
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochromism and molecular logic gate operation of a water-compatible bis-glycosyl diarylethene†
Xianzhi
Chai‡
a,
You-Xin
Fu‡
a,
Tony D.
James
 b,
Junji
Zhang
*a,
Xiao-Peng
He
*a and
He
Tian
a
b,
Junji
Zhang
*a,
Xiao-Peng
He
*a and
He
Tian
a
aKey Laboratory for Advanced Materials & Institute of Fine Chemicals, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai, 200237, P. R. China. E-mail: zhangjunji@ecust.edu.cn; xphe@ecust.edu.cn
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
First published on 1st August 2017
Abstract
We report the synthesis of a water-compatible bis-glycosyl diarylethene using click chemistry that undergoes photochromism and functions as a molecular logic gate.
Photochromic molecules are a family of unique chromophores, which can undergo light-controlled reversible transformations between two isomeric states.1 Because of the sharp difference in physical properties2 such as optical,3 electrochemical4 and geometrical features5 between the two isomers, photochromic compounds can be used to construct a variety of photoswitchable functional materials including fluorescence probes,6 molecular logic gates,7 optical data-storage devices8 and photo-responsive assemblies.9
Of the photochromic compounds reported, diarylethenes are viewed as one of the most promising candidates for photo-electronic applications due to their outstanding fatigue-resistance,10 thermal stability11 and rapid response towards light.12 In recent years, the design and synthesis of diarylethene derivatives incorporating fluorophores have been a decent strategy in the rational design of molecular logic gates based on Förster resonance energy transfer (FRET) or photoinduced electron transfer (PET).13,14
Logic-gate systems are employed in the binary notation, 0 and 1, and the principles of Boolean language15–17 to mimic computing functions of conventional semiconductors. More interestingly, they have also been extended for biochemical applications such as drug delivery, pro-drug activation and medical diagnostics.18,19 Recently, Schiller et al. reported a Mn(I) tricarbonyl complex as a molecular logic “OR” gate with two inputs: light and peroxide and outputs: fluorescence and CO, which can be used to monitor peroxide and track CO in vivo.20 Berreau et al. reported an extended flavonol motif which operates as an AND logic gate in the sensing of cellular thiols and release of CO induced by visible light and O2.21 The requirement of O2 for CO release demonstrates that the molecular logic system is suitable for probing variable cellular levels of O2.
Despite the progress made in the synthesis and application of diarylethene derivatives for molecular logic gates, the majority of the systems developed require the presence of a large portion of organic solvents, which are not suitable for biological applications. As a result, the development of water-compatible photo-switchable logic gates is still a challenging task.22–25 Recently, we have demonstrated that the glycosylation of hydrophobic organic dyes is a promising strategy to overcome the water insolubility of traditional organic dyes, achieving full-aqueous sensing and targeted cell imaging.26 Here we show that the introduction of a carbohydrate group to diarylethenes can lead to the development of a water-compatible reversible photochromic system with logic-gate operation.
A new bis-glycosyl diarylethene 1o (Scheme 1) was synthesized by the Suzuki coupling between a naphthalimide derivative with a piperazine moiety and diarylethene.27 Then, the resulting conjugate was coupled with two β-D-galactosides by a click reaction (Scheme S1, ESI†). The absorbance and fluorescence spectra of 1o were first measured using a 1 × 10−5 mol L−1 compound solution in water at room temperature. As shown in Fig. 1a, the absorbance maxima of 1o in water was observed at 260 nm. Upon irradiation with a UV light of 254 nm, the absorbance at 260 nm decreased and a new band centred at 535 nm appeared. The intensity of 535 nm increased with irradiation time till the photostationary state (PSS) was reached. Meanwhile, the initial yellow colour of the aqueous solution gradually turned red, indicative of a ring-closing process to form the ring-closed isomer 1c.28 The reddish 1c solution could be recovered to the initial yellowish solution when visible light (>500 nm) was used, suggesting a photocycloreversion from 1c to 1o. The photocyclization quantum yield of the ring-open isomer (ϕ254 = 0.176) was much higher than the photocycloreversion quantum yield of ring-closed isomer (ϕ525 = 0.013).
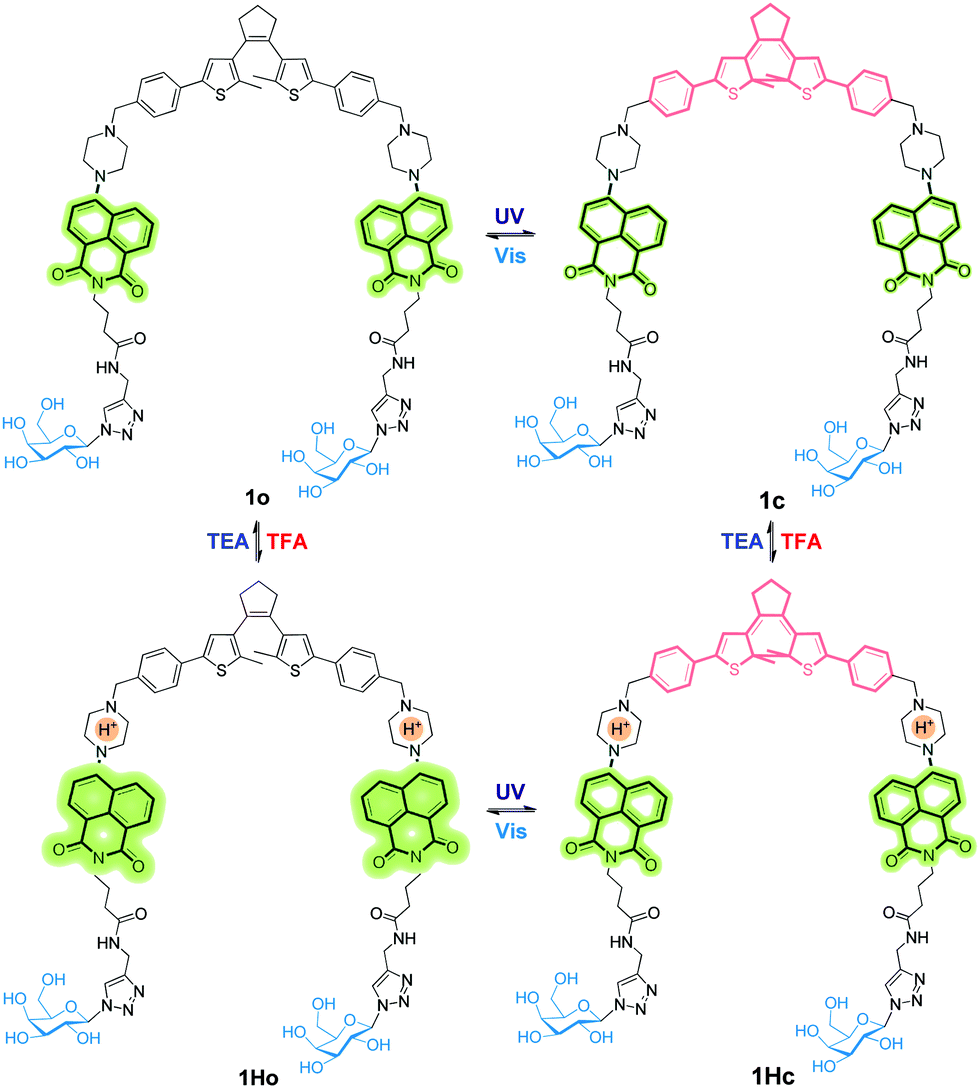 | ||
| Scheme 1 Structural transformations of 1o in response to UV/Vis and TFA/TEA stimuli. | ||
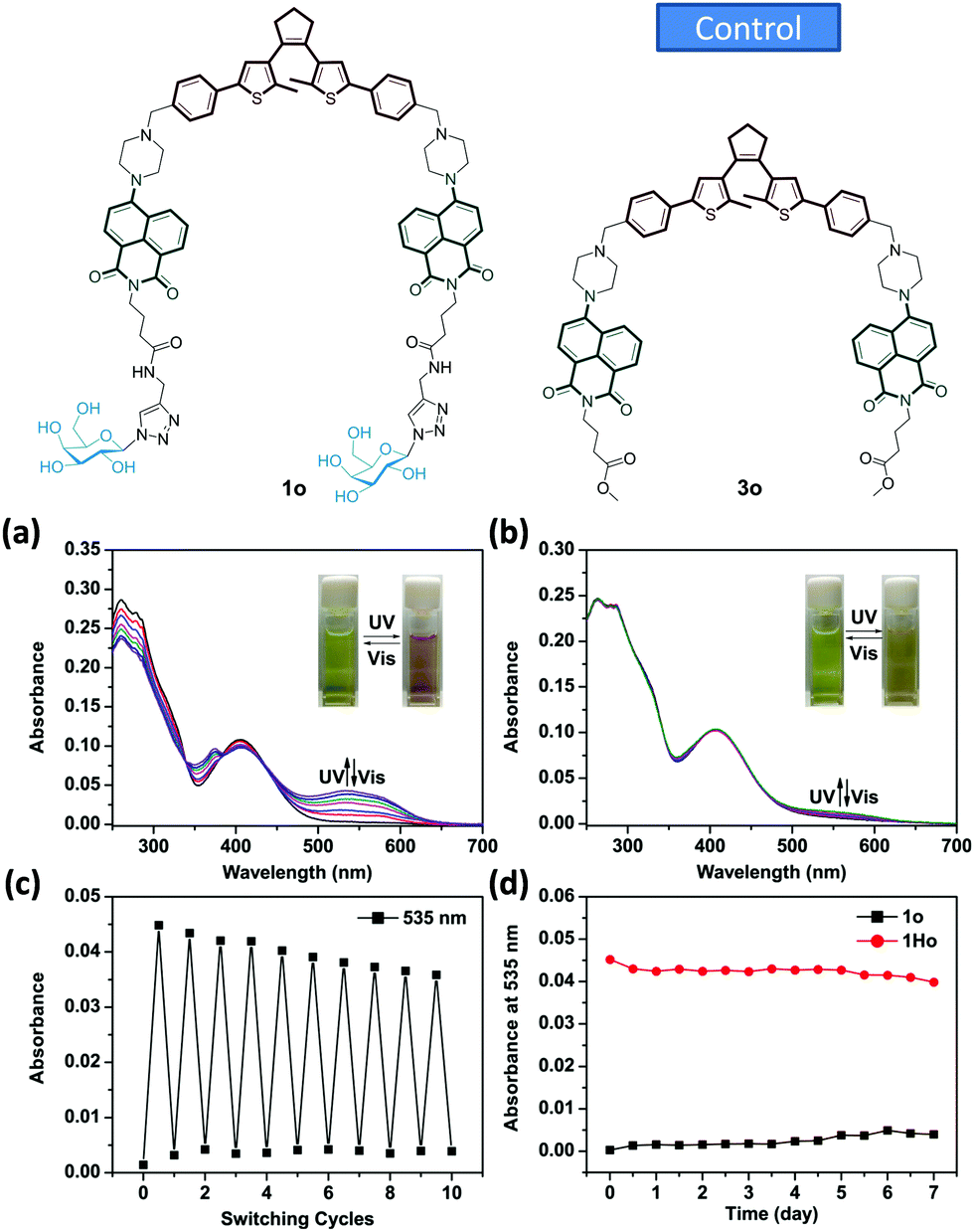 | ||
| Fig. 1 Absorption spectral changes of (a) 1o and (b) 3o (1 × 10−5 mol L−1) in water solution (with 0.25‰ Triton X-100) upon irradiation with UV (254 nm) and visible light (>500 nm). Fatigue resistance of (c) 1o (1 × 10−5 mol L−1) in water solution (with 0.25‰ Triton X-100) upon alternating irradiation with UV and visible light shown in absorption switching cycles. (d) Thermostability of 1o and 1c (1 × 10−5 mol L−1) in water solution (with 0.25‰ Triton X-100) monitored at 535 nm in absorption spectral at 50 °C in the dark. | ||
To verify the importance of the sugar groups for the water-compatible photochromism, a control compound 3o (Fig. 1b and Scheme S1, ESI†) without a bis-galactoside moiety, was synthesized. Despite an obvious photocyclization in organic solvents (Fig. S1, ESI†), photochromism of 3o could hardly be activated by UV light in the same water solution used for 1o (Fig. 1bvs.Fig. 1a). This is probably due to the poor water solubility of 3o. Unlike water-soluble 1o, the absorption spectrum baseline of the ring-opened isomer of 3o fluctuated, suggesting the formation of aggregates.29 The fatigue resistance of 1o was examined using absorbance spectra through an alternate irradiation with UV and visible light at room temperature. The coloration/decoloration cycles of 1o could be repeated 10 times in aqueous solution without obvious degradation (Fig. 1c). The compound also showed a good thermo-stability as the characteristic absorbance at 535 nm for the ring-closed isomer of 1c did not vary after being stored at 323 K in the dark for 7 days (Fig. 1d).
Fig. 2a shows the emission spectral variations of 1o in water (1 × 10−5 mol L−1) under different stimuli at room temperature. Upon excitation with a 408 nm light, the fluorescence emission peak of 1o was observed at 525 nm. Owing to the photo-induced electron transfer from piperazine to 1,8-naphthalimide unit,30 the initial fluorescence of 1o was relatively low (ϕF = 0.02, Table S1, ESI†). The PET effect can be blocked by protonation of piperazine in an acidic environment, thereby recovering the strong emission of 1,8-naphthalimide.31 After addition of 6.0 eq. of TFA to the yellow solution of 1o in water, we observed that the absorbance maxima of 1,8-naphthalimide of 1o (405 nm) blue-shifted to 390 nm in 1Ho (Fig. S2, ESI†) and the solution became colorless. Meanwhile, the fluorescence spectrum exhibited a blue shift from 525 nm to 515 nm and the intensity significantly enhanced by a factor of 3–4 (ϕF = 0.12, Table S1, ESI†). Neutralization of 1Ho to its initial state 1o by addition of triethylamine (TEA) regenerated the low fluorescence. In addition, the bis-glycosyl diarylethene 1o exhibited good fluorescence reversibility in water when adding TFA and TEA alternately for 10 cycles (Fig. 2b). The fluorescence response of 1o at different pH was determined (Fig. S3, ESI†). Under basic and neutral conditions, 1o emitted low fluorescence upon excitation at 408 nm. In contrast, 1o was strongly emissive under acidic pH and the pKa was calculated to be 4.41 ± 0.09 (Fig. S4, ESI†). This value is similar to the pH range of the lysosome (3.8–5.0),32 clearly demonstrating the potential of the system for bio-imaging applications.
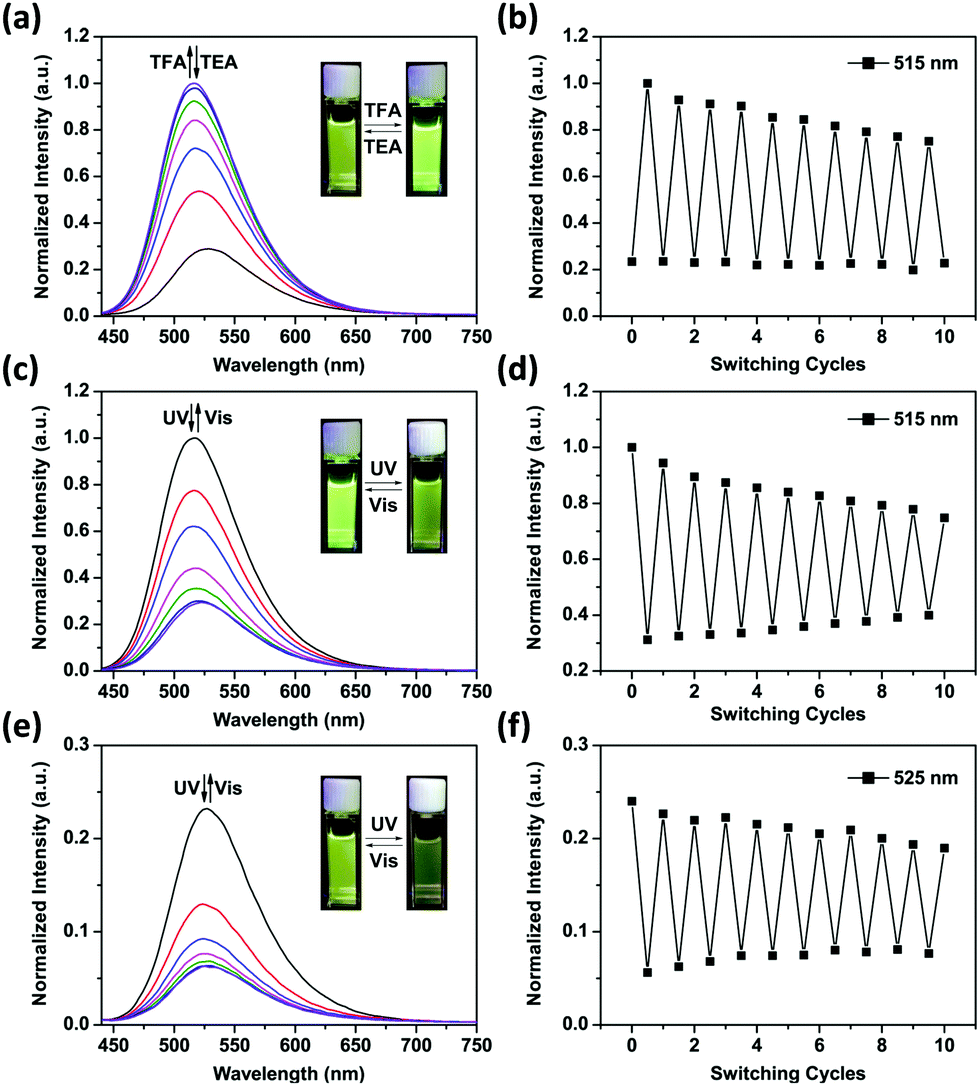 | ||
| Fig. 2 Fluorescence changes (a) and fatigue resistance (b) of 1o (1 × 10−5 mol L−1) in water solution (with 0.25‰ Triton X-100) upon alternating addition with TFA (6.0 eq.) and TEA (6.0 eq.). Fluorescence changes (c) and fatigue resistance (d) of 1Ho upon alternating irradiation with UV (254 nm) and visible light (>500 nm). Fluorescence changes (e) and fatigue resistance (f) of 1o upon alternating irradiation with UV (254 nm) and visible light (>500 nm). All emissions were produced upon excitation at 408 nm. | ||
When the protonated diarylethene 1Ho was irradiated with UV light of 254 nm, a new absorbance band at 540 nm appeared (Fig. S5, ESI†), and the colour of solution turned purple, which was red-shifted with respect to 1c (535 nm). Fig. 2c shows that the emission at 515 nm was remarkably quenched to ca. 30% (ϕF = 0.03, Table S1, ESI†) with respect to the fully protonated state (ϕF = 0.12, Table S1, ESI†). We ascribed this fluorescence quenching to an intramolecular FRET mechanism since the absorbance band of the light-induced product 1Hc overlaps well with the emission band of 1,8-naphthalimide, resulting in the strong FRET effect.33 Furthermore, the fluorescence was recovered by irradiation with visible light (>500 nm). Meanwhile, the fatigue resistance of 1Ho was shown to tolerate 10 switching cycles in water (Fig. 2d). Even with the low emissive 1o state photo-modulated FRET can take place efficiently (Fig. 2e).
The above results suggest that the emission intensity of diarylethene 1o could be independently tuned through stimulation by UV/Vis and TFA/TEA cycling (Fig. S6, ESI†). These results inspired us to employ 1o for the development of an aqueous “INH” logic gate (Fig. 3a) with two inputs: UV light (In1) and TFA (In2). The normalized fluorescence intensity at 520 nm (I520) was treated as the output signal. According to the truth table (Table S2, ESI†), for input, the presence of UV light (In1) or TFA (In2) was defined as the ‘1’ state, and their absence, as the ‘0’ state. For output, I520 value at 0.5 was set as the threshold to define the ‘1’ (> 0.5) and ‘0’ (≤0.5) states. In other words, we can use the fluorescence changes as the output by defining high fluorescence of the solution as ‘1’ state and the low fluorescence of the solution as ‘0’. Either with (1, 1) or without (0, 0) the two inputs will generate low fluorescence states (output ‘0’). When subjected to TFA input (0, 1) alone, the fluorescence of solution could increase sharply, giving an output signal of ‘1’. On the other hand, with only UV light input (1, 0), the I520 was extremely low and the output returned to ‘0’. Thus, an INH logic gate within a single molecule has been achieved.
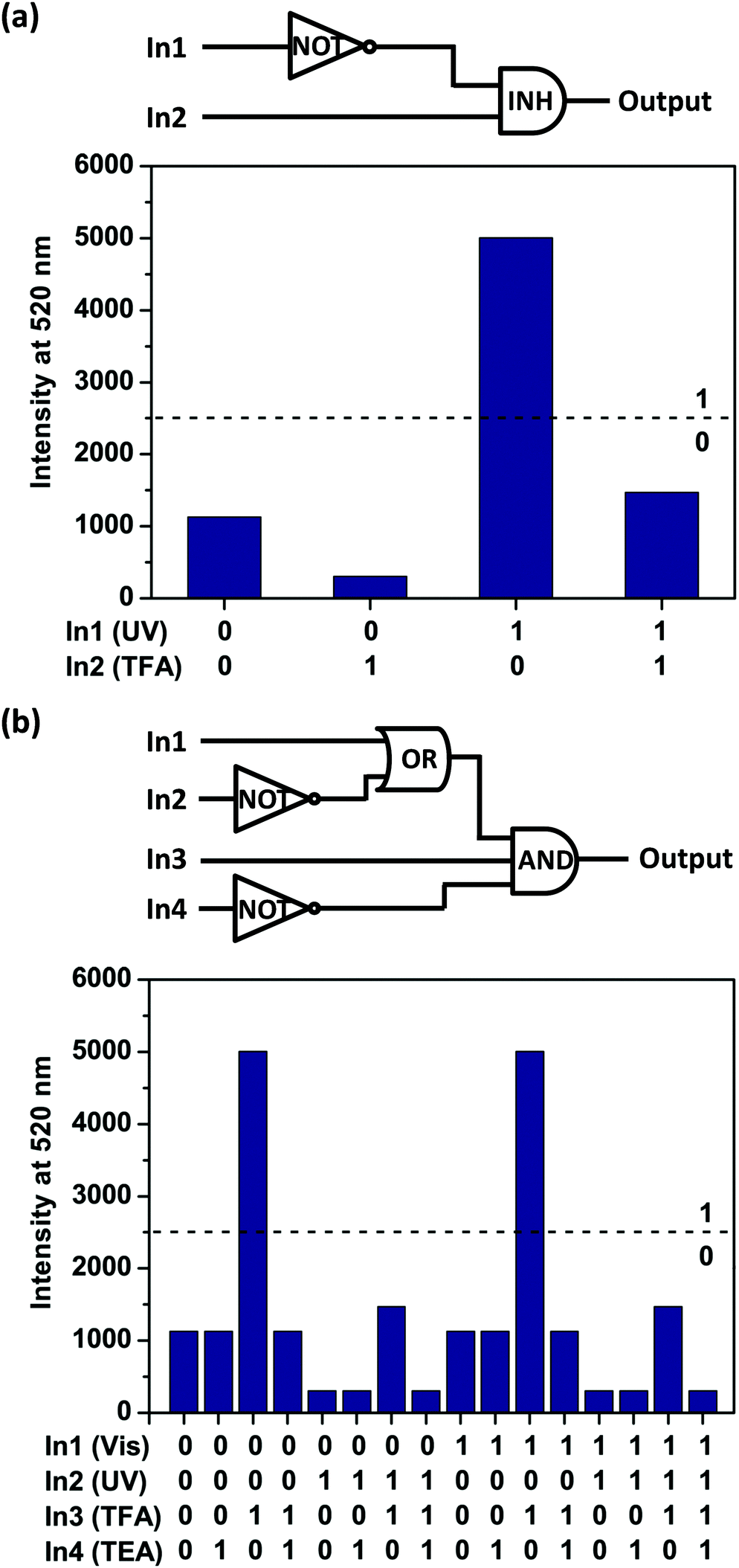 | ||
| Fig. 3 (a) Schematic diagram of aqueous INH logic gate with two inputs: In1 (UV light), In2 (TFA) and a single output: I520 (the normalized fluorescence intensity at 520 nm). The actual output values are shown below the diagram. (b) A more complicated logic circuit designed with four inputs: In1 (visible light), In2 (UV light), In3 (TFA) and In4 (TEA), and a single output: I520 (the normalized fluorescence intensity at 520 nm). The actual output values are shown below the diagram. | ||
Furthermore, to construct a more complicated logic circuit, four stimuli could be used as inputs: visible light (In1), UV light (In2), TFA (In3) and TEA (In4). Similarly, the change of fluorescence intensity at 520 nm (I520) is used as the output signal. The output signal could be regarded as a Boolean value of ‘1’ when I520 ≥ 0.5. Otherwise, it was regarded as a Boolean value of ‘0’. Thus, a logic circuit with AND, OR and NOT gates within a single molecule has been achieved (Fig. 3b) according to the truth table (Table S3, ESI†). Notably, this is a rare logic gate system operated by a water-compatible glycosyl photochromic compound.
To summarize, we have developed a unique water-compatible glycosylated system that can realize photochromic actions and molecular logic gate operation with fluorescence as the output. This research paves the way for the development of photo-controlled diagnostic glycoprobes34–38 for biomedical applications.
This research is supported by the 973 project (2013CB733700), the National Natural Science Foundation of China (21402050, 21572058 and 21576088), the Science and Technology Commission of Shanghai Municipality (15540723800), the Fundamental Research Funds for the Central Universities (222201717003) and the Shanghai Rising-Star Program (16QA1401400) (to X.-P. He). The Catalysis And Sensing for our Environment (CASE) network is thanked for research exchange opportunities. T. D. J. thanks ECUST for a guest professorship.
Notes and references
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- M. Takeshita and H. Jin-nouchi, Chem. Commun., 2010, 46, 3994–3995 RSC.
- M. Herder, M. Utecht, N. Manicke, L. Grubert, M. Pätzel, P. Saalfrank and S. Hecht, Chem. Sci., 2013, 4, 1028–1040 RSC.
- C. B. Fan, L. L. Gong, L. Huang, F. Luo, R. Krishna, X. F. Yi, A. M. Zheng, L. Zhang, S. Z. Pu, X. F. Feng, M. B. Luo and G. C. Guo, Angew. Chem., Int. Ed., 2017, 129, 8008–8014 CrossRef.
- S. Z. Pu, Q. Sun, C.-B. Fan, R. J. Wang and G. Liu, J. Mater. Chem. C, 2016, 4, 3075–3093 RSC.
- J. Andreasson and U. Pischel, Chem. Soc. Rev., 2015, 44, 1053–1069 RSC.
- A. K. Singh, P. K. Yadav, N. Kumari, R. Nagarajan and L. Mishra, J. Mater. Chem. C, 2015, 3, 12123–12129 RSC.
- Z. Yu and S. Hecht, Chem. Commun., 2016, 52, 6639–6653 RSC.
- S. Fredrich, R. Goestl, M. Herder, L. Grubert and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1208–1212 CrossRef CAS PubMed.
- J. C. Chan, W. H. Lam and V. W. Yam, J. Am. Chem. Soc., 2014, 136, 16994–16997 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- Q. Zou, X. Li, J. Zhang, J. Zhou, B. Sun and H. Tian, Chem. Commun., 2012, 48, 2095–2097 RSC.
- S. Wang, X. Li, W. Zhao, X. Chen, J. Zhang, H. Ågren, Q. Zou, L. Zhu and W. Chen, J. Mater. Chem. C, 2017, 5, 282–289 RSC.
- S. Xia, G. Liu and S. Pu, J. Mater. Chem. C, 2015, 3, 4023–4029 RSC.
- Y. Fu, Y. Tu, C. Fan, C. Zheng, G. Liu and S. Pu, New J. Chem., 2016, 40, 8579–8586 RSC.
- H. Ding, B. Li, S. Pu, G. Liu, D. Jia and Y. Zhou, Sens. Actuators, B, 2017, 247, 26–35 CrossRef CAS.
- U. Pischel, J. Andreasson, D. Gust and V. F. Pais, ChemPhysChem, 2013, 14, 28–46 CrossRef CAS PubMed.
- J. Andreasson and U. Pischel, Chem. Soc. Rev., 2015, 44, 1053–1069 RSC.
- U. G. Reddy, J. Axthelm, P. Hoffmann, N. Taye, S. Glaser, H. Gorls, S. L. Hopkins, W. Plass, U. Neugebauer, S. Bonnet and A. Schiller, J. Am. Chem. Soc., 2017, 139, 4991–4994 Search PubMed.
- T. Soboleva, H. J. Esquer, A. D. Benninghoff and L. M. Berreau, J. Am. Chem. Soc., 2017, 139, 9435–9438 CrossRef CAS PubMed.
- B. Roubinet, M. L. Bossi, P. Alt, M. Leutenegger, H. Shojaei, S. Schnorrenberg, S. Nizamov, M. Irie, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 15429–15433 CrossRef CAS PubMed.
- K. Liu, Y. Wen, T. Shi, Y. Li, F. Li, Y. L. Zhao, C. Huang and T. Yi, Chem. Commun., 2014, 50, 9141–9144 RSC.
- Y. Zou, T. Yi, S. Xiao, F. Li, C. Li, X. Gao, J. Wu, M. Yu and C. Huang, J. Am. Chem. Soc., 2008, 130, 15750–15751 CrossRef CAS PubMed.
- A. Mammana, G. T. Carroll, J. Areephong and B. L. Feringa, J. Phys. Chem. B, 2011, 115, 11581–11587 CrossRef CAS PubMed.
- X. P. He, Y. Zang, T. D. James, J. Li, G. R. Chen and J. Xie, Chem. Commun., 2016, 53, 82–90 RSC.
- S. Wang, W. Shen, Y. Feng and H. Tian, Chem. Commun., 2006, 1497–1499 RSC.
- Y. Cai, Z. Guo, J. Chen, W. Li, L. Zhong, Y. Gao, L. Jiang, L. Chi, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2016, 138, 2219–2224 CrossRef CAS PubMed.
- K. Higashiguchi, G. Taira, J. Kitai, T. Hirose and K. Matsuda, J. Am. Chem. Soc., 2015, 137, 2722–2729 CrossRef CAS PubMed.
- X. F. Zhang, T. Zhang, S. L. Shen, J. Y. Miao and B. X. Zhao, J. Mater. Chem. B, 2015, 3, 3260–3266 RSC.
- Y. Fu, J. Zhang, H. Wang, J.-L. Chen, P. Zhao, G.-R. Chen and X.-P. He, Dyes Pigm., 2016, 133, 372–379 CrossRef CAS.
- Q. Wan, S. Chen, W. Shi, L. Li and H. Ma, Angew. Chem., Int. Ed., 2014, 53, 10916–10920 CrossRef CAS PubMed.
- G. Jiang, S. Wang, W. Yuan, L. Jiang, Y. Song, H. Tian and D. B. Zhu, Chem. Mater., 2006, 18, 235–237 CrossRef CAS.
- X.-P. He, Y.-L. Zeng, Y. Zang, J. Li, R. A. Field and G.-R. Chen, Carbohydr. Res., 2016, 429, 1–22 CrossRef CAS PubMed.
- X.-P. He, Y.-L. Zeng, X.-Y. Tang, N. Li, D.-M. Zhou, G.-R. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 13995–13999 CrossRef CAS PubMed.
- K.-B. Li, N. Li, Y. Zang, G.-R. Chen, J. Li, T. D. James, X.-P. He and H. Tian, Chem. Sci., 2016, 7, 6325–6329 RSC.
- J.-X. Song, X.-Y. Tang, D.-M. Zhou, W. Zhang, T. D. James, X.-P. He and H. Tian, Mater. Horiz., 2017, 4, 431–436 RSC.
- D.-T. Shi, D. Zhou, Y. Zang, J. Li, G.-R. Chen, T. D. James, X.-P. He and H. Tian, Chem. Commun., 2015, 51, 3653–3655 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and experimental section. See DOI: 10.1039/c7cc04427e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2017 |