
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA route to small clusters: a twisted half-hexagram-shaped M4(OH)4 cluster and its capacity for hosting closed-shell metals†
I.
Ara
 a,
M. A.
García-Monforte
a,
R.
González
b,
L. R.
Falvello
*b and
M.
Tomás
*a
a,
M. A.
García-Monforte
a,
R.
González
b,
L. R.
Falvello
*b and
M.
Tomás
*a
aDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea, Universidad de Zaragoza-CSIC, E-50009, Zaragoza, Spain. E-mail: milagros@unizar.es
bDepartamento de Química Inorgánica, Instituto de Ciencia de Materiales de Aragón, Universidad de Zaragoza-CSIC, E-50009, Zaragoza, Spain. E-mail: falvello@unizar.es
First published on 20th November 2017
Abstract
By combining different oxidation states, coordination indices and bridging systems, it has been possible to obtain the structurally novel M4(OH)4 cluster core (M = transition metal) found in the organometallic compound (NBu4)2[PtIVPtII3(C6Cl5)8(μ2-OH)2(μ3-OH)2] (1). The cluster is formed by two (μ3-OH) and two (μ2-OH) units that bond platinum atoms in different oxidation states. The cluster core geometry can best be described as a half-hexagram. Compound 1 is an excellent precursor for preparing heterometallic clusters since it can host d10 or s2 Lewis-acid metal centers through Pt→M dative bonds, as demonstrated by its reaction with Ag(I) to produce the heterometallic [Ag2PtIVPtII3(C6Cl5)8(μ2-OH)2(μ3-OH)2] (2), which has four unbridged Pt–Ag bonds.
We report here the preparation of a unique cluster possessing a tetranuclear Pt4(OH)4 core with Pt centers in two different oxidation states and with a core structure that has no precedent in transition-metal hydroxide chemistry. This cluster, further, has hydroxy ligands that bridge two or three Pt atoms − in the latter case the metal centers are in two different oxidation states, in another heretofore unobserved core characteristic; and it can also serve as a stable metalloligand for the formation of new compounds with unsupported Pt→M bonds.
Hydroxy-bridged multinuclear transition metal compounds constitute such a well explored area of chemistry that it may seem injudicious to expect that they have yet to yield products with substantive new features, especially for low-nuclearity systems. The ample body of science in which members of this family of compounds have been explored traverses such diverse contexts as biology and medicine, where hydroxo complexes can be found in redox metalloenzymes1 and in the mechanism of antitumor agents2 or chemical catalytic processes,3 such as water oxidation4 or materials science, where such complexes have appeared as precursors for nanomaterials2a,5 or as nodes for the formation of MOFs,6 and magnetic materials.7
Coordination compounds with M4(OH)4 cores are well known (Chart 1), and particularly so those in which each of the four OH− ligands bridges three metal atoms, [(μ3-OH)4], giving a cubane structure, A in Chart 1.8 Clusters in which each of the four OH− ligands bridges just two metal atoms, (μ2-OH)4, are also known although in lesser numbers;9 these form eight-membered rings (M–OH–M–OH–)2 (B in Chart 1) with differing conformations. Also known are compounds with M–M bonds, such as the molybdenum compound with two double OH bridges and Mo![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Mo bonds (C in Chart 1).10 Clusters with two (μ2-OH) and two (μ3-OH) bridges are known, mainly for M = Cu. These are formed by three fused distorted squares, with the central ring sharing two opposite sides in a staircase or Z-shaped structure (D in Chart 1).11
Mo bonds (C in Chart 1).10 Clusters with two (μ2-OH) and two (μ3-OH) bridges are known, mainly for M = Cu. These are formed by three fused distorted squares, with the central ring sharing two opposite sides in a staircase or Z-shaped structure (D in Chart 1).11
 | ||
| Chart 1 Structure types of “M4(OH)4” clusters. | ||
Platinum hydroxocompounds have also been reported, some of them with interest in C–H activation, C–O bond formation, or the preparation of anticancer complexes.2,12,13 Hydroxocomplexes, though, are not as frequent for Pt as for other transition metals;2b this is especially true for PtII because of its soft-acid character and the hard-base character of the OH− ligand. Polynuclear Pt(OH) compounds are scarce, but reported examples include one of type A in Chart 1, “PtIV4(OH)4”8a with (μ3-OH), and two of type B, with (μ2-OH) joining two PtII atoms.9b,c An additional two platinum hydroxo compounds have been described previously, with the eight-membered ring (type B) and with Hg2+ [PtII4(OH)4(HgIICl)2]14 or Ag+ [PtII4(OH)4Ag4] (3)15 incorporated (Fig. 1a); the d10 metals (Hg2+ or Ag+) appear to exert a template effect. The Ag compound, cyclo-[{Pt(C6Cl5)2(μ2-OH)(μ2-Ag)}4] (3),15 is formed by the reaction of (NBu4)2[{PtII(C6Cl5)2(μ2-OH)}2] (4)16 with Ag+ in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio (Scheme 1, A).
:2 molar ratio (Scheme 1, A).
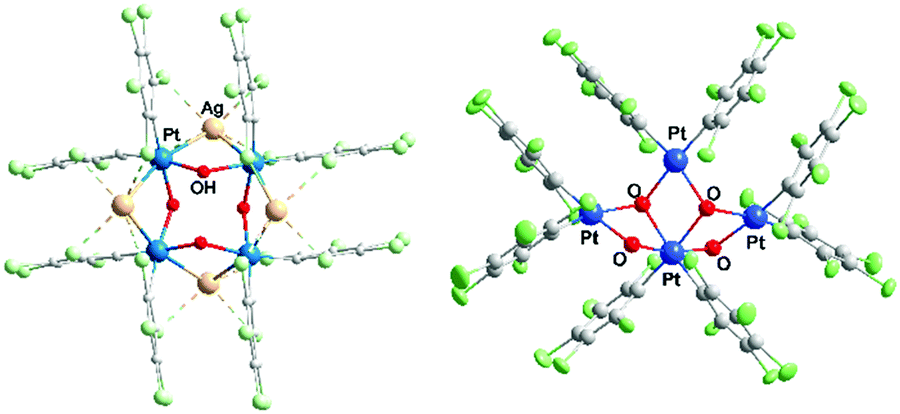 | ||
| Fig. 1 (a) [{PtII(C6Cl5)2(μ2-OH)(μ2-Ag)}4] (3); (b) [PtIVPtII3(C6Cl5)8(μ2-OH)2(μ3-OH)2]2− (1). | ||
 | ||
| Scheme 1 | ||
PtII complexes, especially when anionic or with ligands that promote the dz2 orbital to higher energy levels, can form heteronuclear compounds with Pt–M bonds (M = d10 or s2 Lewis-acid metal centers).17 Since 1985, when the first two pentafluorophenyl compounds with unsupported Pt–Ag bonds were structurally characterized,18 a large number of organometallic compounds with different organic groups and with other acceptor metals such as Tl(I), Cu(I), Au(I), Pb(II), Hg(II), and Cd(II),17,19 have been reported. The commonest acceptor metal is still Ag, and the most frequent organic group is C6F5. X⋯M (X = F, Cl) interactions help in the stabilization of these Pt–M donor–acceptor bonds.15,18 Besides the wide variety of structural types that they present, these compounds are also interesting for the stabilization of unusual oxidation states,20 for their optical properties,15,17,19b–d,21 for the formation of extended structures,19c,d,22 or for their participation in chemical processes such as halide abstraction23 or methyl migration.24 They are also intermediates in important catalytic processes such as inner-sphere oxidation reactions.25
Since PtIV is more suitable than PtII for the formation of hydroxo compounds, while PtII participates readily in Pt→M dative bond formation, and given that the commonest coordination indices of PtIV (six) and PtII (four) are different, we expected that the incorporation of Pt in both oxidation states into the same species would produce novel compounds with interest in the contexts of both hydroxo complexes and Pt→M bonds.
On testing this proposal, we found that the partial oxidation of the above-mentioned dihydroxo compound (NBu4)2[{PtII(C6Cl5)2(μ2-OH)}2] (4) by I2 in the presence of Ag+ (AgNO3) in a 2:1:2 molar ratio produces the anionic cluster [PtIVPtII3(C6Cl5)8(μ2-OH)2(μ3-OH)2]2−, reported here, which is isolated as the NBu4+ salt (1) (Fig. 1b) as the bis-acetone solvate. The synthesis of 1 stands in contrast to the reaction of the same dihydroxo compound 4 with Ag+ in a 1:2 molar ratio, (Scheme 1) which as previously reported15 produces the cyclic “PtII4(OH)4Ag4” core in 3.
Fig. 1b shows the structure of the novel anion in 1 as determined by single crystal X-ray diffraction, which can be compared with the differently-shaped neutral compound 3; Fig. 2 presents a close-up view of the core of 1. In the formation of 1, Ag+ serves to remove the I− produced in the oxidation of PtII (as insoluble AgI), and also perhaps to facilitate the oxidation. The ratio I2:Ag (1:2) is just sufficient to eliminate the I− produced in the reaction, and so compound 1, unlike 3, does not contain silver.
 | ||
| Fig. 2 Core of the anion in 1, with atom labels. For PtIV, distances (Å): Pt–C 2.035(6) and 2.036(7); Pt–O 1.998(5)–2.167(4). Angles (°): cis-74.96(18)–103.2(2); trans-166.81(19)–173.2(2). For PtII, distances (Å): Pt–C 1.947(7)–2.010(7); Pt–O 2.079(5)–2.169(5). Angles (°): cis-73.25(16)–101.9(2); trans-165.75(19)–175.2(2). | ||
The anionic cluster is formed by one PtIV (Pt1) and three PtII centers (Pt2, Pt3, Pt4), bridged by OH groups. PtIV is coordinated by the four OH groups, which is unusual, especially for organometallic compounds;26 and each of the PtII atoms is coordinated by two OH. Also, each Pt center maintains the two terminal, mutually cis C6Cl5 ligands that it has in the starting material. The coordination index is thus six for PtIV and four for the PtII centers, as is usual for these oxidation states. In the only C6Cl5 derivative of PtIV to have been structurally characterized to date, the nominally four-coordinate, homoleptic [Pt(C6Cl5)4],27 the PtIV has two further close contacts, to ortho-Cl atoms of the C6Cl5 ligands [Pt⋯Cl 2.559(2), 2.681(2) Å], and thus resides in what is effectively a very distorted octahedral environment.
The four Pt and the four ligated O atoms in 1 form three distorted rhombi, fused in pairs (Fig. 2), with the platinum atoms at the acute vertices [O–Pt–O range 73.25(16)–79.16(15)°; Pt–O–Pt 98.25(17)–107.1(2)°]. Acute vertices of all three rhombi coincide at the PtIV atom (Pt1), which acts as a lynchpin for the core structure. The central rhombus (Pt1/O2/Pt3/O3) shares one edge with each of its terminal neighbors. This central cycle, with its two O atoms coordinated in a mutually cis fashion to octahedral Pt1, forms dihedral angles of 74.82(11) and 73.72(14)° with the terminal rhombi containing O1 and O4, respectively. The two distal rings form a dihedral angle of 88.57(12)°. If the three rhombi were coplanar, the cluster core could be described as half of a six-point star (hexagram) with the PtII atoms at three points of the star and PtIV at the center. The (μ2-OH) groups at O1 and O4 bridge PtII and PtIV centers, a rare occurrence in hydroxo-platinum chemistry; the other two OH ligands (O2, O3) bridge three platinum atoms (μ3-OH) − two PtII and one PtIV in each case. O2 and O3 in 1 are the first examples of (μ3-OH) groups bridging Pt in different oxidation states. Thus, 1 should be a useful precursor for other PtII or PtIV hydroxo complexes, including mononuclear compounds with terminal OH groups with potential interest in catalytic processes12 or antitumor studies.13
The core geometry in 1 has no precedent in the structures of tetranuclear tetrahydroxo transition metal clusters. At the periphery of the half-hexagram in this anionic, mixed-valence PtIVPtII3 compound, the central PtII atom (Pt3) lies at 3.5589(3) and 3.6101(4) Å from the other PtII atoms (Pt2 and Pt4, respectively) with a Pt2⋯Pt3⋯Pt4 angle of 112.98(1)°. The combination of anionic character and an open stellate periphery make the anion in 1 a potential multi-Pt donor ligand for d10 or s2 Lewis-acid centers such as Ag+, Tl+, Au+, Pb2+, Cd2+ and others. The presence of terminal C6Cl5 ligands also favours such behavior, since these fragments are known to stabilize metal–metal dative bonds.15
Fig. 3(a) shows the “cage-chair” formed by two of the Pt(II) atoms (Pt2 and Pt3) and 4 of the ortho-chlorine atoms of the C6Cl5 groups attached to those platinum atoms. This quite distorted “cage-chair” − whose distances are slightly different from those of the analogous “cage-chair” for Pt3 and Pt4, (Fig. S1, ESI†) shows the relative location of the atoms that may define the potential space for the d10 or s2 Lewis-acid centers.
 | ||
| Fig. 3 (a) Space formed by Pt2, Pt3, Cl22, Cl32, Cl42 and Cl56 in complex 1; (b) space formed by Pt2, Pt3, Cl2, CL14, CL20 and Cl6′ in complex 2 showing the location of Ag. | ||
Upon testing this proposition, we found that the reaction of 1 with AgSO3CF3 produces a unique neutral Pt4Ag2 cluster, [Ag2PtIVPtII3(C6Cl5)8(μ2-OH)2(μ3-OH)2] (2) (Fig. 4). Compound 2 has at its core the anion of 1, to which two Ag+ centers are adjoined, cradled in what was the open space between pairs of peripheral PtII of the half hexagram and between corresponding pairs of their respective C6Cl5 ligands. In the crystal of 2, the two external rhombi are now crystallographically equivalent, related by a twofold axis (0.5 − x, 0.5 − y, z) (absent the bulky NBu4+ cations, the overall structure of neutral 2 is different from that of 1). Two of the PtII atoms (distal Pt2 and its symmetry relative Pt2′) act individually as single Pt→Ag donors [Pt2–Ag1 2.7163(13) Å] while the central PtII atom, Pt3, attaches the two symmetry-related Ag atoms in trans-positions [Pt3–Ag1 2.8855(12) Å, Ag1–Pt3–Ag1′ 175.12(5)°]. Each silver atom accepts two cis-Pt–Ag bonds [Pt2–Ag1–Pt3 79.65(3)°]. The Pt–Ag distances are quite short for both the trans-Ag–Pt–Ag15 and cis-Pt–Ag–Pt28 fragments. Each Ag atom is also involved in short contacts with ortho-Cl atoms from C6Cl5 groups. The shortest of these, [2.630(4) Å], is the shortest Ag⋯Cl–C(ortho) interaction observed to date involving C6Cl5 groups and one of the shortest among all Ag⋯Cl–C interactions;29 indeed, it lies within the range of Ag–Cl distances found for μ2-Cl bridges between silver atoms (2.6–2.7 Å). Fig. 3(b) shows the “cage-chair” formed by Pt2, Pt3 and the four ortho-chlorine atoms in compound 2 with the guest Ag+ within. As can be seen, there has been a change in the shape and dimensions of the initial “cage-chair” [Fig. 3(a)], which shows that it is flexible enough to adapt to the presence of the guest. It is interesting that in order for the Ag+ to reach the inside of the “cage-chair,” it must pass through one of the faces of the cage. The superposition of 2 with the anion of 1, (Fig. S2, ESI†) shows that the “Pt4(OH)4” core does not change and the main difference between 1 and 2 lies in the orientations of the C6Cl5 groups that participate in the Cl···Ag interactions in 2.
 | ||
| Fig. 4 [Ag2PtIVPtII3(C6Cl5)8(μ2-OH)2(μ3-OH)2] (2) showing the Pt–Ag bonds and the Cl⋯Ag interactions. Ag is in yellow. Pt–Ag distances (Å): 2.7163(13) and 2.8855(12); Ag⋯ortho-Cl distances (Å): 2.630(4), 2.694(4), 2.781(5), and 2.939(5). | ||
The crystal of the neutral, nanosized molecule 2 acts as a clathrate, with benzene molecules accommodated in well defined voids. It is also porous, (Fig. S3, ESI†) with channels along the crystallographic a-axis.‡ The formation of channels was also observed in compound 3 and is likely the result of a preferred packing arrangement for the eight C6Cl5 groups of the neutral heteronuclear compounds 2 and 3.
Compound 2 is not the result of a template action by the silver atom, as was the case for the cyclic product 3 (Fig. 1a). Rather, 2 is the result of simple addition of the silver atoms to a stable, previously isolated compound (1), with the formation of four dative Pt→Ag bonds. Compound 1 should be a useful precursor for the formation of new types of clusters with other d10 or s2 metals; and since the incoming metal atoms are assumed into the core of the molecule, they should not significantly affect the size or shape of the resulting neutral compounds, although these compounds may present different physical or chemical properties according to the nature of the guest metal. It is conceivable that such compounds would be crystallographically isomorphous to 2, in which case 1 would also be a useful precursor for molecular solids with both voids and pores.
In conclusion, the combination of compounds with different coordination index (6 and 4 in this case) and hydroxo groups can produce hydroxo clusters of unprecedented chemical and structural characteristics such as compound 1; and this may be a way to obtain new clusters for metals and bridging ligands other than platinum or hydroxide. Compound 1 also raises interesting questions about the reactivity of the μ3-OH bridge to platinum atoms in different oxidation states; about the putative influence of Ag+ in the oxidation of the PtII starting material; and about the behavior of compound 1 as a metalloligand for the preparation of heteropolynuclear compounds with multiple Pt→M dative bonds. The latter question, answered affirmatively by the preparation of 2, in turn raises intriguing possibilities for the formation of new compounds which may display interesting physical properties, or for preparing void-containing systems enclosing organic molecules such as benzene, or for forming porous solids. Also, compound 1 should be a good precursor for new hydroxo compounds of platinum. We are presently investigating some of these possibilities.
Funding was provided by the Ministerio de Ciencia e Innovación (Spain) under grants MAT2015-68200-C2-1-P and CTQ2015-67461-P with cofinancing from the EU Regional Development Fund (FEDER) and additional support from the Diputación General de Aragón (DGA-M4: E21). We thank Prof. Juan Forniés and Dr Babil Menjón for helpful discussions. The use of the Servicio General de Apoyo a la Investigación-SAI, Universidad de Zaragoza is acknowledged.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- G. Yin, Coord. Chem. Rev., 2010, 254, 1826 CrossRef CAS.
- (a) T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436 CrossRef CAS PubMed; (b) B. Lippert and P. J. Sanz Miguel, Coord. Chem. Rev., 2016, 327–328, 333 CrossRef CAS.
- H. W. Roesky, S. Singh, K. K. M. Yusuff, J. A. Maguire and N. S. Hosmane, Chem. Rev., 2006, 106, 3813 CrossRef CAS PubMed; J. R. Fulton, A. W. Holland, D. J. Fox and R. G. Bergman, Acc. Chem. Res., 2002, 35, 44 CrossRef PubMed.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974 CrossRef CAS PubMed; S. Goberna-Ferrón, L. Vigara, J. Soriano-López and J. R. Galán-Mascarós, Inorg. Chem., 2012, 51, 11707 CrossRef PubMed.
- W. Bury, E. Krajewska, M. Dutkiewicz, K. Sokolowski, I. Justyniak, Z. Kaszkur, K. J. Kurzydlowski, T. Plocinski and J. Lewinski, Chem. Commun., 2011, 47, 5467 RSC.
- H. Furukawa, F. Gandara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369 CrossRef CAS PubMed.
- S. K. Ghosh and S. P. Rath, J. Am. Chem. Soc., 2010, 132, 17983 CrossRef CAS PubMed; M. Soler, W. Wernsdorfer, K. Folting, M. Pink and G. Christou, J. Am. Chem. Soc., 2004, 126, 2156 CrossRef PubMed.
- (a) T. G. Spiro, D. H. Templeton and A. Zalkin, Inorg. Chem., 1968, 7, 2165 CrossRef CAS; (b) K. Saruhashi and S. Kobayashi, J. Am. Chem. Soc., 2006, 128, 11232 CrossRef CAS PubMed.
- (a) G. E. Glass, J. Konnert, M. G. Miles, D. Britton and R. S. Tobias, J. Am. Chem. Soc., 1968, 90, 1131 CrossRef CAS; (b) F. D. Rochon, A. Morneau and R. Melanson, Inorg. Chem., 1988, 27, 10 CrossRef CAS; (c) N. M. Weliange, E. Szuromi and P. R. Sharp, J. Am. Chem. Soc., 2009, 131, 8736 CrossRef CAS PubMed.
- F. A. Cotton, Z. Li, C. Y. Liu and C. A. Murillo, Inorg. Chem., 2007, 46, 9294 CrossRef CAS PubMed.
- P.-C. Huang, K. Parthasarathy and C.-H. Cheng, Chem. – Eur. J., 2013, 19, 460 CrossRef CAS PubMed.
- (a) N. M. Weliange and P. R. Sharp, Organometallics, 2012, 31, 6823 CrossRef CAS; (b) J. Khusnutdinova, P. Y. Zavalij and A. N. Vedernikov, Organometallics, 2007, 26, 3466 CrossRef CAS.
- J. J. Wilson and S. J. Lippard, Chem. Rev., 2014, 114, 4470 CrossRef CAS PubMed.
- R. Usón, J. Forniés, L. R. Falvello, I. Ara and I. Usón, Inorg. Chim. Acta, 1993, 212, 105 CrossRef.
- L. R. Falvello, J. Forniés, E. Lalinde, B. Menjón, M. A. García-Monforte, M. T. Moreno and M. Tomás, Chem. Commun., 2007, 3838 RSC.
- G. López, J. Ruiz, G. García, C. Vicente, J. M. Martí, J. A. Hermoso, A. Vegas and M. J. Martínez-Ripoll, J. Chem. Soc., Dalton Trans., 1992, 53 RSC.
- (a) M.-E. Moret, Top. Organomet. Chem., 2011, 35, 157 CrossRef CAS; (b) R. Usón, J. Forniés and M. Tomás, Organomet. Chem., 1988, 358, 525 CrossRef.
- R. Usón, J. Forniés, M. Tomás, J. M. Casas, F. A. Cotton and L. R. Falvello, J. Am. Chem. Soc., 1985, 107, 2556 CrossRef.
- (a) R. Usón, J. Forniés, M. Tomás and I. Usón, Angew. Chem., Int. Ed. Engl., 1990, 29, 1449 CrossRef; (b) J. R. Berenguer, B. Gil, J. Forniés and E. Lalinde, Inorg. Chem., 2009, 48, 5250 CrossRef CAS PubMed; (c) L. R. Falvello, J. Forniés, R. Garde, A. García, E. Lalinde, M. T. Moreno, A. Steiner, M. Tomás and I. Usón, Inorg. Chem., 2006, 45, 2543 CrossRef CAS PubMed; (d) J. Forniés, N. Giménez, S. Ibáñez, E. Lalinde, A. Martín and M. T. Moreno, Inorg. Chem., 2015, 54, 4351 CrossRef PubMed.
- R. Usón, J. Forniés, M. Tomás and R. Garde, J. Am. Chem. Soc., 1995, 117, 1837 CrossRef.
- (a) A. Díez, E. Lalinde and M. T. Moreno, Coord. Chem. Rev., 2011, 255, 2426 CrossRef; (b) Z.-N. Chen, N. Zhao, Y. Fan and J. Ni, Coord. Chem. Rev., 2009, 253, 1 CrossRef CAS.
- (a) R. Usón, J. Forniés, M. Tomás and I. Ara, Inorg. Chem., 1994, 33, 4023 CrossRef; (b) T. Yamaguchi, F. Yamazaki and T. Ito, J. Am. Chem. Soc., 2001, 123, 743 CrossRef CAS PubMed; (c) F. Liu, W. Chen and W. Wang, Dalton Trans., 2006, 3015 RSC.
- J. Forniés, C. Fortuño, S. Ibáñez and A. Martín, Inorg. Chem., 2008, 47, 5978 CrossRef PubMed.
- M.-E. Moret, D. Serra, A. Bach and P. Chen, Angew. Chem., Int. Ed., 2010, 49, 2873 CrossRef CAS PubMed.
- G. J. Arsenault, C. M. Anderson and R. J. Puddephatt, Organometallics, 1988, 7, 2094 CrossRef CAS.
- M. S. Safa, M. C. Jennings and R. J. Puddephatt, Chem. Commun., 2009, 1487 RSC.
- J. Forniés, B. Menjón, R. Sanz-Carrillo, M. Tomás, N. G. Connelly, J. G. Crossley and G. Orpen, J. Am. Chem. Soc., 1995, 117, 4295 CrossRef.
- M. Melnik, P. Mikus and C. E. Holloway, Cent. Eur. J. Chem., 2013, 11, 827 CAS.
- (a) M. R. Colsman, M. D. Noirot, M. M. Miller, O. P. Anderson and S. H. Strauss, J. Am. Chem. Soc., 1988, 110, 6886 CrossRef CAS; (b) A. Decken, C. Knapp, G. B. Nikiforov, J. Passmore, J. M. Rautiainen, X. Wang and X. Zeng, Chem. – Eur. J., 2009, 15, 6504 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 9 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic and crystallographic procedures; figures as cited in the text. CCDC 1461798 and 1461799. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc07712b |
| ‡ Poorly defined electron density was observed in the channels and was modeled as partially occupied, disordered water. (In a separate refinement, not reported here, this electron density was masked using the SQUEEZE algorithm.30) |
| This journal is © The Royal Society of Chemistry 2017 |