DOI:
10.1039/C7AN00875A
(Paper)
Analyst, 2018,
143, 116-122
Detection of single nucleotide polymorphism by measuring extension kinetics with T7 exonuclease mediated isothermal amplification†
Received
27th May 2017
, Accepted 14th October 2017
First published on 17th October 2017
Abstract
In this work, we measured the primer extension kinetics of the Klenow fragment (exo-) to achieve rapid detection of single nucleotide polymorphism (SNP). Both the matching and the single-base mismatching targets were used as the primer in the kinetic measurements to identify the single nucleotide polymorphism. By coupling with the T7 exonuclease-assisted target cycling process, we decreased the detection limit but still maintained a high discrimination factor. After the demonstration of a good discrimination ability with synthetic DNA strands, we applied the method to detect low abundance of epidermal growth factor receptor (EGFR) mutation in human genomic DNA, which was a biomarker of non-small cell lung cancer. The kinetics based SNP detection was performed at room temperature and was robust against photobleaching and other optical interferences for the detection of low abundance of point mutations in human genomic DNA. The detection method is adaptable to a microarray platform for high-throughput and point-of-care detection.
Introduction
Single nucleotide polymorphism (SNP) refers to a single nucleotide variation in the genome. SNP detection is routinely used in genetic association studies, pharmacogenomics, forensics, etc. Many diseases are also associated with SNP, including cancers, β-thalassemia, Alzheimer's disease, cystic fibrosis and sickle-cell anemia.1,2 Therefore, the rapid, sensitive and reliable detection of SNP will be useful in many applications and particularly desirable for the diagnosis of SNP-related diseases.
For SNP detection, the polymerase chain reaction (PCR) is the gold standard method with high sensitivity and low sample consumption because of the efficient amplification process.3,4 However, PCR has the disadvantages of being time consuming and unsuitable for high-throughput analysis because of the required temperature control. PCR-free methods including direct sequencing, molecular beacons, binary probes and three- or four-way DNA junctions have also been developed, most of which are based on the hybridization ability of the DNA sequences.5,6 Hybridization probes possess a broad range of applicability in discriminating SNP and quantifying miRNA. Nonetheless, the difference in thermostability between matching and single-base mismatching DNA targets is often too small to be well discriminated.7–10 Researchers have developed locked nucleic acid probes to enhance both the binding affinity and probe stability to obtain a lower detection limit and a larger linear dynamic range.11 However, the synthesis of the probes is costly while the specificity does not increase much. Toehold exchange probes are applied to improve the specificity in detecting SNP but the sensitivity is not as competitive.12–14 Peptide nucleic acid is designed to achieve both high sensitivity and selectivity through an isocysteine mediated ligation–rearrangement reaction.15 However, the synthesis of the peptide nucleic acids is expensive and the isocysteine mediated reaction is a complicated process due to the requirement for isolating oxygen. Recently, enzyme-assisted target cycling has drawn increasing attention in detecting SNP because of the high sensitivity and simplicity.6,16–18 The main challenges in using the method are the specific labeling location on the DNA sequence and the limited selectivity in allele detection.
Herein, we introduce a new method for SNP detection by measuring the extension kinetics of a DNA polymerase fragment, the Klenow fragment (exo-). The Klenow fragment (exo-) presented different extension rates on various single-base mismatching sequences, which acted as the primer. The kinetics was measured at room temperature with high sensitivity and specificity in discriminating SNP by coupling the primer extension with T7 exonuclease-assisted signal amplification. T7 exonuclease catalyzed the removal of deoxyribonucleoside monophosphate from both the 5′-phosphorylated and the 5′-hydroxylated end to the 3′ end of a DNA sequence. T7 exonuclease preferentially acted on double-stranded DNA with both blunt and recessed ends but was unable to hydrolyze double-stranded DNA with protruding ends.19,20 In the current method, the probe DNA sequence did not need a specific location for modification except the nearby labeling of a fluorophore and a quencher. Compared with the end-point analysis, the current kinetic measurements have many advantages such as short analysis time, insensitivity to variations in the optical setup, light source instability and photobleaching, and high selectivity for multicomponent analysis.21–23 Previously, researchers utilized the kinetics based methods in pyrosequencing or PCR to discriminate SNP with higher specificity compared with the end-point analysis.24,25 In the current method, the kinetics based detection was conducted at room temperature with increased sensitivity and specificity compared with previous methods after coupling with the T7 exonuclease assisted target cycling process.
Method
Reagents
The probes, the matching target and single-base mismatching targets were synthesized by Sangon Biotech and are listed in Table S1.† Human genomic DNA was purchased from Promega. The extension was accomplished in the presence of the Klenow fragment (exo-) (Enzymatics) and dNTPs (Takara) in 1× Blue buffer (Enzymatics). After the extension, the probe–target scaffold with a blunt terminus was hydrolyzed by T7 exonuclease (Affymetrix) in 1× T7 exonuclease buffer (Affymetrix). High vacuum grease (Dow Corning) was utilized to seal the glass capillary containing the reaction mixture.
Detection with the Klenow fragment (exo-) for the matching and the mismatching targets
The probe DNA, the target DNA and the blocker DNA were dissolved and diluted to 18 μM by using 1× Blue buffer. Equal amounts of the probe DNA, the target DNA and the blocker DNA were mixed and annealed to form a double-stranded scaffold (Fig. S1†). We added 5 U μl−1 Klenow fragment (exo-) to the DNA mixture and incubated the mixture at 37 °C for 10 min. The incubated mixture would be subsequently mixed with equal amounts of 1× Blue buffer and 375 μM dNTPs. For the matching target, mixing was accomplished by using a droplet-based microfluidic platform as we previously reported.26 For the mismatching targets, we used a small centrifuge to mix the samples. We used 18 μM, 3.6 μM and 1.8 μM target DNA to perform the reaction and explore the detection limit. In the SNP detection with different abundances of mutations, we purposely mixed different ratios of the matching and the mismatching targets to obtain final abundances of 2%, 1% and 0.2%.
Detection with T7 exonuclease for the optimization of the reaction conditions
We first used a DNA sequence (anti-probe, Table S1†) which was fully complementary to the probe DNA to optimize the concentration of T7 exonuclease and the ratio of [probe]/[anti-probe] (Fig. S2†). We diluted the 18 μM DNA solution to various concentrations by using 1× Blue buffer. After the annealing of the probe and anti-probe DNA, the DNA solutions were mixed with 5× T7 exonuclease buffer and different amounts of 50 U μl−1 T7 exonuclease. The final concentrations in the 10 μl reaction mixture were 500 nM DNA, 1× T7 exonuclease buffer and 4 U μl−1, 5.25 U μl−1, 6.5 U μl−1, 8 U μl−1, 10.5 U μl−1, 13 U μl−1 and 15.5 U μl−1 T7 exonuclease, respectively, for the optimization of the concentration of T7 exonuclease. To optimize the ratio of [probe]/[anti-probe], we used 100 nM anti-probe and different concentrations of probe DNA ranging from 0 nM to 1000 nM.
Detection with the Klenow fragment (exo-) and T7 exonuclease for the matching and the mismatching targets
We utilized 1× Blue buffer to dilute the probe and the target DNA to 30 μM and 6 μM, respectively. We first mixed and annealed the probe and the target DNA to form a double-stranded DNA with a 5′ protruding end on the target (Fig. 1). After the annealing, we added the Klenow fragment (exo-) to the double-stranded DNA and incubated the mixture at 37 °C for 10 min. 375 μM dNTPs were transported to the mixture and incubated at 37 °C for 5 min to extend the primer and form a blunt end. Finally, we injected the T7 exonuclease and 5× T7 exonuclease buffer to mix and detect the increase in fluorescence intensity. We used 6 μM, 1.2 μM, 240 nM, 48 nM, 9.6 nM, 1.92 nM and 384 pM of the target DNA to perform the reaction and explore the detection limit.
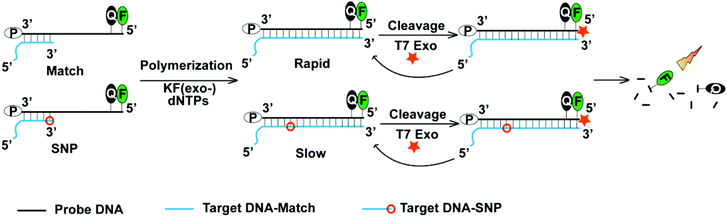 |
| | Fig. 1 The scheme for SNP detection by using the target DNA (blue) as the primer in the DNA extension. Klenow fragment (exo-) assisted primer extension followed by T7 exonuclease-assisted target cycling amplification was used in the process. The red circle highlights the SNP in the target DNA. | |
Handling of the reaction mixture and detection of the fluorescence intensity
The reaction mixture in the centrifuge tube was loaded into a glass capillary. We sealed the glass capillary with high vacuum grease and moved the capillary to the confocal microscope (Nikon C1si). The fluorescence intensity of the mixture was measured at room temperature. We set the excitation and emission wavelengths at 488 nm and 515 nm, respectively.
Results and discussion
Working mechanism of the Klenow fragment (exo-) with the T7 exonuclease-assisted target cycling process for SNP detection
As shown in Fig. 1, the probe DNA was dual-labeled with both a quencher moiety (BHQ-1) and a fluorophore (FAM) on the 5′ terminus and acted as a template. The target DNA had a protruding end on the 5′ terminus to avoid hydrolysis by T7 exonuclease. We used two kinds of targets including the matching target and the targets with a SNP. As for SNP detection, the target DNA was designed to possess a single base mismatch at the 3′ terminus and acted as a primer. The probe DNA and the target DNA were annealed to form a probe–target scaffold. In the presence of the Klenow fragment (exo-) and dNTPs, the primer would be extended to form a double-stranded DNA with a blunt terminus. After the subsequent addition of T7 exonuclease, the probe DNA underwent hydrolysis from the 5′ terminus, leading to the dissociation of the fluorophore from the quencher. Since the probe DNA was in excess, the target DNA could be recycled to hybridize with the remnant probe DNA to realize the signal amplification step. We monitored the increase in fluorescence signal and compared the kinetic parameters of the matching probe–target scaffold and the probe–target scaffolds with various kinds of SNPs.
Validation of the SNP discrimination ability of the Klenow fragment (exo-) in the target extension process
The whole detection process contained a discrimination step of the target extension by the Klenow fragment (exo-) and a signal amplification step of the probe hydrolysis by T7 exonuclease. Because the discrimination mainly arose from the target extension process, we first measured the kinetics of the target extension by the Klenow fragment (exo-) and explored the discriminating ability of the Klenow fragment (exo-) (Fig. S1†). Previously, we observed that the mismatching base pairs at or next to the primer terminus could significantly affect the kinetics of the Klenow fragment (exo-).26 Therefore, we inserted different types of mismatching base pairs at or next to the primer terminus to examine whether the Klenow fragment (exo-) could discriminate SNP with the two positions. As shown in Fig. 2, the kinetic profiles with both matching and single-base mismatching targets possessed the same shape with an initially increasing section followed by a plateau. The matching target presented fast kinetics with an increase of the signal completed in several seconds. By contrast, the single-base mismatching targets at or next to the target terminus showed slow kinetics, with the extension time in the range of hundreds to thousands of seconds. The result validated that the Klenow fragment (exo-) possessed the ability to discriminate the matching and different types of mismatching targets with high discrimination factors (Table S2†). Meanwhile, we found that the Klenow fragment (exo-) showed a better discrimination ability with a mismatch at the target terminus compared with a mismatch next to the target terminus among the three different types of mismatching targets. Hence, we chose the target terminus to be the SNP site for the detections in the following experiment.
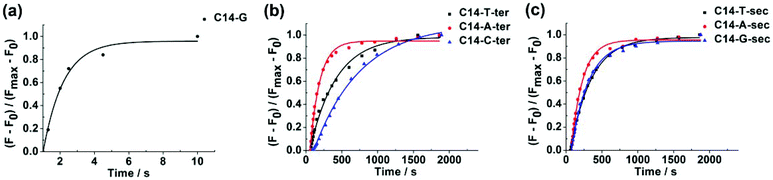 |
| | Fig. 2 Kinetic profiles with the Klenow fragment (exo-). Data points were fitted to a single-exponential model: F = Fmax − A × exp(−kobst). F denotes the fluorescence intensity, t denotes the reaction time, and A is a constant. Fmax and F0 are the maximum and initial fluorescence intensities, respectively, in each process. (a) Kinetic profiles with a matching target. (b) Kinetic profiles with a single-base mismatch at the target terminus. (c) Kinetic profiles with a single-base mismatch next to the target terminus. | |
Next, we explored the specificity and sensitivity of the method without the signal amplification process. V617F(1849 G → T) was a single nucleotide substitution which was closely related to myeloproliferative disorders.27 Herein, we would like to examine whether our method could detect the single nucleotide substitution from guanine to thymine by using the probe and the target sequences listed in Table S1.† Although the target sequence was not the wild type sequence of V617F, we expected that the same method could be applied to the wild type sequence to obtain the same level of specificity as long as the wild type sequence had the same length as the target sequence. We conducted the experiment with the mismatching target in the presence of different abundances of the matching target ranging from 0% to 2%. We observed that the matching target could be discriminated from the mismatching target with an abundance as low as 0.2% (Fig. 3a). As we expected, after the concentration of the target decreased, the fluorescence intensity decreased substantially. The detection limit was determined to be 50 nM (Fig. 3b).
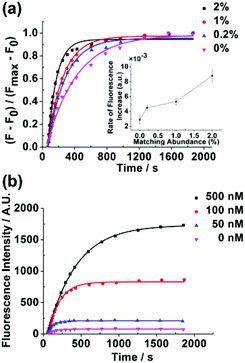 |
| | Fig. 3 Kinetic profiles of the primer extension step by the Klenow fragment (exo-) with different abundances of mutation (a) and different concentrations of the target C14-T-ter (b). Fmax and F0 are the maximum and initial fluorescence intensities, respectively, in each process. The inset shows the increase of the kobs value with the abundance of the matching target ranging from 0% to 2%. The error bars of the kobs values were obtained with more than three trials. | |
Optimization of the reaction conditions of T7 exonuclease-assisted target cycling
In order to decrease the detection limit, we added a T7 exonuclease-assisted target cycling process to amplify the signal (Fig. 1). We applied an “anti-probe” DNA sequence, which was fully complementary to the probe DNA, to optimize the conditions of the signal amplification process (Fig. S2†). After annealing, the anti-probe and the probe DNA formed a double-stranded DNA with a blunt end, which could initiate the hydrolysis of the probe DNA from the 5′ end by T7 exonuclease. An increase in the concentration of T7 exonuclease would lead to an enhancement of the fluorescence intensity with the enzyme concentration ranging from 4 U μl−1 to 8 U μl−1 (Fig. 4a). Increasing the concentration of T7 exonuclease from 8 U μl−1 to 15.5 U μl−1 did not affect the fluorescence intensity. Hence, we chose 8 U μl−1 as the optimal concentration for T7 exonuclease. What's more, the ratio of [probe]/[anti-probe] could also affect the increase of the fluorescence intensity during the hydrolysis of the probe. As demonstrated in Fig. 4b, the increase of the fluorescence intensity reached a plateau after the [probe]/[anti-probe] ratio was larger than 5. Therefore, we controlled the [probe]/[target] ratio and kept the ratio larger than 5 to obtain the maximal fluorescence increase in the following experiment.
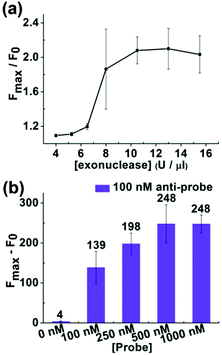 |
| | Fig. 4 Optimization of the reaction conditions of T7 exonuclease-assisted target cycling. F0 is the fluorescence intensity at the initial time point. Fmax is the maximal fluorescence intensity in each process. (a) The increase of the fluorescence intensity with various concentrations of T7 exonuclease. (b) The increase of the fluorescence intensity with different concentrations of the probe. | |
Reaction of the Klenow fragment (exo-) combined with T7 exonuclease-assisted target cycling and the capability of discriminating SNP
After adding the T7 exonuclease-assisted target cycling process, we varied the target concentration from 0 nM to 500 nM (Fig. 5a). The detection limit was diminished to 32 pM, which was a decrease of about 1500 fold compared with the detection containing no signal amplification step. Afterwards, we wanted to verify whether the method could also be applied to discriminate SNP after adding the signal amplification step. The rate of the isothermal amplification reaction in Fig. 5c was calculated from Fig. 5b by using the linear model reported previously (ESI†).28 The matching target could be well distinguished from different kinds of mismatching targets in both the kinetic profiles (Fig. 5b) and the reaction rate (Fig. 5c). The advantages of the kinetics based detection also benefited the discrimination of SNP when there were optical interferences. For example, Fig. 5b showed that the fluorescence signal was fluctuating along the fitting line. The fluctuation of the signal would affect the detection result in end point analysis much more adversely than the kinetics based detection. We then applied the method to detect the low abundance of single nucleotide substitution from guanine to cytosine, the IVSI-5 mutation that accounted for most of the β-thalassemia patients in India and Pakistan,29 by using the probe and the target sequences listed in Table S1.† Again, we expected that the target sequence used here could be replaced by the wild type IVSI-5 sequence to achieve the same specificity as long as the same length of the sequence was used. From Fig. 5d and e, different abundances of the mismatching target could be discriminated in the presence of the matching target ranging from 0% to 2%. The matching target presented a double-exponential kinetics while the mismatch-C presented a single-exponential kinetics. To understand the different kinetic behaviors of the matching and mismatch-C targets, we hypothesized that in the matching targets, almost all the targets were extended to form blunt ends upon extension by the Klenow fragment (exo-). Hence after the addition of T7 exonuclease, there would be a burst phase of the signal increase, which was due to the hydrolysis of the double-stranded DNA and the cycling hybridization of the target to the free probes in the solution for further hydrolysis. The slow signal increasing part of the matching target was corresponding to the extension of the target combined with the hydrolysis of the probes. Therefore, the matching target showed a double exponential model. For the mismatch-C, only a small portion of the targets formed double-stranded DNA during the extension process. After the addition of T7 exonuclease, there would be a slow kinetics limited by the target extension, which presented a single-exponential model.
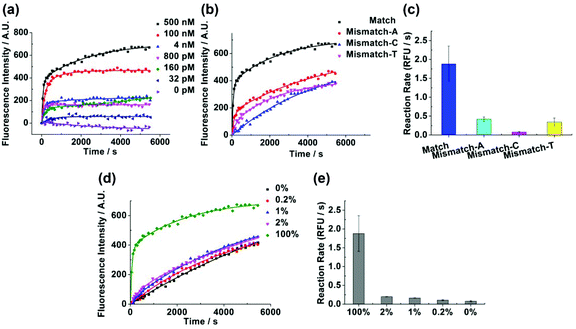 |
| | Fig. 5 The kinetic profiles combining the Klenow fragment (exo-)-assisted primer extension and T7 exonuclease-assisted target cycling processes. (a) The kinetic profiles with different concentrations of the target T-match. Data points were fitted to a double-exponential model: F = Fmax − A1 × exp(−kobs,1t) − A2 × exp(−kobs,2t). (b) The kinetic profiles with matching and various kinds of single nucleotide substituting targets. (c) The isothermal amplification reaction rate derived from the kinetic profiles in (b). (d) The kinetic profiles with different abundances of single nucleotide substitution from G to C. (e) The rate of the isothermal amplification reaction derived from the kinetic profiles in (d). | |
Detection of EGFR mutation in non-small cell lung cancer with human genomic DNA extracted from tumor tissues
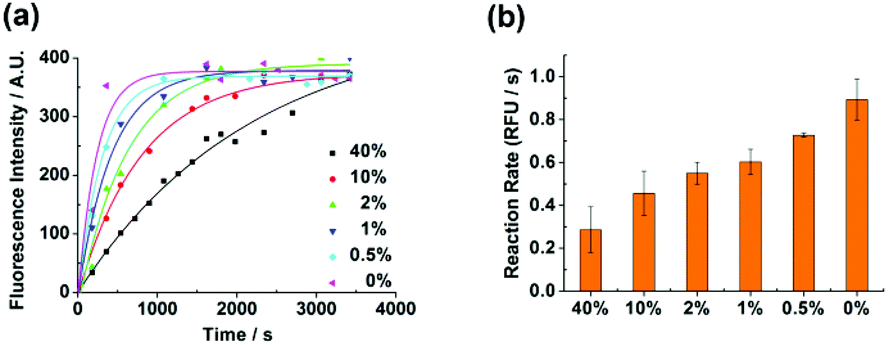
Mutated epidermal growth factor receptor (EGFR) genes are closely associated with non-small cell lung cancer. Herein, we applied the detection method to demonstrate the applicability of the method for detecting EGFR L858R (c. 2573T > G) mutation with human genomic DNA. We purposely mixed different ratios of the wild type and the mutant type targets to obtain the mutation abundance ranging from 0% to 40%. The specific region of human genomic DNA which was associated with the EGFR mutation was cut and used as the target (ESI†). After the treatment of the target, detection was routinely conducted. Different abundances of the mutation could be well discriminated with an abundance as low as 0.5% both in the kinetic profiles (Fig. 6a) and in the isothermal amplification reaction rate (Fig. 6b). The discrimination ability of the method in detecting low abundance mutations suggests that the method is potentially applicable in clinical diagnosis.
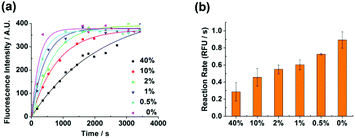 |
| | Fig. 6 Low abundance detection of EGFR mutation in non-small cell lung cancer with human genomic DNA. (a) Fluorescence response of SNP discrimination in detecting different abundances of EGFR L858R (c. 2573T > G) mutation. Data points were fitted to the single-exponential model: F = Fmax − A × exp(−kobst). (b) The rate of the isothermal amplification reaction derived from the kinetic profiles in (a). | |
The detection method in the current work has some attractive features compared to other typical methods of SNP detection. The most widely utilized method for SNP genotyping is sequencing, which is accurate and high throughput.30 Both the sensitivity and the specificity in the current detection method were lower than those in some sequencing methods, especially in single cell sequencing methods with the starting DNA in the mass range of picograms.31 However, the sensitivity and specificity of sequencing methods are critically dependent on the library preparation before the sequencing and on the data filtering after the sequencing, which are labor consuming. Previously reported costs of the sequencing reactions are 2 dollars for SNaPshot minisequencing and 0.83 dollars for pyrosequencing,32 which excluded the costs of the library preparation and the fabrication of sequencing chips. Therefore, although the sensitivity and the specificity of sequencing are higher than those of the current method, sequencing methods were more complex and expensive. Another gold standard method for SNP detection is PCR.33,34 The sensitivity of the PCR method is high with the detection limit as low as several DNA molecules.35 Taking advantage of the temperature cycles and the high fidelity of DNA polymerase, PCR methods also possess high specificity. Meanwhile, PCR methods are more cost effective with the running cost ranging from 0.1 to 0.2 dollars per assay.32 However, the amplification bias might arise from the PCR methods, which could affect the specificity of the SNP detection. Moreover, the precise temperature control during the detection process makes the PCR methods more complicated than the current method, which is performed at room temperature.
Conclusion
In this work, we detected SNP by measuring the primer extension kinetics of the Klenow fragment (exo-). The Klenow fragment (exo-) could discriminate the single nucleotide substituted targets from the matching target with high discrimination factors. After coupling the primer extension with the T7 exonuclease-assisted target cycling process, the method retained a good discrimination ability and the detection limit was decreased to 32 pM. Furthermore, the method could detect low abundance of EGFR mutations in non-small cell lung cancer. The kinetics based detection was performed at room temperature and was robust against photobleaching and other optical interferences. The detection method could be adapted to the microarray platform to achieve high throughput analysis. We expect the kinetics based detection method to be useful in early disease diagnosis as well as in genetic studies.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We thank the Research Grants Council of Hong Kong (GRF14302315) and the Chinese University of Hong Kong (3132691 and 3132692) for financial support.
References
- V. M. Ingram, Nature, 1956, 178, 792–794 CrossRef CAS PubMed.
- A. B. Wolf, R. J. Caselli, E. M. Reiman and J. Valla, Neurobiol. Aging, 2013, 34, 1007–1017 CrossRef CAS PubMed.
- J. P. Schouten, C. J. McElgunn, R. Waaijer, D. Zwijnenburg, F. Diepvens and G. Pals, Nucleic Acids Res., 2002, 30, e57 CrossRef PubMed.
- K. J. Livak, Genet. Anal., 1999, 14, 143–149 CrossRef CAS.
- A.-C. Syvanen, Nat. Rev. Genet., 2001, 2, 930–942 CrossRef CAS PubMed.
- Y. V. Gerasimova and D. M. Kolpashchikov, Chem. Soc. Rev., 2014, 43, 6405–6438 RSC.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed.
- W. Wang, T. Kong, D. Zhang, J. Zhang and G. Cheng, Anal. Chem., 2015, 87, 10822–10829 CrossRef CAS PubMed.
- S. Nakayama, L. Yan and H. O. Sintim, J. Am. Chem. Soc., 2008, 130, 12560–12561 CrossRef CAS PubMed.
- Y. V. Gerasimova, S. Peck and D. M. Kolpashchikov, Chem. Commun., 2010, 46, 8761–8763 RSC.
- H. Dong, X. Meng, W. Dai, Y. Cao, H. Lu, S. Zhou and X. Zhang, Anal. Chem., 2015, 87, 4334–4340 CrossRef CAS PubMed.
- S. X. Chen, D. Y. Zhang and G. Seelig, Nat. Chem., 2013, 5, 782–789 CrossRef CAS PubMed.
- D. Yao, T. Song, X. Sun, S. Xiao, F. Huang and H. Liang, J. Am. Chem. Soc., 2015, 137, 14107–14113 CrossRef CAS PubMed.
- S. X. Chen and G. Seelig, J. Am. Chem. Soc., 2016, 138, 5076–5086 CrossRef CAS PubMed.
- C. Dose, S. Ficht and O. Seitz, Angew. Chem., Int. Ed., 2006, 45, 5369–5373 CrossRef CAS PubMed.
- F. Zhou and B. Li, Anal. Chem., 2015, 87, 7156–7162 CrossRef CAS PubMed.
- L. Cui, Z. Zhu, N. Lin, H. Zhang, Z. Guan and C. J. Yang, Chem. Commun., 2014, 50, 1576–1578 RSC.
- T. Wu, X. Xiao, Z. Zhang and M. Zhao, Chem. Sci., 2015, 6, 1206–1211 RSC.
- C. Kerr and P. D. Sadowski, J. Biol. Chem., 1972, 247, 305–310 CAS.
- K. Shinozaki and O. Tuneko, Nucleic Acids Res., 1978, 5, 4245–4262 CrossRef CAS PubMed.
- S. R. Crouch, J. Chin. Chem. Soc., 1994, 41, 221–229 CrossRef CAS.
- T. O. Tiffany, J. M. Jansen, C. A. Burtis, J. B. Overton and C. D. Scott, Clin. Chem., 1972, 18, 829–840 CAS.
- R. N. Taylor, K. M. Fulford and A. Y. Huong, J. Clin. Microbiol., 1978, 8, 23–27 CAS.
- C. M. Waterfall, R. Eisenthal and B. D. Cobb, Biochem. Biophys. Res. Commun., 2002, 299, 715–722 CrossRef CAS PubMed.
- A. Russom, N. Tooke, H. Andersson and G. Stemme, J. Chromatogr., A, 2003, 1014, 37–45 CrossRef CAS PubMed.
- M. Cui, H. Feng, D. Guo, D. Wang and B. Zheng, Sens. Actuators, B, 2017, 253, 731–737 CrossRef CAS.
- R. Kralovics, F. Passamonti, A. S. Buser, S.-S. Teo, R. Tiedt, J. R. Passweg, A. Tichelli, M. Cazzola and R. C. Skoda, N. Engl. J. Med., 2005, 352, 1779–1790 CrossRef CAS PubMed.
-
T. Beals, 2015, bioRxiv, 017160.
-
J. Old, Genomics and Health in the Developing World, Oxford Medicine Online, United Kingdom, 2012 Search PubMed.
- P. Flaherty, G. Natsoulis, O. Muralidharan, M. Winters, J. Buenrostro, J. Bell, S. Brown, M. Holodniy, N. Zhang and H. P. Ji, Nucleic Acids Res., 2012, 40, e2 CrossRef CAS PubMed.
- T. M. Gierahn, M. H. Wadsworth Ii, T. K. Hughes, B. D. Bryson, A. Butler, R. Satija, S. Fortune, J. C. Love and A. K. Shalek, Nat. Methods, 2017, 14, 395–398 CrossRef CAS PubMed.
- N. Pati, V. Schowinsky, O. Kokanovic, V. Magnuson and S. Ghosh, J. Biochem. Biophys. Methods., 2004, 60, 1–12 CrossRef CAS PubMed.
- G. Schwarz, S. Bäumler, A. Block, F. G. Felsenstein and G. Wenzel, Nucleic Acids Res., 2004, 32, e24 CrossRef PubMed.
- L. Zhang, Q. Cai, R. S. Wiederkehr, M. Fauvart, P. Fiorini, B. Majeed, M. Tsukuda, T. Matsuno and T. Stakenborg, Lab Chip, 2016, 16, 4012–4019 RSC.
- I. A. Nazarenko, S. K. Bhatnagar and R. J. Hohman, Nucleic Acids Res., 1997, 25, 2516–2521 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the methods and principles. See DOI: 10.1039/c7an00875a |
|
| This journal is © The Royal Society of Chemistry 2018 |

 b,
Meiping
Zhao
b,
Meiping
Zhao