
A magnetic micropore chip for rapid (<1 hour) unbiased circulating tumor cell isolation and in situ RNA analysis†
 c
David
Issadore
*ad
c
David
Issadore
*ad
Abstract
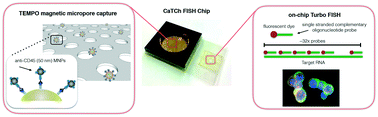
The use of microtechnology for the highly selective isolation and sensitive detection of circulating tumor cells has shown enormous promise. One challenge for this technology is that the small feature sizes – which are the key to this technology's performance – can result in low sample throughput and susceptibility to clogging. Additionally, conventional molecular analysis of CTCs often requires cells to be taken off-chip for sample preparation and purification before analysis, leading to the loss of rare cells. To address these challenges, we have developed a microchip platform that combines fast, magnetic micropore based negative immunomagnetic selection (>10 mL h−1) with rapid on-chip in situ RNA profiling (>100× faster than conventional RNA labeling). This integrated chip can isolate both rare circulating cells and cell clusters directly from whole blood and allow individual cells to be profiled for multiple RNA cancer biomarkers, achieving sample-to-answer in less than 1 hour for 10 mL of whole blood. To demonstrate the power of this approach, we applied our device to the circulating tumor cell based diagnosis of pancreatic cancer. We used a genetically engineered lineage-labeled mouse model of pancreatic cancer (KPCY) to validate the performance of our chip. We show that in a cohort of patient samples (N = 25) that this device can detect and perform in situ RNA analysis on circulating tumor cells in patients with pancreatic cancer, even in those with extremely sparse CTCs (<1 CTC mL−1 of whole blood).
- This article is part of the themed collection: Personalised Medicine: Liquid Biopsy
 Please wait while we load your content...
Please wait while we load your content...

