
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evidence and isolation of tetrahedral intermediates formed upon the addition of lithium carbenoids to Weinreb amides and N-acylpyrroles†
Laura
Castoldi
,
Wolfgang
Holzer
,
Thierry
Langer
and
Vittorio
Pace
 *
*
University of Vienna – Department of Pharmaceutical Chemistry, Althanstrasse, 14, A-1090, Vienna, Austria. E-mail: vittorio.pace@univie.ac.at
First published on 1st August 2017
Abstract
The tetrahedral intermediates generated upon the addition of halolithium carbenoids (LiCH2X and LiCHXY) to Weinreb amides have been intercepted and fully characterized as O-TMS heminals. The commercially available N-trimethylsilyl imidazole is the ideal trapping agent whose employment, combined with a straightforward neutral Alox chromatographic purification, enables the isolation of such labile species. The procedure could be advantageously extended also for obtaining O-TMS heminals from N-acylpyrroles. These intermediates manifest interesting reactivity including as precursors of more complex carbenoids.
The addition reactions of nucleophiles to the amide carbonyl constitute extremely important chemical transformations governing key cascades in biological systems and fundamental synthetic operations.1 Mechanistically, a given nucleophile adds to a C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionality affording a tetrahedral intermediate which, depending on the reaction conditions, could restore the carbonyl group (via the exclusion of a suitable leaving group, i.e. addition–elimination pathway). Conversely – whenever the leaving group could not be eliminated – the overall process upon simple acidolysis affords an hydroxyl-type final product.2 For a long time, the tamed reactivity of amides towards carbon nucleophiles compared to other carboxylic acid derivatives has been rewarded as a limitation, thus considering them reluctant species in analogous processes (Scheme 1).3 Nowadays, the opportune activation of the amide bond with electrophilic reagents represents an excellent and robust tool for enabling the chemoselective addition of organometallic reagents en route to the synthesis of ketones, alcohols, or amines as documented in illuminating recent works by Charette,4 Huang5 and Movassaghi.6 Undoubtedly, the introduction by Nahm and Weinreb of N-methoxy-N-methyl amides in 19817 represented a breakthrough in the field and currently they are routinely employed for accessing carbonyls with high chemocontrol.8 The complexation of the metal of the putative tetrahedral intermediate to form a five-membered cycle is the critical factor justifying the effectiveness in hampering common undesired drawbacks such as overaddition phenomena or low reactivity. Further advancements on N-alkoxy amides pointed out the feasibility of the addition of two different organometallics as showcased recently in elegant works by Chida–Sato,9 Sarpong,10 Helmchen,11 Feringa,12 Schwartz13 and Vincent–Kouklovsky,14 thus, disclosing new concepts for carbonyl and amine synthesis. Additionally, the recent development of impressive catalytic tactics for enabling the reactivity of amide carbonyls with organometallic reagents by the group of Garg15 and Szostak16 opened new avenues for the use of amide as feedstocks in analogous processes.
O functionality affording a tetrahedral intermediate which, depending on the reaction conditions, could restore the carbonyl group (via the exclusion of a suitable leaving group, i.e. addition–elimination pathway). Conversely – whenever the leaving group could not be eliminated – the overall process upon simple acidolysis affords an hydroxyl-type final product.2 For a long time, the tamed reactivity of amides towards carbon nucleophiles compared to other carboxylic acid derivatives has been rewarded as a limitation, thus considering them reluctant species in analogous processes (Scheme 1).3 Nowadays, the opportune activation of the amide bond with electrophilic reagents represents an excellent and robust tool for enabling the chemoselective addition of organometallic reagents en route to the synthesis of ketones, alcohols, or amines as documented in illuminating recent works by Charette,4 Huang5 and Movassaghi.6 Undoubtedly, the introduction by Nahm and Weinreb of N-methoxy-N-methyl amides in 19817 represented a breakthrough in the field and currently they are routinely employed for accessing carbonyls with high chemocontrol.8 The complexation of the metal of the putative tetrahedral intermediate to form a five-membered cycle is the critical factor justifying the effectiveness in hampering common undesired drawbacks such as overaddition phenomena or low reactivity. Further advancements on N-alkoxy amides pointed out the feasibility of the addition of two different organometallics as showcased recently in elegant works by Chida–Sato,9 Sarpong,10 Helmchen,11 Feringa,12 Schwartz13 and Vincent–Kouklovsky,14 thus, disclosing new concepts for carbonyl and amine synthesis. Additionally, the recent development of impressive catalytic tactics for enabling the reactivity of amide carbonyls with organometallic reagents by the group of Garg15 and Szostak16 opened new avenues for the use of amide as feedstocks in analogous processes.
 | ||
| Scheme 1 General context of the presented work. | ||
Recently, we recognized the excellent performance of Weinreb amides as privileged acylating agents for chemoselective homologations with α-substituted methyllithium reagents (LiCH2X).17 Compared to more classical substrates for similar reactions (e.g. esters), Weinreb amides showed an impressive superiority, which we attributed to the chelating effect typical of these species. However, the inherent transient nature of the tetrahedral intermediate18 formed through the addition of a strongly nucleophilic organometallic reagent implies severe difficulties in isolating and identifying via spectroscopic methods these species and, ultimately to fully uncover their synthetic potential.19 With the aim to support these hypotheses, we document herein the isolation and the structural elucidation of O-trimethylsilyl protected hemiaminals obtained via the addition of lithium halocarbenoids (LiCH2X and LiCHXY)20 to variously functionalized Weinreb amides and analogous N-acylpyrroles.21
At the outset of our investigations, we easily recognized the importance of finding suitable reaction conditions and work-up operations enabling not only the safe capture but, more importantly, also preserving the chemical integrity of the labile tetrahedral intermediate. The ideal trapping agent should present a remarkable oxophilicity and contemporaneously provide the sufficient stability to avoid the undesired elimination of the N-methyl-N-methoxy fragment, which would have afforded the corresponding ketone. Cognizant of these critical requirements, Weinreb amide 1 was selected as the model substrate and reacted with LiCH2Cl (Table 1) under our usual Barbier-type conditions.17c,22 Neither through basic aqueous or methanolic work-up could the free hemiaminal be isolated. TMSCl in the presence of pyridine (entry 1) served as an effective trapping agent (3) as deduced from the diagnostic hemiaminal (carbon) 13C (96.0 ppm) and (nitrogen) 15N-NMR (−207.3 ppm) resonances. The employment of imidazole as the base was particularly convenient since the conversion could be increased up to 90% (entry 2). Pleasingly, the commercially available N-TMS-imidazole (Im-TMS) allowed to maximize the conversion up to 95% (entry 3), whereas the more bulky TBDMS and TPS reagents afforded exclusively the corresponding α-chloroketone (entries 4–5), presumably due to steric factors affecting negatively the stability of the intermediate. The work-up and purification technique followed for getting analytically pure 3 deserve particular mention. After basic aqueous treatment (NaHCO3 5%) and extraction of the organic phase with Et2O, we noticed a substantial transformation of the product into ketone 4 during recording of NMR spectra in CDCl3, presumably because of the acidity of this deuterated solvent. Analogously, attempts to purify the crude mixture through silica gel were unsuccessful, even upon prior neutralizing treatment with TEA (10% or neat) or TMSCl (entries 6–8). Excellent purification was achieved by using Brockmann 3 grade neutral alumina (AloxN-BG3), which allowed us to finally isolate 3 in 91% yield (entry 9). Different deactivation grades of Alox were less effective (entries 10–11), while different alumina type phases (acidic or basic) were not suitable for the purpose (not shown).
|
|
||||
|---|---|---|---|---|
| Entrya | Trapping agent (2.0 equiv.) | Conversion/ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4b (%) :4b (%) |
Chromatographic system | Yield of 3c,d (%) |
| a LiCH2Cl was generated from ICH2Cl (3.0 equiv.) and MeLi-LiBr (2.8 equiv.) in THF at −78 °C. b 1H-NMR calculated conversion. c Otherwise stated, yields refer to isolated and purified compounds according to conditions in entry 9 (AloxN-BG3). d For the preparation of AloxN-BG3, see the ESI. | ||||
| 1 | TMSCl-Py | 87/5:1 |
AloxN-BG3 | 71 |
| 2 | TMSCl-Im | 90/20:1 |
AloxN-BG3 | 70 |
| 3 | Im-TMS | 95/25:1 |
AloxN-BG3 | 86 |
| 4 | Im-TBDMS | 86/0:1 |
— | — |
| 5 | Im-TPS | 89/0:1 |
— | — |
| 6 | Im-TMS | 95/25:1 |
SiO2 | 0 |
| 7 | Im-TMS | 95/25:1 |
SiO2-TEA (10%) | 6 |
| 8 | Im-TMS | 95/25:1 |
SiO2-TMSCl (5%) | 12 |
| 9 | Im-TMS | 95/25:1 |
AloxN-BG3 | 91 |
| 10 | Im-TMS | 95/25:1 |
AloxN-BG4 | 78 |
| 11 | Im-TMS | 95/25:1 |
AloxN-BG2 | 70 |

With the optimized conditions in hand, we then applied the methodology to different Weinreb amides (Scheme 2). The corresponding tetrahedral intermediates could be easily isolated and characterized in high to excellent chemical yields. Accordingly, aromatic (3–11) and heteroaromatic (12–13) Weinreb amides provided the O-TMS protected hemiaminals regardless of the electronic behavior of the substituents across the ring. Notably, the presence of an exchangeable iodine atom on the aromatic ring did not have a detrimental effect (10), thus highlighting an excellent chemoselectivity during the carbenoid generation event starting from ICH2Cl. Switching to aliphatic analogues (14–15), no alteration was observed during the trapping–isolation sequence even in the presence of a α-heteroatom (15). Tetrahedral intermediates generated upon the addition of (mixed) dihalocarbenoids23 could be easily accessed (16–20) and could serve as excellent placeholders for forming more complex, synthetically versatile carbenoids (vide infra). Remarkably, C6D6 solutions of the intermediates showed an excellent stability up to 6 months (kept at −20 °C), thus further showcasing the convenience of this solvent for NMR analyses.
 | ||
| Scheme 2 Trapping–isolation sequence of O-TMS protected aminals from Weinreb amides with monohalo- and dihalomethyllithium carbenoids. | ||
The protocol was also amenable for forming the tetrahedral intermediates – under analogous reaction conditions – starting from N-acylpyrroles (Scheme 3). Again the aromatic (21–26) vs. aliphatic (27–28) nature of the materials did not influence the positive outcome of the process, which significantly also proved to be applicable to intermediates that formed upon the addition of LiCH2Br.24 However, the increase of sterical demand on the pyrrole core slightly decreases the isolated yield of the protected hemiaminal (26).
 | ||
| Scheme 3 O-TMS protected hemiaminals from N-acylpyrroles and carbenoids. | ||
Previous reports from Evans25 and Brandänge26 entailed the formation and isolation of hemiaminals from N-acylpyrroles and RLi or RMgX in the native form (i.e. presenting a free OH functionality).27 However, in the cases discussed herein, the plausible intramolecular formation of a halogen bond28 between the putative OH and the halogen would trigger the elimination and, as such, it renders it impossible to obtain analogous hemiaminals (Scheme 4).
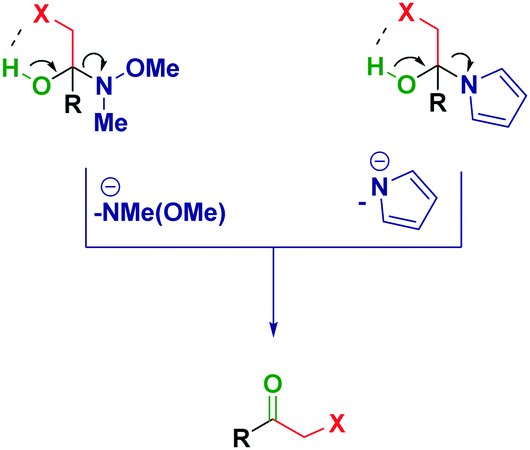 | ||
| Scheme 4 Plausible halogen-bond formation on putative hemiaminals. | ||
The reactivity of the synthesized tetrahedral intermediates was then tested with the aim of investigating the synthetic utility of these species (Scheme 5). The iodo-containing intermediate 10 resulted in an excellent substrate for the Feringa/Fañanas-Mastral Pd-catalyzed coupling of an organolithium.29 Significantly, under the reaction conditions the tetrahedral intermediate could be completely preserved from degradation (29). An O-TMS protected hemiaminal (19) featuring two bromine atoms was amenable to subsequent Li/Br exchange to generate a new, more complex carbenoid (19a) whose stability was increased by the chelating effect of the N-methoxy group. The attack of this carbenoid to benzaldehyde generated an O-lithiated bromohydrin (19b), which upon spontaneous ring-closure and acidic treatment resulted in the formation of the highly valuable and challenging α,β-epoxyketone 30.30 Moreover, Lewis acid treatment (TMSOTf) of the N-acylpyrrole derivative hemiaminal 28 followed by trapping with ethyl iodide afforded the O-ethyl product 31.
 | ||
| Scheme 5 Derivatization of O-TMS protected hemiaminals. | ||
In summary, we have disclosed an effective strategy for obtaining O-trimethylsilyl protected hemiaminals formed through the addition of lithium monohalo- and dihalo-carbenoids to Weinreb amides. The proper combination of Im-TMS (as the trapping agent) and Brockmann grade 3 neutral alumina (as the stationary phase for chromatography) was found to be – together with recording NMR analyses in benzene-d6 – the critical factor for enabling the isolation of such labile tetrahedral intermediates. The method is also amenable for forming analogous protected hemiaminals from N-acylpyrroles. A concise examination of the manipulation these species may undergo is also discussed.
The authors gratefully acknowledge the generous support and a doctoral Uni:docs grant to L.C. from University of Vienna.
Notes and references
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed; (b) The Amide Linkage. Selected Structural Aspects in Chemistry, Biochemistry and Materials Science, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, John Wiley & Sons Inc., New York, 2000 Search PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley-VCH, Weinheim, 5th edn, 2001 Search PubMed.
- Reviews: (a) V. Pace, W. Holzer and B. Olofsson, Adv. Synth. Catal., 2014, 356, 3697 CrossRef CAS; (b) V. Pace and W. Holzer, Aust. J. Chem., 2013, 66, 507 CAS; (c) D. Seebach, Angew. Chem., Int. Ed., 2011, 50, 96 CrossRef CAS PubMed.
- (a) A. B. Charette and M. Grenon, Can. J. Chem., 2001, 79, 1694 CrossRef CAS; (b) W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228 CrossRef CAS PubMed.
- (a) K.-J. Xiao, J.-M. Luo, X.-E. Xia, Y. Wang and P.-Q. Huang, Chem. – Eur. J., 2013, 19, 13075 CrossRef CAS PubMed; (b) K.-J. Xiao, A.-E. Wang, Y.-H. Huang and P.-Q. Huang, Asian J. Org. Chem., 2012, 1, 130 CrossRef CAS; (c) K.-J. Xiao, A.-E. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2012, 51, 8314 CrossRef CAS PubMed; (d) K.-J. Xiao, Y. Wang, K.-Y. Ye and P.-Q. Huang, Chem. – Eur. J., 2010, 16, 12792 CrossRef CAS PubMed; (e) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037 CrossRef CAS PubMed.
- J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2012, 51, 4572 CrossRef CAS PubMed.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815 CrossRef CAS.
- Review: (a) S. Balasubramaniam and I. S. Aidhen, Synthesis, 2008, 3707 CAS ; For a concise overview on N-alkoxyamide, see: ; (b) T. Sato and N. Chida, Org. Biomol. Chem., 2014, 12, 3147 RSC ; For the development of new acylating reagents (N-acylazetidines), see: ; (c) C. Liu, M. Achtenhagen and M. Szostak, Org. Lett., 2016, 18, 2375 CrossRef CAS PubMed.
- (a) K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2010, 49, 6369 CrossRef CAS PubMed; (b) Y. Kurosaki, K. Shirokane, T. Oishi, T. Sato and N. Chida, Org. Lett., 2012, 14, 2098 CrossRef CAS PubMed; (c) Y. Oda, T. Sato and N. Chida, Org. Lett., 2012, 14, 950 CrossRef CAS PubMed; (d) Y. Yanagita, H. Nakamura, K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, Chem. – Eur. J., 2013, 19, 678 CrossRef CAS PubMed; (e) K. Shirokane, T. Wada, M. Yoritate, R. Minamikawa, N. Takayama, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2014, 53, 512 CrossRef CAS PubMed; (f) M. Yoritate, T. Meguro, N. Matsuo, K. Shirokane, T. Sato and N. Chida, Chem. – Eur. J., 2014, 20, 8210 CrossRef CAS PubMed; (g) M. Nakajima, Y. Oda, T. Wada, R. Minamikawa, K. Shirokane, T. Sato and N. Chida, Chem. – Eur. J., 2014, 20, 17565 CrossRef CAS PubMed.
- S. T. Heller, J. N. Newton, T. Fu and R. Sarpong, Angew. Chem., Int. Ed., 2015, 54, 9839 CrossRef CAS PubMed.
- M. Jäkel, J. Qu, T. Schnitzer and G. Helmchen, Chem. – Eur. J., 2013, 19, 16746 CrossRef PubMed.
- M. Giannerini, C. Vila, V. Hornillos and B. L. Feringa, Chem. Commun., 2016, 52, 1206 RSC.
- J. Nugent and B. D. Schwartz, Org. Lett., 2016, 18, 3834 CrossRef CAS PubMed.
- G. Vincent, R. Guillot and C. Kouklovsky, Angew. Chem., Int. Ed., 2011, 50, 1350 CrossRef CAS PubMed.
- (a) J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413 CrossRef CAS PubMed ; For a breakthrough in the field, see: ; (b) L. Hie, N. F. Fine Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79 CrossRef CAS PubMed.
- (a) G. Meng and M. Szostak, Org. Lett., 2015, 17, 4364 CrossRef CAS PubMed; (b) G. Meng, S. Shi and M. Szostak, ACS Catal., 2016, 6, 7335 CrossRef CAS.
- (a) V. Pace, W. Holzer, G. Verniest, A. R. Alcántara and N. De Kimpe, Adv. Synth. Catal., 2013, 355, 919 CrossRef CAS; (b) V. Pace, I. Murgia, S. Westermayer, T. Langer and W. Holzer, Chem. Commun., 2016, 52, 7584 RSC; (c) V. Pace, L. Castoldi and W. Holzer, J. Org. Chem., 2013, 78, 7764 CrossRef CAS PubMed; (d) A. D. Mamuye, L. Castoldi, U. Azzena, W. Holzer and V. Pace, Org. Biomol. Chem., 2015, 13, 1969 RSC ; For accounts, see: ; (e) V. Pace, W. Holzer and N. De Kimpe, Chem. Rec., 2016, 16, 2061 CrossRef CAS PubMed; (f) V. Pace, L. Castoldi, S. Monticelli, M. Rui and S. Collina, Synlett, 2017, 879 CrossRef CAS.
- Review: M. Adler, S. Adler and G. Boche, J. Phys. Org. Chem., 2005, 18, 193 CrossRef CAS.
- The addition of the Ruppert–Prakash reagent (TMSCF3) to Weinreb amides gives trifluoromethyl analogue intermediates: D. M. Rudzinski, C. B. Kelly and N. E. Leadbeater, Chem. Commun., 2012, 48, 9610 RSC.
- Reviews: (a) V. Capriati, Contemporary Carbene Chemistry, John Wiley & Sons, Inc, 2013, p. 325 Search PubMed; (b) V. Capriati and S. Florio, Chem. – Eur. J., 2010, 16, 4152 CrossRef CAS PubMed; (c) V. H. Gessner, Chem. Commun., 2016, 52, 12011 RSC; (d) S. Molitor and V. H. Gessner, Synlett, 2015, 861 CAS ; For recent contributions, see: ; (e) S. Molitor and V. H. Gessner, Angew. Chem., 2016, 128, 7843 CrossRef; (f) S. Molitor, K.-S. Feichtner, C. Kupper and V. H. Gessner, Chem. – Eur. J., 2014, 20, 10752 CrossRef CAS PubMed; (g) S. Molitor, J. Becker and V. H. Gessner, J. Am. Chem. Soc., 2014, 136, 15517 CrossRef CAS PubMed; (h) V. Pace, L. Castoldi, E. Mazzeo, M. Rui, T. Langer and W. Holzer, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201706236.
- Review: A. M. Goldys and C. S. P. McErlean, Eur. J. Org. Chem., 2012, 1877 CrossRef CAS.
- (a) V. Pace, Aust. J. Chem., 2014, 67, 311 CAS ; For an interesting report on the use of LiCH2X under non-Barbier type conditions via flow chemistry, see: ; (b) L. Degennaro, F. Fanelli, A. Giovine and R. Luisi, Adv. Synth. Catal., 2015, 357, 21 CrossRef CAS.
- V. Pace, L. Castoldi, A. D. Mamuye and W. Holzer, Synthesis, 2014, 2897 CrossRef.
- V. Pace, A. Pelosi, D. Antermite, O. Rosati, M. Curini and W. Holzer, Chem. Commun., 2016, 52, 2639 RSC.
- D. A. Evans, G. Borg and K. A. Scheidt, Angew. Chem., Int. Ed., 2002, 41, 3188 CrossRef CAS PubMed.
- S. Brandänge, E. Holmgren, H. Leijonmarck and B. Rodriguez, Acta Chem. Scand., 1995, 49, 922 CrossRef.
- For additional rare cases, see: (a) M. Szostak, L. Yao and J. Aubé, J. Am. Chem. Soc., 2010, 132, 2078 CrossRef CAS PubMed; (b) M. Szostak and J. Aube, Org. Biomol. Chem., 2011, 9, 27 RSC; (c) M. Szostak and J. Aubé, Chem. Rev., 2013, 113, 5701 CrossRef CAS PubMed; (d) A. J. Kirby, I. V. Komarov and N. Feeder, J. Am. Chem. Soc., 1998, 120, 7101 CrossRef CAS; (e) J. Halli, K. Hofman, T. Beisel and G. Manolikakes, Eur. J. Org. Chem., 2015, 4624 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- (a) M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667 CrossRef CAS PubMed For a highlight, see: ; (b) V. Pace and R. Luisi, ChemCatChem, 2014, 6, 1516 CrossRef CAS.
- (a) Q. Ke, B. Zhang, B. Hu, Y. Jin and G. Lu, Chem. Commun., 2015, 51, 1012 RSC; (b) W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc05215d |
| This journal is © The Royal Society of Chemistry 2017 |