
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Challenges and prospects of the role of solid electrolytes in the revitalization of lithium metal batteries
Alberto
Varzi
ab,
Rinaldo
Raccichini
ab,
Stefano
Passerini
 *ab and
Bruno
Scrosati
*ac
*ab and
Bruno
Scrosati
*ac
aHelmholtz Institute Ulm (HIU), Electrochemistry 1, Helmholtzstrasse 11, D-89081 Ulm, Germany. E-mail: stefano.passerini@kit.edu
bKarlsruhe Institute of Technology (KIT), P. O. Box 3640, D-76021, Karlsruhe, Germany
cIstituto Italiano di Tecnologia, Via Morego 30, I-16163 Genova, Italy. E-mail: bruno.scrosati@gmail.com
First published on 4th October 2016
Abstract
The scientific community is continuously committed to the search for new high energy electrochemical storage devices. In this regard, lithium metal batteries, due to their very high electrochemical energy storage capacity, appear to be a highly appealing choice. Unfortunately, the use of lithium metal as the anode may lead to some safety hazards due to its uneven deposition upon charging, resulting in dendrite growth and eventual shorting of the battery. This issue may be successfully addressed by using intrinsically safer electrolytes capable of establishing a physical barrier at the electrode interface. The most promising candidates are solid electrolytes, either polymeric or inorganic. The main purpose of this review is to describe the present status of worldwide research on these electrolyte materials together with a critical discussion of their transport properties and compatibility with metallic lithium, hoping to provide some general guidelines for the development of innovative and safe lithium metal batteries.
1. Introduction
Lithium-ion batteries (LIBs) are, nowadays, the power sources of choice for consumer electronics with a continuously expanding market to meet the increasing sophistication of popular devices, such as smartphones, tablets and so on.1 However, crucial phenomena that affect our lives, such as the severe pollution of urban areas and the gas emissions of gasoline-powered cars, call with urgency for energy renewal, both in terms of the sustainability of energy sources and wider road circulation of low-emission hybrid electric vehicles and, ideally, no-emission full-electric vehicles. The successful evolution of these emerging markets requires the availability of a battery technology suitable to ensure stationary energy storage and electric engine power.2 Unfortunately, despite the continuous improvements from its first commercialization back in 1991, the present LIB technology based on the “Carbon–Lithium Metal Oxide” intercalation chemistry is still not adequate for properly performing these duties.1,3 Accordingly, there have been many efforts to identify and develop alternative battery chemistries with higher energy densities and lower costs. Different approaches are presently considered to reach the goal. In this respect, lithium metal represents the ideal anode material and, indeed, in the initial commercial development of lithium batteries (in the late 80s), lithium was the anode of choice.4 Emerging candidates employing lithium metal are, for example, lithium–sulfur5 and lithium–air batteries.6 However, several unsolved issues make such systems difficult for practical exploitation. One of these is the serious safety hazard associated with the presence of lithium metal, which was the reason behind the commercial withdrawal of the first lithium batteries and motivated the shift to the safer and more reliable LIB technology.4,7Effectively, the use of lithium metal in a practical battery poses serious challenges. Indeed, it readily reacts with most electrolytes and, in particular, with liquid organic solutions commonly adopted in LIBs, experiencing dendritic growth upon charging with associated serious safety concerns (Fig. 1a). This explains why most of the battery manufacturers are still reluctant to move away from the more comfortable LIB design. Interestingly though, there has been lately, in the battery community, a renewed interest in the use of lithium metal in batteries.8 The excitement is obviously motivated by the unique specific capacity values of lithium (i.e., 3.86 A h g−1 and 2.06 A h cm−3) and its low electronegative standard potential of −3.040 V, which result in a very high electrochemical energy equivalent.9 Indeed, a battery using lithium metal would have a considerably higher energy density (both gravimetric and volumetric) than those employing other common anode materials (see Fig. 1b). Certainly, the use of lithium metal in practical batteries may be acceptable only if the safety of operation is totally ensured. This essential condition can be achieved by means of electrolytes providing thermodynamic stability against Li or, alternatively, able to form a stable passivation layer on their surface,10 enabling a smooth and reversible deposition upon cycling.
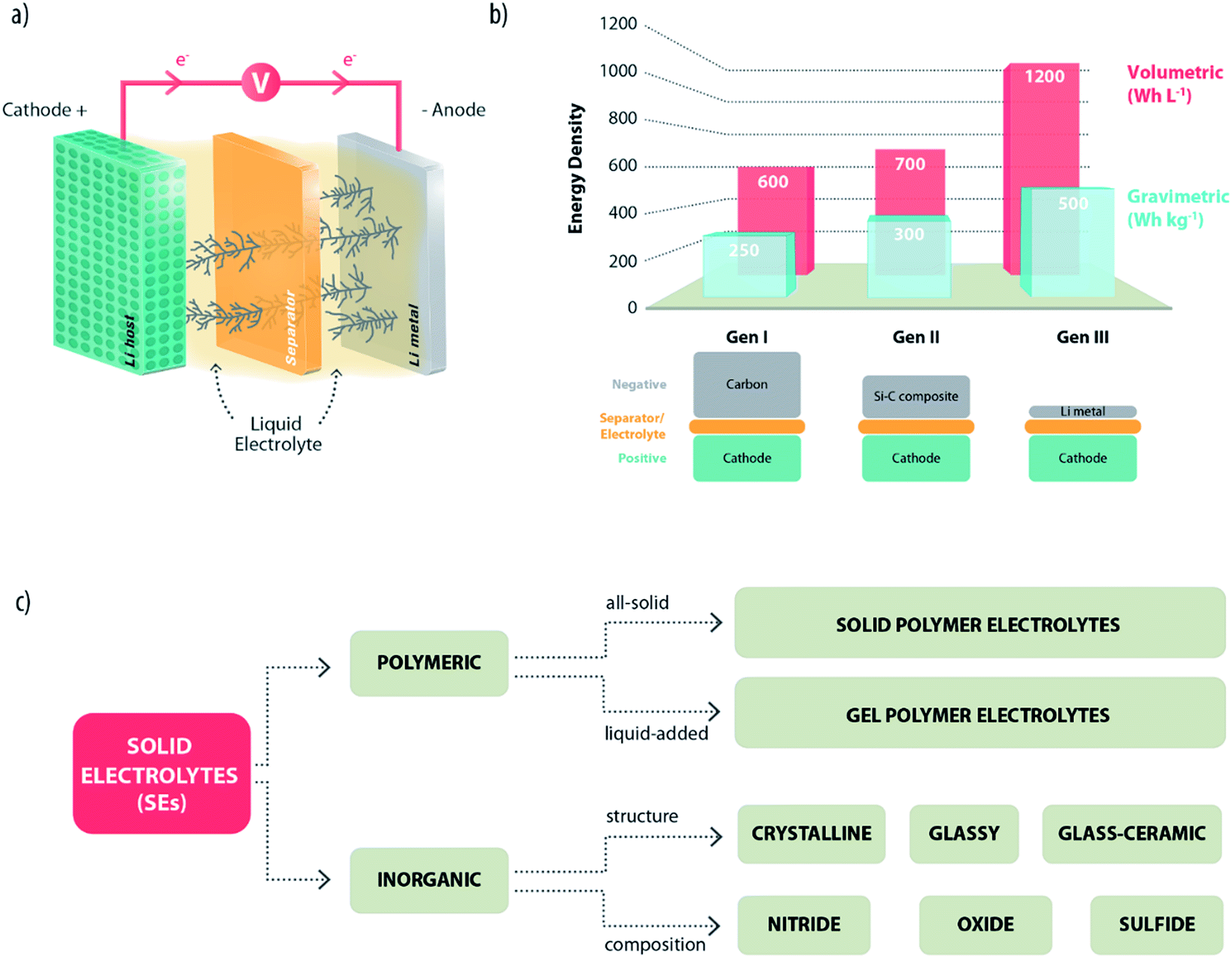 | ||
| Fig. 1 (a) Schematic representation of a lithium metal battery experiencing dendrite growth upon charging. (b) Gravimetric and volumetric energy densities achieved using Li-metal (Gen III) or other common anodes such as graphite (Gen I) and silicon–carbon composites (Gen II). The values are taken from ref. 8 and are presumably obtained with different cathode materials (Gen I: LiCoO2, Gen II: not specified; Gen III: not specified). (c) A possible classification of solid electrolytes which might be employed in lithium metal batteries. | ||
Although the growth of lithium dendrites can be partially suppressed by introducing additives in conventional liquid electrolytes,11,12 this cannot be considered as the ultimate answer to the safety issues of lithium metal batteries. Organic carbonates are, indeed, highly flammable anyway and can easily ignite in LIBs, too. The ideal strategy would involve the complete replacement of conventional electrolytes with safer electrolytic media establishing a physical barrier to the growth of dendrites. The most promising category is constituted by the so-called solid electrolytes (SEs). These are generally defined as electronically insulating solid materials with high mobility and selective transport of charged ionic species within their structure.13 In general, they can be divided into two main categories: polymeric and inorganic. Over the years, then, different classification schemes have been proposed, according to their chemical composition, structure, etc. (Fig. 1c).14 In this review article, we do not aim at a systematic description of all the typologies of SEs, which has been already done in previously published technical reviews.13–18 Our approach will be, instead, a more critical discussion and evaluation of the main features of such materials. Particular attention will be devoted to their compatibility with the lithium metal electrode and the open challenges.
2. Polymer electrolytes
Solid polymer electrolytes (SPEs), first proposed for lithium metal batteries in the late seventies,16,18–20 have the advantage of combining solid-state behaviour with the ease of processing plastic materials. The historically most well-known SPEs are based on poly(ethylene oxide) (PEO) and a lithium salt (LiX; X = ClO4, PF6, N(SO2CF3)2).19–23 In such electrolytic systems, Li+ ions are complexed to the ether oxygens of the PEO chains, and, therefore, their mobility is strongly affected by the motion of the complexing polymer segments. As schematically described in Fig. 2a, the ionic transport is caused by the motion of the complexation sites assisted by the segmental motion of the PEO matrix.18 PEO-based SPEs are highly appealing because of their compatibility with the lithium metal electrode. Recent studies have demonstrated that, similar to conventional organic carbonates, they are not fully thermodynamically stable with lithium metal and a passivation layer is formed at the interface.24 Nevertheless, this allows quite reversible stripping–plating behaviour (see Fig. 2b).22 Furthermore, their plastic nature allows, in principle, the development of safe and flexible lithium polymer batteries (LPBs). Indeed, efficient rechargeable LPBs have been developed starting from the early eighties25 (see a typical configuration in Fig. 2c) and power, nowadays, the full electric vehicles produced by Bolloré.26 The famous Bluecars are a solid reality, which proves that LPBs are safe and reliable, as testified by the 3000 car fleet that has been driven over 10 million miles without issues. Nevertheless, the large excess of Li metal needed to ensure such performance (three-fold excess) definitely leaves much room for improvement in terms of energy density.27 Nowadays, the major drawback of PEO-based electrolytes is their modest ionic conductivity (i.e., <10−5 S cm−1 at RT), which obligates the battery to operate above 60 °C, where it reaches acceptable values (at least 10−3 S cm−1) owing to the crystalline-to-amorphous (highly conducting) phase transition.21 An important contribution to this field was given, in 2013, by Armand et al.28 Their multifunctional single-ion polymer electrolyte, based on polyanionic block copolymers comprising polystyrene segments, demonstrated indeed an unprecedented level of performance. Besides the excellent mechanical properties and the large electrochemical stability window (up to 5 V versus Li+/Li) it showed an ionic conductivity of 1.3 × 10−5 S cm−1 at 60 °C and a lithium-ion transport number close to unity (>0.85). As the decrease of conductivity at the interface is one of the triggers of dendrite formation, this class of electrolytes would be highly promising to increase the safety of LPBs.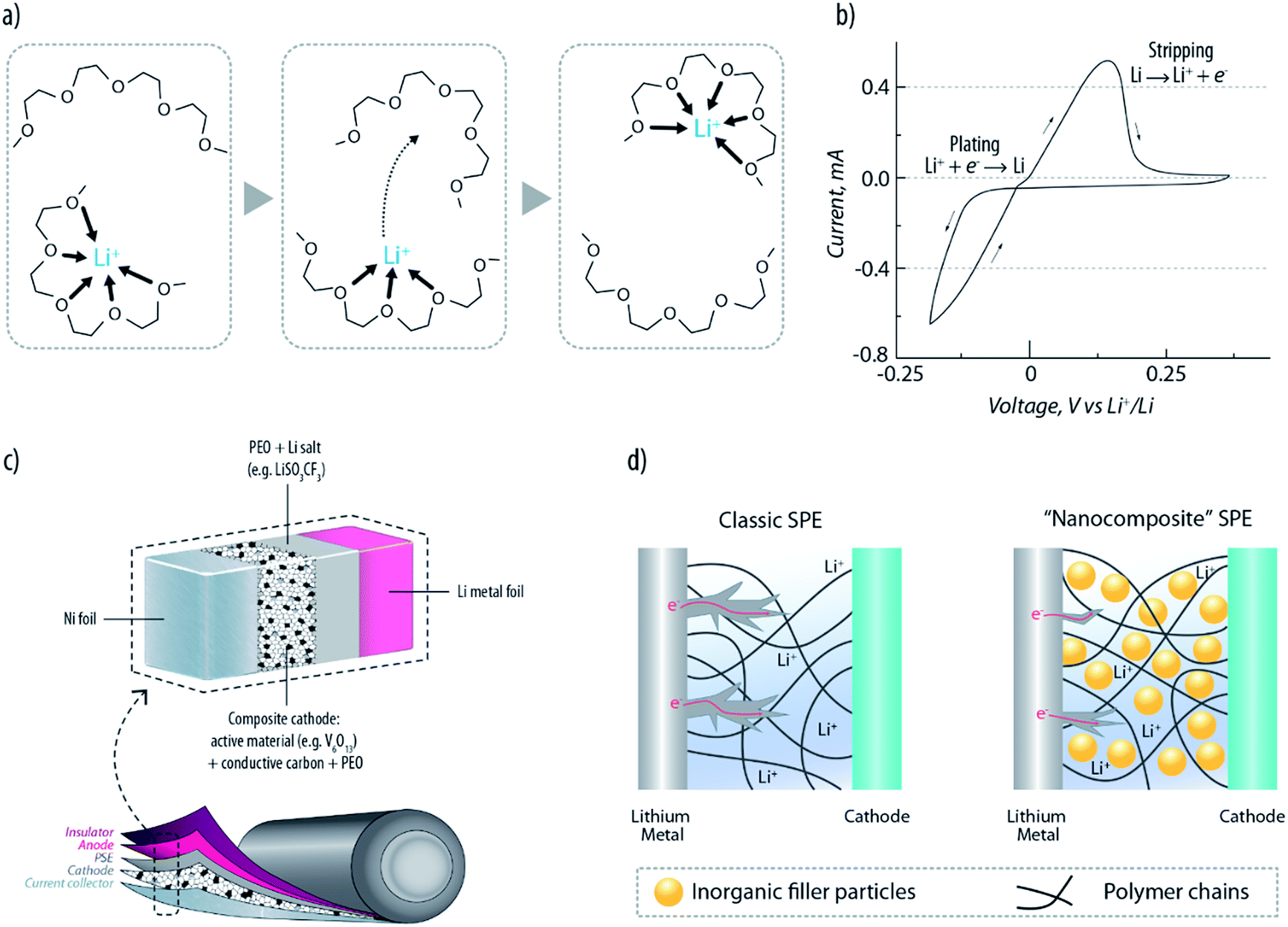 | ||
| Fig. 2 (a) Schematic drawing of Li+ transport in a PEO matrix, assisted by the segmental motion of the polymeric chains.18 (b) Cyclic voltammetry showing the reversible Li stripping–plating achievable with a PEO-based PSE. (c) Example of a typical lithium-polymer battery (LPB) configuration. (d) Schematic representation of dendrite growth suppressed by the nanocomposite SPE.36 | ||
Usually, a common approach to address the ionic conductivity issue relies on the addition of liquid plasticizers (leading to the so-called “gel polymer electrolytes”, i.e. GPEs), such as propylene carbonate (PC) or ethylene carbonate (EC), which, by reducing the crystalline fraction of the polymer matrix, lead to an increase of the conductivity, however, adversely accompanied by the loss of the solid-state configuration (these are indeed gels) and compatibility with the lithium metal electrode.14,29,30 In order to ensure both reasonably high conductivity and compatibility with the lithium electrode, one of the most successful strategies was found to be the addition of selected ionic liquids (ILs). The presence of two salts, one containing the organic cations (i.e., the IL, N-propyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide, named PYR13TFSI) and the other the Li+ (i.e., lithium bis(trifluoromethanesulfonyl)imide, named LiTFSI) in the PEO matrix, enables a relatively high conductivity of 10−4 S cm−1 at RT, with enhanced lithium-ion transfer properties compared to the IL-free PEO–LiTFSI electrolyte.31–34 Additionally, the IL has the beneficial effect of increasing the onset for dendrite growth. Such dendrite suppression is fundamental for ensuring long term stability of the lithium metal anode.
A further approach to inhibit the growth of Li dendrites is the dispersion of solid plasticizers (namely, selected ceramic powders with a sub-micrometric particle size) into the polymeric matrix to form “nanocomposite” (or “hybrid”) polymer electrolytes.35 Experimental results suggest that the uniformly dispersed ceramic filler (e.g., TiO2 or Al2O3) may provide sturdier and more tortuous interfaces hampering the penetration of nucleated dendrites (see Fig. 2d).36 It should be furthermore mentioned that, besides inorganic fillers, cross-linked polymeric chains can also improve the cycling stability of LPBs.37
3. Inorganic solid electrolytes
3.1 Conductivity mechanism and classic examples
Unlike liquids, where the ions move in the medium with their solvation shells, the conductivity in inorganic solid electrolytes (ISEs) occurs via ionic motion across crystalline lattice sites. Furthermore, ISEs are generally single-ion conductors with a high transference number (approaching unity), which represents a considerable advantage over aprotic electrolytes (transference number in the range 0.2–0.5),38 to eliminate concentration polarization at electrode interfaces. ISEs are formed by fixed ionic sub-lattice offering paths (composed of vacant or interstitial sites) along which the mobile ions can diffuse by hopping between adjacent sites. For most of the time, the mobile ions are localized in given lattice sites where they vibrate along their equilibrium positions. Occasionally, the ions leave their stable positions to move (by thermally activated jumps) to adjacent sites, where they reside for a certain time before continuing their motion either to another empty site or back to their initial one (theory of casual motion). To jump across the crystalline sites, the ions have to overcome an activation energy barrier. For solids with highly packed structures (e.g., NaCl and β-AgI), having not well defined motion channels, the activation energy is typically of the order of 1 eV (∼96 kJ) or higher and, consequently, the room temperature (RT) conductivity is modest (i.e., in the order of 10−6 S cm−1 or less). Differently, in solids characterized by open conduction channels, the values of activation energy are considerably lower (e.g., in the order of 0.03 eV) and the conductivity is several orders of magnitude higher.A classic example of the latter is α-AgI16 (see Fig. 3a) where the Ag+ ions, on average two per unit cell, are statistically and randomly dispersed over a great number of interstitial sites having a preferential coordination 4 (where Ag+ ions reside for most of the time) but, also, coordination 3 or 2 (where the ions move during their migration). Unfortunately, the α-AgI phase is stable only at temperatures higher than 147 °C, and this obviously reduces its practical interest. However, by “doping” silver iodide with large ionic radius cations or anions, it is possible to expand the unit cell, and thus to “freeze” down the α-AgI disordered structure to ambient temperature. Typical examples of these stabilizing ions are Rb+, K+ and (CH3)4N+ among the cations and P2O74− and WO42− among the anions.16 The most known electrolyte of this family is silver rubidium iodide, RbAg4I5,39 whose structure is schematically shown in Fig. 3b. This was indeed proposed for battery fabrication.40 However, although proved to have a uniquely long shelf-life,41 these batteries, due to the high cost and low voltage issues of silver, have been of modest practical relevance.
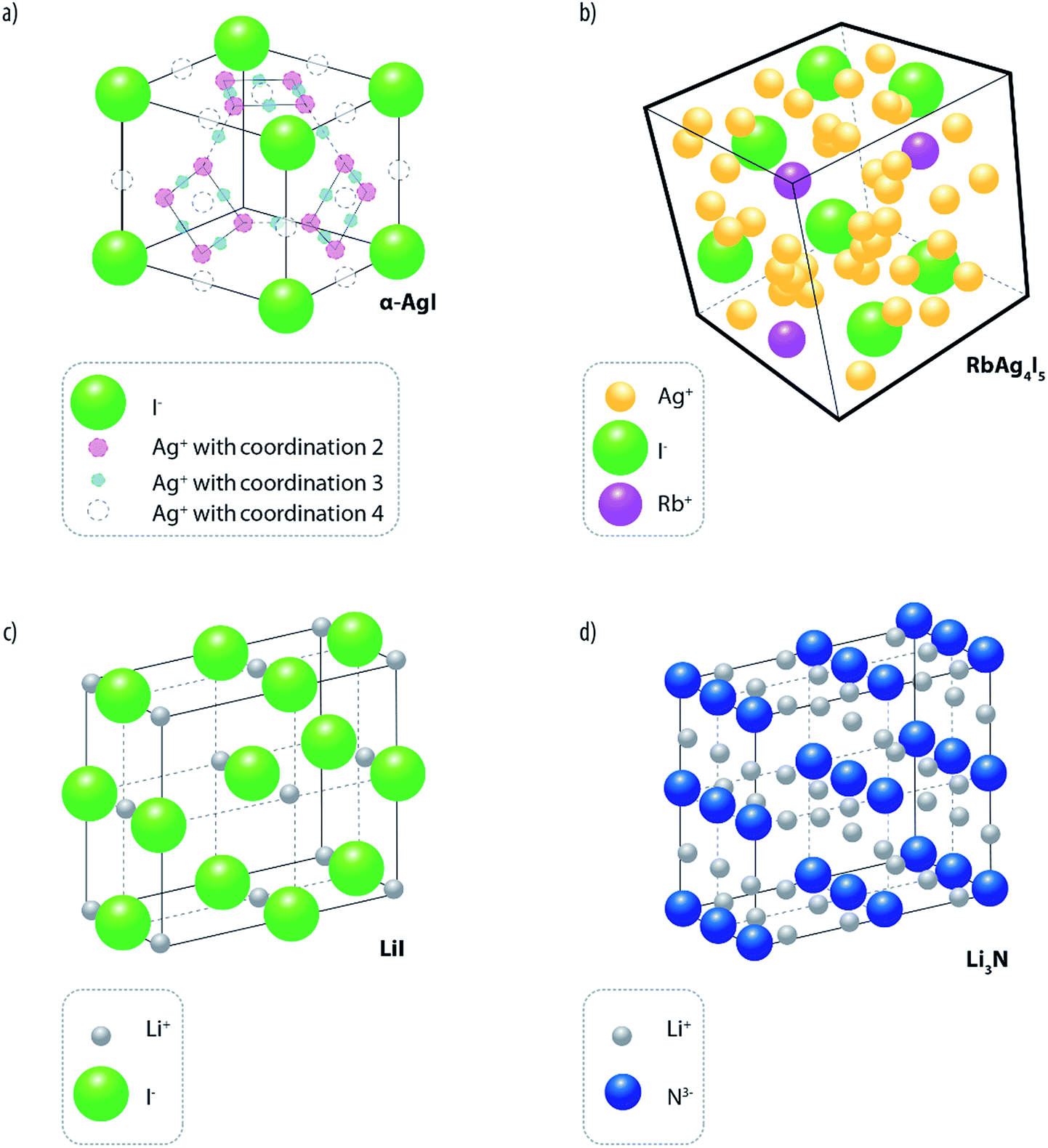 | ||
| Fig. 3 Classical examples of solid ionic conductors and their structures. (a) α-AgI, (b) RbAg4I5, (c) LiI and (d) Li3N. | ||
Lithium-ion conducting inorganic solid electrolytes (LISEs) would encounter considerably more interest since, in principle, they may lead to the development of safer lithium batteries. In fact, compared to conventional liquid electrolytes, LISEs offer a series of key advantages, such as high reliability (no leakage) and enhanced safety (intrinsically not flammable).12,13,42,43 Unfortunately, the effective use of LISEs is still limited mainly by (i) their low ionic mobility, which results in conductivities considerably lower than those of conventional organic liquids, and (ii) reactivity at the interface with the electrodes, especially at low potentials (i.e. vs. Li or graphite). Historically, the first example of a LISE of practical interest is lithium iodide, LiI.16 Although it adopts a NaCl-type compact structure (see Fig. 3c) and possesses a very low lithium ion conductivity at RT (i.e., 10−7 S cm−1), clever manufacturing procedures, aimed at reducing its thickness, enabled the development of thin film lithium metal batteries of great relevance in the medical sector. Indeed, cardiac pacemakers, which require isothermal operation (i.e., 37 °C) at a very low rate (i.e., 10 year rate), all use LiI-based batteries.44 It was also shown that the bulk ionic conductivity of LiI can be considerably enhanced by the incorporation of Al2O3 powders up to values of the order of 10−5 S cm−1. The increase in conductivity was proposed to be promoted by the occurrence of preferential pathways on the surface of the aluminium-based additive.45 In this way, the LiI–Al2O3 conductivity resulted to be sufficient for the development of a solid-state battery, although no substantial market impact was achieved at the time. Another classical LISE is H-doped (1–2%) lithium nitride (Li3N).46 It is constituted by a structure formed by Li2N layers, which are separated by additional bridging of loosely bonded Li+ ions (see Fig. 3d), leading to a conductivity of about 10−3 S cm−2 (at 25 °C),47i.e., a value comparable with that of organic liquid solutions. Unfortunately, the electrochemical stability window (ESW) of Li3N is too narrow for use with high voltage cathodes (ca. 0.45 V), thus, preventing its use for the development of batteries of practical relevance.
3.2 Present developments and ongoing challenges
Due to the renewed interest in high performance solid-state lithium metal batteries (SSLMBs), tremendous research efforts are presently underway in academia and industry to develop advanced solid lithium ion conductors having fast ion transport and a wide ESW.8,48 As testified by some detailed technical reviews recently published, the research is currently focused, mainly, on crystalline and glassy (or glass-ceramic) compounds.13,15,49A promising class of crystalline lithium ion conductors is provided by garnet-like structured compounds with the Li5La3M2O12 general formula, where M = Ta, Nb, Ba or Zr50,51 (see Fig. 4a). Although these materials offer excellent compatibility with metallic lithium and most of the common lithium cathodes, their commonly low RT conductivity (i.e., in the order of 10−5 S cm−1) has so far prevented their use in practical SSLMBs. Nevertheless, it has been recently demonstrated that the ionic conductivity of this class of LISEs can be boosted up to 10−4 to 10−3 S cm−1 at RT, by stabilizing the cubic form of the structure with small amounts of Al.52 Recently, Kanno et al. reported a superionic conductor, i.e., Li10GeP2S12 (LGPS), a solid having mono-dimensional conductive pathways (Fig. 4b) and fast lithium-ion transport, which showed one of the highest ionic conductivity. Indeed, the resulting ionic conductivity above 10−2 S cm−1 at 27 °C (ref. 53 and 54) is sufficient for battery use.55 Although it was experimentally shown that LGPS possesses a wide electrochemical stability window, with no reactions occurring between 0 and 5 V versus Li+/Li,53,56 some computational studies predict potential instability when in contact with both metallic lithium and cathode materials.56,57 The discrepancy between theory and experiments might indicate the presence of large overpotentials at the interfaces. In general, reactions between the electrode materials and the solid electrolyte are still poorly understood, and the formation of interfacial layers causing such overpotential cannot be excluded.56,57 Recently, Kanno's research group58 developed a further superionic conductor (i.e., Li9.54Si1.74P1.44S11.7Cl0.3) which exhibits even higher conductivity up to 25 mS cm−1 at RT. Such a high value is two times higher than that of Li10GeP2S12. The authors attributed the high ionic conduction characteristics of this novel superionic conductor to the 3D structural pathway.
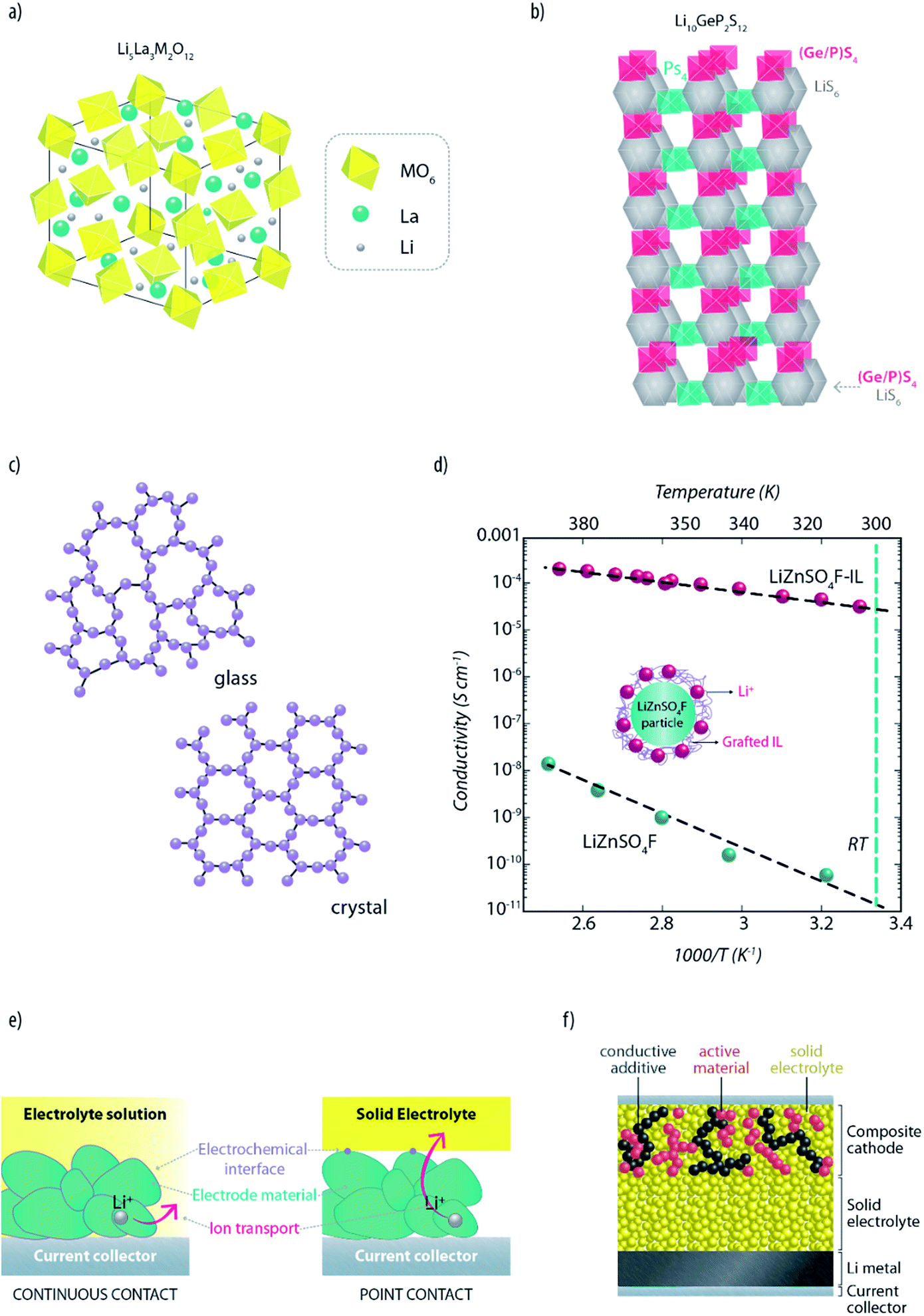 | ||
| Fig. 4 Framework structure of (a) a generic garnet-like lithium ISE (M = Ta, Nb, Ba or Zr) and (b) the recently reported superionic conductor Li10GeP2S12 (re-drawn and re-adapted from the original manuscript of Kamaya et al.53). (c) Schematic description of the differences between glassy and crystalline phases in ISEs. (d) Effect of IL grafting on the ionic conduction properties of the LiZnSO4F ceramic electrolyte (re-drawn and re-adapted from the original manuscript of Barpanda et al.64). (e) Influence of electrolytes on the establishment of the electrode/electrolyte interfaces. Differences between liquid (left panel) and solid electrolytes (right panel) are highlighted. (f) Schematic cross-section of an all-solid-state lithium metal battery showing the importance of creating percolative pathways for ions and electrons through the whole composite electrode depth, in order to allow full utilization of the active materials. | ||
A further interesting ISE is the LISICON-type crystalline phase consisting of a Li2O–Al2O3–SiO2–P2O5–TiO2–GeO2 composite (glass-ceramic), having a RT lithium ion conductivity of about 10−3 to 10−4 S cm−1, which was developed and patented by Ohara Inc. in 1997.59 Being rather stable in moist air makes it very appealing; however, its practical application in the battery field is still hindered by its poor chemical stability against metallic lithium and, in part, high costs. A successful approach to overcome the poor compatibility of ISEs with lithium metal is the introduction of an additional interfacial layer of lithium phosphorous oxynitride, Li2PO2N, commonly known by LIPON.16,60 This popular glass-ceramic ISE, one of the most explored in the last few years,13 in fact, shows negligible reactivity with lithium metal. Furthermore, it possesses a considerably high shear modulus (7.3 times higher than Li), which is a fundamental requirement for suppressing dendrite growth.61 Unfortunately, though, it shows a very low conductivity (i.e., in the order of 10−6 S cm−1 at room temperature). For this reason, its application is still limited to thin-layer solid-state lithium batteries (a few microns thick).60
Among glassy electrolytes, sulfides offer high lithium ion conductivity at room temperature (10−3 to 10−5 S cm−1),13,62,63 due to the large ionic radius and high polarizability of sulfur ions compared to oxide ions. Li2S–P2S5 is, probably, the most famous example of sulfide ISEs. Li2S–P2S5 glasses with high Li+ concentration have shown encouraging conductivities of over 10−4 S cm−1.62 This typology of sulfides can be produced also as glass-ceramic electrolytes (crystallized glasses). Mechanical milling of Li2S–P2S5 can lead to the precipitation of superionic crystals with a structure similar to those of thio-LISICON phases.49 It is known that crystallization usually decreases the conductivity. Glasses generally have, indeed, higher conductivity than the corresponding crystalline phase due to isotropic ionic conduction and their so-called “open structure” (Fig. 4c). However, glass-ceramic compounds are constituted by crystalline domains surrounded by an amorphous phase, resulting in a considerably reduced grain-boundary resistance compared to polycrystalline systems. Despite such encouraging results, the large resistance at grain-boundaries still represents the most serious issue hindering the development of all-solid-state batteries. In fact, it has been proven that there are classes of ISEs which can provide sufficient ionic conduction (bulk conductivity) for practical application. However, the influence of grain-boundaries can decrease the total conductivity by a few orders of magnitude.49,62 Interestingly, Tarascon's group has recently proposed a strategy which might solve such an issue.64 This involves the grafting of ILs on lithium-containing ISE particles leading to an improvement of up to 6 orders of magnitude in conductivity, for example, in ceramic LiZnSO4F (see Fig. 4d). It is not yet clear whether such an improvement is due to the reduction of grain-boundary resistance or, simply, to the IL layer providing a better percolation path for the ions. Nevertheless, such pioneering work is surely opening up new opportunities for the application of ISEs in batteries.
So far, great efforts have been made to increase the conductivity and compatibility of ISEs with lithium. The above mentioned examples prove that materials suited for all-solid-state lithium metal batteries are indeed available, and their performance is improving day-by-day. However, further technological advances are needed to introduce ISEs into bulk-type batteries (which are of more practical interest than thin-film devices). Contrarily to conventional LIBs, where the liquid electrolyte easily penetrates into the electrode, the solid electrolyte, having no fluidity, does not form continuous but point-to-point contacts with the active material (see Fig. 4e).47 As charge transfer only occurs at the contact points, establishing an extended electrode–electrolyte interface is necessary to allow full utilization of the active material. Furthermore, it would be beneficial to ensure an even current distribution in the electrode. In fact, avoiding spots with large current density is essential to suppress the formation of Li dendrites, which could grow in the voids and propagate along the grain boundaries of the ISE.65
In order to increase the contact area, the solid electrolyte needs to percolate throughout the whole electrode depth (see Fig. 4f).
Several strategies are under development to achieve this goal, such as preparation of composites by ball milling, surface coating of the active material and impregnation with supercooled liquid glass.62 Of course, the strategy to apply is highly dependent on the type of ISE used. Although these methods work with glassy materials, they might not be suitable for the processing of crystalline electrolytes.
4. Conclusions
Lithium metal batteries, abandoned for about 40 years because of safety issues associated with uncontrolled and hazardous growth of dendrites upon cycling, are recently seeing a renewed interest.66 They may indeed offer superior electrochemical performance with respect to conventional LIBs, when used in important applications aimed to improve the quality of our life, such as efficient energy use and sustainable road transportation. Undoubtedly, the development of solid-state lithium metal batteries (SSLMBs) is the most promising way to obtain power sources combining high energy with intrinsic safety. Unfortunately, the appropriate way to reach this goal is still practically prevented by a series of issues, the main being the unavailability of a solid electrolyte capable of combining good compatibility with lithium metal as well as high ionic conductivity at medium-low temperature.As discussed in this review paper, in this respect there are two classes of materials under consideration, one involving polymer-based systems and the other inorganic solid compounds. The former is possibly more promising since it has been shown that additives such as ionic liquids and/or ceramic fillers may lead to improvements in the transport properties. Indeed, lithium polymer batteries are currently commercially produced to be used as power sources for electric vehicles. An increase in conductivity can also be obtained by adding liquid plasticizers, but this approach is not totally acceptable since it is accompanied by a decay of the solid-state configuration, as well as by an increase of reactivity with the lithium metal anode.
A real breakthrough in the field may be enabled by inorganic solid electrolytes (ISEs). These benefit from a series of relevant properties, such as non-flammability, robustness (preventing lithium dendrite growth) and a single-ion conduction mechanism (greatly mitigating the problem of concentration polarization under high drain), which make them the most promising electrolyte category for the next generation of lithium metal batteries. However, the main problems of ISEs, i.e., the low ionic conductivity resulting in a high ohmic polarization during cell operation, and the poor interconnection of the solid to solid electrode–electrolyte interface, which does not guarantee the full utilization of the electrode active materials, are not yet fully solved, still hindering their practical use. To achieve high performance, nano-structured electrode/electrolyte interfaces need to be used. This would help to overcome diffusion problems and provide sufficiently good contacts to all particles. Keeping a certain degree of flexibility in the cell would also be very appealing for applications. From this point of view, polymer-based electrolytes may be a more reasonable choice.
Overall, the interest in SSLMBs is continuously increasing and, according to the latest research records, many industrial and academic laboratories are devoting large efforts to address these issues. In this regard, different strategies for reaching this goal are employed. In the case of glassy ISEs, for example, one valid method to increase the conductivity is represented by the electrolyte crystallization to form glass-ceramic compounds. Clever engineering approaches, including thin film construction and/or design of electrode and electrolyte powder mixtures, may help to overcome the interfacial issue. Furthermore, the low intrinsic electronic transport can be tackled by adding a conductive component in the electrode configuration. Indeed, some examples of practical SSLMBs are already available.8,26 The academic and, especially, industrial interest in developing solid-state batteries is increasing at such a fast rate that it is reasonable to forecast important breakthroughs in the near future.
Acknowledgements
A. V., R. R. S. P. and B. S. acknowledge the financial support of Helmholtz Institute Ulm (HIU) and the Karlsruhe Institute of Technology (KIT).References
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2014, 7, 19–29 CrossRef PubMed.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS.
- B. Scrosati, J. Solid State Electrochem., 2011, 15, 1623–1630 CrossRef CAS.
- D. Bresser, S. Passerini and B. Scrosati, Chem. Commun., 2013, 49, 10545–10562 RSC.
- L. Grande, E. Paillard, J. Hassoun, J. Park, Y. Lee, Y. Sun, S. Passerini and B. Scrosati, Adv. Mater., 2015, 27, 784–800 CrossRef CAS PubMed.
- B. Scrosati, K. M. Abraham, W. A. van Schalkwijk and J. Hassoun, in Lithium Batteries-Advanced technologies and Applications, Wiley, 2013, pp. 21–38 Search PubMed.
- Q. Hu, Nature, 2015, 526 Search PubMed , sponsor feature.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. R. Busche, T. Drossel, T. Leichtweiss, D. A. Weber, M. Falk, M. Schneider, M. Reich, H. Sommer, P. Adelhelm and J. Janek, Nat. Chem., 2016, 8, 426–434 CrossRef CAS PubMed.
- Z. Li, J. Huang, B. Yann Liaw, V. Metzler and J. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 CAS.
- P. Knauth, Solid State Ionics, 2009, 180, 911–916 CrossRef CAS.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525 RSC.
- J. G. Kim, B. Son, S. Mukherjee, N. Schuppert, A. Bates, O. Kwon, M. J. Choi, H. Y. Chung and S. Park, J. Power Sources, 2015, 282, 299–322 CrossRef CAS.
- B. B. Owens, J. Power Sources, 2000, 90, 2–8 CrossRef CAS.
- C. Cao, Z. B. Li, X. L. Wang, X. B. Zhao and W. Q. Han, Front. Energy Res., 2014, 2, 1–10 Search PubMed.
- W. H. Meyer, Adv. Mater., 1998, 10, 439–448 CrossRef CAS PubMed.
- M. Armand and M. Duclot, French Patent-FR2442512, 1978, 33.
- M. Armand, J. M. Chabagno and M. Duclot, in Second International Meeting on Solid Electrolytes, St. Andrews, Scotland, 1978 Search PubMed.
- F. Gray, Solid Polymer Electrolytes: Fundamentals and Technological Applications, VCH publishers, New York, 1991 Search PubMed.
- F. Bonino, B. Scrosati and A. Selvaggi, Solid State Ionics, 1986, 18–19, 1050–1053 CrossRef CAS.
- F. Bonino, B. Scrosati, A. Selvaggi and M. Lazzari, J. Electrochem. Soc., 1985, 132, C344 Search PubMed.
- C. Xu, B. Sun, T. Gustafsson, K. Edström, D. Brandell and M. Hahlin, J. Mater. Chem. A, 2014, 2, 7256 CAS.
- A. Hooper and J. M. North, Solid State Ionics, 1983, 9 & 10, 1161–1166 Search PubMed.
- J. Motavalli, Nature, 2015, 526, S96–S97 CrossRef CAS PubMed.
- M. Armand, in International Battery Association (IBA) Meeting, Barcelona, 2013 Search PubMed.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- Y. T. Kim and E. S. Smotkin, Solid State Ionics, 2002, 149, 29–37 CrossRef CAS.
- J. Hassoun and B. Scrosati, J. Electrochem. Soc., 2015, 162, A2582–A2588 CrossRef CAS.
- J. Shin, Electrochem. Commun., 2003, 5, 1016–1020 CrossRef CAS.
- J. H. Shin, W. A. Henderson and S. Passerini, J. Electrochem. Soc., 2005, 152, A978 CrossRef CAS.
- G. B. Appetecchi, G. T. Kim, M. Montanino, F. Alessandrini and S. Passerini, J. Power Sources, 2011, 196, 6703–6709 CrossRef CAS.
- S. Passerini, I. Osada, H. De Vries and B. Scrosati, Angew. Chem., Int. Ed., 2016, 55, 500–513 CrossRef PubMed.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- S. H. Kim, K. H. Choi, S. J. Cho, E. H. Kil and S. Y. Lee, J. Mater. Chem. A, 2013, 1, 4949 CAS.
- W. K. Shin, J. H. Yoo, W. Choi, K. Y. Chung, S. S. Jang and D. W. Kim, J. Mater. Chem. A, 2015, 3, 12163–12170 CAS.
- S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, H. D. Wiemhöfer and H. J. Gores, Electrochim. Acta, 2011, 56, 3926–3933 CrossRef CAS.
- S. Geller, Science, 1967, 157, 310–312 CAS.
- M. Z. A. Munshi, Handbook of Solid State Batteries and Capacitors, Intermedics Inc., USA, 1995 Search PubMed.
- B. B. Owens, P. Reale and B. Scrosati, Electrochem. Commun., 2007, 9, 694–696 CrossRef CAS.
- C. Masquelier, Nat. Mater., 2011, 10, 649–650 CrossRef CAS PubMed.
- J. Li, C. Ma, M. Chi, C. Liang and N. J. Dudney, Adv. Energy Mater., 2015, 5, 1401408 CrossRef.
- C. F. Holmes, in Batteries for Implantable Biomedical Devices, ed. B. B. Owens, Plenum Press, New York, 1986 Search PubMed.
- C. C. Liang, J. Electrochem. Soc., 1973, 120, 1289 CrossRef CAS.
- B. A. Boukamp and R. A. Huggins, Mater. Res. Bull., 1978, 13, 23–32 CrossRef CAS.
- H. Munukata and K. Kanamura, in Advanced Materials for Clean Energy, ed. Q. Xu and T. Kobayashi, CRC Press, 2015, ch. 9, p. 271 Search PubMed.
- E. C. Evarts, Nature, 2015, 526, S93–S95 CrossRef CAS PubMed.
- C. Cao, Z. Bin Li, X. L. Wang, X. B. Zhao and W. Q. Han, Front. Energy Res., 2014, 2, 1–10 Search PubMed.
- V. Thangadurai, H. Kaack and W. J. F. Weppner, J. Am. Ceram. Soc., 2003, 86, 437–440 CrossRef CAS.
- J. Schwenzel, V. Thangadurai and W. Weppner, J. Power Sources, 2006, 154, 232–238 CrossRef CAS.
- D. O. Shin, K. Oh, K. M. Kim, K.-Y. Park, B. Lee, Y.-G. Lee and K. Kang, Sci. Rep., 2015, 5, 18053 CrossRef CAS PubMed.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- J. Hassoun, R. Verrelli, P. Reale, S. Panero, G. Mariotto, S. Greenbaum and B. Scrosati, J. Power Sources, 2013, 229, 117–122 CrossRef CAS.
- G. Oh, M. Hirayama, O. Kwon, K. Suzuki and R. Kanno, Chem. Mater., 2016, 28, 2634–2640 CrossRef CAS.
- J. C. Bachman, S. Muy, A. Grimaud, H. H. Chang, N. Pour, S. F. Lux, O. Paschos, F. Maglia, S. Lupart, P. Lamp, L. Giordano and Y. Shao-Horn, Chem. Rev., 2016, 116, 140–162 CrossRef CAS PubMed.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef.
- J. Fu, 5,702,995, Patent-US5702995A, 1997, 6.
- F. Xu, N. J. Dudney, G. M. Veith, Y. Kim, C. Erdonmez, W. Lai and Y.-M. Chiang, J. Mater. Res., 2010, 25, 1507–1515 CrossRef CAS.
- E. G. Herbert, W. E. Tenhaeff, N. J. Dudney and G. M. Pharr, Thin Solid Films, 2011, 520, 413–418 CrossRef CAS.
- M. Tatsumisago, M. Nagao and A. Hayashi, J. Asian Ceram. Soc., 2013, 1, 17–25 CrossRef.
- R. Kanno, in Encyclopedia of Electrochemical Power Sources, ed. J. Garche, Elsevier, 2009, p. 129 Search PubMed.
- P. Barpanda, J. N. Chotard, C. Delacourt, M. Reynaud, Y. Filinchuk, M. Armand, M. Deschamps and J. M. Tarascon, Angew. Chem., Int. Ed., 2011, 50, 2526–2531 CrossRef CAS PubMed.
- Y. S. Jung, D. Y. Oh, Y. J. Nam and K. H. Park, Isr. J. Chem., 2015, 55, 472–485 CrossRef CAS.
- W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |