
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C–H and H–H activation at a di-titanium centre†‡
Nikolaos
Tsoureas
a,
Jennifer C.
Green
b and
F. Geoffrey N.
Cloke
 *a
*a
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, UK. E-mail: f.g.cloke@sussex.ac.uk
bDepartment of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Road, Oxford OX1 3QR, UK
First published on 25th October 2017
Abstract
The reaction of the bis(pentalene)dititanium complex Ti2(μ:η5,η5-Pn†)2 (Pn† = C8H4(1,4-SiiPr3)2) with the N-heterocyclic carbene 1,3,4,5-tetramethylimidazol-2-ylidene results in intramolecular C–H activation of one of the iPr methyl groups of a Pn† ligand and formation of a “tucked-in” bridging hydride complex. The “tuck-in” process is reversed by the addition of hydrogen, which yields a dihydride featuring terminal and bridging hydrides.
Group IV metallocene chemistry has been instrumental in the development of organometallic chemistry,1 elucidating fundamental aspects of bonding2 and reactivity,3 especially via the synthesis, isolation and study of low valent metallocene complexes.1,4 In this context, the synthesis and isolation of such complexes bearing hydride ligands has been important;5 for example, such low or mixed valence hydride complexes have been shown to promote or be involved in the fixation of N2 to NH3.6 In this communication, we present the first examples of the synthesis of bridging titanium hydrides under non-reducing conditions, via the reaction of bis(pentalene)dititanium complex Ti2(μ:η5,η5-Pn†)2 (Pn† = C8H4(1,4-SiiPr3)2) with an N-heterocyclic carbene and subsequent hydrogenolysis.
We have previously reported on the reactivity of Ti2(μ:η5,η5-Pn†)2 (Pn† = C8H4(1,4-SiiPr3)2) (1) towards a variety of small molecules and π-acceptor ligands.7 In order to gain a better insight into the reactivity of (1), we decided to study its interaction with strong σ-donor ligands. When (1) was treated with an excess of PMe3 no reaction was observed. However, addition of 1,3,4,5-tetramethylimidazol-2-ylidene (2) to (1) in toluene at 0 °C resulted in an immediate colour change from crimson red to a dark pine green (Scheme 1).
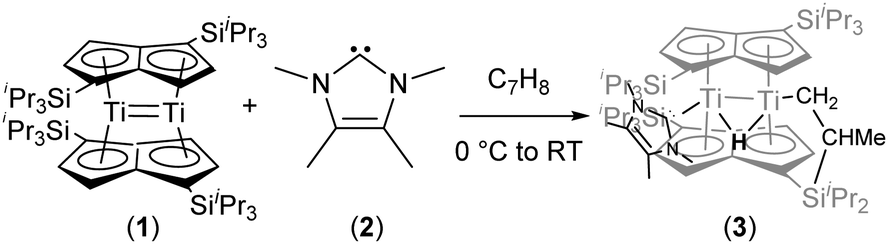 | ||
| Scheme 1 Synthesis of a new syn-bimetallic hydride titanium cluster. | ||
The formulation of the new complex (3) as a “tucked-in” hydride resulting from C–H activation of one of the iPr methyl groups of a Pn† induced by addition of the strongly donating NHC (2) was initially confirmed by NMR. In particular, the 1H-NMR spectrum showed two inequivalent pentalene ring environments, a sharp singlet at −7.91 ppm for the bridging hydride (T1 479 ms), with one of the diastereotopic protons of the “tucked-in” CH2 group appearing as an overlapping dd at −2.91 ppm whilst the other was largely obscured by the complex aliphatic region of the spectrum at ca 1.37 ppm; the coordination of the NHC was confirmed by the observation of a peak at 197.78 ppm in the 13C{1H}-NMR spectrum.
The structure of (3) was confirmed by X-ray diffraction and is shown in Fig. 1. The NHC coordinates to one of the Ti centres (Ti2 in Fig. 1), while one of the methyl groups on a TIPS substituent has been cyclometallated on the other Ti centre (Ti1 in Fig. 1) with concurrent formation of a bridging hydride.8 The Ti–Ti bond has been retained but lengthened to 2.5610(8) Å (from 2.399(2) Å in (1)7e) and is typical of a single bond. The Ti–C(carbene) bond (2.300(2) Å; Ti2–C1 in Fig. 1) is within the range of 2.2–2.35 Å reported for other Ti–NHC complexes.9 The Ti–H bond lengths (i.e. Ti2–H1: 1.72(3) Å, Ti1–H1: 1.79(3) Å) are identical within esd's and are similar to previously reported monomeric10 and dimeric6d,6b,11 titanium hydrides as well as Ti(III) alumino-12 and borohydrides13 (1.7–1.9 Å), although it has to be noted that, probably due to the topology of the hydride ligand in (3), these Ti–H bond distances fall at the shorter end of the known range. Due to this unique topology, the Ti–H–Ti bond angle (93.4(13)°) approaches a right angle and is the most acute ever observed in dimeric titanium hydrides.6b,6d,11
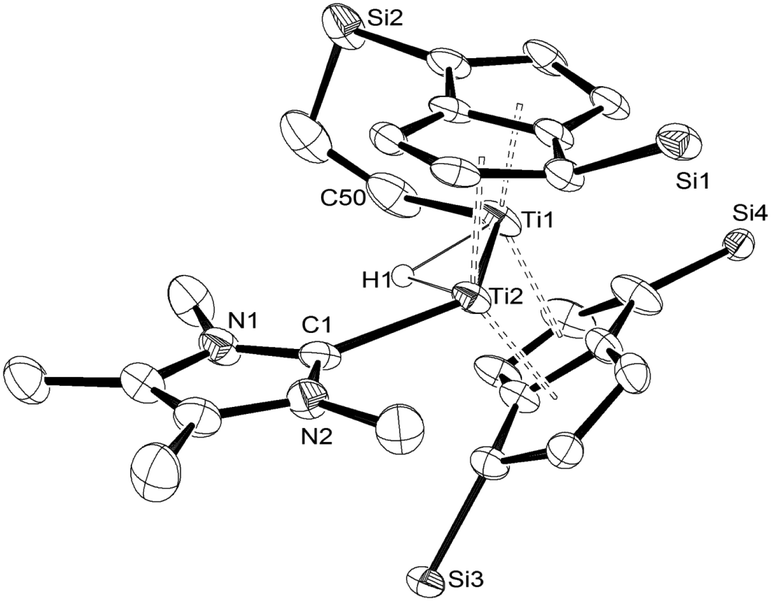 | ||
| Fig. 1 ORTEP diagram of the molecular structure of (3) displaying 50% probability ellipsoids. iPr groups omitted for clarity. | ||
It is also worth noting that the addition of (2) to (1) results in the formal oxidation of the two Ti centres (i.e. from +2 to +3), and employing the CBC model each Ti has a count of 18 e (16 e in (1)).14
With a view to synthesising a new hydride derivative via σ-bond metathesis of the Ti–CH2 bond in (3) with dihydrogen, 2 bar of H2 was added to a C7D8 solution of (3) in an NMR tube This indeed resulted in clean conversion of (3) (100% spectroscopic yield) to a new complex (4) (Scheme 2).
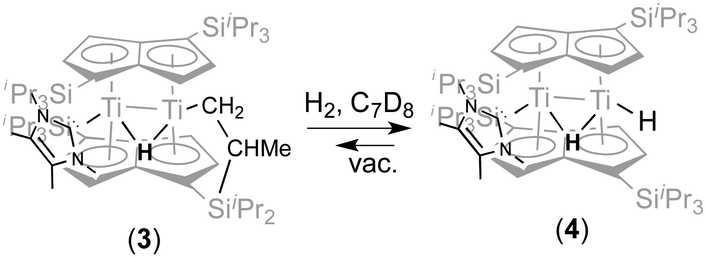 | ||
| Scheme 2 Hydrogenolysis of (3) to afford (4) | ||
Compared to the 1H NMR spectrum of (3), (4) displays a new, broader hydride peak (Δν1/2 = 29 Hz) at −8.82 ppm at room temperature, whilst the signal for the “tucked-in CH2 group has disappeared completely; the NHC is still coordinated (13C{1H} δ 198.27 ppm). Removal of the H2 overpressure by freeze–thaw-degassing showed that (4) is persistent in solution, although some regeneration of (3) was observed (Scheme 2). Addition of H2 to a solution of (3), via a Toepler pump, showed that for the conversion of (3) to (4) to occur quickly (minutes) 5 eq of H2 are required (when 1–2 equivalents of H2 were added, complete conversion to (4) occurred after ca. 1 week). The rate of reaction was also found to be pressure dependent: when (3) was exposed to an atmosphere of 10% H2 in N2 at 1.5 bar but in an amount corresponding to only 1 equivalent of H2 the reaction was again complete in minutes.
Variable temperature 1H NMR studies showed that the broad hydride peak at −8.82 ppm in (4) becomes fully resolved into a doublet at 0 °C (with no further change below that temperature and down to −70 °C) with a T1 of 310 ms, with the concomitent appearance of a second doublet centred at 2.17 ppm (T1 336 ms), which is too broad to be observable at room temperature (Fig. 2); these two signals are related by a coupling constant of JHH = 11 Hz. EXSY spectroscopy (in both the presence and absence of an H2 overpressure) confirmed that these two protons exchange at 30 °C while at 0 °C the process is quenched. Thus the peak at −8.82 ppm is assigned to the bridging hydride in (4) and that at 2.17 ppm to the terminal one (Scheme 2).
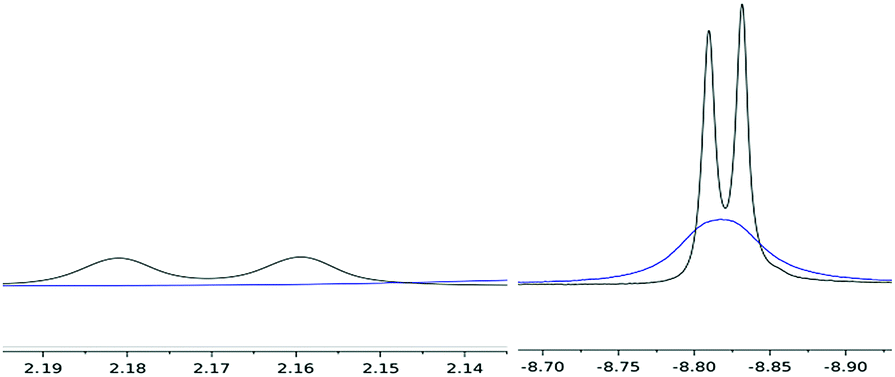 | ||
| Fig. 2 Hydride peaks in (4) at RT (blue) and at 0 °C (black). | ||
Initial attempts to crystallise (4) by standard methods (i.e. removal of volatiles and recrystallisation) were frustrated by the preferential isolation of crystalline (3) (as it is less soluble than (4)) with the mother liquor consisting of a mixture of (3) and (4) (ca. 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 by NMR), due to the partial reversibility of the reaction. However, the solid state molecular structure8,15 of (4) (Fig. 3) was eventually determined from single crystals grown by cooling slowly a freshly prepared solution of (4) at −78 °C under an overpressure (1.5 atm) of H2, and confirms the spectroscopic assignment.
:80 by NMR), due to the partial reversibility of the reaction. However, the solid state molecular structure8,15 of (4) (Fig. 3) was eventually determined from single crystals grown by cooling slowly a freshly prepared solution of (4) at −78 °C under an overpressure (1.5 atm) of H2, and confirms the spectroscopic assignment.
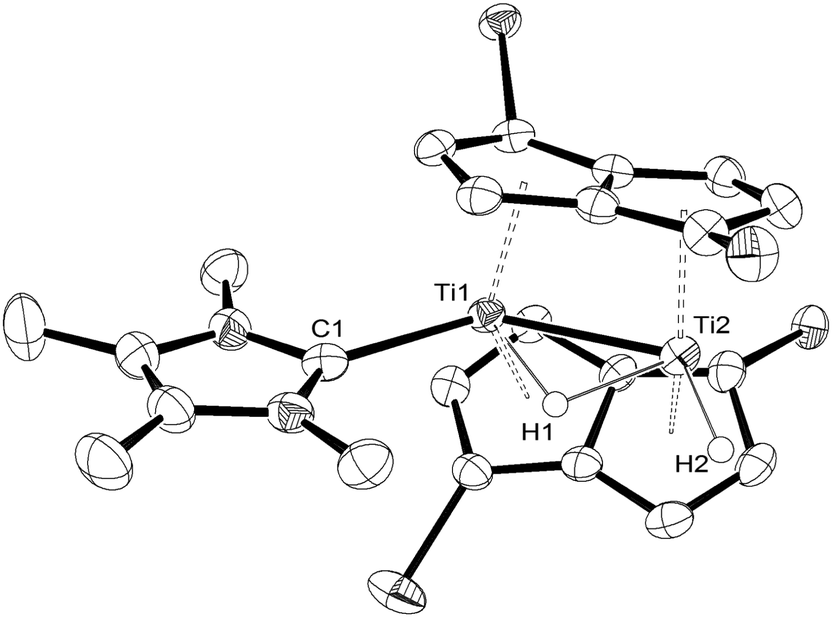 | ||
| Fig. 3 ORTEP diagram of the molecular structure of (4) displaying 50% probability ellipsoids. iPr groups omitted for clarity. | ||
The Ti–C(carbene) bond length in (4) is 2.291(4) Å and is identical to that found in (3). On the other hand, the Ti–Ti bond is slightly shortened in (4) from 2.5610(8) Å in (3) to 2.5413(8) Å possibly due to the negligible steric requirements of the terminal hydride ligand. The Ti–H(bridging) bond distances (Ti1–H1 = 1.84(5) Å; Ti2–H1 = 1.79(5) Å) in (4) are similar within esd's and compare with the ones found in (3); the same applies to the Ti–H(terminal) (i.e. Ti2–H2 = 1.74(4) Å in Fig. 2) bond length. The Ti1–H1–Ti2 bond angle in (4) again approaches 90° (89(2)°) and is very similar to that found in (3).
When (3) was treated with an excess of D2 (5 eq.), the formation of (4-D) was observed, but deuterium was found to be only incorporated in the hydridic positions, and not in the new Me group derived from the previously “tucked-in” CH2 group (confirmed by 2H-NMR, DEPT-135 and gHSQC). Hence the reaction of (3) with H2 to form (4) does not go via σ-bond metathesis (which would lead to D incorporation in the Me group in the reaction with D2). Hence the formation of (4) (and also (3)) was probed computationally (ADF:BP/TZP: details are given in the ESI‡). Preliminary studies suggested that sterics were important in determining the reaction energies. For example energy of binding an NHC to a Ti2Pn2 dimer depended critically on the substituents. Introduction of the methyl substituents on the NHC made very little difference to its binding energy but the bulky SiiPr3 substituents on the pentalene ligands increased the Ti–Ti–C angle forcing the NHC to a less favourable binding position thus decreasing the binding energy significantly (Table 1).
The degree to which the tuck-in reaction was favoured in the absence of an NHC was also investigated. The formation of Ti2Pn†(Pn†-H)(μ-H) from Ti2Pn†2 was calculated to have ΔE = −0.03 eV and ΔG = 11 kJ mol−1. However, the energies of the observed tuck-in reaction with the methylated NHC present were calculated to be ΔE = −0.69 eV and ΔG = 9 kJ mol−1. Thus the presence of the base improves the energetics of the tuck-in reaction. The significant entropy disadvantage in the gas phase would be lessened in solution.
The HOMO of 3 (Fig. 4) shows a Ti–Ti σ-bond. The calculated Ti–Ti distance is 2.56 Å in excellent agreement with experiment. The Ti–H distances are 1.83 Å and the angle at the bridging hydrogen 89°. Such discrepancies from the experimental values are not unusual when comparing distances to bound hydrogen between theory and X-ray diffraction experiments.
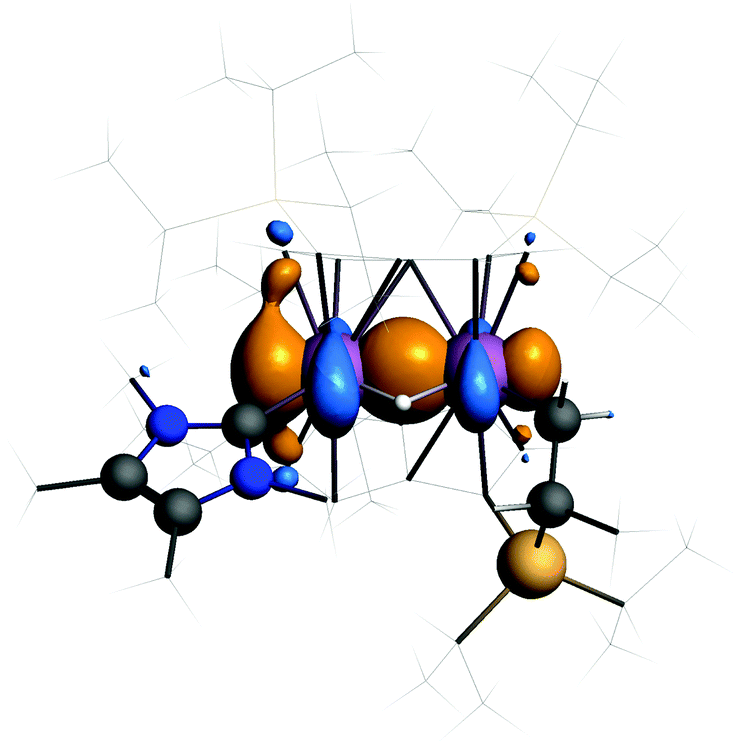 | ||
| Fig. 4 Isosurface for the HOMO of 3. | ||
Addition of H2 to 3 to form 4 is calculated to have reaction energies ΔE = −0.77 eV and ΔG = −31 kJ mol−1. The calculated Ti–Ti distance for 4 is 2.54 Å reproducing the shortening from 3 found experimentally. The Ti–H(terminal) distance is 1.74 Å, the Ti–H(bridging) distances 1.81 and 1.82 Å and the Ti–H–Ti angle unchanged at 89°.
A transition state for this reaction was modelled using just one SiiPr3 substituent on one of the pentalene ligands and C3H4N as the NHC for computational efficiency. The free energy of activation was estimated as 84 kJ mol−1 for such a system. The transition state structure is shown in Fig. 5.
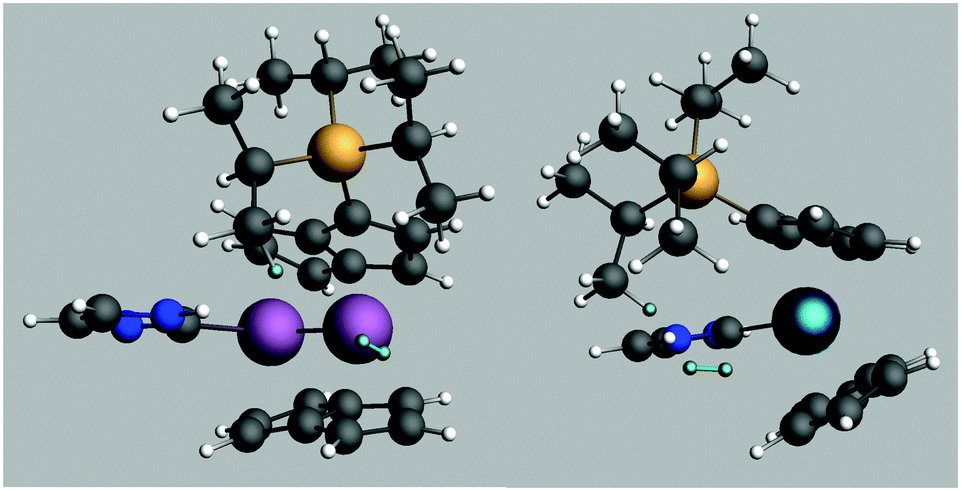 | ||
| Fig. 5 Structure of the calculated transition state for H2 addition. The reacting Hs are highlighted. | ||
The Ti distance to the previously bridging H is 3.53 Å and the Ti distances to the reacting H2 are 2.74 and 3.24 Å, the H–H distance being 0.76 Å. Such a geometry indicates that the tuck-in process is reversed before complete H2 addition, consistent with the lack of deuterium incorporation into iPr groups and the conclusion that σ bond metathesis is not in play. It may be that the steric compression induced by the mere approach of the H2 molecule is sufficient to reverse the tuck-in process, an idea given some credence by the pressure dependence of the reaction of (3) with H2 (vide infra).
In conclusion, we have described the facile preparation of the first example of a syn-bimetallic Ti complex (3) featuring a bridging hydride, originating from the C–H activation of a iPr substituent induced by addition of the strong Lewis base 1,3,4,5-tetramethylimidazol-2-ylidene. Preliminary studies show that this transformation is also effected by other, effectively “planar” Lewis bases, e.g. dimethylaminopyridine (DMAP). The resultant C–H activation product (3) readily reacts with an excess of H2 to produce very cleanly a unique syn-bimetallic di-hydride complex (4) featuring bridging and terminal hydride ligands. Labelling experiments and computational studies strongly suggest that the latter reaction does not proceed via a σ-bond metathesis mechanism.
We thank the EPSRC for funding (N. T.), Dr Graham Tizzard (National Crystallography Service, University of Southampton), and Dr Alexander Kilpatrick (University of Oxford) and Dr Iain Day (University of Sussex) for help with NMR experiments.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. J. Chiric, Organometallics, 2010, 29, 1500 CrossRef.
- (a) G. Wilkinson and A. K. Fischer, J. Inorg. Nucl. Chem., 1956, 2, 149 CrossRef; (b) H. H. Brintzinger and J. E. Bercaw, J. Am. Chem. Soc., 1970, 92, 6182 CrossRef CAS; (c) A. Davison and S. S. Wreford, J. Am. Chem. Soc., 1974, 96, 3017 CrossRef CAS; (d) E. Bercaw, J. Am. Chem. Soc., 1974, 96, 5087 CrossRef; (e) S. I. Troyanov, H. Antropinsova and K. Mach, J. Organomet. Chem., 1992, 427, 49 CrossRef CAS; (f) P. Mosimann and J. J. Salzmann, Helv. Chim. Acta, 1967, 50, 1831 CrossRef; (g) M. D. Walter, C. D. Sofield and R. A. Andersen, Organometallics, 2008, 27, 2959 CrossRef CAS.
- (a) S. A. Cohen, P. R. Auburn and J. E. Bercaw, J. Am. Chem. Soc., 1983, 105, 1136 CrossRef CAS; (b) U. Rosenthal, V. V. Burlacov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2003, 22, 884 CrossRef CAS; (c) I. Pappas and P. J. Chiric, J. Am. Chem. Soc., 2016, 138, 13379 CrossRef CAS PubMed; (d) I. Pappas and P. J. Chiric, J. Am. Chem. Soc., 2015, 137, 3498 CrossRef CAS PubMed; (e) K. T. Tarantino, D. C. Miller, T. A. Callon and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 6440 CrossRef CAS PubMed; (f) M. Mori, Heterocycles, 2009, 78, 281 CrossRef CAS; (g) M. E. Vol’pin and B. V. Shur, Nature, 1966, 209, 1236 CrossRef; (h) G. H. Olivè and G. S. Olivè, Angew. Chem., Int. Ed., 1969, 8, 650 CrossRef.
- (a) M. Horáček, V. Jupfer, U. Thewalt, P. Stepnicka, M. Polasek and K. Mach, Organometallics, 1999, 18, 3572 CrossRef; (b) L. Lukesová, M. Horáček, P. Stepnicka, K. Fejfarová, R. Gyepes, I. Gisorová, J. Kubišta and K. Mach, J. Organomet. Chem., 2002, 663, 134 CrossRef; (c) L. Lukesová, J. Pinkas, M. Horáček, R. Gyepes, I. Gisorová, J. Kubišta and K. Mach, J. Organomet. Chem., 2006, 691, 748 CrossRef; (d) P. B. Hitchcock, F. M. Kerton and G. A. Lawless, J. Am. Chem. Soc., 1998, 120, 10264 CrossRef CAS; (e) T. E. Hanna, E. Lobkovsky and P. J. Chiric, J. Am. Chem. Soc., 2004, 126, 14688 CrossRef CAS PubMed; (f) T. E. Hanna, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 6018 CrossRef CAS PubMed.
- (a) J. E. Bercaw and H. H. Brintzinger, J. Am. Chem. Soc., 1971, 93, 2045 CrossRef; (b) J. E. Bercaw, R. H. Marvich, L. G. Bell and H. H. Brintzinger, J. Am. Chem. Soc., 1972, 94, 1219 CrossRef CAS.
- (a) J. A. Pool, E. Lobkovsky and P. J. Chirik, Nature, 2004, 427, 527 CrossRef CAS PubMed; (b) T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549 CrossRef CAS PubMed; (c) T. E. Hanna, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 6018 CrossRef CAS PubMed; (d) S. P. Semproni, C. Milsman and P. J. Chiric, Organometallics, 2012, 31, 3672 CrossRef CAS.
- (a) A. F. R. Kilpatrick and F. G. N. Cloke, Chem. Commun., 2014, 50, 2769 RSC; (b) A. F. R. Kilpatrick, J. C. Green and F. G. N. Cloke, Organometallics, 2015, 34, 4816 CrossRef CAS PubMed; (c) A. F. R. Kilpatrick, J. C. Green and F. G. N. Cloke, Organometallics, 2015, 34, 4830 CrossRef CAS PubMed; (d) A. F. R. Kilpatrick, J. C. Green and F. G. N. Cloke, Organometallics, 2017, 36, 352 CrossRef CAS; (e) A. F. R. Kilpatrick, J. C. Green, F. G. N. Cloke and N. Tsoureas, Chem. Commun., 2013, 49, 9434 RSC; (f) F. G. N. Cloke, J. C. Green, A. F. R. Kilpatrick and D. O’Hare, Coord. Chem. Rev., 2017, 344, 238 CrossRef CAS.
- The hydride was found in the Fourier difference map and refined freely. We recognize the difficulties associated with the location of hydrogen atoms next to heavy atoms as Fourier ripples can be erroneously misinterpreted for hydrogen atoms due to the sharp cut-off at high angles. Nevertheless, based on the spectroscopic evidence the hydrogen atoms have been included in the supplied models.
- (a) A. Doddi, C. Gemel, R. W. Seidel, M. Winter and R. A. Fischer, Polyhedron, 2013, 52, 1103 CrossRef CAS; (b) G. B. Nikivorov, H. W. Roesky, P. G. Jones, J. Magull, A. Ringe and R. B. Oswald, Inorg. Chem., 2008, 47, 2171 CrossRef PubMed; (c) M. Manβen, C. Adler and R. Beckhaus, Chem. – Eur. J., 2016, 22, 4405 CrossRef PubMed; (d) C. Lorber and L. Vendier, Organometallics, 2008, 27, 2774 CrossRef CAS; (e) J. Li, c. Schulzke, S. Merkel, H. W. Roesky, P. P. Samuel, A. Döring and D. Stalke, Z. Anorg. Allg. Chem., 2010, 636, 511 CrossRef CAS.
- J. M. de Wolf, A. Meetsma and J. H. Teuben, Organometallics, 1995, 14, 5466 CrossRef CAS.
- (a) J. B. Love, H. C. S. Clark, F. G. N. Cloke, J. C. Green and P. B. Hitchcock, J. Am. Chem. Soc., 1999, 121, 6843 CrossRef CAS; (b) L. Hao, J. F. Harrod, A.-M. Lebuis, Y. Mu, R. Shu, E. Samuel and H.-G. Woo, Angew. Chem., Int. Ed., 1998, 37, 3126 CrossRef CAS; (c) S. R. Frecichs, B. K. Stein and J. E. Ellis, J. Am. Chem. Soc., 1987, 109, 559 CrossRef; (d) J. R. Haragorn and M. J. McNevin, Organometallics, 2003, 22, 609 CrossRef; (e) S. I. Troyanov, K. Mach and V. Varga, Organometallics, 1993, 12, 3888 Search PubMed; (f) G. Bai, P. Wei and D. W. Stephan, Organometallics, 2006, 25, 2650 Search PubMed; (g) E. G. Perevalova, I. F. Urazowski and D. A. Lemenovskii, J. Organomet. Chem., 1985, 289, 319 CrossRef CAS.
- (a) E. B. Loblovskii, G. L. Soloveichik, A. I. Sizov and B. M. Bulychev, J. Organomet. Chem., 1985, 280, 53 CrossRef; (b) L. J. Gugenbereger and F. N. Tebbe, J. Am. Chem. Soc., 1973, 95, 7870 CrossRef; (c) F. N. Tebbe and L. J. Gugenberger, J. Chem. Soc., Chem. Commun., 1973, 227 RSC.
- K. H. Melmed, D. Coucouvanis and S. J. Lippard, Inorg. Chem., 1973, 12, 232 CrossRef CAS.
- (a) M. L. H. Green and G. Parkin, J. Chem. Educ., 2014, 91, 807 CrossRef CAS; (b) J. C. Green, M. L. H. Green and G. Parkin, Chem. Commun., 2012, 48, 11481 RSC.
- Data were collected up to 0.82 Å resolution using Cu/Kα as well up to 0.78 Å using Mo/Kα with identical metric parameters.
Footnotes |
| † Dedicated to Phil on the occasion of his 65th birthday- Lá Breithe Sona! |
| ‡ Electronic supplementary information (ESI) available: Full experimental and computational details, and X-ray data. CCDC 1577130–1577132. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc07726b |
| This journal is © The Royal Society of Chemistry 2017 |