
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light and force dual-responsive waterborne polyurethane in multiple states†
Qiuhong Zhang abc,
Yikai Wangabc,
Chong Xingabc,
Yifeng Caiabc,
Kai Xia and
Xudong Jia*abc
abc,
Yikai Wangabc,
Chong Xingabc,
Yifeng Caiabc,
Kai Xia and
Xudong Jia*abc
aState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210023, P. R. China. E-mail: jiaxd@nju.edu.cn
bDepartment of Polymer Science and Engineering, Nanjing University, Nanjing 210023, P. R. China
cNanjing National Laboratory of Microstructures, Nanjing University, Nanjing 210093, P. R. China
First published on 23rd February 2017
Abstract
Light and force-sensitive waterborne polyurethane in multiple states was firstly synthesized and its responsive behaviors were investigated. Di-hydroxyl spiropyran (SP) was prepared and embedded in waterborne polyurethane (WPU) as a chain extender. The SP-WPU showed photo-sensitive properties in both emulsion state and solid state (bulk polymer). The intensity of absorbance at λmax increased as UV (365 nm) irradiation time increased, and decayed when exposed in visible light or in the dark. The SP-WPU emulsion showed a little slower UV activation rate but a much faster decay rate compared with that of SP-WPU film. The SP-WPU emulsion did not show ultrasonic sensitive properties, however, the SP-WPU in the THF/water mixed dispersion showed increasing ultrasonic sensitive properties as water content increased. Moreover, the absorption at λmax of SP-WPU in the solid state increased as elongation increased, especially in the stress stiffening region. The photo-responsive and mechanical responsive waterborne polyurethane could be used as a multi-responsive probe from the solution to the bulk state. It could also provide an environmentally friendly alternative to extend their application in smart coatings and textiles in industry.
1. Introduction
Mechanically responsive polymers1–5 have attracted researchers' interests in the past decades, due to their potential application in mechanochromic force sensors,6,7 mechanically induced catalysis,8 self-reinforcing materials9 and scaffolds for small molecule release.10,11 Spiropyran (SP) is one of the most famous mechanophores due to its sensitive color generation when exposed in light12–15 or applied stress, and the colorless SP experiences a ring-open process and transfers to the colored merocyanine (MC) (Scheme 1). In 2009, Davis et al.16 introduced SP into plastics and elastomers to map the force in polymers. Since then, SP has been chemically covalently bonded to different kinds of materials such as polycyclolactide,17,18 poly(methyl acrylate) and poly(methyl methacrylate),16,19–25 polystyrene26 and polyurethane,27,28 where it plays a role as a mechanically sensitive motif. Among these works, most existing SP embedded polymers are in glassy or semicrystalline states, whose activation requires irreversible plastic deformation or fracture of the polymers. As a result, these polymers cannot fully recover their initial shapes after the first activation, and therefore cannot be activated reversibly or repeatedly.7 SP embedded elastomers are popular because they can recover to their original shape and the mechanical activation can be triggered more than once. Another reason is that functionalized SP is designable in the elastomers. Lee et al.27,28 reported a force responsive polyurethane achieved by embedded SP in the main chain. They introduced SP in both soft segment and hard segment to detect the mechanical response of the SP-PU, acting as a molecular level probe. Craig et al.6 introduced a di-vinyl functionalized SP as a crosslinking agent in a commercial polydimethylsiloxane (PDMS) system, and the SP-PDMS showed mechanically responsive properties with full and repeatable macroscopic shape recovery at room temperature. Later, together with Zhao,29 they applied the SP-PDMS in bio-inspired electro-mechano-chemically responsive elastomers. Further, they applied this system in the soft robots with multi-functionality.30 Weng et al.31 used a di-hydroxyl SP which was chemically covalent into an ureidopyrimidinone (UPy) hydrogen bond linked polyurethane and got a tough and stress sensitive elastomer. They also found that the introduction of UPy based hydrogen bond could enhance the mechanical activation of the SP in the materials.32 Further, they also applied SP as a mechanochromic probe in a physical and chemical dual crosslinked elastomer.33 Most of these mechanically responsive elastomers were produced by solvent process or in bulk reaction. Though the previous literature34 reported mono-functionalized spiropyran was cooperated into waterborne polyurethane with photochromic properties, to the best of our knowledge, there have been no reports of mechanically responsive waterborne polyurethane till now. | ||
| Scheme 1 Equilibrium of SP-MC transformation by external stimuli. | ||
Waterborne polyurethane, an important class of materials, has been widely applied in coatings, biological fluids, drilling muds, food, cosmetics, and pharmaceuticals.35 Due to the low VOC content,36 waterborne polyurethane is exceedingly environment friendly and economic efficient. Waterborne polyurethane has excellent mechanical property with higher stress and larger strain37 because of the hydrogen bond enriched hard segments and microphase separated morphology. Waterborne polyurethane with responsive properties is of great application in industry, such as coatings on the window glass of the car, sun glasses and fabrics.
Ionic waterborne polyurethane often introduces pendant ionized segments like dimethylolpropionic acid (DMPA) and the neutralizer triethylamine (TEA) in the backbone of polymer.38 The amphiphilic structure may induced the formation of micelle or nanoparticles distributed in the aqueous phase. It has been reported that the linear amphiphilic block polymer leads to an enhanced mechanophore activation in the mixed solution.39 The unique waterborne polyurethane may also induce mechanically responsive properties in different states. Inspired by this, we designed and synthesized amphiphilic waterborne polyurethane with di-hydroxyl SP embedded in the main chain, and the light and force sensitive properties in different states were systematically investigated.
2. Experimental section
2.1 Materials
2,3,3-Trimethylindolenine and 2-iodoethanol were acquired from Sigma-Aldrich (USA). 3-Chloromethyl-5-nitrosalicylaldehyde was supplied by Nanjing Chemlin Chemical Industry Co. Ltd., China. All chemicals were used as received without further purification. Stannous octoate was obtained from Shanghai Guoyao Corporation, China. Diphenylmethane diisocyanate (MDI), polytetramethylene glycol (PTMG, Mn = 1000), isophorone diisocyanate (IPDI), dimethylolpropionic acid (DMPA) were all supplied by Sigma-Aldrich (USA) and dried under vacuum before use. THF, acetone and triethylamine (TEA), (Shanghai Guoyao Corporation, China) were dried before use.2.2 Synthesis of di-hydroxyl SP
The di-hydroxyl SP was synthesized by following the method reported in an existing procedure by Raymo et al.40 and McElhanon.18 Details of the synthesis and characterization could be seen in the ESI Scheme S1 and Fig. S1.†2.3 Synthesis of SP-WPU
Di-hydroxyl SP (0.008 g, 0.02 mmol) was put in a Schlenk reactor protected by nitrogen, and an over excess molar diphenylmethane diisocyanate (MDI, 0.16 g, 0.64 mmol) and 4 ml of dry THF were added. The mixture was stirred and refluxed at 70 °C for 2 h to ensure all the hydroxyl group had been terminated by diisocyanate.10.0 g (10 mmol) of polytetramethylene glycol (PTMG, Mn = 1000) was added in another flask which was equipped with a mechanical stirrer and protected by nitrogen, followed by adding isophorone diisocynate (IPDI, 6.446 g, 29.0 mmol), and the mixture was fully reacted at 70 °C for another 3 h. After that, the diisocyanate terminated spiropyran solution was added dropwise into the flask. THF was then removed from the mixture under vacuum. The SP embedded PU prepolymer was synthesized (Scheme 2).
 | ||
| Scheme 2 Synthesis of SP-WPU. | ||
In the next step, 1.34 g (10 mmol) of dimethylolpropionic acid (DMPA) dissolved in 3 ml of dry acetone was added into the prepolymer. After reacted for 2 h, the viscosity of the mixture increased and 50 ml of acetone was added to dissolve the mixture. Meanwhile, 3 ml of triethylamine (TEA) was added to neutralize the polymer. The solution was then added dropwise into the distilled water and stirred vigorously at the same time to get an emulsion. By controlling the temperature and pressure, the acetone and TEA were removed, the SP-WPU emulsion was obtained thereafter. After removing the water, the dispersion turned to be a yellow and transparent film (SP-WPU film).
2.4 UV-vis absorbance spectra
The total reflecting mode was used to collect the signals of the sample. The specimen was stuck on the holder and the test region was marked. After stretching and relaxing of the tensile specimen, the specimen reached a temporary state, and the marked area was then cut to test UV absorbance data by sticking to the holder with barium sulfate as background. The amount of the sample in the collection was kept the same, so all the color change of the samples was tested. The scan range was 800 nm to 200 nm, step 5 nm.
2.5 AFM test
Surface topographies were also characterized by atomic force microscopy (AFM) in tapping mode using silicon tips with Olympus OMCL-AC160TS and controller from Veeco Corporation. Samples were spin-coated on the mica's surface by emulsion or mixed solutions of polymer films (15 mg ml−1).3. Results and discussion
3.1 Photo-sensitive properties of SP-WPU
The product presented to be a slightly yellow emulsion. DLS showed that the size of the polyurethane nanoparticles was about 53 nm in average (ESI Fig. S2†), AFM showed the particle size was around 70 nm (see ESI Fig. S3†). As Fig. 1 showed, it turned to plum purple immediately when exposed in UV (365 nm). The color change of the emulsion could be essentially attributed to the change of the photochromic molecule in the polymer chain triggered by the UV light. As shown in Scheme 3, the SP in the emulsion converted to MC after the UV irradiation. It took over 20 min for the SP-PU emulsion to recover to its original color in visible light (at room temperature). In the dark situation, it took much longer time (10 h) for the purple emulsion to recover to its original color. | ||
| Fig. 1 SP-WPU (emulsion) under UV light and recovery under visible light. | ||
 | ||
| Scheme 3 Transformation of SP-WPU under UV light. | ||
UV-vis absorption spectrum was used to monitor the activation of mechanophore in SP-WPU by the irradiation of UV (365 nm). Fig. 2a showed the UV-vis absorbance spectra of SP-WPU emulsion versus the irradiated time. The black line was the original absorption of the SP-WPU. The intensity of the absorbance peaked at 550 nm grew up as irradiated time increased, indicating more contents of SP transferred to MC. At the time of 1005 s, the intensity of the absorbance reached saturation and kept constant even after further irradiation. For the color decay process in the dark, the absorbance decreased as the time increased (Fig. 2b), suggesting the activated MC transformed to the SP gradually in the dark situation.
 | ||
| Fig. 2 UV-vis absorption measurements of mechanophore activation of SP-WPU emulsion by UV 365 nm (a) and decoloration (b) of the SP-WPU emulsion in the dark. | ||
A transparent SP-WPU film was made by removing the water of the SP-WPU emulsion. The film changed to purple as the irradiated time increased, which showed the same UV-responsive behavior as the emulsion did (Fig. 3). UV-vis absorption spectrum was also performed to measure the activation of mechanophore in SP-WPU film. The absorbance peaked at 567 nm changed as UV irradiated time increased (Fig. 4A). The peak of the absorbance shifted from 550 nm to 567 nm compared with the emulsion state, because of the different circumstance of the merocyaine. The absorption also showed a decreasing trend in the dark as expected (Fig. 4B).
 | ||
| Fig. 3 SP-WPU films irradiated by UV 365 nm at different time, the numbers on top right of each figure represents the irradiated time [seconds]. | ||
 | ||
| Fig. 4 Activation (A) and de-coloration (B) of SP-WPU film vs. time evolution in the dark. | ||
For irradiation process, it could be considered as an opposing reaction, which follows the eqn (1):
 | (1) |
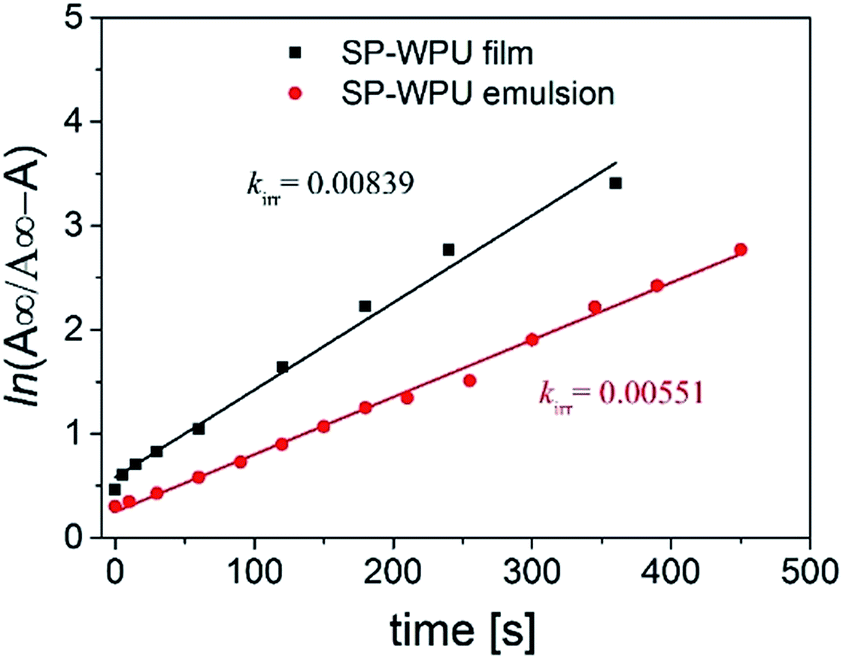 | ||
| Fig. 5 Kinetic mechanophore activation study of SP-WPU emulsion and film under UV (365 nm). | ||
For the decay process in the dark, it had been proven that it was a first order reaction which fit the following eqn (2):
| A = A∞ + (A0 − A∞)e−kdect | (2) |
For the decoloration process (Fig. 6), it could be ascribed to the restriction of molecular transition in the solid state, and the mobility of the merocyanine in the solid state was relatively slow. However, the photochromic molecule in the emulsion state could move more freely, hence it took much less time for the molecular conversion to a much stable structure. This showed that the decoloration rate of the MC in solid state was much slower than that in the emulsion state.
 | ||
| Fig. 6 Kinetic decoloration study of SP-WPU film and SP-WPU emulsion in the dark. | ||
3.2 Mechanically responsive behavior of SP-WPU
The mechanical chemistry often performed in different states by using different methods.41 For the solutions, ultra-sonication was usually introduced due to requirement of small quantities, easy achievement of high strain rates, and rapid response. It is widely accepted that ultra-sonication induced polymer chemistry is the result of solvodynamic shear created by cavitation, which involves the nucleation, growth, and collapse of microbubbles in solution.7 Polymer chains undergo a coil-stretch transition during the ultra-sonication process. It was reported that several systems42 had been applied to ultra-sonication activation tests. However, less test had been applied in SP embedded polyurethane solution or dispersion. | ||
| Fig. 7 AFM images (height and phase) of the SP-WPU mixed dispersions (from left to right: THF, 5 vol% water, 10 vol% water and 15 vol% water, respectively. Three figures on top represent the height figure and other three down represent the phase figure). | ||
There was no obvious color change when the SP-WPU emulsion was performed by sonication experiment over 60 min. The SP-WPU in THF solution showed a slightly color change when it was applied sonication. Different amount of water was added into the solution and formed dispersion with mixed solvent, which were labeled as 5 vol%, 10 vol%, 15 vol% and 20 vol% water, respectively. An obvious color change occurred immediately when the sonication was loaded (Fig. 8), indicating more SP transferred to the MC in the mixed solvent. The sonication activation of mechanophore in SP-WPU dispersion was measured by UV-vis absorption spectra. Taken 15 vol% water as an example (Fig. 9), the absorbance at 550 nm increased as sonication time increased.
 | ||
| Fig. 8 Photos of SP-WPU dispersion (THF/water mixture) before and after sonication. | ||
 | ||
| Fig. 9 UV-vis absorption spectra of SP-WPU mixed solvent (15 vol% water) as sonication time increased. | ||
The UV-vis absorption of samples with different amounts of water peaked at λmax at different sonication time were plotted in Fig. 10. The absorption increased rapidly at first and then slow down to keep constant when the sonication time reached ca. 35 min. It was shown that the maximum absorption of 15 vol% water increased about 2.5 times comparing with that of the pristine THF solution. It was witnessed that the maximum absorbance of SP-WPU mixed solvent increased as water content increased up to 15 vol%. Further increase of water will cause the reduction of activation (e.g. 20 vol% water). When the water content reached 25 vol%, the sample turned turbid. As a result, the system would not be stable, in which the assemblages of SP-WPU in a suspension would be formed in large scale.
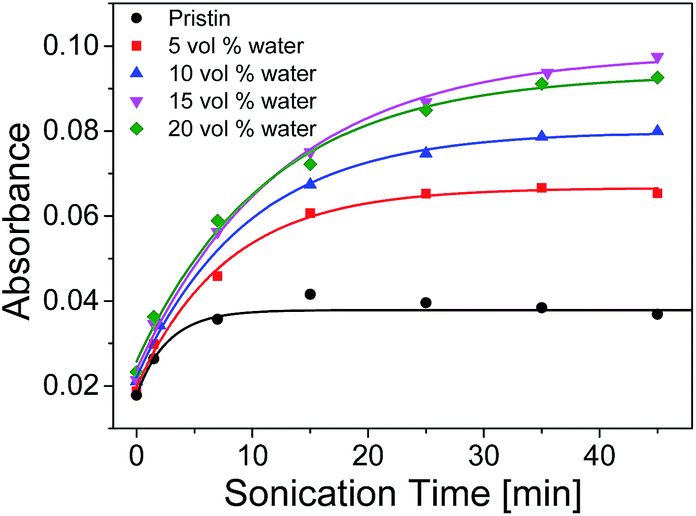 | ||
| Fig. 10 Ultra-sonication activation of SP-WPU in different mixed solvents. | ||
The synergetic effect of both THF and water contributed to the enhanced activation of the mechanophore in SP-WPU (shown in Scheme 4). Firstly, the state of the polymer chain in the solvent is important. If the polymer chain was strongly coiled, then much higher intensity ultra-sonication power should be applied.7 THF makes the chain disaggregate, which is similar with the “swelling effect” as other systems.43 As a result, a much free and extended chain would be formed in the dispersion. During the sonication process, the force will transmit effectively to the mechanophore. At the same time, water is another important solvent providing the polarity surroundings where activated MC can be stabilized. The less polar environment destabilizes the zwitterionic MC, shifting toward the SP form. In other words, by increasing the polarity of mixed solvent, the equilibrium toward MC occurred thereafter. This is further proved by the activation of SP-WPU THF/methanol mixed dispersion, which was between the activations of SP-WPU THF solution and SP-WPU THF/water mixed dispersion (ESI Fig S5†).
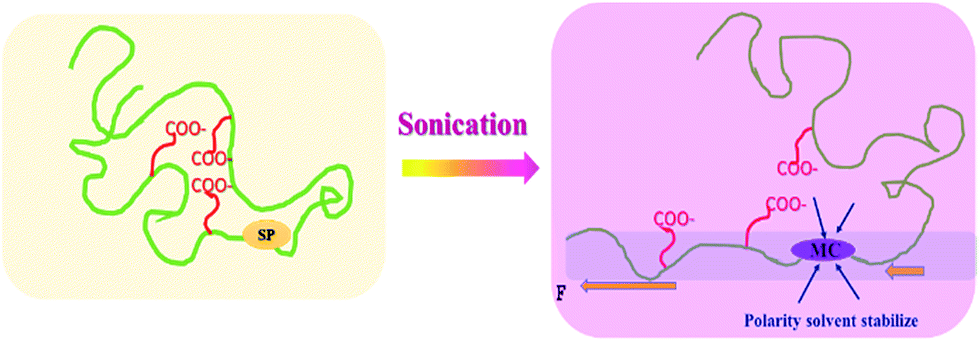 | ||
| Scheme 4 Illustration of the sonication activation of SP-WPU in mixed solution. | ||
SP-WPU emulsion (100 vol% water) did not show any ultrasonication responsive properties. Because the polyurethane chain aggregated together and formed elastic nanoparticles, the strongly coiled chain in the emulsion restricted the force to transmit to the mechanophore. The SP-WPU THF solution (0 vol% water) showed less sensitive activation, because the less polarity of the solvent led the MC to the thermally stable SP. There are two contradictory effects by adding water: the positive polarity effect and the negative size effect. These two effects induced a threshold concentration of water 15 vol%, beyond which the response would decrease. Further increasing the content of water, the SP-WPU would not be stable in the mixed solvent and swelling effect would also decrease, which was witnessed by the decreasing intensity of UV-vis in 20 vol% water.
In addition, we performed ultra-sonication of SP-WPU with different DMPA contents (see ESI, Fig S6†). It showed that the ultra-sonication activation intensity increased as the DMPA content decreased, which was ascribed to the reason that the micellization could make SP in SP-WPU behave higher activity toward sonication.44
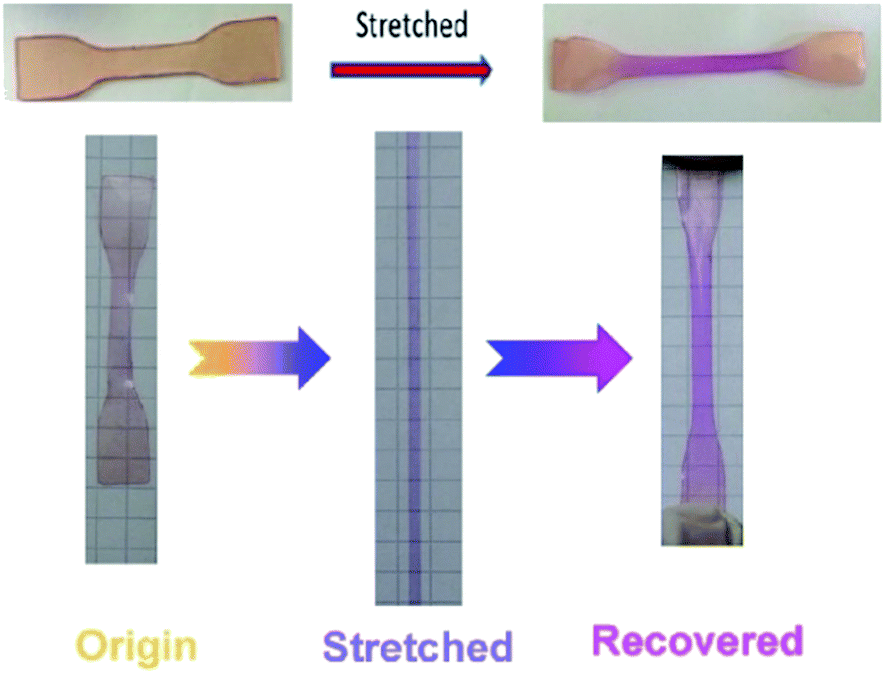 | ||
| Fig. 11 Photos of stretch sensitive SP-WPU film. | ||
In order to get rid of the other effects during tensile, control experiments of the di-hydroxyl SP sample whose structure was shown in the ESI (Scheme S2†) were also performed at the same condition. However, the sample did not show any mechanochromic properties in the tensile experiment, while showed obvious photo-chromic property (ESI Fig. S10†). This suggested that the activation of SP-WPU film was activated by force instead of other stimuli.
The UV-vis absorption spectrum was used to test the activation of spiropyran in the SP-WPU film during stretching (Fig. 12). The SP-WPU film showed an increasing intensity of absorption when it was applied larger strain, indicating more mechanophore was converted from SP to MC. The absorbance was normalized and plotted with the strain (Fig. 13). It could be found that the activation of the mechanophore was obvious when the strain was over 500%. Sufficient chain alignments along the tensile direction, which were formed at the stress stiffening region, is essential to transfer the load to the mechanophores thus to activate the mechanochemical reaction.45
 | ||
| Fig. 12 UV-vis absorption spectra of SP-WPU film as elongation increased. | ||
 | ||
| Fig. 13 Normalized absorption at 560 nm as a function of elongation increased and the corresponding stress–strain curve. | ||
4. Conclusions
The light and force dual-responsive waterborne polyurethane was firstly reported. The UV light triggered color change in the emulsion state was slightly faster than that of the SP-WPU film. The decay rate was quite different in the two states. The decay rate of the emulsion state was rapider than that of the hindered solid state, which was ascribed to the faster molecular motion.The mechanically responsive properties were investigated in two different states, the mixed dispersion and the bulk film by ultra-sonication and tensile test, respectively. The ultra-sonication responsive properties relied on the water content of the mixture. The SP-WPU mixed solution containing of 15 vol% water showed the most sensitive ultra-sonication activation properties due to the synergetic effect of the polarity of the mixed solvents. The SP-WPU film also showed stretch responsive properties, giving the color change when the elongation over 500%, where sufficient chain alignments along the tensile direction were formed. The SP embedded waterborne polyurethane is a smart and environmental friendly material. It is easy to scale up and has the great applications in the light sensitive coatings like sunglasses, damage detection and so on.
Acknowledgements
Funding was provided by the National Natural Science Foundation of China (Grants 21404056) and Natural science foundation of Jiangsu Province (Grants BK20130556). The authors would like to thank the Program for Changjiang Scholars and Innovative Research Team in University for financially supporting this research.References
- M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921–2948 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- H. Zhang, Y. Lin, Y. Xu and W. Weng, in Polymer Mechanochemistry, ed. R. Boulatov, 2015, vol. 369, pp. 135–207 Search PubMed.
- X. Meng, Q. Chen, Z. Wang, S. Xu and Y. Ma, Polym. Bull., 2014, 23–33 Search PubMed.
- A. Pucci, R. Bizzarri and G. Ruggeri, Soft Matter, 2011, 7, 3689–3700 RSC.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, ACS Macro Lett., 2014, 3, 216–219 CrossRef CAS.
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- R. T. M. Jakobs, S. Ma and R. P. Sijbesma, ACS Macro Lett., 2013, 2, 613–616 CrossRef CAS.
- H. Zhang, F. Gao, X. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS PubMed.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2014, 136, 1276–1279 CrossRef CAS PubMed.
- M. B. Larsen and A. J. Boydston, J. Am. Chem. Soc., 2013, 135, 8189–8192 CrossRef CAS PubMed.
- Y. Hirshberg, J. Am. Chem. Soc., 1956, 78, 2304–2312 CrossRef CAS.
- Y. Huang, R. Dong, X. Zhu and D. Yan, Soft Matter, 2014, 10, 6121–6138 RSC.
- R. Perez-Ruiz and D. Diaz Diaz, Soft Matter, 2015, 11, 5180–5187 RSC.
- D. Ramachandran, F. Liu and M. W. Urban, RSC Adv., 2012, 2, 135–143 RSC.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- G. I. Peterson, M. B. Larsen, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2015, 7, 577–583 CAS.
- G. O'Bryan, B. M. Wong and J. R. McElhanon, ACS Appl. Mater. Interfaces, 2010, 2, 1594–1600 Search PubMed.
- B. A. Beiermann, S. L. B. Kramer, P. A. May, J. S. Moore, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2014, 24, 1529–1537 CrossRef CAS.
- C. M. Degen, P. A. May, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2013, 46, 8917–8921 CrossRef CAS.
- B. A. Beiermann, S. L. B. Kramer, J. S. Moore, S. R. White and N. R. Sottos, ACS Macro Lett., 2012, 1, 163–166 CrossRef CAS.
- B. A. Beiermann, D. A. Davis, S. L. B. Kramer, J. S. Moore, N. R. Sottos and S. R. White, J. Mater. Chem., 2011, 21, 8443–8447 RSC.
- C. M. Kingsbury, P. A. May, D. A. Davis, S. R. White, J. S. Moore and N. R. Sottos, J. Mater. Chem., 2011, 21, 8381–8388 RSC.
- A.-D. N. Celestine, B. A. Beiermann, P. A. May, J. S. Moore, N. R. Sottos and S. R. White, Polymer, 2014, 55, 4164–4171 CrossRef CAS.
- M. Li, Q. Zhang and S. P. Zhu, Polymer, 2016, 99, 521–528 CrossRef CAS.
- M. E. Grady, B. A. Beiermann, J. S. Moore and N. R. Sottos, ACS Appl. Mater. Interfaces, 2014, 6, 5350–5355 CAS.
- C. K. Lee, D. A. Davis, S. R. White, J. S. Moore, N. R. Sottos and P. V. Braun, J. Am. Chem. Soc., 2010, 132, 16107–16111 CrossRef CAS PubMed.
- C. K. Lee, B. A. Beiermann, M. N. Silberstein, J. Wang, J. S. Moore, N. R. Sottos and P. V. Braun, Macromolecules, 2013, 46, 3746–3752 CrossRef CAS.
- Q. Wang, G. R. Gossweiler, S. L. Craig and X. Zhao, Nat. Commun., 2014, 5, 4899 CrossRef PubMed.
- G. R. Gossweiler, C. L. Brown, G. B. Hewage, E. Sapiro-Gheiler, W. J. Trautman, G. W. Welshofer and S. L. Craig, ACS Appl. Mater. Interfaces, 2015, 7, 22431–22435 CAS.
- X. Fang, H. Zhang, Y. Chen, Y. Lin, Y. Xu and W. Weng, Macromolecules, 2013, 46, 6566–6574 CrossRef CAS.
- Y. Chen, H. Zhang, X. Fang, Y. Lin, Y. Xu and W. Weng, ACS Macro Lett., 2014, 3, 141–145 CrossRef CAS.
- H. Zhang, Y. Chen, Y. Lin, X. Fang, Y. Xu, Y. Ruan and W. Weng, Macromolecules, 2014, 47, 6783–6790 CrossRef CAS.
- L.-h. Bao, J.-x. Sun and Q. Li, J. Polym. Res., 2014, 21, 575 CrossRef.
- S. A. Madbouly and J. U. Otaigbe, Prog. Polym. Sci., 2009, 34, 1283–1332 CrossRef CAS.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- D. Xu, K. Wu, Q. Zhang, H. Hu, K. Xi, Q. Chen, X. Yu, J. Chen and X. Jia, Polymer, 2010, 51, 1926–1933 CrossRef CAS.
- L. Hou, Y. Ding, Z. Zhang, Z. Sun and Z. Shan, Colloids Surf., A, 2015, 467, 46–56 CrossRef CAS.
- L.-J. Wang, X.-J. Zhou, X.-H. Zhang and B.-Y. Du, Macromolecules, 2016, 49, 98–104 CrossRef CAS.
- F. M. Raymo and S. Giordani, J. Am. Chem. Soc., 2001, 123, 4651–4652 CrossRef CAS PubMed.
- K. M. Wiggins, J. N. Brantley and C. W. Bielawski, Chem. Soc. Rev., 2013, 42, 7130–7147 RSC.
- P. A. May, N. F. Munaretto, M. B. Hamoy, M. J. Robb and J. S. Moore, ACS Macro Lett., 2016, 5, 177–180 CrossRef CAS.
- C. K. Lee, C. E. Diesendruck, E. Lu, A. N. Pickett, P. A. May, J. S. Moore and P. V. Braun, Macromolecules, 2014, 47, 2690–2694 CrossRef CAS.
- L. J. Wang, X. J. Zhou, X. H. Zhang and B. Y. Du, Macromolecules, 2016, 49, 98–104 CrossRef CAS.
- G. N. Hong, H. Zhang, Y. J. Lin, Y. J. Chen, Y. Z. Xu, W. G. Weng and H. P. Xia, Macromolecules, 2013, 46, 8649–8656 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00156h |
| This journal is © The Royal Society of Chemistry 2017 |