
A detection and confirmation strategy for screening of veterinary drugs in honey by liquid chromatography coupled quadrupole time-of-flight mass spectrometry†
Zhibin
Wang
 *a,
Yan
Li
a,
Qiaoying
Chang
ab,
Jian
Kang
c and
Guo-Fang
Pang
ab
*a,
Yan
Li
a,
Qiaoying
Chang
ab,
Jian
Kang
c and
Guo-Fang
Pang
ab
aCollege of Environmental and Chemical Engineering, Hebei Key Laboratory of Applied Chemistry, Yanshan University, Qinhuangdao 066004, China. E-mail: wzb@ysu.edu.cn
bChinese Academy of Inspection and Quarantine, Beijing 100123, China
cAgilent Technol China Co Ltd, 3 Wang Jing Bei Lu, Beijing 100102, People's Republic of China
First published on 23rd November 2017
Abstract
A strategy for the quantification and confirmation of 40 multi-class veterinary drugs in honey by QuEChERS (quick, easy, cheap, effective, rugged, and safe) combined with liquid chromatography coupled quadrupole time-of-flight mass spectrometry (LC-Q-TOF/MS) is described. The veterinary drugs examined belong to four classes: quinolones, sulfonamides, macrolides, and tetracyclines. Before analysis by LC-Q-TOF/MS, the sample was diluted with a solution of Na2EDTA-McIlvaine buffer solution (0.1 M, pH = 4), extracted with 5% acetic acid in acetonitrile, and cleaned up with an NH2 sorbent. The average recoveries for the majority of analytes (86.9%), based on matrix-matched external calibration curves, were between 70% and 120%, and there was no significant difference in the recoveries between different honey matrices. The repeatability and reproducibility of the method expressed as the RSDs were less than 20% for all analytes. The data acquired by LC-Q-TOF/MS were cross-referenced with an accurate mass database of veterinary drugs, and the suspected analytes were finally confirmed based on a full product ion library match. Compared with the low-resolution MS technique, obvious advantages were obtained in terms of confirmation and identification by LC-Q-TOF/MS. The applicability of the method was verified by applying it to 12 different honey samples, and ciprofloxacin residue (at 99.7 μg kg−1) was detected in one sample.
1 Introduction
Honey, which is frequently used as a sweetener and flavoring agent, is produced by bees using the nectar from flowers or honeydew.1 In the process of production, bacteria, fungi, and viruses, derived from the environment and agricultural practices, pose a serious risk to the queen, worker, drone, or larvae.2 Therefore, veterinary drugs have been utilized to control and treat several diseases of honey bees, and consequently, residues such as sulfonamides and tetracyclines have been found in different honey samples.3,4 These residues pose a potential threat to human health, e.g., increased drug resistance and allergic reactions.In order to ensure food safety, the European Union (EU),5 United States (http://www.mrldatabase.com/), Japan (http://www.ffcr.or.jp/zaidan/FFCRHOME.nsf/pages/MRLs-p), and many other countries and international organizations have established maximum residue limits (MRLs) for veterinary drugs in different food matrices of animal origin. Some organizations in the United States, Japan, and Australia have approved the use of certain veterinary drugs in honey. However, no MRLs have been established by the EU, because there is no authority in place for the treatment of honey bees, although some recommended concentrations for sulfonamides (50 μg kg−1) and tetracyclines (20 μg kg−1) can be found in the Community of Reference Laboratories (CRL) Guidance Paper.6
Different sample pretreatment methods have been applied for the effective trace detection of veterinary drugs. The most frequently used sample preparation methods are solid phase extraction (SPE)1,7,8 and the QuEChERS method,9,10 in addition to other methods such as liquid–liquid microextraction11 or solvent extraction.12 Among these, the QuEChERS method is frequently used for the extraction of pesticides from fruits and vegetables.13 Simplicity, high efficiency, and other advantages suggest the applicability of QuEChERS to the determination of veterinary drug residues in chicken breasts, eggs, and muscle tissues.14–16
A review of the literature reveals that LC-MS is a powerful technique to detect and quantify multiclass veterinary drugs because of its high sensitivity and selectivity. Currently, LC-MS methods mainly feature MS/MS,17 ion trap mass spectrometry,18 orbitrap,19,20 time-of-flight mass spectrometry,21,22 and Q-TOF/MS.23 Among these, high-resolution mass spectrometry (HRMS) is suitable for the simultaneous screening of an unlimited number of compounds. Therefore, most published HRMS methods have been widely applied in the screening and confirmation of veterinary drugs in milk,24 meat,25 honey,26 and aquacultured species.27 Different strategies were used in these studies. First, the contaminants were identified based on the MS information; the accurate mass of the precursor ion and the isotope profile were compared to the theoretical values.19,26,28,29 Second, the MS information was combined with the MS/MS information; the additional MS/MS spectra can be used to further confirm the assignment of drug residues.20,27 More information about the structural characterization of the product ions can avoid false positive results.30,31 However, only a few methods based on the second strategy have been reported, particularly for the determination of veterinary drugs in honey.
The purpose of this work is to develop a simple, sensitive, selective, and efficient method for the quantification and confirmation of 40 multiclass veterinary drugs, including macrolides, quinolones, sulfonamides, and tetracyclines, which are usually inspected by the importers in international trade, in honey by LC-Q-TOF/MS. A two-injection screening strategy is proposed here. Firstly, the full MS scan mode was conducted and all potential compounds were cross-referenced with an accurate mass database. Secondly, targeted MS/MS analysis was carried out by using hybrid Q-TOF/MS and full product ion match was made to identify the target compounds. The search parameters (accurate mass error, retention time window, ionization forms) were optimized to avoid false positive or negative results. The accuracy and stability of the searching method were evaluated. Finally, the method was applied to real samples.
2 Materials and methods
2.1 Reagents and chemicals
All veterinary drug standards were purchased from Dr Ehrenstorfer (Ausburg, Germany). Individual stock standard solutions (approximately 1000 mg L−1) were prepared in methanol and stored in the dark below 4 °C. HPLC grade acetonitrile and methanol were purchased from Fisher Scientific (New Jersey, USA). Formic acid was obtained from Duksan Pure Chemicals (Ansan, Korea), and HPLC-grade water was obtained from a Millipore ultrapure water system. Analytical-reagent-grade acetic acid, disodium ethylene diamine tetraacetic acid (Na2EDTA), disodium hydrogen phosphate (Na2HPO4), citric acid (C6H8O7), sodium chloride (NaCl), anhydrous magnesium sulfate (MgSO4), anhydrous sodium sulfate (Na2SO4), and ammonium acetate (NH4OAc) were purchased from Beijing Chemical Co. (Beijing, China). PSA (primary secondary amine) and NH2 sorbents were purchased from Agela (Tianjin, China). Twelve various honey samples were purchased from several local markets in Beijing (China), including 2 vitex, 2 Chinese date, 2 linden, 4 acacia, and 2 clover honeys.2.2 Instruments and software
The analytes were separated using an Agilent 1290 LC system equipped with an autosampler (Agilent, Santa Clara, CA), using a reversed-phase ZORBAX SB-C18 column (2.1 mm × 100 mm and 3.5 μm particle size) from Agilent. Q-TOF-MS detection was performed using an Agilent 6550 system equipped with a Dual JetSpray ionization source (Agilent, Santa Clara, CA). Data were acquired using Agilent MassHunter Workstation Software (version B.05.00). The accurate mass database was established using Excel (2010) in the CSV format.An oscillator (Taitec, SR-2DS, Japan), centrifuge (SIGMA, 3-30K, Germany), nitrogen evaporator (Organomation Associates, EVAP 112, USA), and vortex mixer (AS ONE, TRIO TM-1N, Japan) were used. Water was provided by a Milli-Q high-purity water generator (Milford, MA, USA). The electronic analytical balance used was obtained from Mettler-Toledo (PL602-L, Switzerland).
2.3 Sample preparation
As defined in Fig. 1, honey (1.0 g) was weighed in a 50 mL centrifuge tube, diluted with 6 mL Na2EDTA-McIlvaine buffer solution (0.1 M, pH 4), and stirred vigorously in a vortex. Then, 5% acetic acid in acetonitrile (18 mL) was added, and the sample was mixed by vortex for 30 s. 2.0 g of NaCl and 4.0 g of Na2SO4 were added, and the tube was shaken for 2 min. The mixture was then centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (10397 × g) for 5 min at 10 °C. The resulting supernatant solution (9 mL) was transferred into a 15 mL centrifuge tube containing 200 mg NH2 sorbent, and the tube was shaken for 2 min. The mixture was centrifuged again under the same conditions. 4.5 mL of the resulting supernatant was further transferred into a 10 mL glass tube and evaporated to dryness under a stream of nitrogen at 40 °C. Finally, the residue was re-dissolved in 1.0 mL of 0.1% formic acid in water/acetonitrile (9:1, v/v), stirred in a vortex for 30 s, filtered through a 0.22 μm nylon filter membrane, and subjected to LC-Q-TOF/MS analysis.
000 rpm (10397 × g) for 5 min at 10 °C. The resulting supernatant solution (9 mL) was transferred into a 15 mL centrifuge tube containing 200 mg NH2 sorbent, and the tube was shaken for 2 min. The mixture was centrifuged again under the same conditions. 4.5 mL of the resulting supernatant was further transferred into a 10 mL glass tube and evaporated to dryness under a stream of nitrogen at 40 °C. Finally, the residue was re-dissolved in 1.0 mL of 0.1% formic acid in water/acetonitrile (9:1, v/v), stirred in a vortex for 30 s, filtered through a 0.22 μm nylon filter membrane, and subjected to LC-Q-TOF/MS analysis.
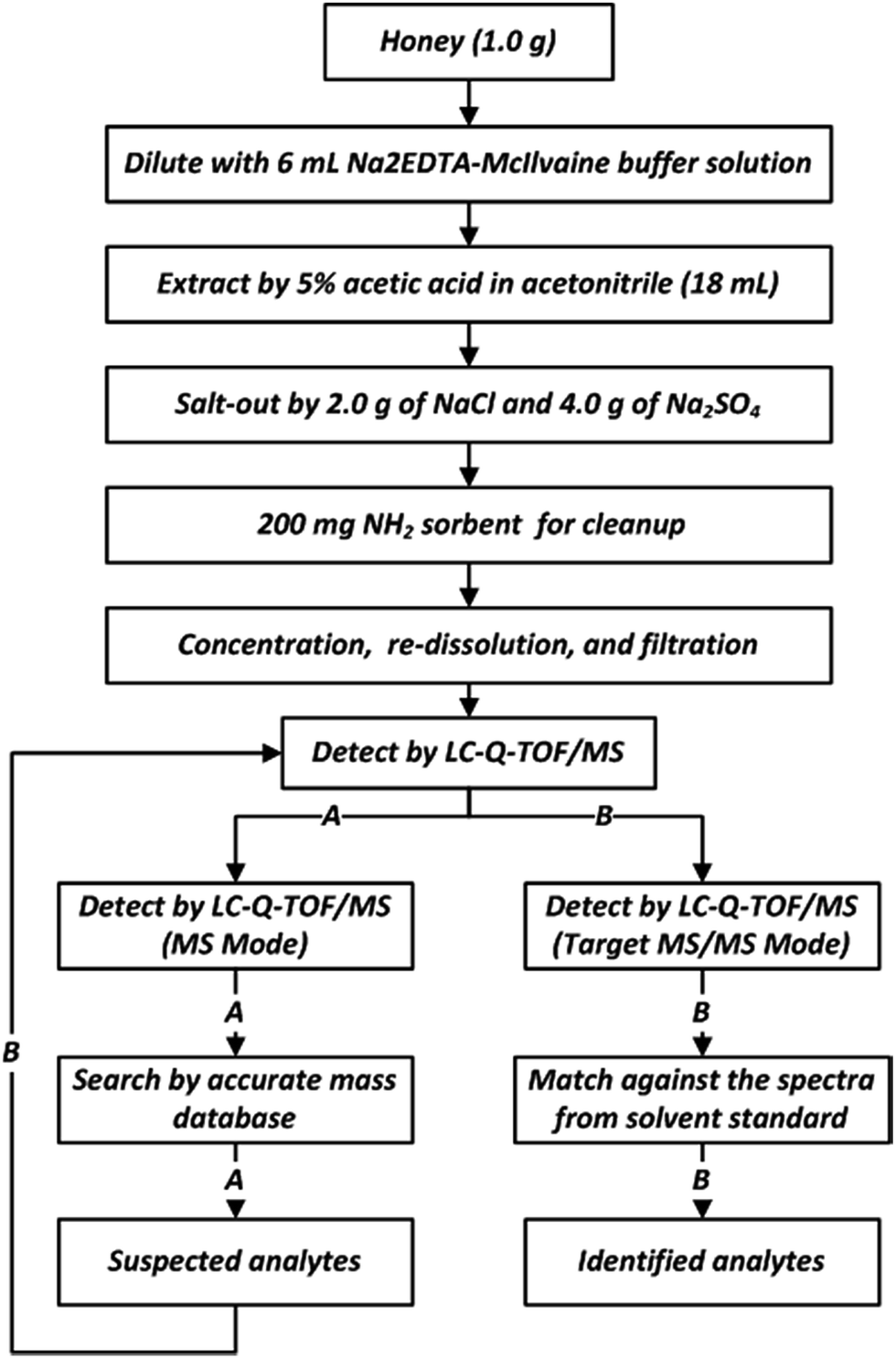 | ||
| Fig. 1 The workflow for sample preparation and screening methodology. | ||
2.4 LC-Q-TOF/MS analysis
Chromatographic analyses were carried out using a gradient profile with mobile phase A (5 mmol L−1 NH4OAc/0.1% formic acid/water) and mobile phase B (acetonitrile). The gradient profile was 0 min: 5% B, 6 min: 15% B, 20 min: 30% B, 26 min: 80% B, 30 min: 100% B, 35 min: 100% B, 36 min: 5% B; the post-run equilibrium time was 4 min, the flow rate was 0.3 mL min−1, the column temperature was controlled at 40 °C, and the injection volume was 10 μL.For Q-TOF/MS detection, electrospray ionization was conducted under positive mode. The capillary voltage was set at 4000 V. The drying gas temperature and flow were controlled at 225 °C and 14 L min−1, respectively. The nebulizer pressure was set at 40 psi. The sheath gas temperature and flow rate were maintained at 325 °C and 11 L min−1, respectively. The skimmer voltage and fragmentor voltage were controlled at 65 V and 400 V, respectively. The nozzle voltage was 1000 V. Reference ions with m/z of 121.0509 and 922.0098 were used for real-time calibration under positive ionization mode. The data were collected in centroid mode. The TOF-MS full scanning range was set within 50–1700 m/z at a rate of 4 spectra per s. The product ion data were acquired in target MS/MS mode under a fixed retention time (tR), precursor ion ([M + H]+), collision energy (CE), and medium isolation width (4 m/z). The parameters for the target MS/MS are shown in Table S1 (ESI† available online).
2.5 Screening methodology
In order to screen the positive results in real samples, two major steps were followed:(1) A search through an accurate mass database (homemade) was performed. The sample was analyzed in MS mode of TOF/MS, and the resultant data cross-referenced with an established database (CSV file) using the “Find by formula” function provided by Qualitative MassHunter software. The search parameters such as the tR window (0.25 min), mass tolerance (10 ppm), and ionization forms (+H) were restricted, and the software calculated the deviation of the accurate mass, tR, and the distribution and proportion of isotopic clusters between the measured value and the theoretical value to obtain a score value of the target compound. When the score of the target compound was more than 70, this compound was identified as a suspected veterinary drug.
(2) To further confirm and identify these suspected veterinary drugs, a second injection of extracts are required to obtain the MS/MS data, they were analyzed under target MS/MS mode of Q-TOF/MS to obtain complete product ions. The obtained data were extracted using the “Find by targeted MS/MS” function, and the resultant spectra were matched against the spectra from the solvent standard. When more than two of the major product ions were matched (mass error lower than 10 ppm, tolerances of relative intensity lower than 20%), this compound was considered to be confirmed. The two major product ions are listed in Table S1.† The workflow of the screening methodology is shown in Fig. 1.
2.6 Method validation
The method was validated in terms of linearity, recovery, repeatability, reproducibility, and limits of confirmation (LOCs). The validation parameters were evaluated by a database search. For optimization of sample preparation, the peak area of the [M + H]+ ion in the spiked sample was used to calculate the concentration or recovery. For matrix effects and linearity, the peak area of the precursor ion generated from the [M + H]+ ion versus concentration was utilized to establish linear regression.Recovery experiments were carried out in a blank acacia honey sample spiked with a mixed standard of 40 veterinary drugs at four levels (5, 20, 50, and 100 μg kg−1), with six replicates at each level. Recovery experiments between different varieties of honey, including vitex, Chinese date, linden, acacia, and clover honey, were also carried out at 50 μg kg−1, with six replicates. The spiked samples were allowed to remain at room temperature for at least 30 min to ensure the appropriate distribution of the analytes. The repeatability (intraday) was evaluated at the same concentrations (n = 6), and the reproducibility (interday) was evaluated at 50 μg kg−1 for three consecutive days. All of the results were determined by matrix-matched external calibration curves.
LOCs represented the capabilities of reliable confirmation of the target compounds. They were estimated by analyzing spiked acacia honey samples at 1–100 μg kg−1 to find the concentrations which would give signal-to-noise ratios (S/N) of 10, and were calculated as the minimum concentration of the target compound according to the confirmation criteria (see Section 2.5).
Matrix effects were evaluated using calibration curves that were established by adding standard solutions into blank matrices at 5–500 μg kg−1. Then, slope ratios (matrix/solvent) were calibrated for each compound.32 Matrix effects between honeys from various sources were also evaluated from the response of the target compounds. Linearity was evaluated by spiking honey samples at the same ranges as matrix effects described above.
3 Results and discussion
3.1 Optimization of sample preparation
Honey is a complex matrix with a large number of sugars, pigments, and other substances. Therefore, extraction, purification, and concentration of the components are necessary before LC-Q-TOF/MS analysis. The blank honey samples were fortified with a mixed standard of 40 veterinary drugs at 100 μg kg−1, and three replicates were carried out to evaluate the extraction conditions.The first step in the pretreatment process is dissolution in a suitable solution or buffer before extraction, because of the viscous nature of honey (a supersaturated sugar solution). Na2EDTA-McIlvaine buffer solution is a good choice for dissolving honey, as it precludes the complexation of macrolides and tetracyclines with metal ions and enhances the recovery.33 It is often used to extract tetracyclines, quinolones and other drugs.34,35 Then, the range of pH (3–8) was further evaluated for this buffer, better recoveries were obtained at pH = 4. Acetonitrile has often been used as an extraction solution in QuEChERS methods, because of the lower co-extraction, and acidified acetonitrile can further improve the extraction efficiency.10,14 The results demonstrated that 5% acetic acid in acetonitrile provided the best recoveries, especially for quinolones and tetracyclines. As shown in Fig. 2A, the recovery increased as the acidity increased (from 0% to 5% acetic acid in acetonitrile), e.g., for tetracycline (from 39.7% to 79.5%) and enoxacin (from 51.7% to 72.1%). Moreover, acid hydrolysis was previously reported to extract the sulfonamides that bind to the sugars.36 However, for this method, the extraction focused only on unbound sulfonamides in honey.
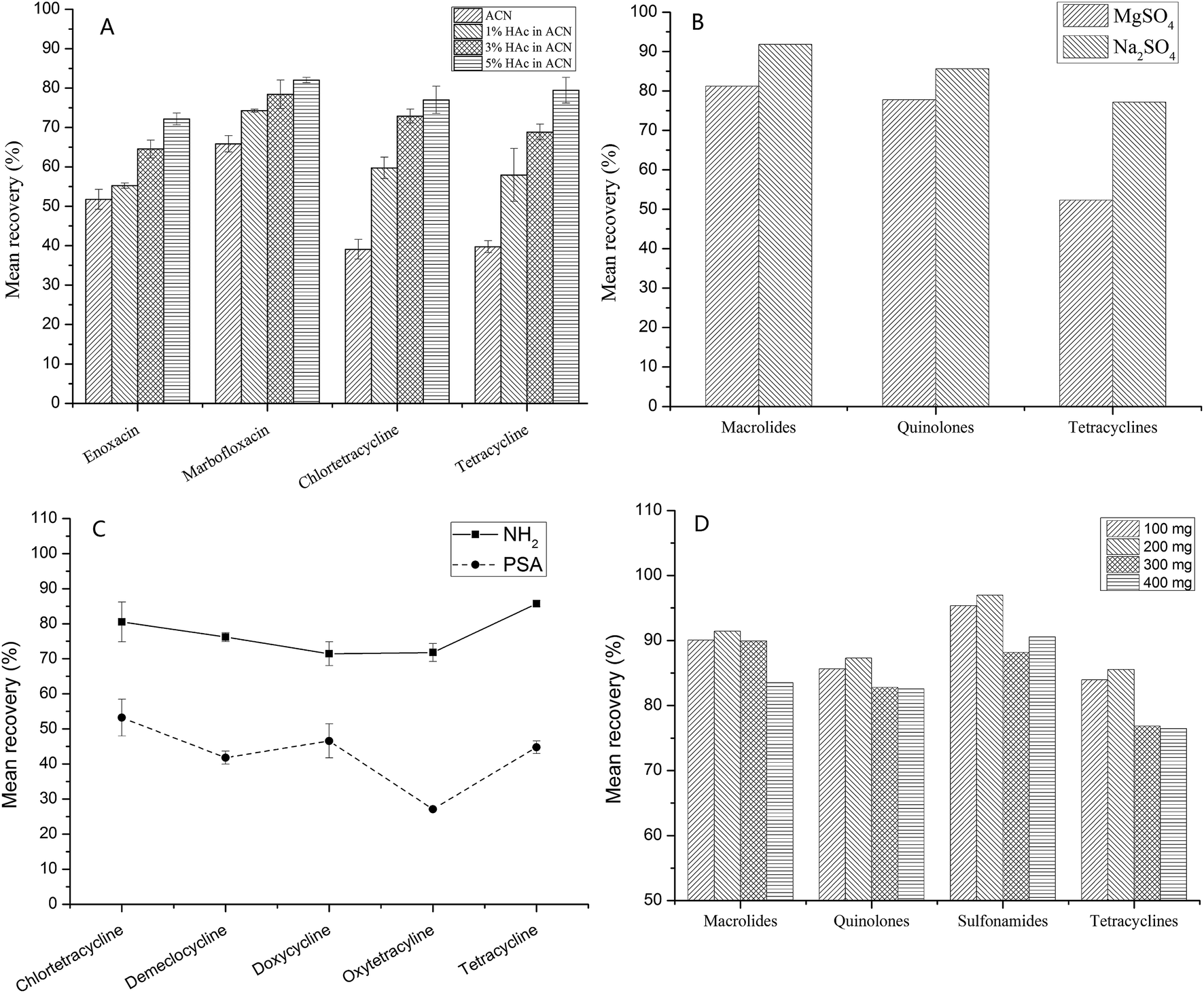 | ||
| Fig. 2 Optimization of sample preparation ((A) effects on recovery under different acidic conditions; (B) effects on recovery under MgSO4 and Na2SO4; (C) effects on recovery under NH2 and PSA; (D) effects on recovery under different amounts of sorbent). | ||
Second, for pesticides, MgSO4 was typically used to bind large amounts of water in QuEChERS procedures.13 However, our results indicated that Na2SO4 achieved a superior recovery compared to MgSO4. As can be seen in Fig. 2B, when using MgSO4, the mean recoveries of tetracyclines, quinolones, and macrolides declined by 24.8%, 7.9%, and 10.6%, respectively.
Finally, the choice of sorbent is based on the matrix, i.e., graphitized carbon black (GCB) has often been used to remove pigments, NH2 and PSA for organic acids and sugars, and C18 for lipids in meat and fish. For this study, sugars are largely responsible for the matrix effect and interference in honey. Therefore, NH2 and PSA were more suitable sorbents for honey. Comparing the two sorbents, better recoveries were observed when NH2 was used. PSA can absorb some drugs, leading to lower recoveries, see Fig. 2C. Moreover, the amount of NH2 sorbent was also considered from 100 mg to 400 mg. As shown in Fig. 2D, the recovery declined as the amount of NH2 sorbent increased, 200 mg NH2 sorbent was suitable for the cleanup process.
3.2 Optimization of search parameters
For the database search, some parameters should be optimized to avoid reporting false positive or negative results and improve the accuracy of the database search.37 These parameters are the mass error window, the limited tR range, and the ionization forms.First, the mass error window was evaluated in blank acacia honey samples at 20, 50, and 100 μg kg−1 (n = 6). The obtained results showed that the mass deviations were below 5 ppm, and 92.5% of the total target compounds were accounted for. The mass errors of all the compounds were less than 10 ppm, except tetracyclines at the level of 20 μg kg−1, because of the low response (see Fig. 3A). Therefore, this value is set to 10 ppm.
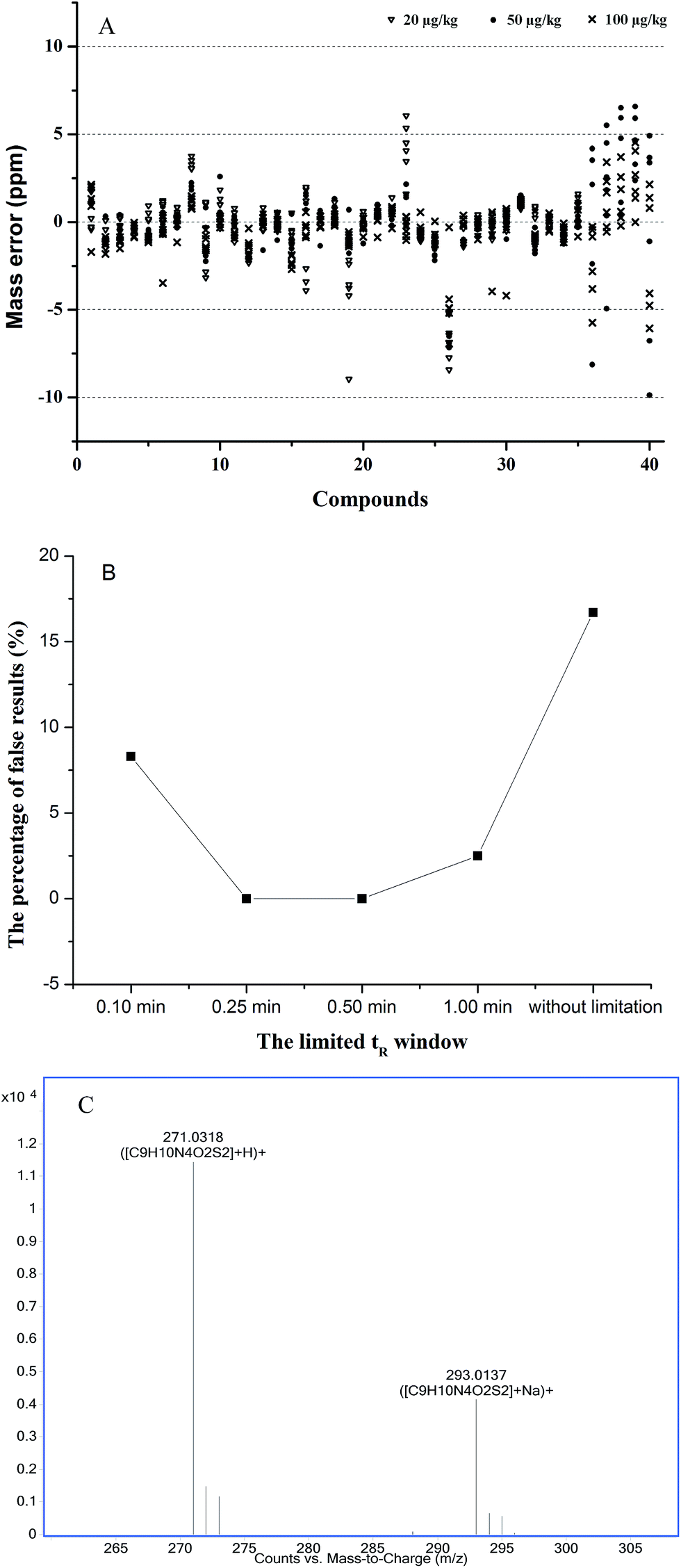 | ||
| Fig. 3 Optimization of the search parameters ((A) distributions of mass error for 40 veterinary drugs in three spiked levels (n = 6), veterinary drug codes are listed in Table 1; (B) the percentage of false results under different tR windows; (C) the ionized forms of sulfamethizole). | ||
Second, the limited tR window was evaluated in the blank acacia honey sample at 0.1, 0.25, 0.5, 1 min and without limitation, the percentages of false results (including positive and negative) were 8.3%, 0.0%, 0.0%, 2.5%, and 16.7%, respectively (see Fig. 3B). Moreover, under the condition of no limited tR window, some isobaric compounds could not be distinguished, such as sulfamonomethoxine and sulfameter, tetracycline and doxycycline. Then, the deviations of tR over a day (n = 6) and from day-to-day over three days (n = 6) were evaluated, and the maximum intra- and interday standard deviations of tR were ±0.14 min and ±0.24 min, respectively. Hence, the tR window should be set to 0.25 min to prevent the false results.
Third, the choice of ionized forms was considered, because of the multiple ionization modes in LC-MS, e.g., [M + H]+, [M + NH4]+ and [M + Na]+. For 40 veterinary drugs, the response of the [M + H]+ ions was the highest among the three. In contrast, excepting sulfonamides, the response of the [M + Na]+ ions was lower than 20% of the base peak ([M + H]+ ion), such as sulfamethizole shown in Fig. 3C. Moreover, the [M + NH4]+ ion peak was hardly found. Therefore, based on the response, the ionized form [M + H]+ was more suitable for the database search.
3.3 Identify confirmation
For this screening method, the confirmation process for the target compounds was based on the score and product ion match. The score was obtained by calculating the difference between the measured value and the theoretical value of the mass error, tR, and the profile of isotopic clusters. The mass error and tR window were restricted in the search parameter above. The profile of isotopic clusters was also considered to be a useful tool for identification purposes. The 40 veterinary drugs included in this study mainly consisted of C, H, O, N, S, F, and Cl. The A+1 signals were mainly provided by 13C, and the A+2 signals were obtained from 34S and 37Cl. Experiments were carried out in blank honey samples at three spiked levels (20, 50, and 100 μg kg−1, n = 6) to evaluate the score. The results showed that the compounds identified in honey samples with scores ≥70 and ≥90 accounted for 96.5% and 78.9% of the total amount of drug, respectively. RSDs of the score less than 10% and 20% accounted for 90.5% and 96.5%, respectively. However, for tetracyclines, a score over 70 was not obtained at a low spike level (20 μg kg−1) because of the low response.Moreover, all of 40 target residues (vitex, Chinese date, linden, acacia, and clover honey samples with six replicates) were detected in samples fortified at the level of 50 μg kg−1. No false positive results have been found when optimized search parameters were used. However, false negative results have been found in linden and vitex honey at a manageable level (total of 12 residues, or approximately 1%), these were attributed to the existence of interfering ions from the matrix. Although no false positive results have been found in honey samples, basing the confirmation only on the score of TOF/MS does not satisfy the confirmation criteria of four identification points (IPs) from EC/2002/657.38 Therefore, to improve the accuracy of qualitative methods, product ion match was applied in the screening method for final confirmation. The product ion data of the target compounds were acquired under targeted MS/MS of Q-TOF/MS; the [M + H]+ ion was selected to be the precursor ion, and the choice of CE was based on the maximum number of product ions with a relative intensity ≥10% of the base peak. The resultant spectra of the target compounds were compared to the accurate mass and the relative abundances observed in solvent standards. According to the confirmation criteria, when the product ions are matched, this compound is considered to be confirmed. Examples of the product ions obtained in a solvent standard and spiked honey are shown in Fig. 4. Moreover, the IPs of the 40 target compounds were calculated according to EC/2002/657 (see Table S1†). The results demonstrated that 70.0% of the veterinary drugs earned over 10 IPs at the optimized CE. Moreover, over 30 IPs could be reached for some veterinary drugs such as sparfloxacin. Although IPs of 4–8 were obtained for some veterinary drugs such as flumequine and sulfabenzamide, this still satisfies the confirmation criteria. Compared with the low resolution MS technique, this method offers obvious advantages in terms of confirmation and identification, because of much more IPs obtained. Additionally, TOF-MS/Q-TOF-MS, as full scan HRMS analyzers, together with the data mining strategy, have gained increasing popularity in non-targeted analysis and data tracing.39–42
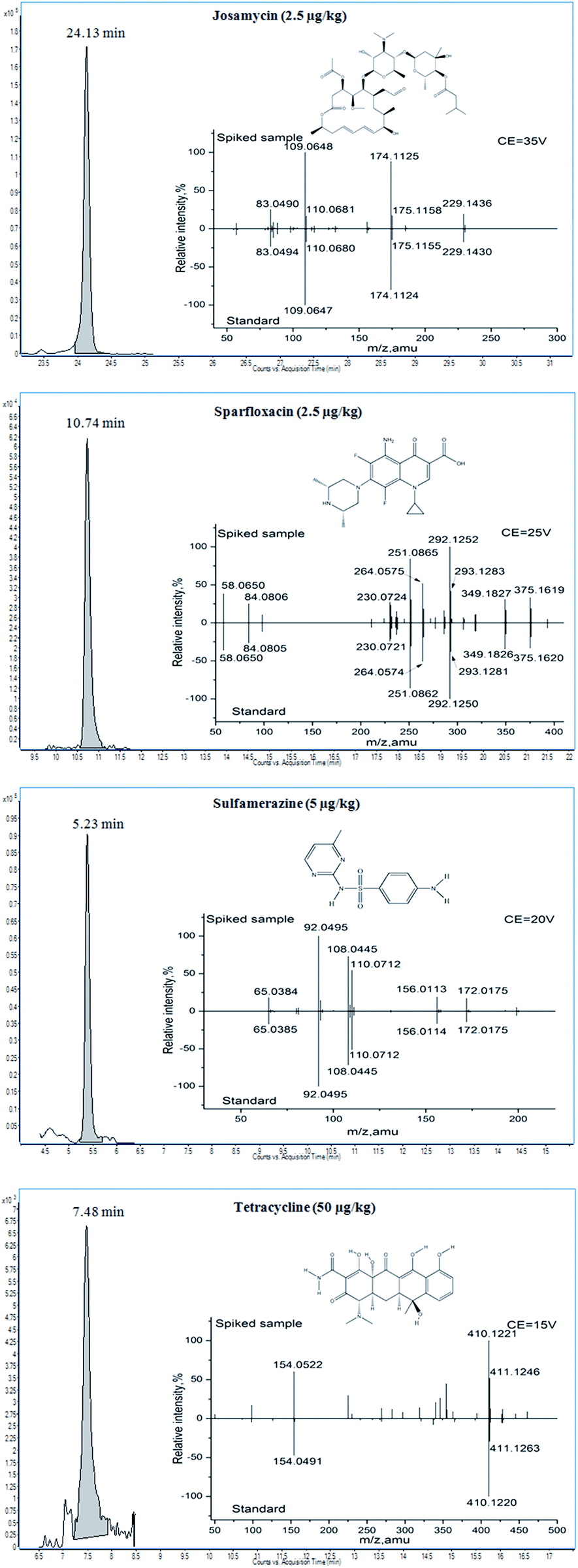 | ||
| Fig. 4 The example EIC chromatograms and product ion spectra at the LOC-spiked level in honey. | ||
3.4 Quantification of the target compounds
During LC-MS analysis, matrix effects were inevitable. For the 40 veterinary drugs, the obtained results show that only 92.5% of the compounds experienced a soft matrix effect; the slope ratio ranged from 0.8 to 1.2, except for pipemidic acid (0.74), clindamycin (0.75), and oxytetracyline (0.78) (see Table 1). There were no obvious differences in the matrix effect between various honeys (vitex, Chinese date, linden, acacia, and clover honey), and the RSD values for the response of the target compounds were always lower than 20%, except for tylosin (22.5%). These results suggested that the pretreatment was quite effective at reducing the interference. Although the matrix effects were not obvious, a matrix-matched standard calibration curve was still used for the quantification to minimize the effects.| No. | Compound | Linear range (μg kg−1) | R 2 | SRa | LOC (μg kg−1) | Intraday | Interday | Between various honeys | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Recoveryb (rs)c (%) | Recovery (Rs)d (%) | Recovery (Rsv)e (%) | |||||||||
| 5 (μg kg−1) | 20 (μg kg−1) | 50 (μg kg−1) | 100 (μg kg−1) | 50 (μg kg−1) | 50 (μg kg−1) | ||||||
| a SR: slope ratios (matrix/solvent). b Recovery: the mean recovery of the spiked sample expressed as percentage (n = 6). c rs: repeatability expressed as the relative standard deviation in the spiked sample (n = 6) on 1 day. d Rs: reproducibility expressed as the relative standard deviation in the spiked sample (n = 6) on 3 days. e Rsv: reproducibility expressed as the relative standard deviation in the spiked sample (n = 6) on 5 various honey samples (vitex, Chinese date, linden, acacia, and clover honey). f N.D.: not detected in the spiked sample. | |||||||||||
| 1 | Clindamycin | 5–500 | 0.9997 | 0.75 | 1 | 85.2(2.0) | 83.7(7.5) | 77.9(3.8) | 75.1(3.3) | 78.2(7.1) | 78.2(3.9) |
| 2 | Josamycin | 5–500 | 0.9985 | 0.97 | 2.5 | 92.1(1.2) | 95.0(1.3) | 91.9(2.5) | 97.7(2.7) | 94.9(4.5) | 99.9(7.4) |
| 3 | Roxithromycin | 5–500 | 0.9998 | 0.99 | 1 | 97.1(2.1) | 95.4(1.4) | 95.4(1.5) | 89.9(2.2) | 91.3(3.7) | 93.0(4.0) |
| 4 | Tiamulin | 5–200 | 0.9987 | 0.93 | 2.5 | 93.0(2.1) | 94.3(0.9) | 93.9(2.5) | 91.3(1.5) | 94.0(2.3) | 95.4(2.8) |
| 5 | Tylosin | 5–500 | 0.9999 | 1.04 | 2.5 | 88.7(3.7) | 100.0(1.9) | 90.0(2.9) | 101.8(3.1) | 97.2(7.7) | 101.2(11.9) |
| 6 | Ciprofloxacin | 5–500 | 0.9997 | 0.91 | 5 | 74.8(4.4) | 77.1(2.3) | 80.7(0.8) | 81.2(1.9) | 79.1(8.5) | 90.5(8.0) |
| 7 | Danofloxacin | 5–500 | 0.9999 | 0.99 | 5 | 87.0(2.4) | 85.1(2.5) | 85.1(1.1) | 82.0(2.6) | 85.4(5.1) | 89.2(2.7) |
| 8 | Difloxacin | 5–500 | 0.9997 | 0.97 | 5 | 87.4(11.3) | 103.5(6.8) | 92.3(3.4) | 92.4(3.2) | 94.0(3.9) | 100.5(12.5) |
| 9 | Enoxacin | 5–500 | 0.9996 | 0.97 | 2.5 | 75.0(2.4) | 73.9(4.6) | 77.7(1.8) | 73.8(2.2) | 77.7(9.8) | 79.1(4.0) |
| 10 | Enrofloxacin | 5–500 | 0.9998 | 0.98 | 1 | 87.1(4.9) | 85.8(3.0) | 85.7(2.6) | 90.4(3.9) | 95.5(11.4) | 97.4(6.9) |
| 11 | Fleroxacin | 5–500 | 0.9975 | 0.95 | 2.5 | 85.2(2.0) | 88.3(0.6) | 87.2(3.2) | 86.5(8.9) | 89.6(6.2) | 89.1(10.3) |
| 12 | Flumequine | 10–200 | 0.9930 | 1.08 | 10 | N.D.f | 102.4(7.0) | 95.1(1.0) | 93.6(3.4) | 94.8(4.0) | 95.8(2.5) |
| 13 | Lomefloxacin | 5–500 | 0.9992 | 0.94 | 5 | 87.5(3.4) | 84.7(4.2) | 84.7(10.5) | 91.1(5.9) | 88.5(5.3) | 90.7(3.7) |
| 14 | Marbofloxacin | 5–500 | 0.9969 | 0.91 | 2.5 | 88.4(2.7) | 84.0(2.8) | 83.0(2.9) | 83.5(2.3) | 84.4(5.4) | 88.1(4.1) |
| 15 | Nalidixic acid | 5–500 | 1.0000 | 1.04 | 2.5 | 88.7(4.8) | 103.7(8.2) | 82.9(5.9) | 94.6(9.5) | 95.8(8.9) | 96.6(3.5) |
| 16 | Norfloxacin | 5–500 | 0.9999 | 0.97 | 5 | 79.9(3.5) | 69.8(4.2) | 75.0(3.4) | 74.8(3.8) | 76.5(11.4) | 80.9(3.9) |
| 17 | Ofloxacin | 5–500 | 0.9999 | 0.95 | 2.5 | 82.9(2.4) | 90.6(4.3) | 89.3(1.7) | 86.2(2.8) | 89.6(4.1) | 89.7(4.0) |
| 18 | Orbifloxacin | 5–500 | 0.9998 | 0.95 | 1 | 90.8(1.3) | 91.8(1.7) | 90.7(2.9) | 89.5(2.3) | 91.6(4.2) | 93.5(2.7) |
| 19 | Pipemidic acid | 10–500 | 0.9999 | 0.74 | 10 | N.D. | 67.6(1.1) | 70.1(1.1) | 66.4(2.6) | 71.9(9.8) | 72.6(9.6) |
| 20 | Sparfloxacin | 5–500 | 0.9989 | 0.91 | 2.5 | 91.0(3.5) | 94.5(6.3) | 85.0(2.0) | 88.0(3.6) | 88.5(4.1) | 93.6(3.8) |
| 21 | Sulfabenzamide | 5–500 | 0.9991 | 0.89 | 5 | 73.5(23.2) | 116.7(16.4) | 81.5(3.0) | 92.7(6.6) | 93.9(10.3) | 93.0(19.7) |
| 22 | Sulfachloropyridazine | 5–500 | 0.9998 | 0.94 | 5 | 89.4(5.2) | 110.7(16.0) | 81.3(3.0) | 88.9(6.9) | 90.8(10.4) | 91.1(15.9) |
| 23 | Sulfadiazine | 20–500 | 0.9999 | 0.96 | 20 | N.D. | 114.9(15.0) | 78.0(2.8) | 81.5(6.0) | 86.7(11.6) | 85.9(14.9) |
| 24 | Sulfadimethoxine | 5–200 | 0.9999 | 0.94 | 5 | 83.8(4.6) | 76.6(13.8) | 72.9(8.4) | 96(17.9) | 95.3(19.6) | 89.7(14.1) |
| 25 | Sulfamerazine | 5–500 | 0.9995 | 0.96 | 5 | 84.9(5.6) | 121.2(18.7) | 74.2(3.8) | 80.7(8.0) | 86.4(14.5) | 86.4(15.5) |
| 26 | Sulfameter | 10–500 | 1.0000 | 0.98 | 10 | N.D. | 110.5(15.5) | 82.1(3.5) | 88.6(5.7) | 86.9(12.2) | 86.8(16.1) |
| 27 | Sulfamethazine | 5–500 | 0.9964 | 0.95 | 5 | 86.9(3.8) | 119.6(19.2) | 73.9(7.7) | 79.5(7.7) | 85.4(16.9) | 88.3(14.4) |
| 28 | Sulfamethizole | 5–500 | 1.0000 | 0.96 | 5 | 72.4(12.1) | 115.3(17.2) | 76.4(3.3) | 86.6(8.7) | 88.6(12.2) | 91.7(19.1) |
| 29 | Sulfamethoxazole | 5–500 | 0.9997 | 0.86 | 5 | 89.5(8.1) | 108.5(14.5) | 86.9(3.0) | 88.4(6.0) | 93.8(8.7) | 88.3(17.1) |
| 30 | Sulfamonomethoxine | 20–500 | 1.0000 | 0.95 | 20 | N.D. | 83.8(11.3) | 79.8(7.2) | 83.8(10.9) | 96.6(19.2) | 89.3(14.8) |
| 31 | Sulfaphenazole | 5–500 | 0.9997 | 0.84 | 5 | 87.2(5.0) | 117.7(16.8) | 80.1(2.4) | 96.8(8.5) | 96.1(12.5) | 90.2(18.2) |
| 32 | Sulfapyridine | 10–500 | 0.9998 | 1.04 | 10 | N.D. | 71.8(7.9) | 70.6(4.7) | 77.4(8.9) | 82.9(15.7) | 88.0(15.6) |
| 33 | Sulfaquinoxaline | 5–500 | 0.9977 | 0.88 | 5 | 86.6(7.3) | 112.9(18.9) | 79.9(3.2) | 91.6(9.1) | 94.6(12.7) | 90.3(16.1) |
| 34 | Sulfathiazole | 5–500 | 1.0000 | 0.94 | 5 | 77.6(6.7) | 115.8(19.7) | 73.5(5.8) | 78.9(9.2) | 85.4(14.0) | 86.5(18.8) |
| 35 | Sulfisoxazole | 5–500 | 0.9998 | 0.89 | 5 | 91.1(4.0) | 116.3(16.0) | 79.1(3.6) | 89.0(6.8) | 90.5(10.4) | 90.4(14.6) |
| 36 | Chlortetracycline | 50–500 | 0.9987 | 0.81 | 50 | N.D. | N.D. | 67.8(3.4) | 83.6(7.9) | 101.2(12.3) | 84.3(9.0) |
| 37 | Demeclocycline | 50–500 | 0.9993 | 1.11 | 50 | N.D. | N.D. | 75.5(4.8) | 84.4(6.2) | 84.0(16.1) | 80.7(11.2) |
| 38 | Doxycycline | 50–500 | 0.9987 | 0.87 | 50 | N.D. | N.D. | 77.1(15.6) | 73.2(12.8) | 83.4(18.0) | 76.8(7.7) |
| 39 | Oxytetracyline | 50–500 | 0.9966 | 0.78 | 50 | N.D. | N.D. | 73.6(4.3) | 79.0(15.6) | 75.4(10.3) | 71.6(6.0) |
| 40 | Tetracycline | 50–500 | 0.9910 | 0.89 | 50 | N.D. | N.D. | 85.6(5.4) | 85.1(4.6) | 77.8(8.8) | 77.7(10.5) |
Matrix-matched external calibrations were established for honey by adding standard solutions into blank acacia honey matrices at 5–500 μg kg−1. Linear correlation coefficients (R2) of 0.99 or above were observed for all studied drugs. For some drugs, such as tetracyclines, good linear correlation coefficients at low concentrations could not be obtained because of the low response. Moreover, the linear range of flumequine, sulfadimethoxine, and tiamulin was 5–200 μg kg−1, because of the low linear dynamic range. The linear range and R2 values for the 40 veterinary drugs are listed in Table 1.
The recovery experiments were carried out in blank acacia honey samples at four spiked levels. Statistical analysis demonstrated that the percentage of compounds with a recovery between 70% and 120% was 86.9% (see Table 1). At 5 μg kg−1, 11 compounds could not be found, such as tetracyclines and part of sulfonamides, because of the low response. All the compounds were found at other three spiked levels, except tetracyclines at 20 μg kg−1, because of the same reason. Moreover, the recoveries of some compounds were lower than 70%, such as chlorotetracycline at 50 μg kg−1 (67.8%) and norfloxacin at 20 μg kg−1 (69.8%). For different varieties of honey, the recovery experiments were also carried out in vitex, Chinese date, linden, and clover honey samples at 50 μg kg−1. The results demonstrated that the differences of recoveries were not obvious, recoveries ranged from 71.6% to 101.2%, and the RSDs between different varieties of honey were less than 20%. But, for Chinese date honey, the mean recoveries of sulfonamides were slightly lower than other honey matrices, close to 70%. The precision of the method was studied in terms of its repeatability (intraday) and reproducibility (interday), and the RSDs (n = 6) of all the compounds were less than 20%.
The LOCs of the target compounds were estimated by analyzing the spiked acacia honey samples. Using the optimized screening method and criteria, the minimum concentration of the target compound that could be identified was taken as the LOC. As shown in Table 1, the LOCs of the compounds in honey ranged from 1 to 50 μg kg−1, and most of them (82.5%) were at a low concentration (≤10 μg kg−1). Unfortunately, because of the low response, tetracyclines could not be confirmed at low concentrations. Although the tetracycline LOCs meet the MRLs from Japan and Australia (300 μg kg−1), they do not reach the recommended concentrations from the EU (20 μg kg−1). Sample EIC chromatograms and product ion spectra of an LOC-spiked sample are shown in Fig. 4.
3.5 Analysis of real samples
The developed method was applied to the screening of veterinary drugs in twelve honey samples, including 2 vitex, 2 Chinese date, 2 linden, 4 acacia, and 2 clover honeys. Real samples were purchased randomly from a local market. In order to ensure the quality of the results, internal quality controls were carried out, including a matrix-matched external calibration, a reagent blank, and a spiked blank sample (50 μg kg−1). After the samples were pretreated, they were analyzed by LC-Q-TOF/MS. A positive result of ciprofloxacin was found in one honey sample; the confirmation and concentration data are shown in Fig. 5 and Table 2. Using LC-Q-TOF/MS, the suspected ciprofloxacin residue was first found by TOF/MS with a score of 96.2, and a full product ion match was used for further confirmation. The mass errors of the product ions were satisfactory, between −4.1 ppm and −0.6 ppm; the tolerances of the relative intensity were within ±20% (between −13.4% and 14.5%), when calibrated by the data from the standard as the reference. However, we also found that the response of isotopic signals (C17H16FN3O2, A+1) was higher than the theoretical response (19.7%); this was possibly due to ion interference. The concentration was determined to be 99.7 μg kg−1. LC-Q-TOF/MS had obvious advantages in the confirmation, because of the 10 IPs obtained for ciprofloxacin.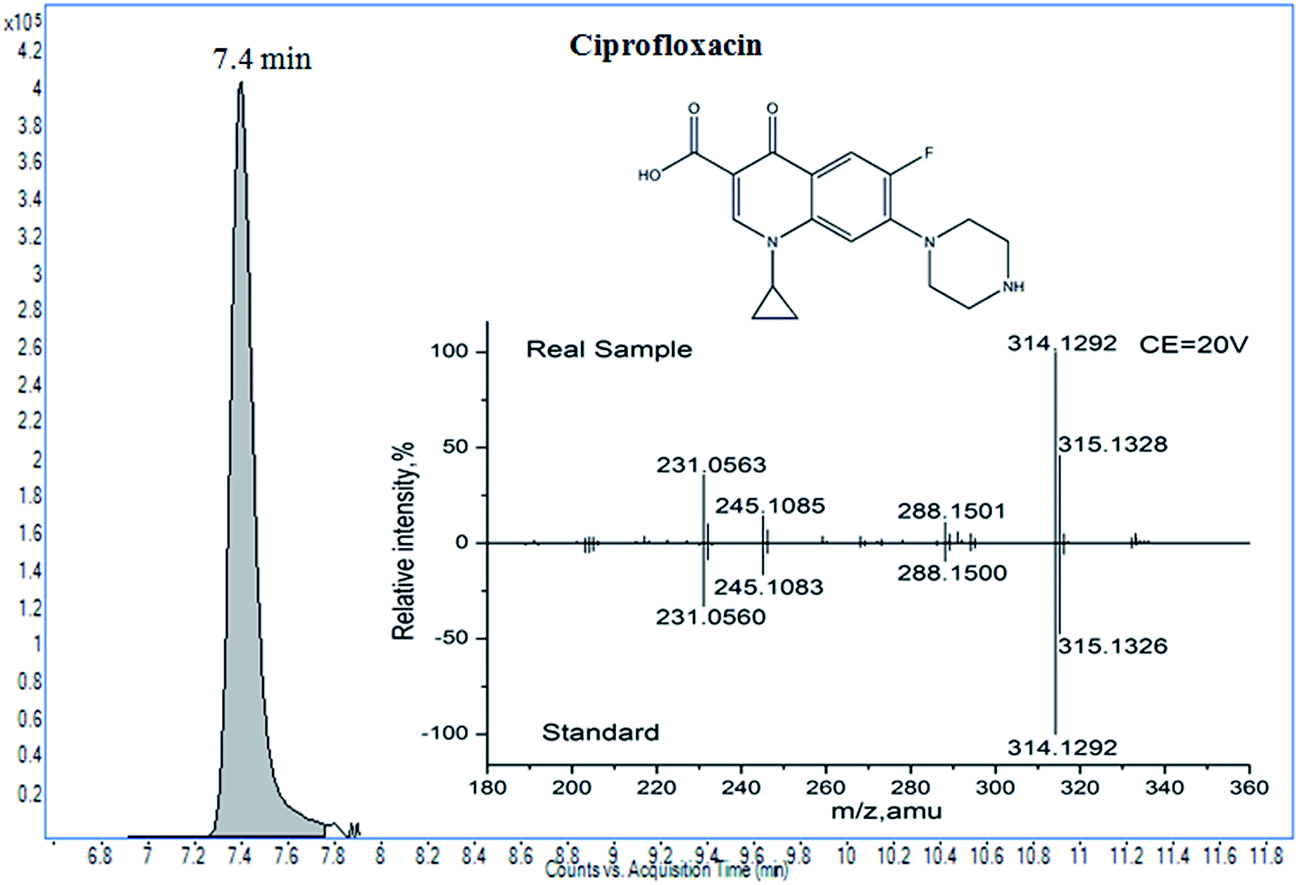 | ||
| Fig. 5 The results of ciprofloxacin in real samples of honey confirmed by LC-Q-TOF/MS. | ||
| Compound | Precursor (CEa) | Theory mass | Elemental composition | Measured mass | Relative intensity (% of base peak) | Conc.b (μg kg−1) | |||
|---|---|---|---|---|---|---|---|---|---|
| Solution (error, ppm) | Real sample (error, ppm) | Solution | Real sample | Tolerances (%) | |||||
| a CE: collision energy (eV). b Conc.: concentration of the compound. | |||||||||
| Ciprofloxacin | 332.1405 (20) | 314.1299 | C17H16FN3O2 | 314.1293(−1.9) | 314.1292(−2.3) | 100.0 | 100.0 | 0.0 | 99.7 |
| 315.1330 | C17H16FN3O2 (A+1) | 315.1326(−1.3) | 315.1328(−0.6) | 47.4 | 45.9 | −3.2 | |||
| 231.0570 | C12H8FN2O2 | 231.0560(−4.3) | 231.0563(−3.0) | 33.0 | 35.8 | 8.6 | |||
| 245.1085 | C14H13FN2O | 245.1083(−0.8) | 245.1075(−4.1) | 16.5 | 14.3 | −13.3 | |||
| 288.1507 | C16H18FN3O | 288.1501(−2.1) | 288.1500(−2.3) | 9.5 | 10.9 | 14.5 | |||
4 Conclusion
In this work, a rapid method was established using LC-Q-TOF/MS to screen 40 veterinary drugs in honey. An accurate mass database was first searched to find suspected compounds, and a full product ion match was applied as a final confirmation. The search parameters were optimized (the mass window, the limited range tR window, the ionization form) to improve the accuracy of the screening method and reduce false positive or negative results. The stability and sensitivity of the screening method were also evaluated by testing blank honey samples with different spiked levels, and the results demonstrated that the screening method was stable and reliable. The LOCs were lower than 10 μg kg−1 for most of the veterinary drugs. The established method is simple, fast, accurate, and reliable, and could be applied for routine determination. Compared with low-resolution mass spectrometry, LC-Q-TOF/MS has an obvious advantage in confirmation and identification.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Key Technology R&D Program (No. 2012BAD29B01) and the Special Program for Basic Research (No. 2015FY111200) of the Ministry of Science and Technology, China, China Postdoctoral Science Foundation (No. 2014M561203), and the Doctoral Foundation of Yanshan University (No. 8190010 & No. B853). The authors also acknowledge Agilent Technologies for the technology support of this study.Notes and references
- M. W. Kujawski and J. Namieśnik, Trends Anal. Chem., 2008, 27, 785–793 CrossRef CAS.
- Y. A. Hammel, R. Mohamed, E. Gremaud, M. H. LeBreton and P. A. Guy, J. Chromatogr. A, 2008, 1177, 58–76 CrossRef CAS PubMed.
- L. Verzegnassi, M. C. Savoy-Perroud and R. H. Stadler, J. Chromatogr. A, 2002, 977, 77–87 CrossRef CAS PubMed.
- G. H. Wan, H. Cui, H. S. Zheng, J. Zhou, L. J. Liu and X. F. Yu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 824, 57–64 CrossRef CAS PubMed.
- Commission Regulation (EU) No. 37/2010 of 22 December 2009, Off. J. Eur. Commun., 2010, L15, 1–72.
- CRL Guidance Paper, 7 December, 2007, http://www.rivm.nl/bibliotheek/digitaaldepot/crlguidance2007.pdf, accessed Oct. 2017.
- J. L. M. Vidal, M. D. M. Aguilera-Luiz, R. Romero-González and A. G. Frenich, J. Agric. Food Chem., 2009, 57, 1760–1767 CrossRef PubMed.
- M. I. Lopez, J. S. Pettis, I. B. Smith and P. S. Chu, J. Agric. Food Chem., 2008, 56, 1553–1559 CrossRef CAS PubMed.
- L. Wiest, A. Buleté, B. Giroud, C. Fratta, S. Amic, O. Lambert, H. Pouliquen and C. Arnaudguilhem, J. Chromatogr. A, 2011, 1218, 5743–5756 CrossRef CAS PubMed.
- M. Lombardo-Agüí, A. M. García-Campaña, L. Gámiz-Gracia and C. Cruces-Blanco, Talanta, 2012, 93, 193–199 CrossRef PubMed.
- P. Jovanov, V. Guzsvány, M. Franko, S. Lazić, M. Sakač, B. Šarić and V. Banjac, Talanta, 2013, 111, 125–133 CrossRef CAS PubMed.
- H. G. J. Mol, P. Plaza-Bolaños, P. Zomer, T. C. de Rijk, A. A. M. Stolker and P. P. J. Mulder, Anal. Chem., 2008, 80, 9450–9459 CrossRef CAS PubMed.
- M. Anastassiades, S. J. Lehotay, D. Stajnbaher and F. J. Schenck, J. AOAC Int., 2003, 86, 412–431 CAS.
- G. Stubbings and T. Bigwood, Anal. Chim. Acta, 2009, 637, 68–78 CrossRef CAS PubMed.
- A. G. Frenich, M. D. M. Aguilera-Luiz, J. L. M. Vidal and R. Romero-González, Anal. Chim. Acta, 2010, 661, 150–160 CrossRef PubMed.
- C. Blasco, A. Masia, F. G. Morillas and Y. Pico, J. AOAC Int., 2011, 94, 991–1003 CAS.
- M. Kanda, T. Sasamoto, K. Takeba, H. Hayashi, T. Kusano, Y. Matsushima, T. Nakajima, S. Kanai and I. Takano, J. AOAC Int., 2012, 95, 923–931 CrossRef CAS PubMed.
- J. Bernal, M. T. Martín, L. Toribio, R. Martín-Hernández, M. Higes, J. L. Bernal and M. J. Nozal, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 1596–1604 CrossRef CAS PubMed.
- A. Kaufmann, P. Butcher, K. Maden, S. Walker and M. Widmer, Anal. Chim. Acta, 2011, 700, 86–94 CrossRef CAS PubMed.
- M. d. M. Aguilera-Luiz, R. Romero-González, P. Plaza-Bolaños, J. L. M. Vidal and A. G. Frenich, J. Agric. Food Chem., 2013, 61, 829–839 CrossRef CAS PubMed.
- A. Carrasco-Pancorbo, S. Casado-Terrones, A. Segura-Carretero and A. Fernández-Gutiérrez, J. Chromatogr. A, 2008, 1195, 107–116 CrossRef CAS PubMed.
- A. Staub Sporri, P. Jan, E. Cognard, D. Ortelli and P. Edder, Food Addit. Contam., Part A: Chem., Anal., Control, Exposure Risk Assess., 2014, 31, 806–816 CrossRef CAS PubMed.
- S. Grimalt, J. V. Sancho, Ó. J. Pozo and F. Hernández, J. Mass Spectrom., 2010, 45, 421–436 CAS.
- S. B. Turnipseed, J. M. Storey, S. B. Clark and K. E. Miller, J. Agric. Food Chem., 2011, 59, 7569–7581 CrossRef CAS PubMed.
- A. Kaufmann, P. Butcher, K. Maden and M. Widmer, J. Chromatogr. A, 2008, 1194, 66–79 CrossRef CAS PubMed.
- J. Wang and D. Leung, Drug Test. Anal., 2012, 4, 103–111 CrossRef CAS PubMed.
- S. B. Turnipseed, S. B. Clark, J. M. Storey and J. R. Carr, J. Agric. Food Chem., 2012, 60, 4430–4439 CrossRef CAS PubMed.
- D. Ortelli, E. Cognard, P. Jan and P. Edder, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 2363–2374 CrossRef CAS PubMed.
- M. L. G. Pérez, P. P. Bolanos, R. Romero-González, J. L. M. Vidal and A. G. Frenich, J. Chromatogr. A, 2012, 1248, 130–138 CrossRef PubMed.
- J. Sancho, Ó. Pozo, M. Ibáñez and F. Hernández, Anal. Bioanal. Chem., 2006, 386, 987–997 CrossRef CAS PubMed.
- B. Gilbert-López, L. Jaén-Martos, J. F. García-Reyes, M. Villar-Pulido, L. Polgar, N. Ramos-Martos and A. Molina-Díaz, Food Control, 2012, 26, 341–346 CrossRef.
- M. L. G. Pérez, R. Romero-González, J. L. M. Vidal and A. G. Frenich, J. Sep. Sci., 2013, 36, 1223–1230 CrossRef PubMed.
- S. Yang, J. Cha and K. Carlson, Anal. Bioanal. Chem., 2006, 385, 623–636 CrossRef CAS PubMed.
- V. F. Samanidou, K. I. Nikolaidou and I. N. Papadoyannis, J. Sep. Sci., 2005, 28, 2247–2258 CrossRef CAS PubMed.
- M. Kanda, T. Kusano, S. Kanai, H. Hayashi, Y. Matushima, T. Nakajima, K. Takeba, T. Sasamoto and T. Nagayma, J. AOAC Int., 2010, 93, 1331–1339 CAS.
- J. Bernal, M. J. Nozal, J. J. Jiménez, M. T. Martín and E. Sanz, J. Chromatogr. A, 2009, 1216, 7275–7280 CrossRef CAS PubMed.
- M. Mezcua, O. Malato, M. A. Martinez-Uroz, A. Lozano, A. Agüera and A. R. Fernández-Alba, J. AOAC Int., 2011, 94, 1674–1684 CrossRef CAS PubMed.
- EC Decision 2002/657, Off. J. Eur. Commun., 2002, L221, 8.
- L. Vaclavik, O. Lacina, J. Hajslova and J. Zweigenbaum, Anal. Chim. Acta, 2011, 685, 45–51 CrossRef CAS PubMed.
- J. Zweigenbaum, Agro Food Ind. Hi-Tech, 2011, 22, 4–6 Search PubMed.
- J. Cotton, F. Leroux, S. Broudin, M. Marie, B. Corman, J. C. Tabet, C. Ducruix and C. Junot, J. Agric. Food Chem., 2014, 62, 11335–11345 CrossRef CAS PubMed.
- C. Coscolla, N. Leon, A. Pastor and V. Yusa, J. Chromatogr. A, 2014, 1368, 132–142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ay02440a |
| This journal is © The Royal Society of Chemistry 2018 |