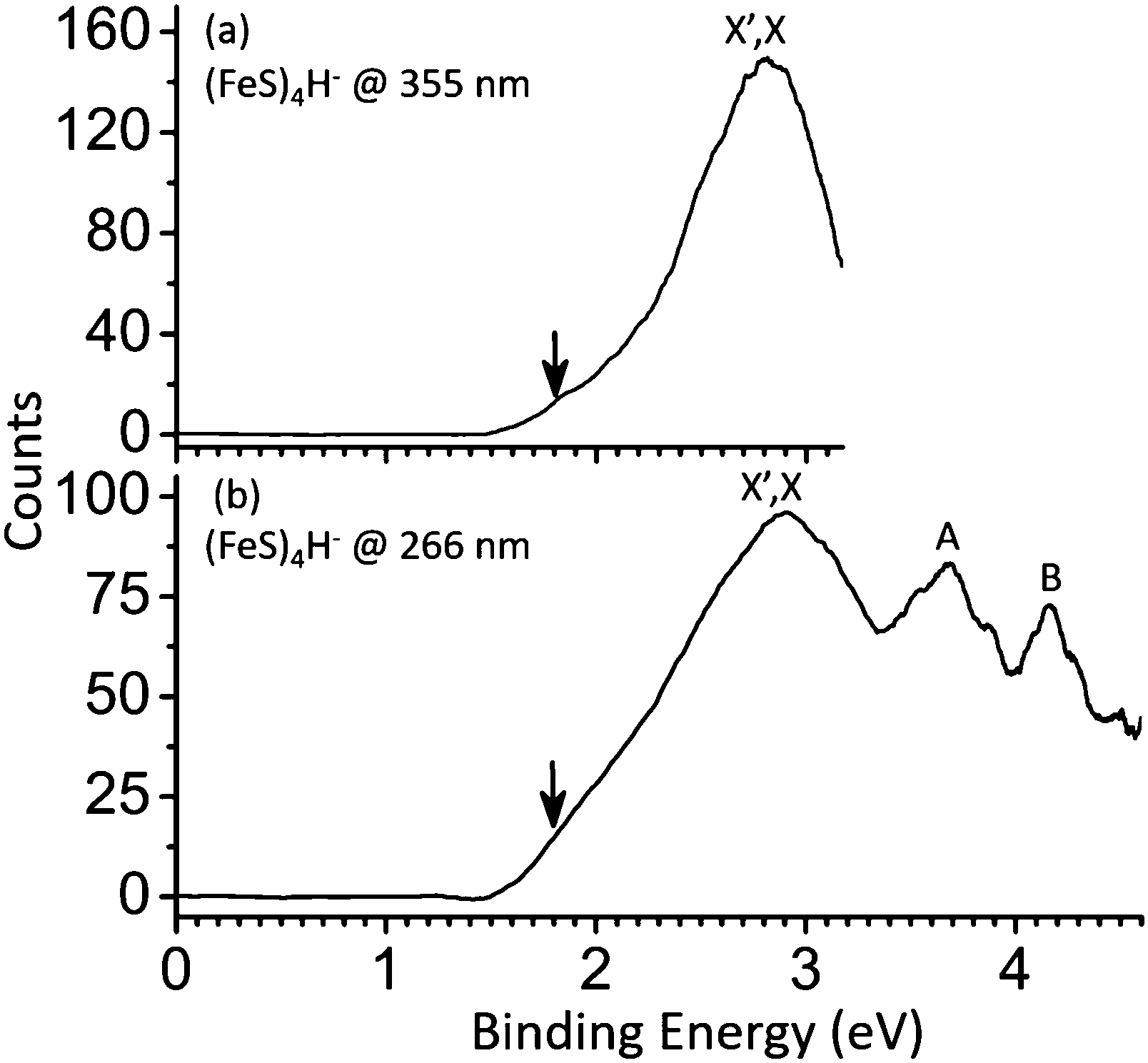 | ||
| Fig. 3 Photoelectron spectra of (FeS)4H− cluster anions at 355 nm and 266 nm. X and X′ label the ground state transition peaks of isomer I and II, respectively, presented in Fig. 4 respectively. A and B label the first and second low-lying transition peaks at high VDE. The downward pointing arrow (↓) in the figure indicates the value of the assigned EA (the slope of the first onset is linearly extrapolated to the base line of signal to assign the experimental EA). | ||
Cluster geometrical structure is an important determination for all cluster, since this cluster property is the basis for the description of all other cluster characteristics (e.g., spin, electronic structure, electron density, charge and spin densities, etc.). Various structural isomers of (FeS)2–4H− are investigated, and different spin multiplicities from low to high are considered for each isomer. ΔE between different structural isomers with different spin multiplicities are calculated and compared to evaluate their relative stability. To evaluate and pursue a good theoretical method to study these (FeS)2–4H− cluster anions, different reasonable functionals (B3LYP and BPW91) coupled with different basis sets (TZVP, 6-311+G(d), and aug-cc-PVQZ) are selected to calculate ΔEs and the first VDE of the (FeS)2H− structural isomers with different spin multiplicities as a test. The two lowest relative energy spin multiplicity states of each isomer are presented in Table 1, and the first VDE for each isomer is calculated. ΔEs for different spin multiplicities are corrected for zero point energies. ΔE = 0.00 eV means that the energy of the given spin multiplicity isomer is the lowest one among all possible spin multiplicities of all possible structural isomers.
| (FeS)2H− | Spin (M) | B3lyp (eV) | BPW91 (eV) | Exp. VDE (eV) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| TZVP | 6-311++G(d,p) | aug-cc-PVQZ | Avg. | TZVP | 6-311++G(d,p) | aug-cc-PVQZ | Avg. | ||||
Isomer II

|
7 | ΔE | 1.06 | 1.21 | 0.66 | 0.89 | 0.85 | 0.85 | 1.85 | ||
| VDE | 1.61 | 1.76 | 2.15 | 1.84 | 1.76 | 2.14 | 2.12 | 2.01 | |||
| 9 | ΔE | 0.71 | 0.51 | 0.44 | 0.99 | 0.58 | 0.56 | ||||
| VDE | 2.59 | 2.28 | 2.17 | 2.35 | 1.70 | 1.91 | 1.88 | 1.83 | |||
Isomer I
|
7 | ΔE | 0.77 | 0.66 | 0.51 | 0.30 | 0.25 | 0.19 | 2.84 | ||
| VDE | 2.45 | 2.68 | 2.64 | 2.59 | 2.49 | 2.69 | 2.70 | 2.63 | |||
| 9 | ΔE | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | ||||
| VDE | 3.08 | 3.18 | 3.14 | 3.13 | 2.75 | 2.92 | 2.85 | 2.84 | |||
Calculational results for (FeS)2H−, obtained employing the BPW91 functional coupled with different basis sets (Table 1) show 1. the spin state M = 9 is the lowest energy state for isomer I, 2. the lowest energy spin state for (FeS)2H− isomer II is also M = 9, and 3. the calculated first VDEs of isomer I (M = 9) and isomer II (M = 9) are within ∼0.2 eV of each other employing BPW91 and any of selected basis sets. This performance suggests that the calculated properties of (FeS)2H− cluster anion are insensitive to the employed basis sets, so the average ΔE and first VDE values for the different basis sets are given in Table 1 for discussion convenience. Calculation results employing B3LYP functional coupled with different basis sets are obtained for comparison with these. The results displayed in Table 1 agree with BPW91 results discussed above: (1) the spin state M = 9 is the lowest energy state for isomer I; (2) the lowest energy spin state for (FeS)2H− isomer II is also M = 9; and (3) the calculated results obtained with the B3LYP DFT functional are insensitive to the employed basis sets. The calculated VDEs for spin state M = 9 of isomers I and II (3.13 eV and 2.35 eV, average values) employing a B3LYP functional are higher than those of M = 9 for isomers I and II (2.84 eV and 1.83 eV, average values) obtained using a BPW91 functional. The BPW91 functional results are in better agreement with the experimental results (2.84 eV and 1.85 eV, see Fig. 1 and Table 1), although reasonable assignments are obtained based on the B3LYP functional results as well.
In summary, comparing calculated VDEs with respect to experimental measurements for (FeS)2H− clusters at a number of different DFT levels, two generalizations can be extracted: (1) calculated properties are insensitive to the basis sets employed; and (2) the BPW91 density functional performs better than the B3LYP density functional for these VDE calculations. Therefore, the BPW91 functional is suggested for the theoretical studies for these single hydrogen containing iron hydrosulfide clusters, and it is thereby selected for calculations employed in the following sections relating with (FeS)3,4H0/−1 cluster anions and neutrals. ΔE between different structural isomers with different spin multiplicities for (FeS)2–4H− cluster anions at BPW91/TZVP level are displayed in Fig. S1 to S3 in the ESI,† respectively.
To estimate the theoretical first VDEs of these open shell (FeS)mH− (m = 2–4) cluster anions (with unpaired electrons), one electron is removed from the highest singly occupied molecular orbital (HSOMO) of the (FeS)mH− cluster with the optimized geometry. Therefore, studies of HSOMOs properties of these (FeS)mH− clusters are helpful to understand their different first VDEs. Furthermore, partial atomic charges are suggested to play a decisive role in determining core electron binding energy in small molecules.60 The partial charge of the HSOMO localized site in these iron hydrosulfide cluster anions may to some extent affect the energy (VDE) required to remove an electron from such clusters through a “charge effect”: a small negative charge number of the site means less electron density distribution on that site, and therefore removal of an electron from that site may require more energy than otherwise estimated based simply on the NBO/HSOMO distribution for that site. Removal of an electron from a site with a positive charge should require still more energy than removal of an electron from a site with a negative charge, and removal of an electron from a site with a large positive charge may require even more energy. With the above ideas, potential effects of the HSOMOs properties and partial atomic charges on HSOMO localized sites to the first VDEs of the iron sulfide/hydrosulfide cluster anions are discussed in following Sections B to E of the Results and discussion.
In Fig. 1–3, the adiabatic electron affinity (EA = Eoptimizedneutral − Eoptimizedanion) for ground state (FeS)2–4H neutral cluster can be estimated. To assign the experimental EA, the slope of the first onset is linearly extrapolated to the base line of signal. The values of EA are indicated by a downward pointing arrow in the figures. EA for ground state of (FeS)2–4H neutral clusters are calculated and summarized in Table 2. Their experimental EAs are also listed there for comparison. Note that the obtained experimental EAs of these iron hydrosulfide clusters are possibly affected by their anion vibrational hot bands due to their high temperature generation conditions (∼3 mJ pulse−1 ablation laser energy) and potential transition state barriers. Thus, experimental EAs might be underestimated in this work.
| Cluster | Spin multiplicity | EA (eV) | |
|---|---|---|---|
| Calculated | Experimental | ||
| (FeS)2H | 8 | 1.58 (isomer II) | 1.3 |
| (FeS)3H | 12 | 2.27 (isomer III) | 1.5 |
| (FeS)4H |
16
8 |
2.35 (isomer II)
2.39 (isomer I) |
1.8 |
B. Photoelectron spectra and DFT studies of the (FeS)2H− cluster anions
The obtained PE spectra for the (FeS)2H− cluster anion at different photon energies are shown in Fig. 1. As the PE spectra of (FeS)2H− clusters shown in Fig. 1a, two features are observed at 355 nm, and their measured VDEs are 1.85 (X′) and 2.84 (X) eV, respectively. One broader peak is observed for the higher transition labeled feature A (3.59 eV) at 266 nm photon energy (Fig. 1b). | ||
| Fig. 4 DFT optimized structures of (FeS)mH− (m = 2–4) at the BPW91/TZVP level. The lowest relative energy spin state geometry of each isomer is displayed in this figure. Geometries of other spin states for each cluster are generally similar to the one shown but with slightly different bond lengths and angles. Bond lengths (in angstroms), relative energies for each isomer (in kcal mol−1), point group symmetry, and spin multiplicity M [M(FeS)mH−] are indicated below the structures. | ||
The calculated first VDEs of spin state M = 7 of isomer I (2.63 eV, average value) and M = 7 isomer II (2.01 eV, average value) are also close to the experimental VDEs associated with features X (2.84 eV) and X′ (1.85 eV). Their average ΔEs are calculated to be lower than 1 eV, so spin state M = 7 of isomers I and II of (FeS)2H− clusters probably also exist in the experimental system, and should be considered when evaluating the properties and behavior of these iron hydrosulfide clusters in real chemical and biological systems.
The intensity of X′ (isomer II, SH-type) peak is lower than that of X (isomer I, FeH-type) peak, as displayed in Fig. 1 (see structures of isomers in Fig. 4a). This may be due to the higher relative energy of isomer II (M = 9) than that of isomer I (M = 9) (see Table 1): the higher ΔE implies a less stable state and a lower population of the isomer II than isomer I in the experiment cooled molecular beam. Nonetheless, both isomers I and II exist as structural isomers for the (FeS)2H− cluster anion. The single hydrogen of isomer II is bonded to the sulfur site (SH-type), and the single hydrogen of isomer I is bonded to the iron site (FeH-type). Their first VDEs are very different, however. The experimental first VDE of the FeH-type (FeS)2H− (isomer I) is 2.84 eV (X peak in Fig. 1), about 1 eV higher than that of SH-type (FeS)2H− (isomer II), which is 1.85 eV (X′ peak in Fig. 1). From our previous study of the (FeS)m− species,61 the experimental first VDE of the (FeS)2− cluster is observed to be 2.34 eV (see Table S1 in ESI†). Different bonding sites for the single hydrogen in the (FeS)2H− cluster have significantly different effects on its first VDE. The experimental first VDE of FeH-type (FeS)2H− is 0.5 eV higher, and the experimental first VDE of SH-type (FeS)2H− is 0.49 eV lower than that of the (FeS)2− cluster, due to the different bonding types and sites for the single H addition to a pure iron sulfur (FeS)2− cluster.
 | ||
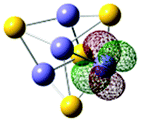
| Fig. 5 NBO plots showing the highest singly occupied molecular orbital (HSOMO) of (FeS)mH0,1− (m = 2–4) cluster anions. The spin multiplicity (M) is listed as MFemSm−. The Mulliken charges for important atoms are given in the figure. Only the lowest energy spin states of each isomer are displayed. | ||
Mulliken charge distributions are calculated for the FeH-type (isomer I) of (FeS)2H−, and the (FeS)2− cluster anions (whose NBO/HSOMOs are of a similar nature) to explore the “charge effect” (proposed in Section A) for their different first VDEs. The partial charge numbers of the S sites, on which the NBO/HSOMO is localized, are −0.635 for the (FeS)2− cluster and −0.553 for the FeH-type (FeS)2H− (isomer I) cluster (see Fig. 5b). Since the S atom is more electronegative than the iron atom, the negative charge number at the S site in these iron sulfide/hydrosulfide clusters is not unreasonable. The observed higher first VDE for the FeH-type (FeS)2H− is consistent with the smaller negative charge number of the S site, on which the NBO/HSOMO is localized (−0.553), compared to that of the (FeS)2− cluster (−0.635).
| Cluster | Observed feature | Experimental VDEs (eV) | Calculated VDEs (eV) | |
|---|---|---|---|---|
| TDDFT | OVGF | |||
| (FeS)2H− | X′ | 1.85 | 1.70 (isomer II, M = 9) | |
| X | 2.84 | 2.75 (isomer I, M = 9) | ||
| A | 3.59 | 3.26 (isomer I, M = 9) | 4.31 (isomer I, M = 9) | |
| (FeS)3H− | X′ | 2.38 | 2.43 (isomer III, M = 11) | |
| X,X′′ | 3.33 |
3.14 (isomer II, M = 9)
3.00 (isomer I, M = 5) |
||
| (FeS)4H− | X′,X | 2.90 |
2.59 (isomer II, M = 15)
2.62 (isomer I, M = 9) |
|
| A | 3.70 |
3.18 (isomer II, M = 15)
3.19 (isomer I, M = 9) |
3.40 (isomer II, M = 15)
3.39 (isomer I, M = 9) |
|
| B | 4.17 |
3.38 (isomer II, M = 15)
3.44 (isomer I, M = 9) |
4.28 (isomer II, M = 15)
3.80 (isomer I, M = 9) |
|
In summary, two types of structural isomers (SH-type, II and FeH-type, I) are found for the (FeS)2H− cluster. Comparing properties of these isomers with the related pure iron sulfur cluster anion, the first VDE of the SH-type (FeS)2H− (I) is found to be ∼0.5 eV lower, and the first VDE of FeH-type (FeS)2H− (II) is found to be ∼0.5 eV higher than that of the (FeS)2− cluster anion (2.34 eV). Such available different SH- and FeH-type structural isomers need to be considered when evaluating the properties and behavior of single hydrogen containing iron hydrosulfide clusters in real chemical and biological systems. Compared with their relatively pure iron sulfur (FeS)2− cluster, the single hydrogen atoms in (FeS)2H− cluster anions probably affect its first VDEs through the different structure types, the changing nature of the NBO/HSOMOs, and differing partial charge numbers of the important sites.
C. Photoelectron spectra and DFT studies of the (FeS)3H− cluster anions
In Fig. 2, the obtained PE spectra for (FeS)3H− clusters at different photon energies are presented. Only one feature is observed at 355 nm, and its measured VDE is 2.38 eV (X′ peak in Fig. 2a). The next broad peak (X,X′′ peak, VDE = 3.33 eV) is observed at 266 nm (Fig. 2b). A BPW91 functional coupled with TZVP and 6-311+G(d) basis sets are adopted to calculate ΔEs and the first VDEs of different (FeS)3H− structural isomers with different spin multiplicities (see Fig. S2 in the ESI† for other different spin state energies of various (FeS)3H− species).| (FeS)3H− | Spin (M) | BPW91 (eV) | Exp. VDE (eV) | ||
|---|---|---|---|---|---|
| TZVP | 6-311++G(d,p) | ||||
Isomer III

|
9 | ΔE | 0.37 | 0.19 | 2.38 |
| VDE | 2.40 | 2.65 | |||
| 11 | ΔE | 0.31 | 0.12 | ||
| VDE | 2.43 | 2.67 | |||
Isomer II

|
3 | ΔE | 0.39 | 0.35 | 3.33 |
| VDE | 3.29 | 3.31 | |||
| 9 | ΔE | 0.12 | 0.06 | ||
| VDE | 3.14 | 3.29 | |||
| 11 | ΔE | 0.21 | 0.16 | ||
| VDE | 3.03 | 3.20 | |||
| 13 | ΔE | 0.58 | 0.57 | ||
| VDE | 2.93 | 3.03 | |||
Isomer I

|
3 | ΔE | 0.17 | 0.09 | 3.33 |
| VDE | 2.93 | 3.07 | |||
| 5 | ΔE | 0.00 | 0.08 | ||
| VDE | 3.00 | 2.87 | |||
| 9 | ΔE | 0.09 | 0.01 | ||
| VDE | 2.82 | 3.03 | |||
| 11 | ΔE | 0.04 | 0.00 | ||
| VDE | 2.97 | 3.16 | |||
The experimental first VDE of FeH-type (FeS)3H− (isomer II) and Fe3H-type (FeS)3H− (isomer I) is 3.33 eV (not distinguishable, as X,X′′ peak displayed in Fig. 2), about 1 eV higher than that of the SH-type (FeS)3H− (isomer III), which is 2.38 eV (X′ peak in Fig. 2). The experimental first VDE of (FeS)3− cluster is observed to be 3.57 eV in our previous study (see Table S1 in ESI†61). We found the experimental first VDE of SH-type (FeS)3H− is ∼1.2 eV, much lower than that of the (FeS)3− cluster, while the experimental first VDEs of the FeH-type and Fe3H-type (FeS)3H− are close to that of (FeS)3− cluster.
Mulliken charge distributions are considered for these cluster anions (see Fig. 5c), for which the nature of NBO/HSOMOs are the same, to explore the “charge effect” for their first VDEs. The charge number of the mainly NBO/HSOMO localized Fe site is 0.175 for the (FeS)3− cluster, which is close to that for the FeH-type (isomer II) (FeS)3H− cluster (0.180). Their close partial charge number on Fe sites can relate to their close VDEs. These results suggest that the “charge effect” proposed in Section A still make sense.
In sum, the first VDE difference between (FeS)3H− and (FeS)3− noted can be related to the different electron distribution (wave function) properties of NBO/HSOMOs of each cluster anion. Of particular note is the changing nature of their HSOMO from a p orbital on an Fe atom to a p orbital on an S atom, due to the SH-type bonding single hydrogen. Thus, these evolving NBO representations of the HSOMO electronic distribution for (FeS)3H− compared with that of (FeS)3− clusters are quite reasonable, and correlate and explicate the observed different effect to anion cluster first VDEs with various hydrogen bonding types.
D. Photoelectron spectra and DFT studies of the (FeS)4H− cluster anion
The PE spectra for (FeS)4H− clusters at different photon energies are obtained and presented in Fig. 3. One broad peak is observed by 355 nm, and its measured VDE is 2.90 eV (X′,X peak in Fig. 3a). The next two broad peaks (A peak, VDE = 3.70 eV; and B peak, VDE = 4.17 eV) are observed at 266 nm (Fig. 2b). BPW91 functional coupled with TZVP and 6-311+G(d) basis sets are employed to calculate ΔEs and the first VDEs of different (FeS)4H− structural isomers with different spin multiplicities.| (FeS)4H− | Spin (M) | BPW91 (eV) | Exp. VDE (eV) | ||
|---|---|---|---|---|---|
| TZVP | 6-311++G(d,p) | ||||
Isomer II

|
15 | ΔE | 0.31 | 0.24 | 2.90 |
| VDE | 2.59 | 2.78 | |||
Isomer I

|
3 | ΔE | 0.27 | 0.22 | 2.90 |
| VDE | 2.43 | 2.72 | |||
| 9 | ΔE | 0.00 | 0.00 | ||
| VDE | 2.62 | 2.76 | |||
| 13 | ΔE | 0.37 | 0.35 | ||
| VDE | 2.84 | 2.98 | |||
| 15 | ΔE | 0.14 | 0.13 | ||
| VDE | 2.93 | 3.10 | |||
| 17 | ΔE | 0.36 | 0.43 | ||
| VDE | 2.79 | 2.94 | |||
Therefore, BPW91/TZVP results is selected for discussions in below. The spin state M = 9 isomer I of (FeS)4H− is suggested to be the ground state for this cluster due to its lowest ΔE: its calculated first VDE is 2.62 eV, which is in good agreement with the experimental value of X′,X labeled feature (2.90 eV) in Fig. 3. For the other spin states and isomers listed in Table 5, ΔEs are lower than 0.37 eV, and calculated first VDEs are in the range 2.43 eV to 2.93 eV. These states can probably exist in the experimental system, and can thereby contribute to the PES spectra of the (FeS)4H− cluster anions. Such a spin/isomer mixture should be considered when evaluating (FeS)4H properties and behavior in real chemical and biological systems.
The calculated first VDEs for the SH-type (FeS)4H− (isomer II) and the FeH-type (FeS)4H− (isomer I) are close, within ∼0.3 eV (see Table 5), so the broad peak observed at ∼2.90 eV (labeled, X′,X for both isomers in Fig. 3) is assigned to the ground state transition of both SH-type and FeH-type (FeS)4H− clusters.
61). To understand this behavior, NBOs are calculated for SH-type (isomer II), FeH-type (isomer I) (FeS)4H−, and (FeS)4− cluster anions (see Fig. 5d). As might be anticipated, the NBO/HSOMOs of SH-type (isomer II), FeH-type (isomer I) (FeS)4H−, and (FeS)4− are similar: localized d orbitals of Fe sites. These electron wave function plots enable such VDE values to be explained and understood. The similar first VDEs for (FeS)4H− and (FeS)4− can be associated with the properties of their NBO/HSOMOs: the (FeS)4H− and (FeS)4− clusters have the same electron distribution of their NBO/HSOMOs as a d orbital on Fe site. Note that this discussion of orbital nature for the two different cluster anions not only explains the cluster VDEs similarities observed, but also evaluates the importance and applicability of the NBO/HSOMO localization.
Mulliken charge distributions are also investigated for these (FeS)4H− and (FeS)4− cluster anions (Fig. 5d) to test if the “charge effect” discussed above is still valid or useful for these larger clusters even though their similar first VDEs are already well described and understood to be related to the similar electron distribution properties of their NBO/HSOMOs. The charge numbers of the Fe sites, on which the NBO/HSOMO is localized, are 0.247 for the (FeS)4− cluster, 0.111 for the SH-type (FeS)4H− (isomer II) cluster, and 0.185 for the FeH-type (FeS)4H− (isomer I) cluster. The first VDEs of these three cluster anions are similar (within 0.2 eV) and well understood based on the NBO/HSOMOs discussed above. Thus, the effect of different partial charge values at the respective Fe sites do not appear to be a major contributing factor for the observed first VDE similarity in this instance.
In summary, the first VDEs of SH-type (isomer II) and the FeH-type (isomer I) (FeS)4H− clusters are found to be the same (X′,X peak at ∼2.90 eV in Fig. 3), and close to that of the (FeS)4− cluster. The similar first VDEs of (FeS)4H0,1− clusters are suggested to be mainly related to their indistinguishable NBO/HSOMOs properties (a d orbital on Fe site), and the “charge effect” is found not to be a major contributing factor for the observed close first VDEs of these large iron sulfide/hydrosulfide cluster anions.
E. Comparison of (FeS)1–4H0,1− cluster anions: VDEs vs. NBO/HSOMOs and the “charge effect”
This section provides a further exploration of the correlation of the NBO/HSOMO detailed NAO compositions and properties and the partial charge numbers (employing various calculation methods) of the NBO/HSOMO localized site, in particular as they relate to the first VDEs of iron sulfide/hydrosulfide cluster anions, (FeS)mH0,1− (m = 1–4). The trend of experimental VDEs of (FeS)mH0,1− (m = 1–4) cluster anions as a function of number m is plotted in Fig. 6. Detailed atomic hybrid orbital composition of the NBO/HSOMOs, partial charges (Mulliken, CHELPG, and NBO charge) of the NBO/HSOMOs localized sites, and experimental and calculated first VDEs for these cluster anions are summarized and listed in Table 6 for comparison. The first VDEs for (FeS)m− (m = 1–4) and FeSH− are taken from our previously published work.43,61 The red line in Fig. 6 shows that the experimental first VDEs for SH-type (FeS)mH− (m = 1–4) clusters are observed to increase as cluster size m increases. The experimental first VDEs for FeH-type (FeS)mH− (m = 2–4) clusters are emphasized by the blue line, and for pure iron sulfur (FeS)m− (m = 1–4) clusters are emphasized by the black line. Five general categories of NBO/HSOMOs are identified by calculations for (FeS)mH0,1− (m = 1–4) cluster anions: (1) a dipole bound and valence electron mixed character orbital (“DB&V”); (2) a valence p orbital on S (“p on S”); (3) a valence d orbital on Fe (“d on Fe”); (4) a valence p orbital on Fe (“p on Fe”); and (5) an Fe–Fe delocalized valence bonding orbital (“Fe–Fe BD”). These are highlighted in different color areas in Fig. 6, and grouped in Table 6. Based on results and conclusions discussed in above sections, understanding of the first VDE trends of these SH-type, FeH-type (FeS)mH− and (FeS)m− cluster anions can be expected from NBO/HSOMO properties and the “charge effect” proposed in Section A. | ||
| Fig. 6 Observed experimental electron binding energies (the first VDEs) of (FeS)mH0,1− (m = 1–4) as a function of number m. The first VDE for (FeS)m− (m = 1–4) and FeSH− are taken from ref. 43 and 61. DB&V: a dipole bound and valence electron mixed character orbital; p on S: a valence p orbital on S; d on Fe: a valence d orbital on Fe; and Fe–Fe BD: an Fe–Fe delocalized valence bonding orbital. | ||
| Group based on HSOMO property | HSOMO plot for cluster | Atomic hybrid orbital composition of HSOMO | Atom localized | Partial charge (BPW91/TZVP) | VDEcal. (eV) | VDEexp. (eV) | |||
|---|---|---|---|---|---|---|---|---|---|
| Mulliken | CHELPG | NBO | |||||||
| a The number in parentheses shows percentage of s-character, p-character, and d-character of each hybrid orbital.2CB: 2-center bond. LP: 1-center valence lone pair. DB&V: a dipole bound and valence electron mixed character orbital. p on S: a valence p orbital on S. d on Fe: a valence d orbital on Fe. p on Fe: a valence p orbital on Fe. Fe–Fe BD: an Fe–Fe delocalized valence bonding orbital. | |||||||||
| DB&V |
5FeSH−

|
s (72.07%)
p (5.26%) d (22.67%) |
Fe (LP) | −0.503 | −0.509 | −0.503 | 1.13 | 1.63 | |
9(FeS)2H− isomer II

|
s (80.76%)
p (6.66%) d (12.58%) |
Fe (LP) | −0.232 | −0.241 | 0.048 | 1.70 | 1.85 | ||
| p on S |
4FeS−

|
s (0.00%)
p (99.98%) d (0.02%) |
S (LP) | −0.751 | −0.815 | −0.899 | 1.12 | 1.85 | |
8(FeS)2−

|
s (0.00%)
p (99.98%) d (0.02%) |
S (LP) | −0.635 | −0.744 | −0.823 | 2.04 | 2.34 | ||
11(FeS)3H− isomer III

|
s (0.27%)
p (99.71%) d (0.02%) |
S (LP) | −0.573 | −0.669 | −0.695 | 2.43 | 2.38 | ||
9(FeS)2H− isomer I

|
s (0.79%)
p (99.19%) d (0.02%) |
S (LP) | −0.553 | −0.667 | −0.693 | 2.75 | 2.84 | ||
| d on Fe |
15(FeS)4H− isomer II

|
s (0.00%)
p (0.16%) d (99.84%) |
Fe (LP) | 0.111 | 0.256 | 0.281 | 2.59 | 2.90 | |
9(FeS)4H− isomer I

|
s (3.75%)
p (0.63%) d (95.62%) |
Fe (LP) | 0.185 | 0.369 | 0.171 | 2.62 | 2.90 | ||
16(FeS)4−
|
s (0.00%)
p (0.11%) d (99.89%) |
Fe (LP) | 0.247 | 0.462 | 0.363 | 2.87 | 2.71 | ||
| p on Fe or Fe–Fe BD |
10(FeS)3−

|
s (0.00%)
p (93.36%) d (6.64%) |
Fe (LP) | 0.175 | 0.232 | 0.253 | 3.24 | 3.57 | |
9(FeS)3H− isomer II

|
s (0.04%)
p (93.41%) d (6.55%) |
Fe (LP) | 0.180 | 0.236 | 0.311 | 3.14 | 3.33 | ||
5(FeS)3H− isomer I

|
s (6.01%)
p (48.43%) d (45.55%) |
s (14.73%)
p (14.52%) d (70.75%) |
Fe–Fe (2CB) | 0.131–0.121 | 0.304–0.235 | 0.067–0.089 | 3.00 | 3.33 | |
The NBO/HSOMOs of 4FeS−, 8(FeS)2−, 11(FeS)3H− (isomer III, SH-type), and 9(FeS)2H− (isomer I, FeH-type) clusters are localized on S sites with a valence lone pair orbital: their compositions are similar to large p orbital components (>99%) to the NBO/HSOMO wave functions “p on S”. Their sulfur p orbital localized NBO/HSOMOs are probably responsible for their low first VDEs, EAs, and the high reactivity of their respective neutral clusters, associated with a “sulfur radical” electronic nature.34,43 The experimental first VDEs of these four clusters are in the range from 1.85 to 2.84 eV (yellow area in Fig. 6), which are higher than those of 5FeSH− and 9(FeS)2H− (isomer II, SH) clusters (with “DB&V” NBO/HSOMOs).
For 16(FeS)4−, 15(FeS)4H− (isomer II), and 9(FeS)4H− (isomer I, FeH) clusters, their NBO/HSOMOs are localized on Fe sites as a valence lone pair orbital with large d orbital components to the orbital wave functions “d on Fe”, and their experimental first VDEs are 2.71 and 2.90 eV (blue area in Fig. 6). These latter first VDE values are even higher than those for most of the clusters with “p on S” NBO/HSOMOs [4FeS−, 8(FeS)2−, 11(FeS)3H− (isomer III), and except 9(FeS)2H− (isomer I)].
The experimental first VDE of 10(FeS)3− is found to be the highest (3.57 eV, see green area in Fig. 6) among all (FeS)mH0,1− (m = 1–4) clusters. Its NBO/HSOMOs is localized on Fe sites as a valence lone pair orbital with large NAO p orbital components (93.36%) to the NBO wavefunctions “p on Fe”. The calculated first VDEs of 9(FeS)3H− (isomer II, FeH) and 5(FeS)3H− (isomer I, FeHFe) are similar, 3.14 and 3.00 eV, respectively. One broad PES peak is observed at ∼3.33 eV and assigned for their ground state transitions. The NBO/HSOMO nature of 9(FeS)3H− (isomer II) is the same as that for 10(FeS)3−: “p on Fe”, while the NBO/HSOMO nature of 5(FeS)3H− (isomer I) is a two Fe center bonding orbital “Fe–Fe BD”. These results suggest that the first VDEs of clusters with “p on Fe” and “Fe–Fe BD” types NBO/HSOMOs may be close, and are the highest among those of all discussed iron sulfide and single hydrogen containing iron hydrosulfide cluster anions.
In sum, the properties of the NBO/HSOMOs are found to play a key role with regard to the properties of all the anions discussed in this work. The change of cluster first VDE from low to high is related with the changing nature of their NBO/HSOMO from a “DB&V” orbital, to a “p on S” orbital, to a “d on Fe” orbital, and to a “p on Fe” orbital or “Fe–Fe BD” orbital.
For the “p on S” NBO/HSOMO group clusters, the order of Mulliken, CHELPG, and NBO charge numbers on S sites of 4FeS−, 8(FeS)2−, 11(FeS)3H− (isomer III), and 9(FeS)2H− (isomer I) clusters is the same. For example, the CHELPG charge on the S site of the 4FeS− cluster is −0.815, of the 8(FeS)2− cluster is −0.744, and of the 9(FeS)2H− (isomer I) cluster is −0.667. An ∼0.08 systematic difference of partial charge numbers on S sites of these clusters is noted. The experimental first VDE of the 4FeS− cluster is 1.85 eV, of the 8(FeS)2− cluster is 2.34 eV, and of the 9(FeS)2H− (isomer I) cluster is 2.84 eV. Their experimental first VDEs increase about 0.5 eV as their partial charge numbers on an S site increase ∼0.08 positive charge number. These results suggest the proposed “charge effect” still contributes to the experimental first VDEs of these “p on S” NBO/HSOMO clusters. Note that the NBO and Mulliken charges here yield very similar trends, if not absolute values. The 11(FeS)3H− (isomer III) cluster is apparently special in this group, however. Its partial charge number on an S site is close that of the 9(FeS)2H− (isomer I) cluster, but its experimental first VDE is ∼0.4 eV smaller than that of the 9(FeS)2H− (isomer I) cluster. The special behavior of the 11(FeS)3H− (isomer III) cluster may be due to an unexplored cluster size effect on its experimental first VDE.
The Mulliken and CHELPG charge numbers on Fe sites (NBO/HSOMO localized) of 10(FeS)3− and 9(FeS)3H− (isomer II) clusters are barely distinguishable (≤0.005 difference), and their experimental first VDEs are also close (∼7% difference). This behavior is in agreement with the suggested “charge effect”. The NBO charge numbers for these two clusters at the appropriate NBO/HSOMO localized Fe sites, however, differ by almost a factor 10 (∼0.06 difference). These results demonstrate that the relative partial charge trends for large clusters, such as (FeS)3H0,1−, may be different if different calculational methods are employed to obtain the partial charges. For the (FeS)4H0,1− clusters listed in the “d on Fe” NBO/HSOMO group, the proposed “charge effect” is apparently not a dominant factor, and the experimental first VDEs of these clusters are mainly determined by the nature of their “d on Fe” NBO/HSOMOs as discussed in above Section D. Note here too, that the different partial calculational methods do not express the same consistency trends as those discussed above, which may also be an indication that the “charge effect” is not a dominant factor for the first VDEs of these clusters.
In sum, for clusters having the same NBO/HSOMOs composition, the partial charge value of the NBO/HSOMO localized sites are also factors affecting their first VDEs: the less negative or more positive site charge value is correlated with a higher first VDE, especially for small size (FeS)mH0,1− (m = 1, 2) cluster anions. Employing multiple methods to calculate and evaluate partial charge distributions for the (FeS)mH0,1− (m = 1–4) cluster anions is probably reasonable and valuable especially for the grouped cluster anions with the same NBO/HSOMOs composition. If these NBO/HSOMO grouped clusters display consistent partial charge trends for the three different charge calculation methods, a “charge effect” on the first VDE trend of such clusters seems to be a reliable and valuable correlation to note.
Based on above comparisons and analyses, the first VDEs of (FeS)mH0,1− (m = 1–4) cluster anions are found to be principally related to/dependent upon the intrinsic properties of the NBO/HSOMO compositions. Depending on the changing nature of cluster NBO/HSOMO in the order, “DB&V” orbital → “p on S” orbital → “d on Fe” orbital → “p on Fe” or “Fe–Fe BD” orbital, the cluster first VDE predominantly increases from low to high. Additionally, a “charge effect” on the first VDEs of clusters with the same NBO/HSOMO properties can be qualitatively characterized if the partial charge distributions evaluated by multiple calculational methods display similar trends and overall behavior. This hypothesis is probably advantageous for the approximate estimation of the first VDE of the larger, more complex iron sulfide/hydrosulfide cluster anions. More studies are needed on larger and multiple hydrogen containing iron hydrosulfide clusters to explore and support these systematics and trends further.
Conclusions
Single hydrogen containing iron hydrosulfide (FeS)mH− (m = 2–4) cluster anions are studied by PES at 3.492 eV (355 nm) and 4.661 eV (266 nm) photon energies, and by DFT calculations. The structural properties, relative energies of different spin states, and first calculated VDEs of different spin states for these (FeS)mH− (m = 2–4) cluster anions are investigated at various reasonable theory levels. The most probable structures and ground state spin multiplicities for these clusters are assigned by comparing their theoretical and experiment first VDE values. Calculated and experimental adiabatic electron affinities of these ground state neutral clusters are also reported. Two types structural isomers are found for these (FeS)mH− (m = 2–4) clusters. One type has the single hydrogen bonded to a sulfur site (SH-type), and the other type has the single hydrogen bonded to an iron site (FeH-type). Compared to the relative pure iron sulfur cluster, the first VDE of SH-type (FeS)2H− is found to be ∼0.5 eV lower than that of the (FeS)2− cluster. A dipole bound and valence electron mixed character of the NBO/HSOMO of the SH-type (FeS)2H− cluster is observed and is probably responsible for the decrease of its first VDE with respect to that of the (FeS)2− cluster. The first VDE of the FeH-type (FeS)2H− is found to be ∼0.5 eV higher than that of the (FeS)2− cluster. The NBO/HSOMOs of the FeH-type (FeS)2H− and (FeS)2− clusters both appear to be localized p orbitals the S site. The observed increase of the first VDE of FeH-type (FeS)2H− is probably due to the decrease of the negative partial charge number (Mulliken) of the S site, from −0.635 on the S site of (FeS)2− to −0.553 on the S site of FeH-type (FeS)2H− (isomer I) (Fig. 6).The first VDE of SH-type (FeS)3H− (isomer III) is found to be ∼1.2 eV lower than that of the (FeS)3− cluster, and the first VDE of FeH-type (FeS)3H− (isomer II) is found to be similar to that of (FeS)3− cluster (within ∼0.2 eV). These distinctly different first VDEs can be related to the different electron distribution (wave function) properties of the NBO/HSOMOs of each cluster anion; in particular, the nature of these NBO/HSOMO wave functions changes from a p orbital on an Fe atom [(FeS)3− and FeH-type (FeS)3H− (isomer II)] to a p orbital on an S atom. For (FeS)4H0,1− clusters, the first VDEs of both SH- and FeH-type (FeS)4H− clusters are close to that of (FeS)4− cluster (within ∼0.2 eV difference), and their NBO/HSOMOs are all found to be localized d orbitals of Fe sites. The theoretical VDEs of SH- and FeH-type isomers of these (FeS)mH− (m = 2–4) clusters are in good agreement with their experimental VDE values. Therefore, these two geometry types of single hydrogen containing iron sulfide (FeS)mH− (m = 2–4) cluster anion likely co-exist under the present experimental conditions. Such available different SH- and FeH-type structural isomers must be considered when evaluating the properties and behavior of these single hydrogen containing iron hydrosulfide clusters in real chemical and biological systems. The single hydrogen in these (FeS)mH− (m = 2–4) cluster anions is suggested to affect their first VDEs through the different structure types (SH- or FeH-), the changing nature of the site localized NBO/HSOMOs, and the changing partial charge number of the localized NBO/HSOMO site.
In order to explore further the relationship between NBO/HSOMO properties, the “charge effect”, and the first VDEs for iron sulfide/hydrosulfide cluster anions, the NBO/HSOMO compositions and properties, partial charge numbers (employing various calculation methods) of the NBO/HSOMO localized site, and the first VDEs of (FeS)mH0,1− (m = 1–4) clusters are compared and contrasted. The first VDEs of diverse type (FeS)mH0,1− (m = 1–4) clusters are found to be rationalized by 1. the different electron distribution properties of NBO/HSOMO, and 2. the partial charge distribution on the NBO/HSOMO localized sites of each cluster anion. Generally, the nature of the NBO/HSOMOs play a key role with regard to the properties of all the anions. The change of cluster VDE from low to high is related to the changing nature of their NBO/HSOMO from a dipole bound and valence electron mixed character orbital, to a valence p orbital on S, to a valence d orbital on Fe, and to a valence p orbital on Fe or an Fe–Fe delocalized valence bonding orbital. For clusters having the same properties of NBO/HSOMOs, the partial charge distributions at the NBO/HSOMO localized sites additionally affect their VDEs: a more negative (less positive) localize charge distribution is correlated with a lower first VDE. In order to test such a NBO/HSOMO localized site partial charge trend for cluster anions with similar NBO/HSOMO properties, multiple atomic partial charge calculational methods must be considered; for example, Mulliken population analysis, Breneman's CHELPG algorithm, and NBO analysis are suggested. If the same trend of partial charges vis a vis first VDEs is obtained by the different calculational methods, a predictive “charge effect” can probably be considered for the first VDE trend of these specifically identified and grouped clusters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by a grant from the US Air Force Office of Scientific Research (AFOSR) through grant number FA9550-10-1-0454, the National Science Foundation (NSF) ERC for Extreme Ultraviolet Science and Technology under NSF Award No. 0310717, the Army Research Office (ARO, Grant No. FA9550-10-1-0454 and W911-NF13-10192), and a DoD DURIP grant (W911NF-13-1-0192).References
- R. Cammack, Advances in Inorganic Chemistry, Academic Press, New York, 1992, vol. 38, p. 281 Search PubMed.
- M. S. Nurmaganbetova, M. I. Baikenov, M. G. Meiramov, A. A. Mukhtar, A. T. Ordabaeva and V. A. Khrupov, Catalytic hydrogenation of anthracene on modified iron sulfide catalysts, Pet. Chem., 2001, 41, 26–29 Search PubMed.
- E. Munck and E. L. Bominaar, Chemistry – bringing stability to highly reduced iron–sulfur clusters, Science, 2008, 321, 1452–1453 CrossRef CAS PubMed.
- D. C. Rees and J. B. Howard, The interface between the biological and inorganic worlds: iron–sulfur metalloclusters, Science, 2003, 300, 929–931 CrossRef CAS PubMed.
- R. D. Bryant, F. V. Kloeke and E. J. Laishley, Regulation of the Periplasmic Fe Hydrogenase by Ferrous Iron in Desulfovibrio-Vulgaris (Hildenborough), Appl. Environ. Microbiol., 1993, 59, 491–495 CAS.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Structural and Functional Aspects of Metal Sites in Biology, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS PubMed.
- S. B. Jang, L. C. Seefeldt and J. W. Peters, Insights into Nucleotide Signal Transduction in Nitrogenase: Structure of an Iron Protein with MgADP Bound, Biochemistry, 2000, 39, 14745–14752 CrossRef CAS PubMed.
- O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Nitrogenase MoFe–Protein at 1.16 Å Resolution: A Central Ligand in the FeMo-Cofactor, Science, 2002, 297, 1696–1700 CrossRef CAS PubMed.
- T. I. Doukov, T. M. Iverson, J. Seravalli, S. W. Ragsdale and C. L. Drennan, A Ni–Fe–Cu Center in a Bifunctional Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase, Science, 2002, 298, 567–572 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, X-ray Crystal Structure of the Fe-Only Hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Angstrom Resolution, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- F. Berkovitch, Y. Nicolet, J. T. Wan, J. T. Jarrett and C. L. Drennan, Crystal Structure of Biotin Synthase, an S-Adenosylmethionine-Dependent Radical Enzyme, Science, 2004, 303, 76–79 CrossRef CAS PubMed.
- O. A. Lukianova and S. S. David, A role for iron–sulfur clusters in DNA repair, Curr. Opin. Chem. Biol., 2005, 9, 145–151 CrossRef CAS PubMed.
- P. J. Kiley and H. Beinert, The role of Fe–S proteins in sensing and regulation in bacteria, Curr. Opin. Microbiol., 2003, 6, 181–185 CrossRef CAS PubMed.
- J. A. Zuris, D. A. Halim, A. R. Conlan, E. C. Abresch, R. Nechushtai, M. L. Paddock and P. A. Jennings, Engineering the Redox Potential over a Wide Range within a New Class of FeS Proteins, J. Am. Chem. Soc., 2010, 132, 13120–13122 CrossRef CAS PubMed.
- E. N. Brown, R. Friemann, A. Karlsson, J. V. Parales, M. M.-J. Couture, L. D. Eltis and S. Ramaswamy, Determining Rieske cluster reduction potentials, JBIC, J. Biol. Inorg. Chem., 2008, 13, 1301–1313 CrossRef CAS PubMed.
- L. M. Hunsicker-Wang, A. Heine, Y. Chen, E. P. Luna, T. Todaro, Y. M. Zhang, P. A. Williams, D. E. McRee, J. Hirst, C. D. Stout and J. A. Fee, High-Resolution Structure of the Soluble, Respiratory-Type Rieske Protein from Thermus thermophilus: Analysis and Comparison, Biochemistry, 2003, 42, 7303–7317 CrossRef CAS PubMed.
- Y. Zu, M. M. J. Couture, D. R. J. Kolling, A. R. Crofts, L. D. Eltis, J. A. Fee and J. Hirst, Reduction Potentials of Rieske Clusters: Importance of the Coupling between Oxidation State and Histidine Protonation State, Biochemistry, 2003, 42, 12400–12408 CrossRef CAS PubMed.
- W. Lovenberg, Iron–sulfur Proteins: Biological Properties, Academic Press, 1973 Search PubMed.
- F. G. Hopkins and E. J. Morgan, The influence of thiol-groups in the activity of dehydrogenases, Biochem. J., 1938, 32, 611–620 CrossRef CAS PubMed.
- A. D. Vinogradov, E. V. Gavrikova and V. V. Zuevsky, Reactivity of the Sulfhydryl Groups of Soluble Succinate Dehydrogenase, Eur. J. Biochem., 1976, 63, 365–371 CrossRef CAS PubMed.
- T. A. Fedotcheva, N. L. Shimanovskii, A. G. Kruglov, V. V. Teplova and N. I. Fedotcheva, Role of mitochondrial thiols of different localization in the generation of reactive oxygen species, Biochem. (Moscow) Suppl. Ser. A: Membr. Cell Biol., 2012, 6, 92–99 CrossRef.
- I. S. Gostimskaya, G. Cecchini and A. D. Vinogradov, Topography and chemical reactivity of the active–inactive transition-sensitive SH-group in the mitochondrial NADH:ubiquinone oxidoreductase (Complex I), Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 1155–1161 CrossRef CAS PubMed.
- R. Stern, M. DeLuca, A. H. Mehler and W. D. McElroy, Role of Sulfhydryl Groups in Activating Enzymes. Properties of Escherichia coli Lysine-Transfer Ribonucleic Acid Synthetase, Biochemistry, 1966, 5, 126–130 CrossRef CAS PubMed.
- A. T. Dinkova-Kostova, W. D. Holtzclaw, R. N. Cole, K. Itoh, N. Wakabayashi, Y. Katoh, M. Yamamoto and P. Talalay, Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11908–11913 CrossRef CAS PubMed.
- B. Mikami, K. Nomura and Y. Morita, Two Sulfhydryl Groups Near the Active Site of Soybean β-Amylase, Biosci., Biotechnol., Biochem., 1994, 58, 126–132 CrossRef CAS PubMed.
- H. Beinert, R. H. Holm and E. Münck, Iron–Sulfur Clusters: Nature's Modular, Multipurpose Structures, Science, 1997, 277, 653–659 CrossRef CAS PubMed.
- H. Ogino, S. Inomata and H. Tobita, Abiological Iron–Sulfur Clusters, Chem. Rev., 1998, 98, 2093–2122 CrossRef CAS PubMed.
- H. Beinert, Recent developments in the field of iron–sulfur proteins, FASEB J., 1990, 4, 2483–2491 CAS.
- G. W. Brudvig, W. F. Beck and J. Paula, Mechanism of photosynthetic water oxidation, Annu. Rev. Biophys. Biophys. Chem., 1989, 18, 25–46 CrossRef CAS PubMed.
- P. Mitchell, The correlation of chemical and osmotic forces in biochemistry, J. Biochem., 1985, 97, 1–18 CrossRef CAS PubMed.
- K. Koszinowski, D. Schröder and H. Schwarz, Formation and reactivity of gaseous iron–sulfur clusters, Eur. J. Inorg. Chem., 2004, 44–50 CrossRef CAS.
- K. Koszinowski, D. Schröder, H. Schwarz, R. Liyanage and P. B. Armentrout, Thermochemistry of small cationic iron–sulfur clusters, J. Chem. Phys., 2002, 117, 10039–10056 CrossRef CAS.
- R. L. Whetten, D. M. Cox, D. J. Trevor and A. Kaldor, Free iron clusters react readily with oxygen and hydrogen sulfide, but are inert toward methane, J. Phys. Chem., 1985, 89, 566–569 CrossRef CAS.
- S. Yin, Z. C. Wang and E. R. Bernstein, Formaldehyde and methanol formation from reaction of carbon monoxide and hydrogen on neutral Fe2S2 clusters in the gas phase, Phys. Chem. Chem. Phys., 2013, 15, 4699–4706 RSC.
- H. J. Zhai, B. Kiran and L. S. Wang, Electronic and structural evolution of monoiron sulfur clusters, FeSn− and FeSn (n = 1–6), from anion photoelectron spectroscopy, J. Phys. Chem. A, 2003, 107, 2821–2828 CrossRef CAS.
- A. Nakajima, T. Hayase, F. Hayakawa and K. Kaya, Study on iron–sulfur cluster in gas phase: electronic structure and reactivity, Chem. Phys. Lett., 1997, 280, 381–389 CrossRef CAS.
- N. Zhang, T. Hayase, H. Kawamata, K. Nakao, A. Nakajima and K. Kaya, Photoelectron spectroscopy of iron–sulfur cluster anions, J. Chem. Phys., 1996, 104, 3413–3419 CrossRef CAS.
- Y. J. Fu, X. Yang, X. B. Wang and L. S. Wang, Probing the electronic structure of 2Fe–2S clusters with three coordinate iron sites by use of photoelectron spectroscopy, J. Phys. Chem. A, 2005, 109, 1815–1820 CrossRef CAS PubMed.
- Y. J. Fu, J. Laskin and L. S. Wang, Electronic Structure and Fragmentation Properties of [Fe4S4(SEt)4–x(SSEt)x]2−, Int. J. Mass Spectrom., 2007, 263, 260–266 CrossRef CAS.
- Y. J. Fu, J. Laskin and L. S. Wang, Collision-Induced Dissociation of [4Fe–4S] Cubane Cluster Complexes: Fe4S4Cl4−x(SC2H5)x2−/1− (x = 0–4), Int. J. Mass Spectrom., 2006, 255, 102–110 CrossRef.
- X. Yang, S. Q. Niu, T. Ichiye and L. S. Wang, Direct measurement of the hydrogen-bonding effect on the intrinsic redox potentials of 4Fe–4S cubane complexes, J. Am. Chem. Soc., 2004, 126, 15790–15794 CrossRef CAS PubMed.
- J. El Nakat, K. J. Fisher, I. G. Dance and G. D. Willett, Gas Phase Metal Chalcogenide Cluster Ions: A New [CoxSy]− Series up to [Co38S24]− and Two Iron–Sulfur [FexSy]− Series, Inorg. Chem., 1993, 32, 1931–1940 CrossRef CAS.
- S. Yin and E. R. Bernstein, Properties of iron sulfide, hydrosulfide, and mixed sulfide/hydrosulfide cluster anions through photoelectron spectroscopy and density functional theory calculations, J. Chem. Phys., 2016, 145, 154302 CrossRef PubMed.
- Z. Zeng and E. R. Bernstein, Photoelectron spectroscopy and density functional theory studies of N-rich energetic materials, J. Chem. Phys., 2016, 145, 164302 CrossRef PubMed.
- H. Wu, S. R. Desai and L.-S. Wang, Chemical Bonding between Cu and Oxygen Copper Oxides vs. O2 Complexes: A Study of CuOx (x = 0–6) Species by Anion Photoelectron Spectroscopy, J. Phys. Chem. A, 1997, 101, 2103–2111 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- J. P. Perdew and Y. Wang, Accurate and Simple Analytic Representation of the Electron-Gas Correlation-Energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- A. D. Becke, Density-Functional Thermochemistry 3. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, 6-31G* basis set for atoms K through Zn, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: characterization and correction of the time-dependent local density approximation ionization threshold, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- L. Cederbaum, One-bodyGreen's function for atoms and molecules: theory and application, J. Phys. B: At., Mol. Opt. Phys., 1975, 8, 290 CrossRef CAS.
- P. A. Hunt, B. Kirchner and T. Welton, Characterising the Electronic Structure of Ionic Liquids: An Examination of the 1-Butyl-3-Methylimidazolium Chloride Ion Pair, Chem. – Eur. J., 2006, 12, 6762–6775 CrossRef CAS PubMed.
- C. M. Breneman and K. B. Wiberg, Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- J. I. Manchester, M. D. Paulsen and R. L. Ornstein, Calculation of Atom-Centered Partial Charges for Heme, in Modelling of Biomolecular Structures and Mechanisms: Proceedings of the Twenty-Seventh Jerusalem Symposium on Quantum Chemistry and Biochemistry Held in Jerusalem, Israel, May 23–26, 1994, ed. A. Pullman, J. Jortner and B. Pullman, Springer Netherlands, Dordrecht, 1995, pp. 181–188 Search PubMed.
- K. Siegbahn, ESCA applied to free molecules, North-Holland, Amsterdam, 1969 Search PubMed.
- S. Yin and E. R. Bernstein, Photoelectron Spectroscopy and Density Functional Theory Studies of Iron Sulfur (FeS)m− (m = 2–8) Cluster Anions: Coexisting Multiple Spin States, J. Phys. Chem. A, 2017, 121, 7362–7373 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The following results are supplied as additional detailed information for these studies: (1) the relative energy of (FeS)2–4H− cluster anions with different spin multiplicities at BPW91/TZVP level in Fig. S1 to S3, respectively; and (2) the first experimental and calculated VDEs (in eV) for (FeS)m− (m = 2–8) cluster anions. See DOI: 10.1039/c7cp07012h |
| This journal is © the Owner Societies 2018 |