
Label-free ferrohydrodynamic cell separation of circulating tumor cells†
Wujun
Zhao
 a,
Rui
Cheng
b,
Brittany D.
Jenkins
c,
Taotao
Zhu
a,
Nneoma E.
Okonkwo
d,
Courtney E.
Jones
e,
Melissa B.
Davis
c,
Sravan K.
Kavuri
f,
Zhonglin
Hao
g,
Carsten
Schroeder
h and
Leidong
Mao
*b
a,
Rui
Cheng
b,
Brittany D.
Jenkins
c,
Taotao
Zhu
a,
Nneoma E.
Okonkwo
d,
Courtney E.
Jones
e,
Melissa B.
Davis
c,
Sravan K.
Kavuri
f,
Zhonglin
Hao
g,
Carsten
Schroeder
h and
Leidong
Mao
*b
aDepartment of Chemistry, The University of Georgia, Athens, GA 30602, USA
bSchool of Electrical and Computer Engineering, College of Engineering, The University of Georgia, Athens, GA 30602, USA. E-mail: mao@uga.edu
cDepartment of Genetics, The University of Georgia, Athens, GA 30602, USA
dDepartment of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
eCollege of Engineering and Computer Science, Syracuse University, Syracuse, NY 13210, USA
fDepartment of Pathology, Augusta University, Augusta, GA 30912, USA
gDepartment of Medicine, Augusta University, Augusta, GA 30912, USA
hDepartment of Surgery, Augusta University, Augusta, GA 30912, USA
First published on 4th August 2017
Abstract
Circulating tumor cells (CTCs) have significant implications in both basic cancer research and clinical applications. To address the limited availability of viable CTCs for fundamental and clinical investigations, effective separation of extremely rare CTCs from blood is critical. Ferrohydrodynamic cell separation (FCS), a label-free method that conducted cell sorting based on cell size difference in biocompatible ferrofluids, has thus far not been able to enrich low-concentration CTCs from cancer patients' blood because of technical challenges associated with processing clinical samples. In this study, we demonstrated the development of a laminar-flow microfluidic FCS device that was capable of enriching rare CTCs from patients' blood in a biocompatible manner with a high throughput (6 mL h−1) and a high rate of recovery (92.9%). Systematic optimization of the FCS devices through a validated analytical model was performed to determine optimal magnetic field and its gradient, ferrofluid properties, and cell throughput that could process clinically relevant amount of blood. We first validated the capability of the FCS devices by successfully separating low-concentration (∼100 cells per mL) cancer cells using six cultured cell lines from undiluted white blood cells (WBCs), with an average 92.9% cancer cell recovery rate and an average 11.7% purity of separated cancer cells, at a throughput of 6 mL per hour. Specifically, at ∼100 cancer cells per mL spike ratio, the recovery rates of cancer cells were 92.3 ± 3.6% (H1299 lung cancer), 88.3 ± 5.5% (A549 lung cancer), 93.7 ± 5.5% (H3122 lung cancer), 95.3 ± 6.0% (PC-3 prostate cancer), 94.7 ± 4.0% (MCF-7 breast cancer), and 93.0 ± 5.3% (HCC1806 breast cancer), and the corresponding purities of separated cancer cells were 11.1 ± 1.2% (H1299 lung cancer), 10.1 ± 1.7% (A549 lung cancer), 12.1 ± 2.1% (H3122 lung cancer), 12.8 ± 1.6% (PC-3 prostate cancer), 11.9 ± 1.8% (MCF-7 breast cancer), and 12.2 ± 1.6% (HCC1806 breast cancer). Biocompatibility study on H1299 cell line and HCC1806 cell line showed that separated cancer cells had excellent short-term viability, normal proliferation and unaffected key biomarker expressions. We then demonstrated the enrichment of CTCs in blood samples obtained from two patients with newly diagnosed advanced non-small cell lung cancer (NSCLC). While still at its early stage of development, FCS could become a complementary tool for CTC separation for its high recovery rate and excellent biocompatibility, as well as its potential for further optimization and integration with other separation methods.
Introduction
Circulating tumor cells (CTCs) are cancer cells that are detached from primary solid tumors and carried through the vasculature to potentially seed distant site metastases in vital organs – the main cause of death by cancer.1,2 Molecular assessments of CTCs not only could benefit basic cancer research, but also might eventually lead to a more effective cancer treatment.3–5 However, one major limitation of CTCs in cancer research and its clinical applications has been the limited availability of viable CTCs for investigations, due in part to the small patient blood volumes that are allowable for research, which usually yielded less than 100 CTCs from 1 mL of whole blood.5–7 As a result, technologies are needed in order to separate these rare cells from blood, and important performance criteria for these technologies include the ability to process a significant amount of blood quickly (e.g., throughput ∼7.5 mL h−1), a high recovery rate of CTCs, a reasonable purity of isolated cancer cells, and cell integrity for further characterization.8CTCs represent the composition of the primary tumor, including the heterogeneity of tumors.5,9 While CTCs initially express same biological or physical markers as the primary tumor epithelial cells, once in circulation they may undergo morphological and gene expression changes, which could determine what distant site will become the new niche for a metastatic tumor. Enriching the whole CTC population, instead of just the ones responding to specific biological or physical markers, can allow basic investigations such as CTC heterogeneity, and may lead to a more precise prognosis of undetected metastasis and recurrence risk for cancer patients.10 Label-based CTC separation technologies were developed to selectively enrich a subset of CTCs from blood, primarily through the use of specific biological markers including epithelial cell adhesion molecule (EpCAM).11–13 These antigen-based labels were a rate-limiting factor in effective CTC separation, as the inherent heterogeneity of CTCs might render these technologies ineffective for general use. The vast array of various biomarkers that might or might not be expressed, and which could not be predicted to remain expressed in CTCs undergoing epithelial-to-mesenchymal transitions (EMT) would be cumbersome and confounding in these label-based methods. Furthermore, most label-based technologies did not conveniently enable comprehensive molecular analysis of separated CTCs because they were either dead or immobilized to a surface.14 On the other hand, a variety of label-free methods including those based on filtration,15 acoustophoresis,16 dielectrophoresis,17–19 dean flow,20–22 and vortex technology23–25 were developed recently to exploit specific physical markers in order to deplete non-CTCs in blood therefore enrich cancer cells. They were not affected by the heterogeneity of biological marker expressions and could permit enrichment of nearly all CTCs that were above a predetermined threshold of a physical marker, for example, the size of CTCs. Most CTCs of epithelial origin have a size range between 15 μm and 25 μm, and are larger than red blood cells (RBCs, 6–9 μm), and the majority of white blood cells (WBCs, 8–14 μm).8 However, CTCs of smaller sizes were found in blood circulation.26,27 The existence of large WBCs such as monocytes that may have overlapping sizes with CTCs could further complicate label-free separation methods.7,14,28 Both label-based and label-free methods had their limitations; more sophisticated strategies including novel sorting methods such as acoustophoresis16 and vortex technology,23–25 or a combination of two or more methods to enrich rare cells based on multiple biological or physical markers could potentially improve the overall performance of CTC separation.29–33 One successful device is the CTC-iChip that integrated both label-based and label-free separation methods. This device first used deterministic lateral displacement to deplete smaller RBCs from patient blood based on their size, then applied inertial force to focus remaining cells into a narrow stream, and eventually separated WBCs that were coated with anti-CD45 and anti-CD66b magnetic beads from CTCs for a high-throughput and high-recovery separation.29,30 While each of these three methods alone might have its own limitation in rare cell separation, their integration were critical to the overall success of CTC-iChip. There is a need to develop new and high-performance CTC separation method that not only performs well on its own, but also can be easily integrated with other methods to achieve high-throughput, high-recovery, high-purity separation of intact CTCs. A frequently used method in CTC or rare cell separation was functionalizing magnetic particles to target and pull cells of interest through magnetic force or “magnetophoresis” towards a magnetic field maxima, as illustrated in Fig. 1A. Magnetophoresis, when used for CTC separation, has achieved high-throughput and high-specificity isolation of cancer cells from blood.13,34–41 On the other hand, it is a label-based method and requires time-consuming and laborious sample preparation.
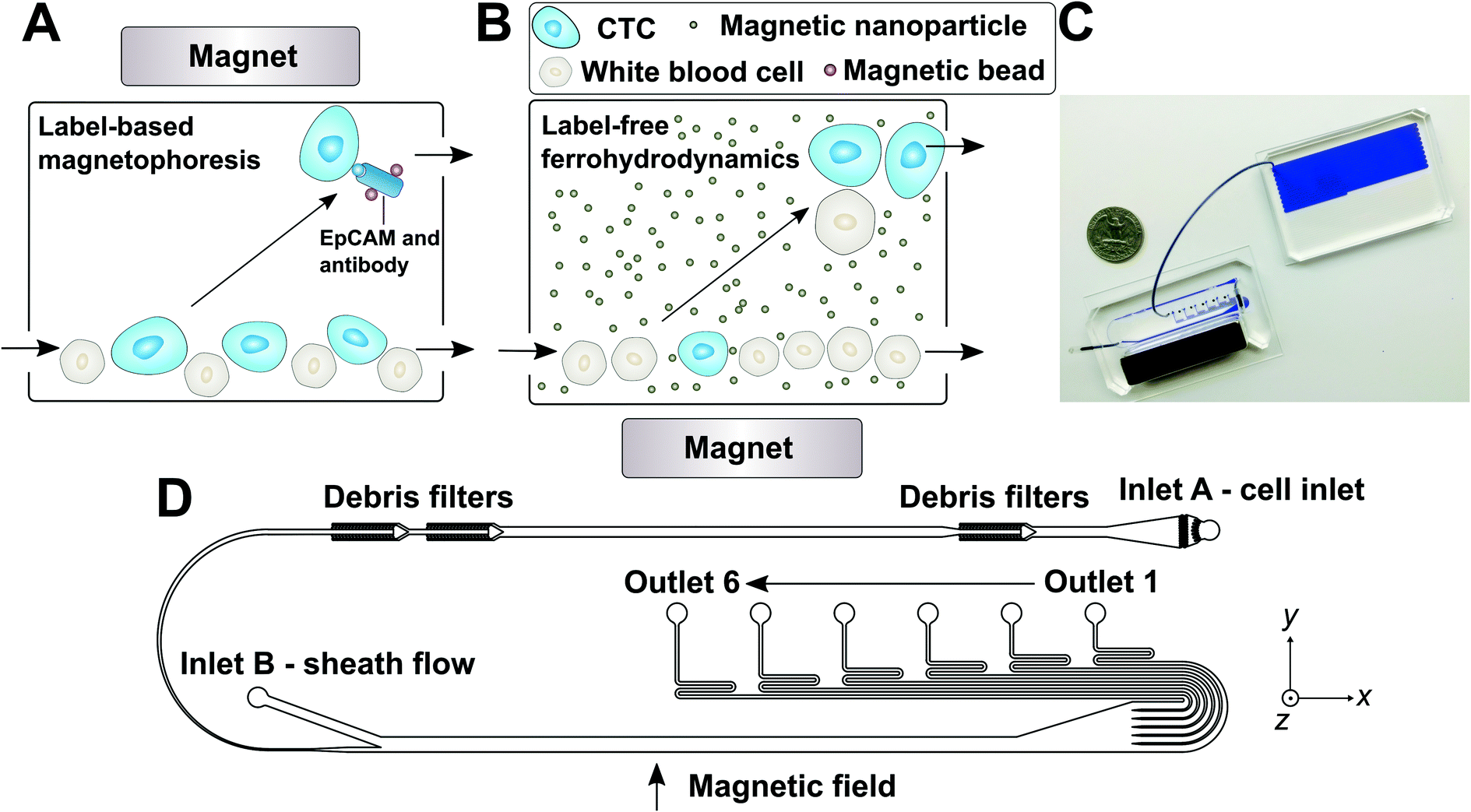 | ||
| Fig. 1 (A) Schematic illustration of traditional and frequently used label-based magnetophoresis for CTC separation, in which rare cells were targeted via specific biomarkers such as epithelial cell adhesion molecule (EpCAM) through functionalized magnetic particles in order to pull these cells through magnetic force towards magnetic field maxima in a continuous-flow manner. (B) Schematic illustration of a label-free ferrohydrodynamic cell separation (FCS) for CTCs. In FCS, RBC-lysed blood and biocompatible ferrofluids (colloidal suspensions of magnetic nanoparticles) were processed in continuous flow within a FCS device, such as the one shown in (C) and (D). Cells in blood were first filtered to remove debris, then focused by a ferrofluid sheath flow from inlet B. After entering the channel region that was on top of a permanent magnet, large cells including CTCs and some WBCs experienced more size-dependent magnetic buoyance force than smaller WBCs, resulting in a spatial separation between them at the outlets of the FCS device. (C) A photo of a prototype FCS device (left) consisted of a PDMS microchannel and a permanent magnet. The FCS device was connected to a serpentine PDMS collection chamber (right) that was used to accurately count cancer cells or WBCs during FCS calibration experiments using cultured cancer cells. A U.S. quarter was shown for size comparison. Blue dye was used to visualize the channel. (D) Top-view of the FCS device with labels of inlets, debris filters and outlets. A total of 6 outlets were fabricated in order to account for the broad size distributions of cells (see ESI,† Fig. S11B). The arrow indicates the direction of magnetic field during device operation. Dimensions of the FCS device and magnet can be found in ESI.† | ||
In this paper, we reported a new ferrohydrodynamic cell separation (FCS) method that still used magnetic buoyance force for size-based CTC separation, but was label-free, biocompatible and enriched rare CTCs from patient blood with a high throughput and a high rate of recovery. We demonstrated that FCS could separate a variety of low-concentration cancer cells of cell culture lines from RBC-lysed blood at a throughput of 6 mL h−1, with an average cancer cell recovery rate of 92.9% and an average cancer cell purity of 11.7% after separation. CTCs were successfully enriched from blood samples of two non-small cell lung cancer (NSCLC) patients using FCS devices. We envision that FCS could offer the potential to serve as a complementary tool in CTC separation because of its excellent biocompatibility and label-free operation. FCS could also be integrated with other separation methods such as magnetophoresis for a more comprehensive isolation of rare cells. The working principle of ferrohydrodynamic cell separation is “negative magnetophoresis” in biocompatible ferrofluids, as illustrated in Fig. 1B.42 Cells including CTCs and WBCs immersed inside an uniformly magnetic media (ferrofluids) can be considered as “magnetic holes”.43 A non-uniform magnetic field gradient induces an imaginary dipole moment in these “magnetic holes”, and generates a size-dependent magnetic body force, also referred to as magnetic buoyancy force that pushes the cells away to a magnetic field minima.44 Forces on the cells can therefore sort them based on their size difference in a continuous ferrofluid flow. In practice, a mixture of RBC-lysed blood and ferrofluids was injected into the inlet A of a FCS device such as the one shown in Fig. 1C. Cells in blood were filtered then focused by a sheath flow from inlet B. After entering the channel region that was on top of a permanent magnet, large cells including CTCs and some WBCs experienced more size-dependent magnetic buoyance force than smaller WBCs, resulting in a spatial separation between them at the outlets of the device. Although ferrohydrodynamic cell separation was demonstrated before,45–49 its application in CTCs was challenging in the past for the following reasons. First, rarity of CTC necessitates a blood-processing throughput of close to 7.5 mL h−1 and recovery rate of at least 80% in low concentration (<100 cells per mL) conditions.8 Previous applications of ferrohydrodynamic cell separation mostly focused on sorting of bacteria and yeast cells,45,46 bacteria and red blood cells,47 and cancer cells of cultured cell lines from blood.48,49 The throughputs of these studies were lower than what was required of CTC separation, and the target cells were mostly spiked at a much higher concentration (e.g., 105–106 cells per mL) than CTCs.45–48 Second, ferrofluids, as a colloidal suspension of magnetic nanoparticles with diameters of approximately 10 nm, need to be rendered biocompatible for CTC separation. Cancer cells should remain alive and their normal functions should be kept intact during and after the separation for post-separation characterization. It is therefore critical to systematically optimize FCS and ferrofluid design so that the throughput and recovery rate of separation are comparable to those needed for CTC separation, and the separated cells are viable and their normal functions are intact.
We overcame these challenges associated with ferrohydrodynamic cell sorting of CTCs, and demonstrated a 92.9% recovery rate and an 11.7% purity of low-concentration (∼100 cells per mL) cancer cells with a blood-processing throughput of 6 mL of blood per hour, and validated the technology using blood from NSCLC patients. We performed systematic parametric studies of key factors influencing the performance of FCS and determined parameters for high-throughput, high recovery rate and biocompatible CTC separation. We then tested and validated the performance of the method with cancer cells from 6 cultured cancer cell lines and 3 different types of cancer. The mean recovery rate of cancer cells from RBC-lysed blood using this technology is 92.9%, a value much better than currently reported an average of 82%.8 Separated cancer cells had excellent short-term viability, unaffected biological marker expressions, and intact capability to proliferate to confluence. Finally, we applied the FCS method to successfully enrich CTCs from blood samples of two stage IVB NSCLC patients, and discussed the advantages and limitations of this method and potential ways to improve.
Experimental section
Modeling of FCS and its calibration
The model used in this study to simulate cell trajectories in three-dimensional (3D) manner was previously described.50,51 We modified the analytical model (see ESI†) for this study, which could predict the 3D transport of diamagnetic cancer cells and WBCs in ferrofluids inside a microfluidic channel coupled with permanent magnets. The magnets produced a spatially non-uniform magnetic field that led to a magnetic buoyancy force on the cells. Trajectories of the cells in the device were obtained by (1) calculating the 3D magnetic buoyancy force via an experimentally verified and analytical distribution of magnetic fields as well as their gradients, together with a nonlinear Langevin magnetization model of the ferrofluid, (2) deriving the hydrodynamic viscous drag force with an velocity profile of the channel obtained from COMSOL Multiphysics (Version 3.5, COMSOL Inc., Burlington, MA), (3) solving governing equations of motion using analytical expressions of magnetic buoyancy force and hydrodynamic viscous drag force in MATLAB (MathWorks Inc., Natick, MA). The parameters of simulation (device dimension and geometry, fluid and cell properties, and magnetic fields) reflected exact experimental conditions.Polystyrene microparticles (Polysciences, Inc., Warminster, PA) with diameters of 15.7 μm were mixed together with WBCs at the concentration of 1 × 104 particles per mL for model calibration. Microparticle and cell mixtures were injected into inlet A of a FCS device with a flow rate of 1.2–6 mL h−1. The flow rate of inlet B was fixed at 6 mL h−1 for all experiments. The magnet was placed 1 mm away from the channel, which corresponded to magnetic field strengths 443 mT and magnetic field gradients 56.2 T m−1 (ESI,† Fig. S1). A ferrofluid with a concentration of 0.26% (v/v) were used in calibration experiments.
Custom-made biocompatible ferrofluids
A water-based ferrofluid with maghemite nanoparticle was synthesized by a chemical co-precipitation method and made biocompatible following a protocol previously described.48,49 Details of the ferrofluid synthesis and functionalization are listed in ESI.† Size and morphology of the maghemite nanoparticles were characterized via transmission electron microscopy (TEM; FEI Corp., Eindhoven, the Netherlands). Magnetic properties of the resulting biocompatible ferrofluid were measured at room temperature using a vibrating sample magnetometer (VSM; MicroSense, LLC, Lowell, MA). Briefly, particle size distribution of the custom-made ferrofluid was 11.24 ± 2.52 nm. Saturation magnetization of the as-synthesized ferrofluid was 0.96 kA m−1, corresponding to an estimated 0.26% volume fraction of magnetic content. This ferrofluid was colloidally stable for up to 10 months' storage, did not show particle agglomeration during microfluidic operations, and was made to be isotonic and have a 7.0 pH and neutral surfactant for biocompatible cell separation.Cell culture and sample preparation
Six cancer cell lines (ATCC, Manassas, VA) including three lung cancer cell lines (H1299, A549 and H3122), one prostate cancer cell line (PC-3), and two breast cancer cell lines (MCF-7 and HCC1806) were used in this study. H1299, A549, H3122, PC-3, and HCC1806 cells were cultured in RPMI-1640 medium (Mediatech, Inc., Manassas, VA) supplemented with 10% (v/v) fetal bovine serum (FBS; Life Technologies, Carlsbad, CA) and 1% (v/v) penicillin/streptomycin solution (Mediatech, Inc., Manassas, VA). MCF-7 cells were cultured in Dulbecco's modified eagle medium (DMEM; Life Technologies, Carlsbad, CA) supplemented with 10% (v/v) FBS, 1% (v/v) penicillin/streptomycin solution and 0.1 mM MEM non-essential amino acid (NEAA; Life Technologies, Carlsbad, CA). All cell cultures were maintained at 37 °C under a humidified atmosphere of 5% CO2. Cell lines were released through incubation with 0.05% trypsin–EDTA solution (Life Technologies, Carlsbad, CA) at 37 °C for 5–10 minutes before each use.Cancer cells were fluorescently stained by incubation with 2 μM CellTracker Green (Life Technologies, Carlsbad, CA) for 30 minutes before each use. Probe solution was replaced with culture medium by centrifuging at 200 × g for 5 minutes. Cells were counted with a hemocytometer (Hausser Scientific, Horsham, PA) and serially diluted in culture medium to achieve a solution with approximately 1 × 104 cells per mL. Cells were then counted with a Nageotte counting chamber (Hausser Scientific, Horsham, PA) to determine the exact number of cells per μL. Desired number of cancer cells (50, 100, 200, 500, 1000, or 2000) were spiked into 1 mL of WBCs (RBC-lysed whole blood). The number of cancer cells spiked was determined by the average of two counts, with an average of 5.2% difference between the counts. We chose to focus on separating cancer cells from WBCs because of the size of WBCs (8–14 μm) were much closer to cancer cells (15–25 μm) than RBCs (6–9 μm).
Human whole blood from healthy subjects (Zen-Bio, Research Triangle Park, NC) was lysed by RBC lysis buffer (eBioscience, San Diego, CA) with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 for 5 minutes at room temperature. Cell mixtures were centrifuged at 800 × g for 5 minutes and the pellet was suspended in the same volume of ferrofluid containing 0.1% (v/v) Pluronic F-68 non-ionic surfactant (Thermo Fisher Scientific, Waltham, MA). WBCs were fixed by 4% (w/v) paraformaldehyde (PFA; Santa Cruz Biotechnology, Dallas, TX) at 4 °C for 30 minutes for long-term use.
:10 for 5 minutes at room temperature. Cell mixtures were centrifuged at 800 × g for 5 minutes and the pellet was suspended in the same volume of ferrofluid containing 0.1% (v/v) Pluronic F-68 non-ionic surfactant (Thermo Fisher Scientific, Waltham, MA). WBCs were fixed by 4% (w/v) paraformaldehyde (PFA; Santa Cruz Biotechnology, Dallas, TX) at 4 °C for 30 minutes for long-term use.
Biocompatibility study of FCS
Short-term cell viability after FCS was examined using a Live/Dead assay (Life Technologies, Carlsbad, CA). 1 × 106 H1299 cancer cells suspended in 1 mL of ferrofluids were injected into inlet A of a FCS device at a flow rate of 6 mL h−1. After separation, cells from outlet 6 were collected and washed with phosphate buffered saline (PBS; Life Technologies, Carlsbad, CA) three times. Cells were then incubated with working solution (2 μM calcein-AM and 4 μM ethidium homodimer-1 (EthD-1)) for 30 minutes at room temperature. After the solution was removed and washed with PBS, labeled cells were observed under a fluorescence microscope (Carl Zeiss, Germany) for counting.For long-term proliferation, separated H1299 cells from a FCS device were collected into a centrifuge tube and washed three times with culture medium to remove the nanoparticles, and then the cells were suspended in culture medium and seeded into a 24-well plate (Corning Inc., Corning, NY). Cells were then cultured at 37 °C under a humidified atmosphere of 5% CO2, the medium was refreshed every 24 h during the first 3 days. Cellular morphology was inspected every 24 hours.
Surface biomarker expression change was studied by immunofluorescence staining of cancer cells with EpCAM and cytokeratin antibodies. HCC1806 cancer cells were collected after FCS and seeded on a coverslip. After 24 h incubation, cells were fixed with 4% (w/v) PFA for 30 minutes and subsequently permeabilized with 0.2% (v/v) Triton X-100 (Sigma-Aldrich, St. Louis, MO) in PBS for 10 minutes. Cells were then blocked by 0.5% (w/v) bovine serum albumin (BSA; Miltenyi Biotec, San Diego, CA) in PBS for 20 minutes. After blocking nonspecific binding sites, cells were immunostained with primary antibodies, anti-cytokeratin 8/18/19 (Abcam, Cambridge, MA), human EpCAM/TROP-1 (R&D System, Minneapolis, MN). Appropriately matched secondary Alexa Fluor-conjugated antibodies (Life Technologies, Carlsbad, CA) were used to identify cells. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies, Carlsbad, CA). After immunofluorescence staining, cells were washed with PBS and stored at 4 °C or imaged with a fluorescence microscope.
FCS device fabrication and cell separation
Microfluidic devices were made of polydimethylsiloxane (PDMS) using standard soft lithography techniques. The thickness of the microfluidic channel was measured to be 52 μm by a profilometer (Veeco Instruments, Chadds Ford, PA). One NdFeB permanent magnet (K&J Magnetics, Pipersville, PA) was embedded into the PDMS channel with their magnetization direction vertical to the channel during the curing stage. The magnet is 5.08 cm in length, 1.27 cm in both width and thickness. Flux density at the center of magnet's surface was measured to be 0.5 T by a Gauss meter (Sypris, Orlando, FL) and an axial probe with 0.381 mm diameter of circular active area. Detailed geometries of device setup can be found in ESI,† Fig. S2. Fabricated devices were first flushed by 70% ethanol for 10 minutes at the flow rate of 6 mL h−1 and then primed with 1× PBS supplemented with 0.5% (w/v) BSA and 2 mM EDTA (Thermo Fisher Scientific, Waltham, MA) for 10 minutes at the flow rate of 6 mL h−1 before each use.22During a typical experiment, a microfluidic device was placed on the stage of an inverted microscope (Carl Zeiss, Germany) for observation and recording. Two fluid inputs were controlled by individual syringe pumps (Chemyx, Stafford, TX) at tunable flow rates. Blood samples were injected into inlet A of a FCS device, sheath flow (ferrofluids) was injected into inlet B. Images and videos of microparticles and cells were recorded with a high-resolution CCD camera (Carl Zeiss, Germany). After separation, cells were collected in a serpentine collection chamber for cell counting.
NSCLC patient blood processing
De-identified blood samples were obtained from newly diagnosed advanced NSCLC patients before treatment with informed consents according to a protocol approved by Institutional Review Board (IRB) at Augusta University. All blood samples were collected into vacutainer tubes (BD, Franklin Lakes, NJ,) containing the anticoagulant K2EDTA and were processed within 3 hours of blood draw. In a typical process, every 1 mL of whole blood was lysed by 10 mL of RBC lysis buffer for 5 minutes at room temperature. WBCs were then collected by spinning down the solution at 800 × g for 5 minutes and the pellet was suspended in 1 mL of ferrofluid containing 0.1% (v/v) Pluronic F-68. The sample was then loaded into a 10 mL syringe (BD, Franklin Lakes, NJ,) followed by processing with the FCS device at a flow rate of 6 mL h−1. A stainless-steel sphere (BC Precision, Chattanooga, TN) with a diameter of 1.6 mm was also loaded into a syringe. A magnet was used to gently agitate the sphere to prevent blood cells from settling down every 5–10 minutes. After separation, the FCS device was flushed by PBS or ThinPrep PreservCyt solution (Hologic, Marlborough, MA) at 30 mL h−1 for 20 minutes to remove any cells in outlet reservoir. During the separation, the cells from outlet 6 of a FCS device were directly preserved in ThinPrep PreservCyt solution for further analysis.CTC identification
After processing of blood with a FCS device, collected cells were preserved in ThinPrep PreservCyt solution. Samples collected in ThinPrep vials were directly loaded into ThinPrep 2000 processor (Hologic, Marlborough, MA), which is an automated slide-processing instrument that was routinely used in cytology laboratory for preparing gynecologic and non-gynecologic samples. The instrument transferred diagnostic cells in the sample to a slide that was then immersed in cell fixative bath ready for staining. Papanicolaou (Pap) staining of the slides was performed using Shandon Gemini stainer (Thermo Fisher Scientific, Waltham, MA) followed by cover-slipping using permount. ThinPrep slides were afterwards evaluated by a cytopathologist using light microscopy to identify and count the number of CTCs. Collected cells were also fixed with 4% (w/v) PFA for 30 minutes and subsequently permeabilized with 0.2% (v/v) Triton X-100 in PBS for 10 minutes. Cells were then blocked by 0.5% (w/v) BSA in PBS for 20 minutes. After blocking nonspecific binding sites, cells were immunostained with primary antibodies, anti-cytokeratin 8/18/19, human EpCAM/TROP-1, and anti-CD45 (Abcam, Cambridge, MA). Following, the appropriately matched secondary Alexa Fluor-conjugated antibodies (Life Technologies, Carlsbad, CA) were used to identify cells. After immunofluorescence staining, cells were washed with PBS and stored at 4 °C or imaged with a fluorescence microscope.Results and discussion
Optimization of FCS for high-throughput, high-recovery and biocompatible CTC separation
Previous ferrohydrodynamic cell sorting devices were developed to process cells at low throughput and high spike ratios,45,47–49 therefore cannot be realistically used to separate CTCs from blood. CTCs are extremely rare in the blood circulation, occurring usually at a concentration of less than 100 CTCs per mL of blood.5–7 These cells are dispersed in a background of billions of RBCs and millions of WBCs, making the separation of CTCs a significant challenge. For any CTC separation method, it is necessary for it to be able to process several milliliters of blood within one hour with a high CTC recovery rate to enrich sufficient numbers of viable CTCs. Thus, high-throughput, high recovery rate, reasonable purity and biocompatible separation of viable CTCs are four criteria for any separation method targeting clinical applications. For ferrohydrodynamic cell separation (FCS) method, the parameters that will affect the above-mentioned criteria include device geometry, magnetic field and its gradient, flow rate of cells, and ferrofluid properties (i.e., magnetic volume fraction or concentration, pH, tonicity, materials and surfactants of nanoparticles, colloidal stability). These parameters are highly coupled with each other and for this reason an effective model was needed for systematic device optimization. To search for parameters for high-throughput, high recovery rate, reasonable purity and biocompatible CTC separation, we first started with a device geometry depicted in Fig. 1D and Fig. S2† that operated in low Reynolds number laminar flow region when its cell flow rates were from 1.2 to 7.2 mL h−1. The corresponding Reynolds numbers were from 0.5 to 3.1, and the upper limit of this flow rate range was close to the clinically relevant throughput in typical CTC separation. We then created an analytical model that could predict three-dimensional (3D) trajectories of cancer cells and blood cells in ferrofluids inside this device coupled with a permanent magnet. We considered both magnetic buoyancy force and hydrodynamic drag force in simulating the cell trajectories. The detailed description of this 3D analytical model is described below and also in ESI.†The dominant magnetic force in ferrohydrodynamic cell sorting (FCS) is a magnetic buoyancy force generated on diamagnetic cells immersed in ferrofluids. Particles immersed in ferrofluids experience this force under a non-uniform magnetic field,44
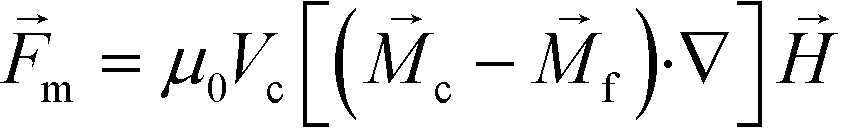 | (1) |
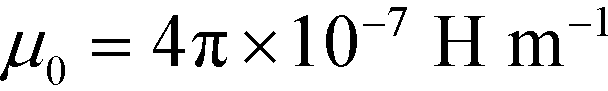 is the permeability of free space, Vc is the volume of the magnetized body, in this case a cell,
is the permeability of free space, Vc is the volume of the magnetized body, in this case a cell, 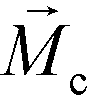 is its magnetization (close to zero for most cells),
is its magnetization (close to zero for most cells), 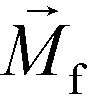 is magnetization of the ferrofluid surrounding the body, and
is magnetization of the ferrofluid surrounding the body, and 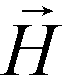 is magnetic field strength at the center of the body.44 For cell separation in ferrofluids under a strong magnetic field, magnetization of the ferrofluid with superparamagnetic particles in it can be modeled via Langevin function,44
is magnetic field strength at the center of the body.44 For cell separation in ferrofluids under a strong magnetic field, magnetization of the ferrofluid with superparamagnetic particles in it can be modeled via Langevin function,44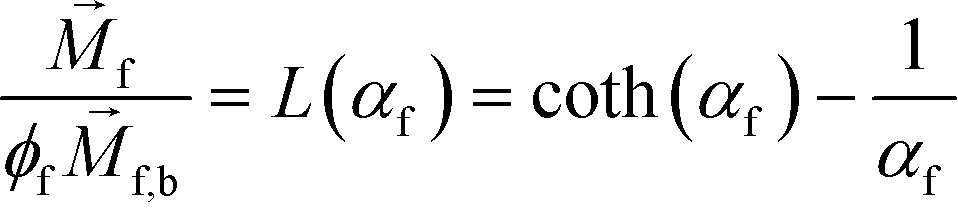 | (2) |
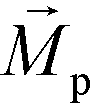 is less than its surrounding magnetic liquid
is less than its surrounding magnetic liquid 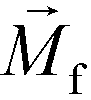 , and the direction of the magnetic force
, and the direction of the magnetic force 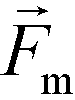 on the cell is pointing towards magnetic field minima.
on the cell is pointing towards magnetic field minima.
The hydrodynamic viscous drag force exerted on diamagnetic cell takes the form,
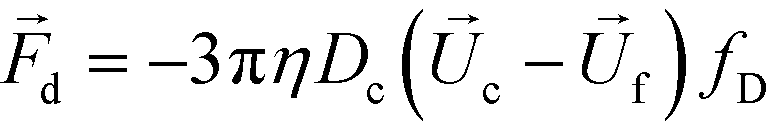 | (3) |
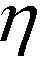 is the viscosity of ferrofluids, Dc is the diameter of the cell,
is the viscosity of ferrofluids, Dc is the diameter of the cell, 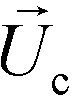 and
and 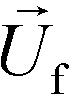 are the velocity vectors of the cell and ferrofluids respectively, fD is the hydrodynamic drag force coefficient for a cell moving near a solid surface, often referred to as the “wall effect”.52–54 Because of the low Reynolds number in FCS devices, inertial effects on the cell were neglected and motion of cells in ferrofluids could be determined by the balance of hydrodynamic viscous drag force and magnetic buoyancy force. From eqn (1)–(3), it can be seen that cells with different volumes experience different magnitudes of magnetic buoyancy force, which can result in the separation of these cells in ferrofluids in a continuous-flow manner.
are the velocity vectors of the cell and ferrofluids respectively, fD is the hydrodynamic drag force coefficient for a cell moving near a solid surface, often referred to as the “wall effect”.52–54 Because of the low Reynolds number in FCS devices, inertial effects on the cell were neglected and motion of cells in ferrofluids could be determined by the balance of hydrodynamic viscous drag force and magnetic buoyancy force. From eqn (1)–(3), it can be seen that cells with different volumes experience different magnitudes of magnetic buoyancy force, which can result in the separation of these cells in ferrofluids in a continuous-flow manner.
We first confirmed the validity of the model by comparing simulated trajectories (ESI,† Fig. S3) with experimental ones (ESI,† Fig. S4) that were obtained from imaging 15.6 μm-diameter polystyrene beads and 11.1 μm-diameter WBCs in a FCS device, as shown in ESI,† Fig. S5. We then used the model to optimize the FCS device for CTC separation. The optimization was focused on the study of separating cancer cells from WBCs, because of their subtle size difference. Briefly, we allowed cancer cells and WBCs (H1299 lung cancer cells with a mean diameter of 16.9 μm, and WBCs with a mean diameter of 11.1 μm) to enter the channel and simulated their trajectories in ferrofluids under external magnetic fields. From their simulated trajectories, we calculated two outputs – a deflection in the y-direction (see Fig. 1 and S2† for coordinates) for cancer cells, denoted as YC, and a separation distance between the two types of cells, denoted as ΔY (ESI,† Fig. S3). Both outputs were optimized using parameters including flow rates of cell inlet (1.2–7.2 mL h−1), magnetic fields and gradients (field: 471–415 mT; gradient: 57.1–54.6 T m−1, as shown in ESI,† Fig. S1), and ferrofluid concentrations (up to 1% v/v). The goal here was to achieve high cell flow rate, cancer cell recovery rate and recovered cancer cell purity, which translated to maximizing both YC and ΔY simultaneously. Fig. 2A shows when the magnetic field gradient increased, the deflection distance of cancer cells YC increased monotonically for all flow rates. This was because the driving force, magnetic buoyancy force on cells, was proportional to the magnitude of magnetic field gradient. As the cell inlet flow rate increased, YC decreases due to reduced time in the channel. Fig. 2B shows similar trend of separation distance ΔY increasing as the field gradient increased when flow rates are 4.8, 6.0 and 7.2 mL h−1. Interestingly, when cell input flow rates are smaller (e.g., 1.2, 2.4 and 3.6 mL h−1), the separation distance ΔY between two cell types had different trends. This was due to the fact that both cell types at slower flow rates reached their maximum deflections very quickly, resulting in a mixing rather than separation of the two types. For practical CTC separation, we chose a cell flow rate of 6 mL h−1 and a magnetic field gradient of 56.2 T m−1 that could be generated realistically through magnet and channel integration in a FCS device to achieve high-throughput and high recovery rate cell separation. It should be noted here that the optimization was conducted on a single-channel device, and higher cell flow rates and throughputs were possible with device scale-up or multiplexing.
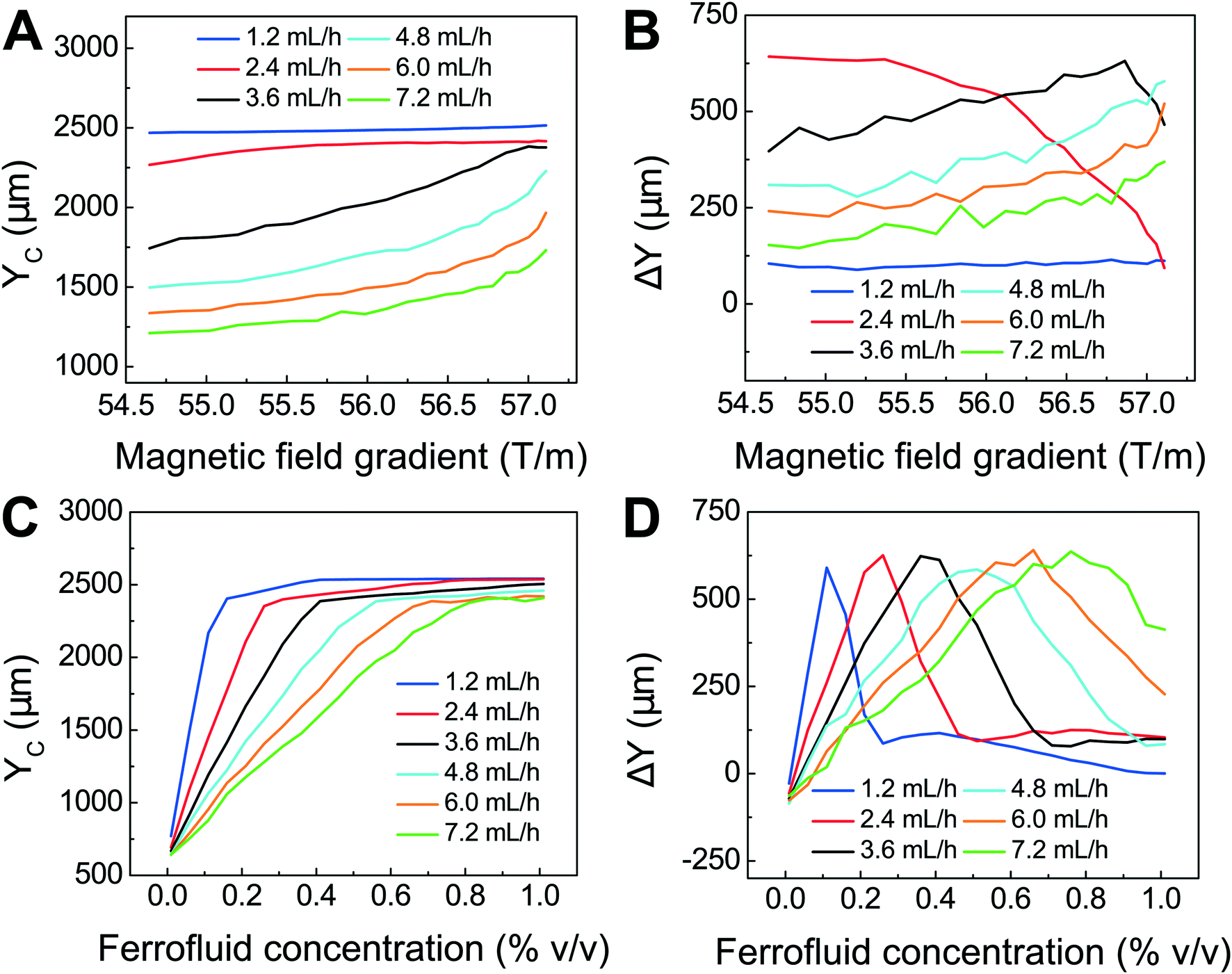 | ||
| Fig. 2 Optimization of FCS devices with their device geometry shown in Fig. 1 for high-throughput, high-recovery and biocompatible CTC separation. A 3D analytical model considering magnetic buoyancy force, hydrodynamic drag force, laminar flow profiles and cancer/blood cell physical properties was developed to guide the optimization. The validity of the model was confirmed by comparing its simulated trajectories with experimental ones, which was described in ESI.† Numerical optimization of deflection distance YC and separation distance ΔY (corresponding to recovery rate and purity) at the end of the FCS device was conducted with parameters including: (A) & (B) magnetic field gradient, and (C) & (D) ferrofluid concentration at flow rates between 1.2 and 7.2 mL h−1. Ferrofluid concentration was fixed at 0.26% (v/v) for (A) & (B). Magnetic field was fixed at 443 mT and its gradient was fixed at 56.2 T m−1 for (C) & (D). | ||
After optimizing flow rate and magnetic field gradient, another critical parameter that still needs to be optimized is the ferrofluid itself. Ideally, the ferrofluid needs to possess properties that are not only biocompatible to CTCs but also enable its colloidal stability under high flow rates and strong magnetic fields. Therefore, its pH value, tonicity, materials and surfactants of nanoparticles need to be optimized as a biocompatible medium for cells, while at the same time the overall colloidal stability of the ferrofluid will have to be well maintained. Based on our previous work,48,49 we have developed a water-based ferrofluid with maghemite nanoparticles in it that was tested to be biocompatible for cancer cells from cultured cells lines. The particles had a mean diameter of 11.24 nm with a standard deviation of 2.52 nm (ESI,† Fig. S6). The diameter of the nanoparticles was chosen to preserve the colloidal stability of ferrofluids against agglomeration due to gravitational settling and magnetic dipole–dipole attraction. As a result, our ferrofluids remained colloidally stable after at least 10 months' storage (ESI,† Fig. S7). The nanoparticles were functionalized with a graft copolymer as surfactants to prevent them from coming too close to one another when there was a magnetic field. The volume fraction of the magnetic content of the ferrofluid is 0.26%. This low volume fraction of the ferrofluid not only leaded to excellent biocompatibility for cell sorting, but also enabled us to observe cell motion in microchannel directly with bright-field microscopy, which was difficult with opaque ferrofluids of high solid volume fractions. The ferrofluid was made to be isotonic and its pH was adjusted to 7.0 for biocompatible cell separation. The outcomes of ferrofluid characterization are listed in ESI,† Fig. S6. We further optimized the ferrofluid concentration for high-throughput and high recovery separation. From eqn (1), the magnetic buoyancy force depends on the magnetization of the ferrofluid and affects the cell separation outcome. Therefore, the concentration of ferrofluid had an impact on the process of cell separation. A higher concentration could lead to a higher magnitude of magnetic buoyancy force on cells and a larger deflection YC (Fig. 2C), but not necessarily a larger ΔY (Fig. 2D). Fig. 2D shows there was an optimal ferrofluid concentration close to 0.6% (v/v) at 6.0 mL h−1 flow rate for ΔY. Concentrations higher than 0.6% (v/v) resulted in larger YC but smaller ΔY. This again was because both cell types achieved sufficient deflections in a strongly magnetized ferrofluid, resulting in mixing rather than separation of the two. In addition, ferrofluid biocompatibility could be compromised as its nanoparticle concentration increases.49 Based on these considerations, we chose a 0.26% (v/v) ferrofluid concentration to strike a balance between high-recovery and biocompatible cell separation at a flow rate of 6 mL h−1.
Verification of FCS for high-throughput and high-recovery spiked cancer cells separation
We performed experimental verification of high-throughput, high-recovery and biocompatible separation of spiked cancer cells of cultured cell lines from WBCs based on the optimal parameters obtained from simulation and calibration. During separation experiments, a permanent magnet was placed 1 mm away from the channel (magnetic field: 443 mT, magnetic field gradient: 56.2 T m−1), and ferrofluids with a concentration of 0.26% (v/v) were used. We first studied the CTC recovery rate at different flow rates using spiked H1299 lung cancer cells in WBCs. The concentration of WBCs was 3–7 × 106 cells per mL; CTCs were simulated by spiking ∼100 CellTracker Green stained H1299 cancer cells into 1 mL of WBCs. The cells were loaded into a FCS device at variable flow rates of 1.2–6 mL h−1 for recovery rate evaluation. Fig. 3 shows a typical cancer cell (Lung cancer H1299) separation process in the FCS device. When the magnetic field was not present, all cell types including cancer cells and WBCs were flowing near the bottom sidewall of the channel and exiting through outlets 1 and 2 (Fig. 3A). When the magnetic field was present, a separation between cancer cells and WBCs was visible. Magnetic buoyancy forces deflected larger H1299 cancer cells with a mean diameter of 16.9 μm from the cell mixture toward outlets 5 and 6, as shown in Fig. 3B–D. Meanwhile, magnetic buoyancy forces on WBCs were insufficient to deflect them above outlet 5, resulting in a spatial separation of the cell mixtures at the end of the channel. Cells from outlets 5 and 6 after separation were collected into a serpentine collection chamber as illustrated in ESI,† Fig. S8, which was used to accurately enumerate fluorescently labeled cancer cells. Representative images for outlet 6 reservoir and collection chambers are shown in ESI,† Fig. S9. The recovery rate was defined as the ratio of the number of identified cancer cells collected from outlets 5 and 6 of the FCS device over the total number of spiked cancer cells from outlets 1–6.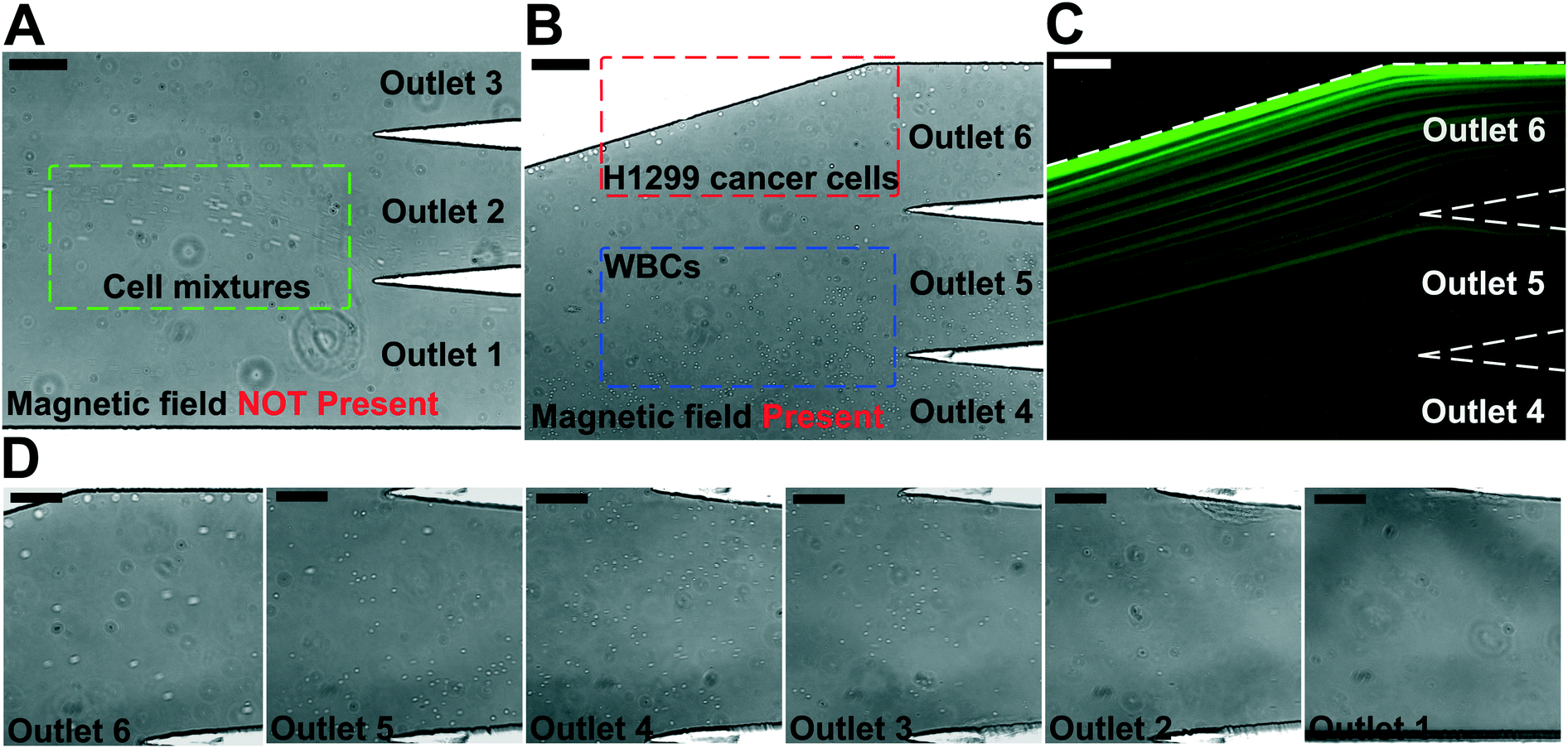 | ||
| Fig. 3 Micrographs of spiked cancer cells of cell culture lines and undiluted WBCs separation process in a FCS device. In order to image the separation process, 1 × 105 cells H1299 lung cancer cells were spiked into 1 mL of undiluted WBCs to increase the cancer cell concentration so that their fluorescent signals were visible. The cell mixture was processed at the flow rate of 6 mL h−1. A ferrofluid with its concentration of 0.26% (v/v) was used; magnetic field was fixed at 443 mT and its gradient was fixed at 56.2 T m−1. (A) In absence of magnetic fields, cell mixtures exited the channel through outlets 1 and 2. Scale bar: 200 μm. (B) When magnetic fields were present, larger H1299 lung cancer cells and some WBCs were deflected and exited through outlets 5 and 6 (collection outlets), while smaller WBCs exited through lower outlets (outlets 1–4, waste outlets). Scale bar: 200 μm. (C) Fluorescence image of spiked H1299 lung cancer cell streams during the separation process when magnetic fields were present. H1299 cells were stained by CellTracker Green. Scale bar: 200 μm. (D) Zoomed-in bright-field images of outlets 1–6 when the magnetic fields were present. Scale bars: 100 μm. | ||
Fig. 4A shows the relationship between cancer cell recovery rates and flow rates for H1299 cancer cells. As flow rates increased from 1.2 mL h−1 to 6 mL h−1, recovery rates decreased from 98.6 ± 5.0% to 92.3 ± 3.6%. An average recovery rate of 92.3% was achieved for current FCS devices with a throughput of 6 mL h−1 when ∼100 H1299 cancer cells were spiked into 1 mL of WBCs. To validate that the device has the potential to process clinically relevant blood samples, a series of spike-in experiments in which a certain number of H1299 cells (50, 100, 200, 500, 1000, and 2000) were spiked into 1 mL of WBCs. As shown in Fig. 4B, an average recovery rate of 91.9% was achieved in the FCS device for this particular lung cancer cell line. Fig. 4C shows the relationship between removal rates of WBCs and cell input flow rates. As the flow rate increased, more WBCs were removed during the separation process. For example, 99.92 ± 2.2% of WBCs were removed at the flow rate of 6 mL h−1 when ∼100 H1299 cancer cells were spiked into 1 mL of WBCs. The corresponding purity of separated cancer cells was 11.1 ± 1.2%. The purities of separated cancer cells in other spike-in experiments were 4.8–67.4% (4.8 ± 1.6%, 20.3 ± 2.8%, 31.2 ± 4.7%, 41.7 ± 4.9%, and 67.4 ± 3.3% when 50, 200, 500, 1000, and 2000 H1299 cancer cells were spiked into 1 mL of WBCs). The purity was defined as the number of identified cancer cells over the total number of cells from FCS device's collection outlets. As the number of spiked cells increased, the number of separated cancer cells also increased, which leaded to a higher purity value. The cell type distribution in each outlet is illustrated in ESI,† Fig. S10.
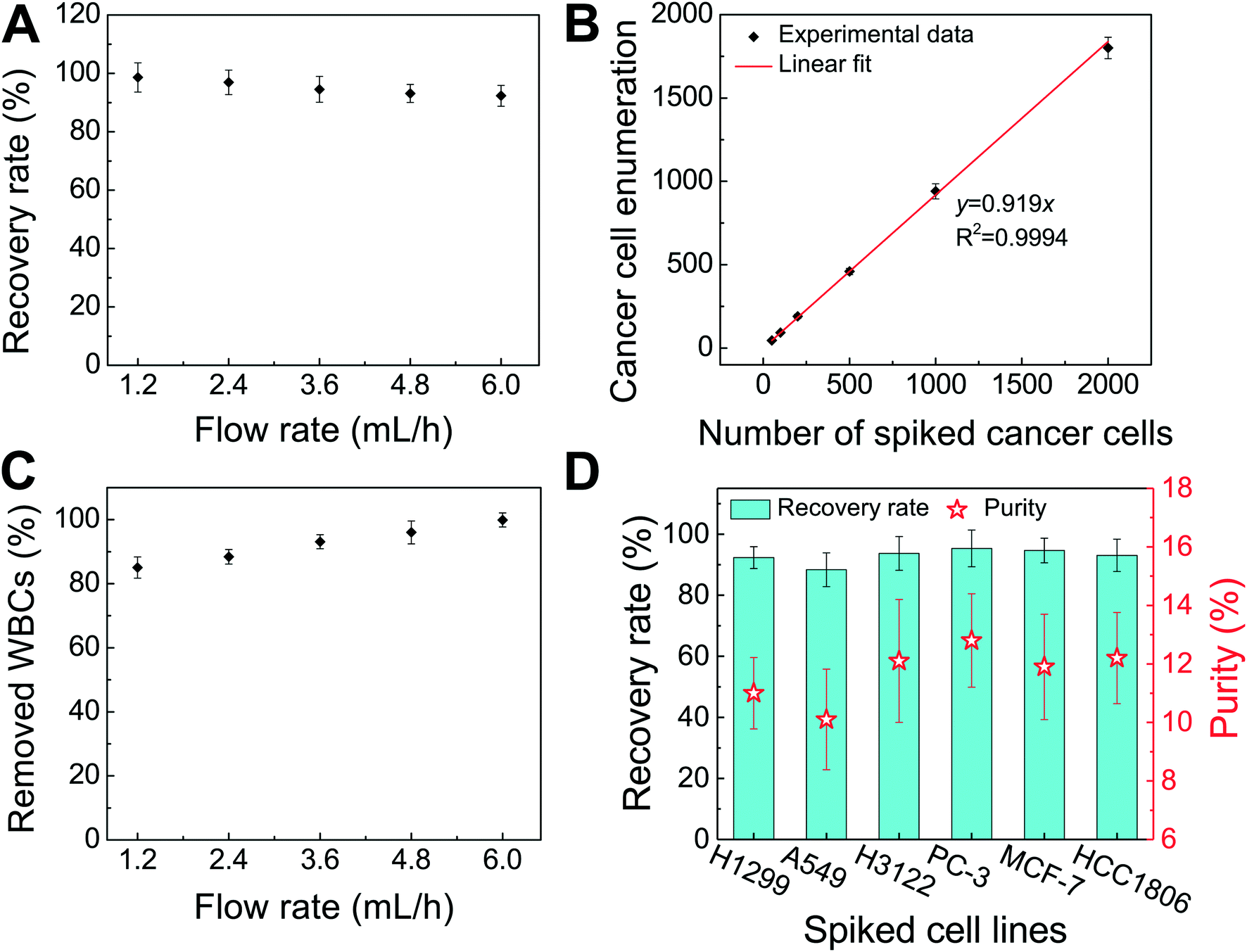 | ||
| Fig. 4 Verification of FCS devices for high-throughput and high-recovery spiked cancer cells separation. (A) Recovery rates of spiked H1299 lung cancer cells from undiluted WBCs at flow rates from 1.2 mL h−1 to 6.0 mL h−1. ∼100 H1299 cancer cells were spiked into 1 mL of undiluted WBCs. Recovery rates decreased from 98.6 ± 5.0% to 92.3 ± 3.6% when flow rate increased from 1.2 mL h−1 to 6.0 mL h−1. (B) A series of spike-in separation experiments in which a certain number (50, 100, 200, 500, 1000, and 2000) of H1299 cells were spiked into 1 mL of undiluted WBCs to simulate clinically relevant CTC concentration at the flow rate of 6.0 mL h−1. An average recovery rate of 91.9% (linear fit, the coefficient of determination R2 = 0.9994 was calculated between the number of cells counted and the number of cells spiked) was achieved for H1299 lung cancer cells. (C) The removal rate of WBCs increased with the flow rate. 99.92 ± 2.2% of WBCs were removed at a flow rate of 6 mL h−1. ∼100 H1299 cancer cells were spiked into 1 mL of undiluted WBCs. (D) Recovery rates and purity of separated cancer cells (∼100 cells per mL) for different cancer cell lines at the flow rate of 6 mL h−1. Recovery rates of 92.3 ± 3.6%, 88.3 ± 5.5%, 93.7 ± 5.5%, 95.3 ± 6.0%, 94.7 ± 4.0%, and 93.0 ± 5.3% were achieved for H1299 (lung cancer), A549 (lung cancer), H3122 (lung cancer), PC-3 (prostate cancer), MCF-7 (breast cancer), and HCC1806 (breast cancer) cell lines, respectively. The corresponding purities of cancer cells of each cell line are 11.1 ± 1.2% (H1299), 10.1 ± 1.7% (A549), 12.1 ± 2.1% (H3122), 12.8 ± 1.6% (PC-3), 11.9 ± 1.8 (MCF-7), and 12.2 ± 1.6% (HCC1806), respectively. For all experiments above, a ferrofluid with its concentration of 0.26% (v/v) was used; magnetic field was fixed at 443 mT and its gradient was fixed at 56.2 T m−1. Error bars indicate standard deviation (s.d.), n = 3. | ||
After successfully demonstrating low-concentration cancer cell separation using H1299 lung cancer cell line, we also characterized the FCS device with 5 other types of cancer cells lines. Size distribution of CTCs from clinical samples is unknown, it is therefore important to characterize the performance of FCS devices with cancer cell culture lines with different sizes. For this purpose, lung cancer, prostate cancer, and breast cancer cell culture lines were used to characterize the cancer cell recovery rates at 6 mL h−1 throughput with a ∼100 cells per mL spike ratio. As shown in Fig. 4D, the average recovery rates of 88.3 ± 5.5%, 93.7 ± 5.5%, 95.3 ± 6.0%, 94.7 ± 4.0%, and 93.0 ± 5.3% were achieved for A549 (lung cancer), H3122 (lung cancer), PC-3 (prostate cancer), MCF-7 (breast cancer), and HCC1806 (breast cancer) cell lines, respectively. The corresponding purities of separated cancer cells for each cell line were 10.1 ± 1.7% (A549), 12.1 ± 2.1% (H3122), 12.8 ± 1.6% (PC-3), 11.9 ± 1.8% (MCF-7), and 12.2 ± 1.6% (HCC1806), confirming the robustness of the FCS device for cancer cell separation. The recovery rate increased as the mean cell size of cancer cells increased (Table 1 and ESI,† Fig. S11), which was expected as FCS was based on size difference of cell types. In summary, we experimentally verified that the optimized FCS device was capable of separating cancer cells from WBCs with a flow rate of 6 mL h−1, with a cancer cell recovery rate of 92.9% and a separated cancer cell purity of 11.7% averaged from all 6 cancer cell lines at ∼100 cells per mL spike ratio, which allowed us to use the devices to process the clinical samples.
| Cancer cell line | Cancer cell type | Measured average cell diameter (μm) | No. of spiked cancer cells | No. of cells (collection outlets) | No. of cells (waste outlets) | Recovery rate | Purity |
|---|---|---|---|---|---|---|---|
| A549 | Lung | 15.5 | 99 ± 2 | 89 ± 4 | 10 ± 6 | 88.3 ± 5.5% | 10.1 ± 1.7% |
| H1299 | Lung | 16.9 | 99 ± 3 | 91 ± 1 | 8 ± 4 | 92.3 ± 3.6% | 11.1 ± 1.2% |
| HCC1806 | Breast | 17.6 | 100 ± 4 | 93 ± 4 | 7 ± 4 | 93.0 ± 5.3% | 12.2 ± 1.6% |
| H3122 | Lung | 17.8 | 101 ± 4 | 92 ± 6 | 9 ± 4 | 93.7 ± 5.5% | 12.1 ± 2.1% |
| MCF-7 | Breast | 18.7 | 100 ± 3 | 94 ± 3 | 6 ± 3 | 94.7 ± 4.0% | 11.9 ± 1.8% |
| PC-3 | Prostate | 18.9 | 100 ± 7 | 95 ± 7 | 5 ± 7 | 95.3 ± 6.0% | 12.8 ± 1.6% |
Effect of FCS on cancer cell viability, proliferation and biomarker expressions
As discussed above, the operating parameters of the FCS device need to preserve cell integrity during its cell separation process. To investigate the impact of ferrofluids and current separation conditions on cell integrity, we examined short-term cell viability, long-term cell proliferation, as well as biomarker expression of cancer cells following the separation process.The short-term viability of cancer cells in ferrofluids was first evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay for 12 h incubation with different concentrations of ferrofluids. The results show that H1299 lung cancer cells had a cell viability of 80.8 ± 2.4% after 12 h incubation with 0.26% (v/v) ferrofluids as shown in ESI,† Fig. S7. Next, we investigated the short-term cell viability after ferrohydrodynamic cell separation using a Live/Dead assay. Cells in 1 mL of ferrofluids (1 × 106 H1299 cells) were processed by the FCS device at a flow rate of 6 mL h−1. The device-operating parameters were chosen to be the same as those used in aforementioned cancer cell separation experiments. After running the cell sample through the device, cancer cells collected from outlet 6 were stained with 2 μM calcein-AM and 4 μM EthD-1 for 30 minutes at room temperature to determine their viability. Cells with a calcein-AM+/EthD-1− staining pattern were counted as live cells, whereas cells with calcein-AM−/EhD-1+ staining patterns were counted as dead cells. As shown in Fig. 5A, cell viability of H1299 cells before and after separation groups were determined to be 98.9 ± 0.9% and 96.3 ± 0.9%, respectively, indicating a very slight decrease in cell viability before and after the ferrohydrodynamic separation process. Representative fluorescence images of cells are shown in Fig. 5B.
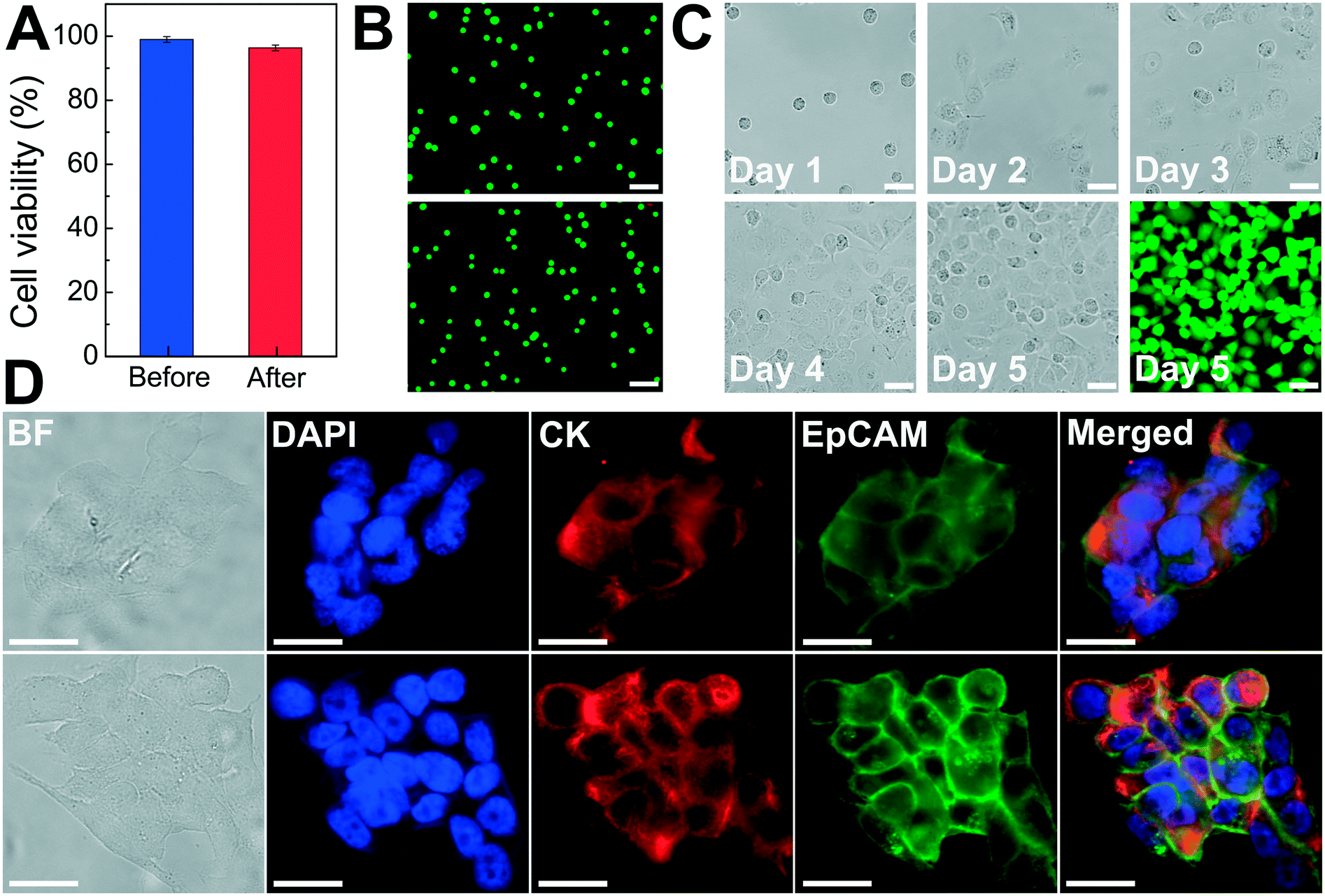 | ||
| Fig. 5 Effect of FCS on cancer cell viability, proliferation and biomarker expressions. (A) Short-term cell viability comparison before and after FCS process using a Live/Dead assay. Cell viabilities of H1299 lung cancer cells before and after separation process were determined to be 98.9 ± 0.9% and 96.3 ± 0.9%, respectively. Error bars indicate standard deviation (s.d.), n = 3. (B) Representative images of Live/Dead cell staining for before (top) and after (bottom) separation groups. Calcein AM (green, live cells) and EhD-1 (red, dead cells) channels were merged. Scale bars: 100 μm. (C) Bright field images of cultured H1299 cells collected after separation from day 1 to day 5. A Live/Dead staining of the cultured cells on day 5 showed excellent cell viability. Scale bars: 50 μm. (D) Comparison of expressions of two key biomarkers (epithelial cell adhesion molecule-EpCAM and cytokeratin-CK) on HCC1806 breast cancer cells before (top) and after (bottom) separation. They showed qualitatively similar EpCAM and CK fluorescence. Scale bars: 20 μm. | ||
After determining short-term cell viability, we examined whether separated cancer cells continued to proliferate normally after the separation process. To simulate the actual separation conditions, 1 × 106 H1299 cells were spiked into 1 mL of ferrofluids and passed through the FCS device. The flow rate and ferrofluid concentration were chosen to be the same as those used in cancer cell separation experiments. Following cell collection, the recovered H1299 cells were washed with culture medium to remove maghemite nanoparticles and transferred to an incubator. Cells were cultured at 37 °C under a humidified atmosphere of 5% CO2. Fig. 5C shows the images of the cultured H1299 cells over a 5 day period. These cells were able to proliferate to confluence and maintain their morphologies after the ferrohydrodynamic separation process. Fluorescence image in Fig. 5C also confirms that cells were viable after the 5 day culture.
In order to determine whether the FCS process would alter the expression of cell surface biomarkers, we looked for changes in biomarker expression using immunofluorescence staining. Specifically, we compared expressions of epithelial cell adhesion molecule (EpCAM) and cytokeratin (CK), two key biomarkers in CTC studies, in paired sets of pre- and post-FCS process. Results shown in Fig. 5D indicate there was no visible change in either EpCAM or CK expression on HCC1806 breast cancer cells because of the FCS process. Collectively, the short-term viability, long-term cell proliferation and biomarker studies presented here demonstrated that the FCS method was biocompatible for cancer cell separation and could enable downstream characterization of separated CTCs.
Enrichment of CTCs from NSCLC patient blood using FCS
There was a large variance in reported numbers of captured CTCs for advanced metastatic cancer patients.5 The exact reasons for this variance are still an area of active research. Nonetheless, most CTC separation methods chose to use blood from advanced metastatic patients for technology validation.11,16,20–25,29,30 As a clinical validation of this method, we validated FCS devices with blood samples obtained from two patients with advanced NSCLC. Peripheral blood was collected from patients with newly diagnosed NSCLC (stage IVB) before initiation of treatment. Blood was lysed to remove RBCs and then processed with FCS devices within 3 hours of blood draw. 6.5 mL of blood was processed from patient A, and 5.6 mL of blood was processed from patient B. After separation, cells from FCS device's outlet 6 were directly preserved in ThinPrep PreservCyt solution. These enriched cells were concentrated and stained using the Pap stain, which was commonly used for cytopathology analysis of clinical samples. Enriched cells were then inspected by a cytopathologist and CTCs were enumerated. Criteria used to identify CTC were as follows: (1) large cells with high nuclear to cytoplasmic ratio; (2) cells with irregular chromatin distribution and nuclear contours; (3) cells that are 4–5 times the size of a WBC. Fig. 6A and ESI,† Fig. S12 show a few Pap-stained CTCs and WBCs separated from two NSCLC patients. Both patients showed high CTC counts through cytopathology: 1165 and 369 CTCs were identified from 6.5 and 5.6 mL of blood samples, respectively. Purity of CTCs (defined as the number of identified CTCs over the total number of cells from FCS device's collection outlets) from these two patients was 17.0 ± 7.8%. Additionally, Immunofluorescent staining of CK8/18/19, EpCAM, and leukocyte marker CD45 was also used to confirm the presence CTCs separated from patient B's blood. Cells were identified as CTCs if the staining pattern is CK+/CD45− or EpCAM+/CD45− or CK+/EpCAM+/CD45−, otherwise, cells were identified as WBCs. Typical fluorescent images are shown in Fig. 6B based on this immunostaining detection criteria.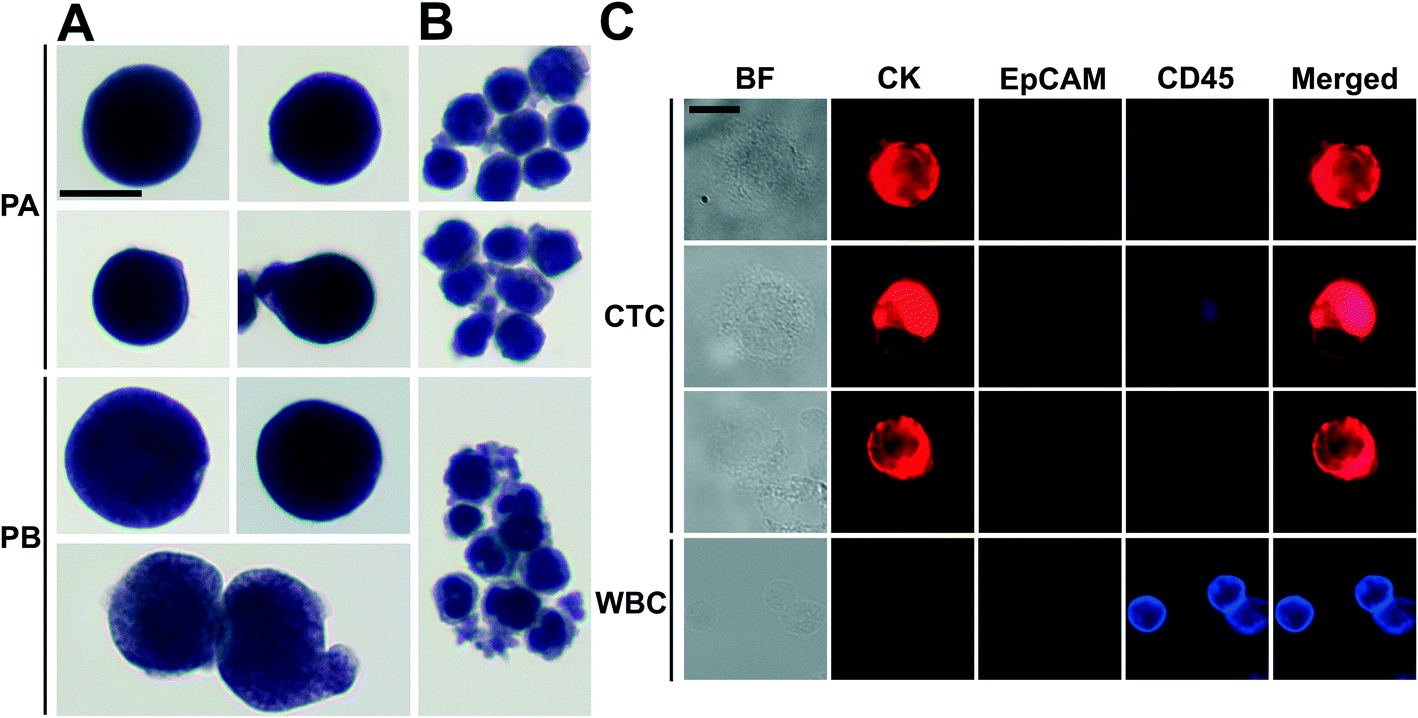 | ||
| Fig. 6 Enrichment of CTCs from NSCLC patient blood using FCS devices, and CTC identification with cytopathology and immunofluorescent staining. CTCs (A) and WBCs (B) from the blood of two NSCLC patients (PA and PB) were enriched by FCS devices and stained with Papanicolaou procedure, then identified by a cytopathologist. (C) Immunofluorescence images of enriched cells from blood samples from patient B. Three channels including CK, EpCAM and CD45 were examined. Cells were identified as CTCs if the staining pattern is CK+/CD45− or EpCAM+/CD45− or CK+/EpCAM+/CD45−, WBC were identified as CK−/EpCAM−/CD45+. Scale bars: 10 μm. | ||
Discussion
In this paper, we developed a ferrohydrodynamic cell separation (FCS) method for CTC separation and its devices that were capable of high-throughput (6 mL h−1), high recovery rate (92.9%, an average from 6 cancer cell lines at ∼100 cells per mL spike ratio) and biocompatible enrichment of cancer cells from RBC-lysed blood with an average 11.7% purity, by systematically investigating the device operating parameters on its separation performance. The FCS process involved multiple parameters that could affect the cell separation performance, including cell flow rates, magnetic fields and its gradient, ferrofluid concentrations and compositions. All of these parameters were highly coupled with each other and required an effective model for device optimization. We have developed and validated such an analytical model that considered magnetic buoyancy force, hydrodynamic drag force, laminar flow profiles and cancer/blood cell physical properties to guide the optimization and design of a high-throughput, high recovery rate FCS devices. We also considered the chemical makeup of the ferrofluids, including its nanoparticle concentration, pH value, nanoparticle size and surfactant, tonicity to optimize a colloidally stable and biocompatible ferrofluid suitable for cancer cell separation. After systematic optimization, we demonstrated that FCS devices were capable of separating various types of low-concentration cancer cells of cultured cell lines (∼100 cells per mL) from WBCs under a flow rate of 6 mL h−1. The recovery rates of spiked cancer cells were on average 92.9% from all tested cell lines at clinically relevant CTC occurrence rates. The recovered cancer cells were viable, could proliferate to confluence and expressions of a few key biomarker remained unaffected. These results indicated the practical use of this method in separating CTCs from patient blood were feasible. We further demonstrated FCS devices worked well with clinical samples by successfully separating and identifying CTCs from blood samples of two late-stage (IVB) non-small cell lung cancer patients.While current FCS devices demonstrated a high-recovery and biocompatible separation of rare cancer cells at a clinically relevant throughput, and was validated with NSCLC patient blood, it was still at its early stage of development and could benefit from further system optimization or integration with other methods in order to achieve high-throughput, high-recovery, high-purity separation of intact CTCs. When comparing FCS performance to other size-based label-free CTC separation methods, its rate of recovery of cancer cells was higher than the current average reported value of 82%,8 including methods based on standing surface acoustic wave (>83%),16 dean flow (>85%),20–22 vortex technology (up to 83%),23–25 and deterministic lateral displacement (>85%).55 Although the throughput of current FCS device (6 mL h−1) was sufficiently high to process clinically relevant amount of blood, it was slower than a few hydrodynamics-based methods that had extremely high flow rates, including the dean flow (56.25 mL h−1),20–22 the vortex technology (48 mL h−1),23–25 and DLD (10 mL min−1).55 Further system optimization, scale-up or multiplexing of FCS devices should be conducted in order to process more blood quickly. The average purity of separated cancer cells in current FCS devices was 11.7%. Reported purity values varied dramatically from 0.1% to 90% in label-free methods,16–25 as most of them focused on improving recovery instead of purification of rare cells. Nonetheless, hydrodynamics-based methods including the dean flow (50%)20–22 and the vortex technology (57–94%)23–25 reported significantly higher purity of cancer cells in their collection outputs than FCS. Low cancer cell purity due to WBC or other cell contamination could interfere with subsequent CTC characterization. It is therefore necessary for future FCS devices to further deplete these contamination cells.
FCS currently distinguished cells primarily based on their size difference. For cancer cells that have similar size as WBCs, this method will result in lower separated cancer cell purity than label-based method. Additional cell characteristics or methods could be integrated with FCS to further improve the purity of separated cancer cells. One possible strategy is for future FCS devices to exploit both size and magnetic labels of cells for CTC separation.56 For example, WBCs in blood can be labeled with sufficient number of anti-CD45 magnetic beads so that the overall magnetization of the WBC-bead complex 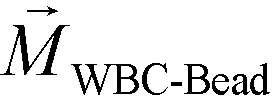 is larger than its surrounding ferrofluids
is larger than its surrounding ferrofluids 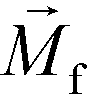 . The direction of magnetic force on the complex is then pointing towards magnetic field maxima. On the other hand, magnetization of the non-labeled CTCs
. The direction of magnetic force on the complex is then pointing towards magnetic field maxima. On the other hand, magnetization of the non-labeled CTCs 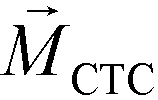 is zero and less than its surrounding ferrofluids
is zero and less than its surrounding ferrofluids 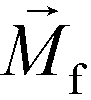 , the direction of magnetic force on CTCs is therefore pointing towards magnetic field minima. In this scenario, both label-based magnetophoresis and size-based FCS co-exist in one system, i.e.,
, the direction of magnetic force on CTCs is therefore pointing towards magnetic field minima. In this scenario, both label-based magnetophoresis and size-based FCS co-exist in one system, i.e., 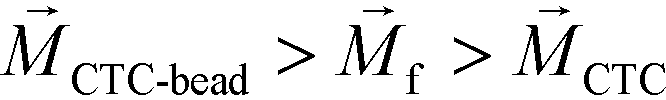 , magnetic force will attract WBC-bead complex towards field maxima while pushes CTCs towards field minima.
, magnetic force will attract WBC-bead complex towards field maxima while pushes CTCs towards field minima.
Conclusions
In this study, we reported a label-free ferrohydrodynamic cell separation (FCS) method that used magnetic buoyance force for size-based CTC separation, which was biocompatible and could enrich rare CTCs from patient blood with a high throughput and a high rate of recovery. We performed systematic optimization of this method and determined parameters in a laminar flow microfluidic device that achieved an average 92.9% recovery rate and an average 11.7% purity of low-concentration (∼100 cells per mL) cancer cells using six different cultured cell lines from undiluted WBCs, with a clinically relevant processing throughput of 6 mL per hour. These parameters include magnetic field and its gradient (magnetic field: 443 mT, magnetic field gradient: 56.2 T m−1), and ferrofluid concentration (0.26%, v/v). Specifically, for each cell lines at ∼100 cells per mL spike ratio, the recovery rates of cancer cells were 92.3 ± 3.6% (H1299 lung cancer), 88.3 ± 5.5% (A549 lung cancer), 93.7 ± 5.5% (H3122 lung cancer), 95.3 ± 6.0% (PC-3 prostate cancer), 94.7 ± 4.0% (MCF-7 breast cancer), and 93.0 ± 5.3% (HCC1806 breast cancer), and the corresponding purities of separated cancer cells were 11.1 ± 1.2% (H1299 lung cancer), 10.1 ± 1.7% (A549 lung cancer), 12.1 ± 2.1% (H3122 lung cancer), 12.8 ± 1.6% (PC-3 prostate cancer), 11.9 ± 1.8% (MCF-7 breast cancer), and 12.2 ± 1.6% (HCC1806 breast cancer). Separated H1299 lung cancer cells from FCS showed a short-term viability of 96.3 ± 0.9%, and they were successfully cultured and demonstrated normal proliferation to the confluence. Separated HCC1806 breast cancer cells from FCS showed unchanged expressions of two key biomarkers including EpCAM and CK. FCS devices were validated with blood samples obtained from two patients with advanced NSCLC. 1165 CTCs were enriched and identified from 6.5 mL of blood samples from one patient, while 369 CTCs were enriched and identified from 5.6 mL of blood samples from the other patient. Although FCS is still at its early stage of development, it could be a complementary tool for rare cell separations because of its high recovery rate and excellent biocompatibility, as well as its potential for further optimization and integration with other compatible methods.Conflicts of interest
University of Georgia filed patent protection for FCS technology.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. 1150042 and 1359095; by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. R21GM104528; and by a seed grant program between University of Georgia and Augusta University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- N. Aceto, M. Toner, S. Maheswaran and D. A. Haber, Trends Cancer, 2015, 1, 44–52 CrossRef PubMed.
- J. Massague and A. C. Obenauf, Nature, 2016, 529, 298–306 CrossRef CAS PubMed.
- M. Yu, A. Bardia, N. Aceto, F. Bersani, M. W. Madden, M. C. Donaldson, R. Desai, H. L. Zhu, V. Comaills, Z. L. Zheng, B. S. Wittner, P. Stojanov, E. Brachtel, D. Sgroi, R. Kapur, T. Shioda, D. T. Ting, S. Ramaswamy, G. Getz, A. J. Iafrate, C. Benes, M. Toner, S. Maheswaran and D. A. Haber, Science, 2014, 345, 216–220 CrossRef CAS PubMed.
- M. Kalinich, I. Bhan, T. T. Kwan, D. T. Miyamoto, S. Javaid, J. A. LiCausi, J. D. Milner, X. Hong, L. Goyal, S. Sil, M. Choz, U. Ho, R. Kapur, A. Muzikansky, H. D. Zhang, D. A. Weitz, L. V. Sequist, D. P. Ryan, R. T. Chung, A. X. Zhu, K. J. Isselbacher, D. T. Ting, M. Toner, S. Maheswaran and D. A. Haber, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1123–1128 CrossRef CAS PubMed.
- C. Alix-Panabieres and K. Pantel, Nat. Rev. Cancer, 2014, 14, 623–631 CrossRef CAS PubMed.
- M. Yu, S. Stott, M. Toner, S. Maheswaran and D. A. Haber, J. Cell Biol., 2011, 192, 373–382 CrossRef CAS PubMed.
- C. Alix-Panabieres and K. Pantel, Clin. Chem., 2013, 59, 110–118 CAS.
- Y. C. Chen, P. Li, P. H. Huang, Y. L. Xie, J. D. Mai, L. Wang, N. T. Nguyen and T. J. Huang, Lab Chip, 2014, 14, 626–645 RSC.
- D. T. Miyamoto, D. T. Ting, M. Toner, S. Maheswaran and D. A. Haber, Cold Spring Harbor Symp. Quant. Biol., 2016, 81, 269–274 CrossRef PubMed.
- M. Poudineh, P. Aldridge, S. Ahmed, B. J. Green, L. Kermanshah, V. Nguyen, C. Tu, R. M. Mohamadi, R. K. Nam, A. Hansen, S. S. Sridhar, A. Finelli, N. E. Fleshner, A. M. Joshua, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2017, 12, 274–281 CrossRef CAS PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- S. L. Stott, C. H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. M. Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd, A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397 CrossRef CAS PubMed.
- A. H. Talasaz, A. A. Powell, D. E. Huber, J. G. Berbee, K. H. Roh, W. Yu, W. Z. Xiao, M. M. Davis, R. F. Pease, M. N. Mindrinos, S. S. Jeffrey and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3970–3975 CrossRef CAS PubMed.
- V. Murlidhar, L. Rivera-Baez and S. Nagrath, Small, 2016, 12, 4450–4463 CrossRef CAS PubMed.
- H. K. Lin, S. Y. Zheng, A. J. Williams, M. Balic, S. Groshen, H. I. Scher, M. Fleisher, W. Stadler, R. H. Datar, Y. C. Tai and R. J. Cote, Clin. Cancer Res., 2010, 16, 5011–5018 CrossRef CAS PubMed.
- P. Li, Z. M. Mao, Z. L. Peng, L. L. Zhou, Y. C. Chen, P. H. Huang, C. I. Truica, J. J. Drabick, W. S. El-Deiry, M. Dao, S. Suresh and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4970–4975 CrossRef CAS PubMed.
- S. B. Huang, M. H. Wu, Y. H. Lin, C. H. Hsieh, C. L. Yang, H. C. Lin, C. P. Tseng and G. B. Lee, Lab Chip, 2013, 13, 1371–1383 RSC.
- P. R. C. Gascoyne, J. Noshari, T. J. Anderson and F. F. Becker, Electrophoresis, 2009, 30, 1388–1398 CrossRef CAS PubMed.
- H. S. Moon, K. Kwon, S. I. Kim, H. Han, J. Sohn, S. Lee and H. I. Jung, Lab Chip, 2011, 11, 1118–1125 RSC.
- H. W. Hou, M. E. Warkiani, B. L. Khoo, Z. R. Li, R. A. Soo, D. S. W. Tan, W. T. Lim, J. Han, A. A. S. Bhagat and C. T. Lim, Sci. Rep., 2013, 3, 1259 CrossRef PubMed.
- M. E. Warkiani, G. F. Guan, K. B. Luan, W. C. Lee, A. A. S. Bhagat, P. K. Chaudhuri, D. S. W. Tan, W. T. Lim, S. C. Lee, P. C. Y. Chen, C. T. Lim and J. Han, Lab Chip, 2014, 14, 128–137 RSC.
- M. E. Warkiani, B. L. Khoo, L. D. Wu, A. K. P. Tay, A. A. S. Bhagat, J. Han and C. T. Lim, Nat. Protoc., 2016, 11, 134–148 CrossRef CAS PubMed.
- J. Che, V. Yu, M. Dhar, C. Renier, M. Matsumoto, K. Heirich, E. B. Garon, J. Goldman, J. Y. Rao, G. W. Sledge, M. D. Pegram, S. Sheth, S. S. Jeffrey, R. P. Kulkarni, E. Sollier and D. Di Carlo, Oncotarget, 2016, 7, 12748–12760 Search PubMed.
- M. Dhar, E. Pao, C. Renier, D. E. Go, J. Che, R. Montoya, R. Conrad, M. Matsumoto, K. Heirich, M. Triboulet, J. Y. Rao, S. S. Jeffrey, E. B. Garon, J. Goldman, N. P. Rao, R. Kulkarni, E. Sollier-Christen and D. Di Carlo, Sci. Rep., 2016, 6, 35474 CrossRef CAS PubMed.
- E. Sollier, D. E. Go, J. Che, D. R. Gossett, S. O'Byrne, W. M. Weaver, N. Kummer, M. Rettig, J. Goldman, N. Nickols, S. McCloskey, R. P. Kulkarni and D. Di Carlo, Lab Chip, 2014, 14, 63–77 RSC.
- D. Marrinucci, K. Bethel, D. Lazar, J. Fisher, E. Huynh, P. Clark, R. Bruce, J. Nieva and P. Kuhn, J. Oncol., 2010, 2010, 861341 Search PubMed.
- W. J. Allard, J. Matera, M. C. Miller, M. Repollet, M. C. Connelly, C. Rao, A. G. J. Tibbe, J. W. Uhr and L. W. M. M. Terstappen, Clin. Cancer Res., 2004, 10, 6897–6904 CrossRef PubMed.
- Y. Dong, A. M. Skelley, K. D. Merdek, K. M. Sprott, C. Jiang, W. E. Pierceall, J. Lin, M. Stocum, W. P. Carney and D. A. Smirnov, J. Mol. Diagn., 2013, 15, 149–157 CrossRef PubMed.
- N. M. Karabacak, P. S. Spuhler, F. Fachin, E. J. Lim, V. Pai, E. Ozkumur, J. M. Martel, N. Kojic, K. Smith, P. I. Chen, J. Yang, H. Hwang, B. Morgan, J. Trautwein, T. A. Barber, S. L. Stott, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Nat. Protoc., 2014, 9, 694–710 CrossRef CAS PubMed.
- E. Ozkumur, A. M. Shah, J. C. Ciciliano, B. L. Emmink, D. T. Miyamoto, E. Brachtel, M. Yu, P. I. Chen, B. Morgan, J. Trautwein, A. Kimura, S. Sengupta, S. L. Stott, N. M. Karabacak, T. A. Barber, J. R. Walsh, K. Smith, P. S. Spuhler, J. P. Sullivan, R. J. Lee, D. T. Ting, X. Luo, A. T. Shaw, A. Bardia, L. V. Sequist, D. N. Louis, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Sci. Transl. Med., 2013, 5, 179ra147 Search PubMed.
- C. W. Shields, J. L. Wang, K. A. Ohiri, E. D. Essoyan, B. B. Yellen, A. J. Armstrong and G. P. Lopez, Lab Chip, 2016, 16, 3833–3844 RSC.
- C. Huang, H. Liu, N. H. Bander and B. J. Kirby, Biomed. Microdevices, 2013, 15, 941–948 CrossRef CAS PubMed.
- Z. B. Liu, W. Zhang, F. Huang, H. T. Feng, W. L. Shu, X. P. Xu and Y. Chen, Biosens. Bioelectron., 2013, 47, 113–119 CrossRef CAS PubMed.
- J. Kim, H. Cho, S. I. Han and K. H. Han, Anal. Chem., 2016, 88, 4857–4863 CrossRef CAS PubMed.
- B. D. Plouffe, M. Mahalanabis, L. H. Lewis, C. M. Klapperich and S. K. Murthy, Anal. Chem., 2012, 84, 1336–1344 CrossRef CAS PubMed.
- C. M. Earhart, C. E. Hughes, R. S. Gaster, C. C. Ooi, R. J. Wilson, L. Y. Zhou, E. W. Humke, L. Y. Xu, D. J. Wong, S. B. Willingham, E. J. Schwartz, I. L. Weissman, S. S. Jeffrey, J. W. Neal, R. Rohatgi, H. A. Wakeleebe and S. X. Wang, Lab Chip, 2014, 14, 78–88 RSC.
- J. H. Kang, S. Krause, H. Tobin, A. Mammoto, M. Kanapathipillai and D. E. Ingber, Lab Chip, 2012, 12, 2175–2181 RSC.
- K. Hoshino, Y. Y. Huang, N. Lane, M. Huebschman, J. W. Uhr, E. P. Frenkel and X. J. Zhang, Lab Chip, 2011, 11, 3449–3457 RSC.
- A. E. Saliba, L. Saias, E. Psychari, N. Minc, D. Simon, F. C. Bidard, C. Mathiot, J. Y. Pierga, V. Fraisier, J. Salamero, V. Saada, F. Farace, P. Vielh, L. Malaquin and J. L. Viovy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14524–14529 CrossRef CAS PubMed.
- L. Y. Yang, J. C. Lang, P. Balasubramanian, K. R. Jatana, D. Schuller, A. Agrawal, M. Zborowski and J. J. Chalmers, Biotechnol. Bioeng., 2009, 102, 521–534 CrossRef CAS PubMed.
- Y. Y. Huang, P. Chen, C. H. Wu, K. Hoshino, K. Sokolov, N. Lane, H. Y. Liu, M. Huebschman, E. Frenkel and J. X. J. Zhang, Sci. Rep., 2015, 5, 16047 CrossRef PubMed.
- W. Zhao, R. Cheng, J. R. Miller and L. Mao, Adv. Funct. Mater., 2016, 26, 3916–3932 CrossRef CAS PubMed.
- A. T. Skjeltorp, Phys. Rev. Lett., 1983, 51, 2306–2309 CrossRef CAS.
- R. E. Rosensweig, Ferrohydrodynamics, Cambridge University Press, Cambridge, 1985 Search PubMed.
- T. T. Zhu, R. Cheng, S. A. Lee, E. Rajaraman, M. A. Eiteman, T. D. Querec, E. R. Unger and L. D. Mao, Microfluid. Nanofluid., 2012, 13, 645–654 CrossRef CAS PubMed.
- J. Zeng, Y. X. Deng, P. Vedantam, T. R. Tzeng and X. C. Xuan, J. Magn. Magn. Mater., 2013, 346, 118–123 CrossRef CAS.
- A. R. Kose, B. Fischer, L. Mao and H. Koser, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21478–21483 CrossRef CAS PubMed.
- W. Zhao, T. Zhu, R. Cheng, Y. Liu, J. He, H. Qiu, L. Wang, T. Nagy, T. D. Querec, E. R. Unger and L. Mao, Adv. Funct. Mater., 2016, 26, 3990–3998 CrossRef CAS PubMed.
- W. Zhao, R. Cheng, S. H. Lim, J. R. Miller, W. Zhang, W. Tang, J. Xie and L. Mao, Lab Chip, 2017, 17, 2243–2255 RSC.
- R. Cheng, T. T. Zhu and L. D. Mao, Microfluid. Nanofluid., 2014, 16, 1143–1154 CrossRef CAS.
- T. T. Zhu, D. J. Lichlyter, M. A. Haidekker and L. D. Mao, Microfluid. Nanofluid., 2011, 10, 1233–1245 CrossRef CAS.
- P. Ganatos, S. Weinbaum and R. Pfeffer, J. Fluid Mech., 1980, 99, 739–753 CrossRef.
- G. P. Krishnan and D. T. Leighton, Phys. Fluids, 1995, 7, 2538–2545 CrossRef CAS.
- M. E. Staben, A. Z. Zinchenko and R. H. Davis, Phys. Fluids, 2003, 15, 1711–1733 CrossRef CAS.
- K. Loutherback, J. D'Silva, L. Y. Liu, A. Wu, R. H. Austin and J. C. Sturm, AIP Adv., 2012, 2, 042107 CrossRef PubMed.
- T. T. Zhu, R. Cheng, Y. F. Liu, J. He and L. D. Mao, Microfluid. Nanofluid., 2014, 17, 973–982 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7lc00680b |
| This journal is © The Royal Society of Chemistry 2017 |