
Integrated catalytic sequences for catalytic upgrading of bio-derived carboxylic acids to fuels, lubricants and chemical feedstocks†
Sankaranarayanapillai
Shylesh
ab,
Amit A.
Gokhale
 ac,
Keyang
Sun
b,
Adam M.
Grippo
a,
Deepak
Jadhav
a,
Alice
Yeh
a,
Christopher R.
Ho
b and
Alexis T.
Bell
*ab
ac,
Keyang
Sun
b,
Adam M.
Grippo
a,
Deepak
Jadhav
a,
Alice
Yeh
a,
Christopher R.
Ho
b and
Alexis T.
Bell
*ab
aEnergy Biosciences Institute, University of California, 2151 Berkeley Way, Berkeley, CA 94720, USA. E-mail: alexbell@berkeley.edu
bDepartment of Chemical and Biomolecular Engineering, University of California, Berkeley, CA 94720, USA
cBASF Corporation, 33 Wood Avenue South, Iselin, NJ 07076, USA
First published on 30th August 2017
Abstract
In the late 1850s, Charles Friedel's dry distillation of calcium acetate gave the world a novel route to the commercial production of acetone, a process that would later be referred to as decarboxylative dehydration (ketonization). While the subsequent development of the petrochemical industry made this route to acetone uncompetitive, today there is considerable interest in ketonization as means for converting biomass-derived fatty acids to produce longer-chained ketones, which could serve as precursors to fuels and lubricants. However, the lack of strategies beyond direct hydrogenation of the ketones into hydrocarbons has limited the practical application of ketonization for producing biofuels. We describe here integrated catalytic sequences for converting a range of biomass-derived carboxylic acids, sourced through fermentation of sugars, hydrolysis of lipids, or biomass pyrolysis, to compounds that are fully compatible with the existing energy infrastructure and require minimal hydrogen input.
Growing public concern with climate change caused by the combustion of petroleum-based fuels combined with the rising worldwide demand for such fuels has created an urgent need to develop sustainable alternatives to fossil fuels.1–3 Nearly 86% of all energy used globally comes from fossil reserves (petroleum, natural gas, and coal) of which nearly 28% is used by the transportation sector; consequently, replacing even a part of the petroleum barrel with biomass could have a positive effect on climate change.4 Of the various bio-derived intermediates, carboxylic acids, especially light carboxylic acids (C2–C6) obtained by biomass fermentation or pyrolysis and mid- (C8–C14) and long-chained fatty acids (C16–C20) produced by hydrolysis of plant, animal, and algal oils (Scheme S1, ESI†) are of particular interest.3,5–8 We note that light carboxylic acids (C2–C3) are present in bio-oils in high concentration. Butyric acid, for example, can be generated in high titer (>60 g L−1) from Clostridium tyrobutyricum by the anaerobic fermentation of glucose.9 Valeric acid (pentanoic acid) can be derived by the reduction of levulinic acid, which in turn is produced from γ-valerolactone (GVL) through hydrolysis of 5-hydroxy-methylfurfural (HMF).10 Because of their low energy density, poor low-temperature properties, high water solubility, relatively high oxygen content, and acidity fatty acids cannot be used as fuels and need to be upgraded to remove oxygen.11–15 Though decarboxylation/decarbonylation is an attractive means for removing oxygen, these reactions do not enlarge the carbon chain length, promote C–C cleavage and usually requires large amounts of hydrogen obtained by steam reforming of methane, which coproduce CO2. Here, we outline upgrading catalytic sequences based on self- and cross-ketonic decarboxylation (ketonization), a process that condenses two molecules of carboxylic acid to produce linear ketones together with CO2, and H2O.12 The product ketones have an acidic CH group at the α position and possess both electrophilic and nucleophilic functionality, allowing them to be used as building blocks for producing petrol, jet, diesel, and lubricants.16–22 This approach utilizes the reactivity of biomass-derived molecules to deoxygenate them via intermolecular dehydration or decarboxylation and enables the production of longer chained hydrocarbons suitable as drop-in fuels and lubricants, while also using reducing the overall hydrogen demand for the process.
Our strategies for processing biomass-derived C2–C5 carboxylic acids and C8–C16 fatty acids to produce linear and branched alkanes and alkylated aromatic-cycloalkane compounds that can function as blending stocks for liquid fuels and as lubricants is summarized in Fig. 1. Step 2 in this scheme involves ketonic decarboxylation. While others have suggested hydrogenating such linear ketones to give fuels,8,17 we choose the dimerization of these ketones over acidic Nb2O5 followed by hydrodeoxygenation (HDO) over NbOPO4-supported Pt. This approach gives access to C10–C18 acyclic alkanes. By controlling the relative proportions of the various acids, it is easy to match the typical product profile for diesel and jet fuels starting with relatively cheap carboxylic acids. In the past, ketonic decarboxylation followed by hydrodeoxygenation has been carried out with relatively expensive, vegetable-oil sourced, longer chained fatty acids (C8–C16) to produce linear alkanes for diesel blends.11 While the C15–C17 hydrocarbons are ideal diesel components, the lack of branching on some of the higher molecular weight compounds makes them undesirable in fuels due to their poor cold flow properties. A more value-added strategy, therefore, is to dimerize the ketone intermediates to produce C30+ branched compounds, suitable for use as industrial lubricants and greases, since the price of these products is significantly higher than that of diesel (∼1700 per mt for group III base oils vs. $950 per mt for ULSD).20 Similarly, cross-ketonic decarboxylation of acetic acid with carboxylic acids of various chain-lengths offers a route for producing alkyl methyl ketones.23 Subsequent condensation of these ketones over acidic silica-supported tantalum oxide produces alkylated aromatic compounds, while reaction over an acid–base Mg/Al mixed oxide produces cyclic enones, compounds that upon hydrodeoxygenation give cycloalkane derivatives ideally suited as fuels or lubricants depending on their molecular weight.
 | ||
| Fig. 1 Various reaction pathways for converting biomass-derived, short-chain carboxylic acids and fatty acids to liquid alkanes suitable for use as fuels and lubricants. Step (1): alcohols produced via fermentation of mixtures of sugars and/or by the hydrogenation of carboxylic acids. Step (2): self- and cross-ketonic decarboxylation of various carboxylic acids to the respective internal and methyl ketones. Step (3): formation of ketones by mono/dialkylation of alcohols with acetone. Step (4): aldol-type condensation of ketones to form respective dimer/trimer enones and alkylated aromatics. Step (5): hydrodeoxygenation of acyclic/cyclic enones, ketones, and alkylated aromatics to produce linear, branched and cyclic alkanes suitable for use as fuels and lubricants. | ||
Since ketonic decarboxylation is the gateway reaction for all of our catalytic schemes, self- and cross-ketonic decarboxylation of C2–C5 carboxylic acids were carried out over calcined zirconia at 573 K to produce a distribution of symmetric and asymmetric C3–C9 ketones (Fig. 2 and S1–S2, ESI†). The calcination temperature was found to have a strong effect on the catalyst activity. Calcination at 823 K produced t-ZrO2 which was about twice as active (per gram) as m-ZrO2, produced by pre-treating zirconia sample at 1073 K. The higher activity of ZrO2@823 K vs. ZrO2@1073 K is ascribable to the higher concentration of basic sites for m-ZrO2 compared to t-ZrO2, 5.09 μmol CO2 per m2vs. 2.72 μmol CO2 per m2, as determined from IR spectra of adsorbed CO2.
 | ||
Fig. 2 Distribution of C5–C9 ketones produced by cross-ketonic decarboxylation of C3–C5 carboxylic acids over a calcined ZrO2 catalyst. Feed composition: (A) C3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C4:C5 = 1:1:1, (B) C3:C4:C5 = 0.5:0.5:2, (C) C3:C4:C5 = 2:0.5:0.5, (D) cross-ketonization scheme of C3–C5 carboxylic acids to C5–C9 ketones. Reaction conditions: T = 573 K, carboxylic acids = 3 mmol, solvent = 7 mL, t = 6 h, Mcat = 0.2 g. All reactions were carried out in a stirred autoclave. :C4:C5 = 1:1:1, (B) C3:C4:C5 = 0.5:0.5:2, (C) C3:C4:C5 = 2:0.5:0.5, (D) cross-ketonization scheme of C3–C5 carboxylic acids to C5–C9 ketones. Reaction conditions: T = 573 K, carboxylic acids = 3 mmol, solvent = 7 mL, t = 6 h, Mcat = 0.2 g. All reactions were carried out in a stirred autoclave. | ||
The C3–C9 internal ketones produced by ketonic decarboxylation are useful building blocks for further C–C bond forming reactions. To this end, various acid, base, and acid-base catalysts were investigated to establish their effectiveness for the selective dimerization of heptan-4-one. While calcined hydrotalcite (Mg(Al)O) is an ideal catalyst for the trimerization of methyl ketones,20 this catalyst and other basic and acid–base condensation catalysts such as MgO and hydroxyapatite (Ca–HAP, Ca/P = 1.6) exhibited low activity (<5% yield) for self-condensation of heptan-4-one at 453 K (Fig. S3, ESI†). By contrast, acidic Nb2O5 pre-treated at 523 K, was found to be highly active and selective because Lewis acidic on Nb2O5 are effective in stabilizing negatively charged enolate intermediates of central ketones. Using this catalyst, we obtained nearly 75% yield to the C14 acyclic enone dimers even under neat conditions (Table 1).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Ketone (1) | Catalyst | Conv. 1 (%) | Yield of condensates (%)* | ||||
| Cn | R | R1 | 2 | 3 | 4 | |||
|
a Ketone (1, 2 mmol), catalyst (200 mg), T (453 K), t (6 h for Mg(Al)O and 24 h for Nb2O5 and Ta/SiO2 catalyst) and toluene (3 mL) were heated in a sealed Q-tube reactor. *Mixture of positional and stereoisomers. +Produced as a mixture of dimer and trimer isomers in a 3:1 ratio. Values in parenthesis show the reusability of the catalysts after third reuse. Cyclic enone trimers and aromatic compounds constitute major side products. (Mg/Al)O represents Mg–Al oxides with an Mg/Al molar ratio of 3, and Ta/SiO2 represent silica-supported Ta2O5 with a Ta/Si molar ratio of 0.2.
|
||||||||
| 1 | C7 | Et | Et | Nb2O5 | 88 | 73 | 0 | 0 |
| 2 | C6 | Et | Me | Nb2O5 | 93 (91) | 83 | 5 | 0 |
| 3+ | C5 | Me | Me | Nb2O5 | 89 | 79 | 1 | 0 |
| 4 | C9 | nPr | nPr | Nb2O5 | 80 | 81 | 3 | 0 |
| 5 | C9 | nBu | Et | Nb2O5 | 81 | 70 | 0 | 0 |
| 6 | C5 | nPr | H | Mg(Al)O | 98 (96) | 0 | 93 | 0 |
| 7 | C6 | nPr | H | Ta/SiO2 | 96 (93) | 0 | 4 | 71 |
Pre-treatment of Nb2O5 was found to be crucial for obtaining high dimer yield. For example, Nb2O5 calcined at 573 K (Nb2O5@573 K), resulted in a nearly two-fold higher conversion of heptan-4-one and a higher selectivity to the dimer product than Nb2O5 pre-treated at 773 K (Nb2O5@773 K). The lower activity of Nb2O5@773 K is attributed to the formation of the crystalline orthorhombic (T) phase, a decrease in acid site concentration (ASC), as well as a decrease in surface area (Fig. S4, ESI†).24 Similarly, nonan-5-one, produced by ketonic decarboxylation of pentanoic acid, could be dimerized very selectively to a C18 acyclic enone, whereas pentan-3-one, obtained by the ketonic decarboxylation of propionic acid, produced a mixture of dimer and trimer acyclic enones in a nearly 3:1 ratio. The C5–C9 ketones produced by the cross-ketonic decarboxylation of C3–C5 carboxylic acids were subsequently cross-condensed over Nb2O5 to produce a distribution of C10–C18 acyclic ketones with varying degrees of branching. Finally, hydrodeoxygenation of the acyclic enone dimers over a Pt supported on niobium phosphate (2 wt% Pt/NbOPO4) produced C10–C18 acyclic alkanes with varying degrees of branching, products that are ideal drop-in replacements for fossil-derived jet and diesel (Fig. S5, ESI†).22 The hydrogen required for these upgrading deoxygenation reactions can be obtained by aqueous reforming of biomass, as reported by Dumesic and co-workers.25 The Nb2O5 and the Pt/NbOPO4 catalysts could be recycled three times without any loss in activity (Table 1).
Ketonic decarboxylation of carboxylic acids can also be used to produce fuels and lubricants from renewable sources. For example, it is possible to produce the high-octane gasoline additive 2,4-dimethyl pentane via ketonic decarboxylation of isobutyric acid followed by hydrodeoxygenation over Pt/NbOPO4 (Fig. S2, ESI†). Similarly, C8–C16 fatty acids obtained from vegetable oils can be converted to their respective ketones with yields >80% at 593 K, using t-ZrO2 as the catalyst. Decarboxylation of fatty acids, a side reaction, reduces the yield but gives linear alkanes, which are still useable for blending into diesel.15 Linear alkanes suitable as diesel and lubricants can also be obtained by hydrodeoxygenation of the C15–C31 internal ketones obtained by ketonic decarboxylation of fatty acids over Pt/NbOPO4 catalysts (Fig. 3A). The C15–C23 linear alkanes produced by this sequence are drop-in diesel fuels having high cetane numbers (CN > 90) while, the corresponding C27–C35 linear alkanes are excellent lubricants that have high viscosity indices and thermal stability.26 It is noted, though, that straight chain alkanes tend to increase the cloud point to between −20 °C and 5 °C; consequently, isomerization of straight chain alkanes is necessary to avoid wax formation for low-temperature applications.14 On the other hand, high quality base-oils containing >30 carbons can be synthesized by dimerization and hydrodeoxygenation of ketones produced by ketonic decarboxylation of fatty acids in yields >50% (Fig. 3B, Scheme S3, ESI†).
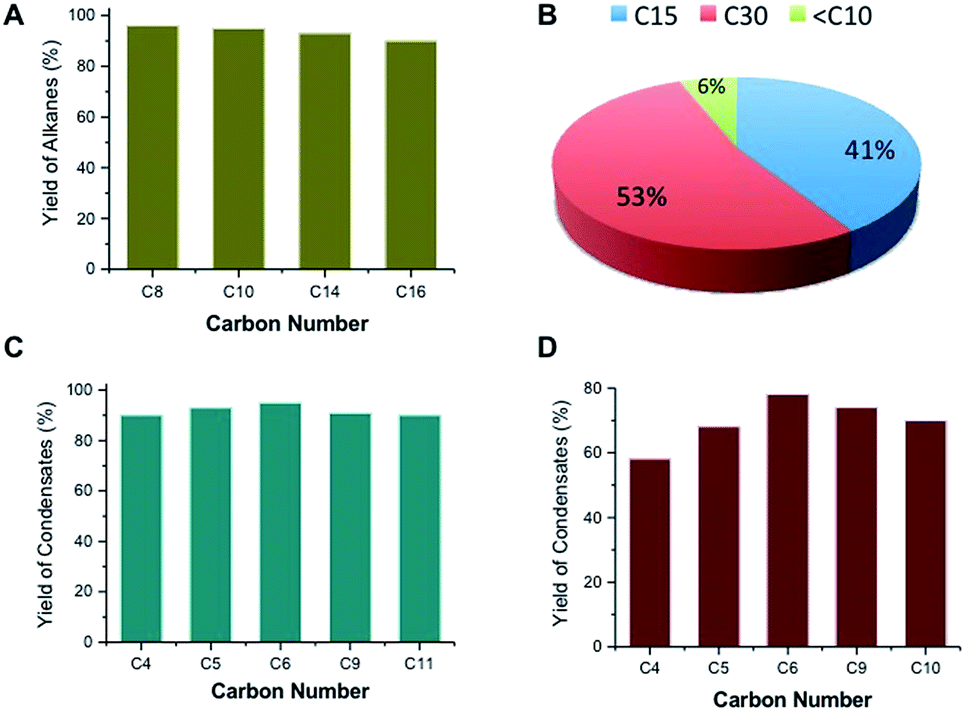 | ||
| Fig. 3 (A) Yield of C15–C31 alkanes after ketonic decarboxylation and hydrodeoxygenation of C8–C16 fatty acids over a ZrO2 and 2 wt% Pt/NbOPO4, respectively. (B) Production of diesel range C15 linear alkanes and synthetic lubricant-type C30 alkanes by ketonic decarboxylation of C8 carboxylic acid and subsequent condensation-hydrodeoxygenation. The small amount of <C10 products are lower molecular weight alkanes produced during hydrodeoxygenation. (C) Catalytic trimerization of C4–C11 methyl ketones to their respective C12–C33 cycloalkane compounds over Mg/Al oxides and hydrodeoxygenation over a 2 wt% Pt/NbOPO4. (D) Catalytic trimerization of C4–C10 methyl ketones to the respective C12–C30 alkylated aromatics over a Lewis-acidic Ta/SiO2 at 453 K. | ||
We investigated avenues for replacing cyclic alkanes and aromatics derived from petroleum.27,28 Such compounds are present in petrol, which contains 5–10% cycloalkanes and 20–50% aromatics, and jet, which contains 10–30% cycloalkanes and aromatics.29 The hydroprocessing of linear alkanes over zeolites has been proposed to produce aromatics and cyclics; however this approach requires relatively large amounts of hydrogen and frequent catalyst regeneration to counteract high coking rates.28 We have found that alkylated cyclic alkanes and aromatics can be produced from alkyl methyl ketones (C4–C11) (Table 1), obtained by the cross-ketonic decarboxylation of carboxylic acid with acetic acid (Fig. S6†) or by monoalkylation of acetone with various alcohols (Scheme S2, ESI†). Screening studies revealed that a γ-Al2O3-supported MgO or a calcined hydrotalcite (Mg/Al = 3) is a very selective catalysts for trimerizing C4–C11 alkyl methyl ketones to their respective cyclic enones. Hydrodeoxygenation of these compounds over Pt/NbOPO4 produced cycloalkane derivatives in >95% yields (Fig. 3C, Table 1). Cycloalkane derivatives made by trimerization of C4–C6 ketones are suitable for jet/diesel, whereas condensates made from C9–C11 ketones are suitable as lubricants.20,22 For instance, the derived cetane number (DCN) of C12–C18 cycloalkanes was found to be 48 while the C33 cycloalkane derivatives have an excellent pour point (PP = −69 °C), viscosity index (VI = 123) and volatility (TGA Noack = 2.58%), properties that are comparable to those of PAOs (PP = −72 °C, VI = 124 and TGA Noack = 9.64%) and significantly better than those of mineral base oils (PP = −10 to −20 °C and VI = 80–120).20 We have extended this approach in order to identify heterogeneous catalysts and appropriate reaction conditions for the self-condensation of C4–C10 methyl ketones to their respective alkylated aromatic compounds (Fig. S7, ESI†). Of the various acidic catalysts screened, we found that silica-supported Ta2O5 (Ta2O5/SiO2) to be ideal for catalyzing the trimerization of alkyl methyl ketones to chemical feedstocks and fuel-lubricant range alkylated aromatic compounds in high overall yields (Fig. 3D, Table 1). Presumably, this catalyst has the appropriate Lewis acidity to effectively convert the methyl ketones to the alkylated aromatics and during this transformation oxygen is removed in the form of water without utilizing hydrogen (Fig. S8, ESI†). However, the Ta2O5/SiO2 catalysts needs to be calcined at 823 K before they are reused due to the adsorption of cyclic enones on the acidic sites, as suggested by IR studies (Fig. S9, ESI†).
The integrated catalytic approaches reported here allow the processing of carboxylic acids of various chain length to linear and branched alkanes as well as aromatics and cycloalkane derivatives that are compatible with existing transportation fuels. Producing fuels, chemicals and lubricants from renewable sources is mainly controlled by the availability of synthons and processing costs, which is closely linked to the number of reaction steps associated with the transformation of highly oxygenated molecules to the oxygen-free liquid alkanes. The integrated catalytic sequences illustrated in this study, which are based on relatively inexpensive carboxylic acids as feedstocks; the use of highly selective, hydrothermally stable catalysts; and the minimal demand for hydrogen make the whole process economically attractive and ideal for commercialization. Interestingly, both ketonic decarboxylation and the ketone condensation can increase the carbon chain length and lower the oxygen content relative to that in the reactant without the need for a hydrogen assisted deoxygenation reaction pathway. For example, utilizing a ketonic decarboxylation-aldol condensation pathway, butyric acid with a C/O ratio of 2 can be enhanced significantly to a C/O ratio of 14 by the removal of CO2 and H2O, without utilizing hydrogen. From a process perspective, it may be theoretically possible to structure a reaction sequence involving the conversion of carboxylic acids into ketones via ketonization followed by ketone condensation and hydrodeoxygenation to alkanes of the desired molecular range without introducing intermediate separation steps. In such a scheme, complete conversion of carboxylic acids to ketones or alkanes would be crucial since the base catalyst used for ketone condensation is highly susceptible to poisoning by the acids. Nevertheless, a cascade of the liquid phase reactions described here could be integrated into a process to convert bio-derived carboxylic acids to renewable fuels and other chemical feedstocks.
Conclusions
In summary, we have shown that a sequence of catalyzed reactions can be envisioned for converting carboxylic acids to linear and branched alkanes as well as aromatics and cycloalkane derivatives that are compatible with the existing transportation fuels. This synthetic strategy is also applicable to the production of high-quality lubricants. We also note that both ketonic decarboxylation and ketone condensation increase the carbon chain length and lower the oxygen content relative to that in the reactant without the need for extensive hydrodeoxygenation. Contrary to previously reported methods for producing jet-diesel range linear alkanes,1,18,30 the processes presented here consumes a maximum of three moles of hydrogen per mole of product for the production of branched alkanes from carboxylic acids, and alkylated aromatics can be produced without any hydrogen input. Therefore, we conclude that the integrated catalytic sequences discussed in this article hold promise because they use relatively inexpensive carboxylic acids and catalysts, and require minimal consumption of hydrogen to produce a broad range of fuels, fuel additives, and lubricants in high overall yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Energy Bioscience Institute funded by BP. We gratefully acknowledge the contributions of Lipeng Wu, and Lin Louie to the experimental section of the manuscript.References
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS PubMed.
- Y. Roman-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- A. Pulido, B. Oliver-Thomas, M. Renz, M. Boronat and A. Corma, ChemSusChem, 2013, 6, 141–151 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- C. Aiello-Mazzarri, F. K. Agbogbo and M. T. Holtzapple, Bioresour. Technol., 2006, 97, 47–56 CrossRef CAS PubMed.
- J.-M. Lee, P. P. Upare, J.-S. Chang, Y. K. Hwang, J. H. Lee, D. W. Hwang, D.-Y. Hong, S. H. Lee, M.-G. Jeong, Y. D. Kim and Y.-U. Kwon, ChemSusChem, 2014, 7, 2998–3001 CrossRef CAS PubMed.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS PubMed.
- A. Corma, M. Renz and C. Schaverien, ChemSusChem, 2008, 1, 739–741 CrossRef CAS PubMed.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- B. Peng, Y. Yao, C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 2072–2075 CrossRef CAS PubMed.
- G. W. Huber, P. O'Connor and A. Corma, Appl. Catal., A, 2007, 329, 120–129 CrossRef CAS.
- E. Santillan-Jimenez and M. Crocker, J. Chem. Technol. Biotechnol., 2012, 87, 1041–1050 CrossRef CAS.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 CAS.
- A. Corma, B. Oliver-Thomas, M. Renz and I. L. Simakova, J. Mol. Catal. A: Chem., 2014, 388–389, 116–122 CrossRef CAS.
- A. Corma, O. de la Torre and M. Renz, Energy Environ. Sci., 2012, 5, 6328–6344 CAS.
- P. Anbarasan, Z. C. Baer, S. Sreekumar, E. Gross, J. Binder, H. W. Blanch, D. S. Clark and F. D. Toste, Nature, 2012, 491, 235–239 CrossRef CAS PubMed.
- M. Balakrishnan, E. R. Sacia, S. Sreekumar, G. Gunbas, A. A. Gokhale, C. D. Scown, F. D. Toste and A. T. Bell, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7645–7649 CrossRef CAS PubMed.
- M. T. Holtzapple, R. R. Davison, M. K. Ross, S. Aldrett-Lee, M. Nagwani, C.-M. Lee, S. Adelson, W. Kaar, D. Gaskim, H. Shirage, N.-S. Chang, V. S. Chang and M. E. Loescher, Appl. Biochem. Biotechnol., 1999, 77–79, 609–631 CrossRef CAS PubMed.
- E. R. Sacia, M. Balakrishnan, M. H. Deaner, K. Goulas, F. D. Toste and A. T. Bell, ChemSusChem, 2015, 8, 1726–1736 CrossRef CAS PubMed.
- C. A. Gaertner, J. C. Serrano Ruiz, D. J. Braden and J. A. Dumesic, Ind. Eng. Chem. Res., 2010, 49, 6027–6033 CrossRef CAS.
- G. S. Nair, A. A. Alsalme, I. V. Kozhevnikov, D. J. Cooke, D. R. Brown and N. R. Shiju, Catal. Sci. Technol., 2012, 2, 1173–1179 CAS.
- R. D. Cotright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef PubMed.
- T. G. Smagala, E. Christensen, K. M. Christison, R. E. Mohler, E. Gjersing and R. L. McCormick, Energy Fuels, 2013, 27, 237–246 CrossRef CAS.
- T. P. Vispute, H. Zhang, A. Sanna, R. Xiao and G. W. Huber, Science, 2010, 330, 1222–1227 CrossRef CAS PubMed.
- J. D. Adjaye and N. N. Bakhshi, Fuel Process. Technol., 1995, 45, 185–202 CrossRef CAS.
- R. W. Jenkins, C. M. Moore, T. A. Semelsberger, C. J. Chuck, J. C. Gordon and A. D. Sutton, ChemSusChem, 2016, 9, 922–931 CrossRef CAS PubMed.
- L. X. Li, E. Coppola, J. Rine, J. L. Miller and D. Walker, Energy Fuels, 2010, 24, 1305–1315 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization of catalysts and reaction study details of ketonization, aldol condensation and hydrodeoxygenation catalysts. See DOI: 10.1039/c7se00359e |
| This journal is © The Royal Society of Chemistry 2017 |