
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective dearomatization of isoquinolines by anion-binding catalysis en route to cyclic α-aminophosphonates†
Abhijnan
Ray Choudhury
 and
Santanu
Mukherjee
*
and
Santanu
Mukherjee
*
Department of Organic Chemistry, Indian Institute of Science, Bangalore 560 012, India. E-mail: sm@orgchem.iisc.ernet.in; Fax: +91-80-2360-0529; Tel: +91-80-2293-2850
First published on 19th August 2016
Abstract
An enantioselective dearomatization of isoquinolines has been developed using chiral anion-binding catalysis. This transformation, catalyzed by a simple and easy to prepare tert-leucine-based thiourea derivative, makes use of silyl phosphite as a nucleophile and generates cyclic α-aminophosphonates. This is the first time asymmetric anion-binding catalysis has been applied to the synthesis of α-aminophosphonates.
Introduction
α-Aminophosphonates and the related α-aminophosphonic acid derivatives are used extensively in medicinal and pharmaceutical sciences as surrogates for α-amino acids.1 The strong correlation between the biological activities of compounds containing α-aminophosphonic acids and their absolute configuration2 renders the enantioselective synthesis of α-aminophosphonates imperative to such studies. Considerable advancement has taken place in the catalytic enantioselective synthesis of acyclic α-aminophosphonates using various strategies.3,4 In contrast, enantioselective synthesis of cyclic α-aminophosphonates remains elusive,5 despite their prominent abundance in biologically active molecules (Fig. 1).6 | ||
| Fig. 1 Biologically relevant cyclic α-aminophosphonic acids and their derivatives. | ||
Among various methods to produce enantioenriched α-aminophosphonates, possibly the most straightforward approach involves the catalytic asymmetric addition of dialkylphosphites to imines (Scheme 1A).7 In fact, the first example of a catalytic enantioselective synthesis of cyclic α-aminophosphonates consisted of an asymmetric hydrophosphonylation of cyclic imines, catalyzed by chiral heterobimetallic lanthanoid complexes.8,9
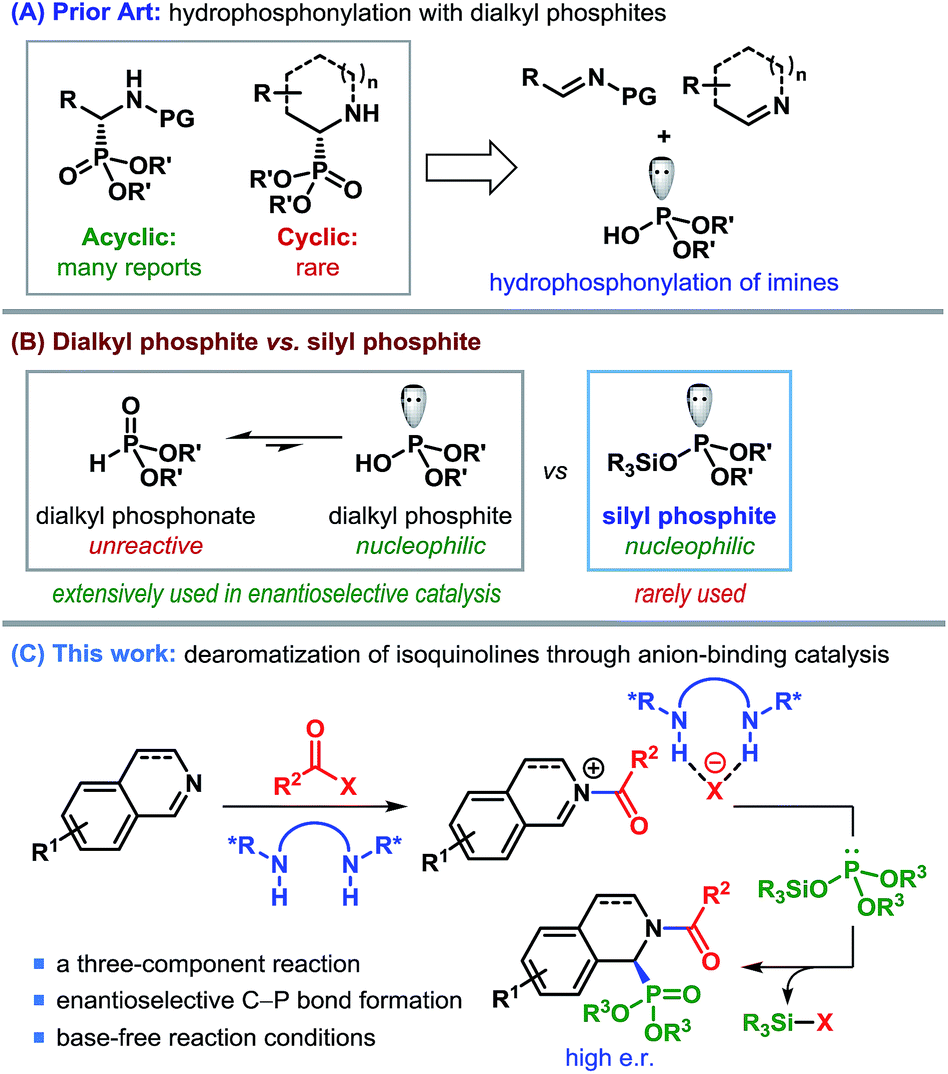 | ||
| Scheme 1 Catalytic enantioselective routes to cyclic α-aminophosphonic acids. | ||
Regardless of the popularity of dialkylphosphites as a nucleophile, the unfavorable equilibrium of unreactive phosphonate over reactive phosphite10 (Scheme 1B) often causes these reactions to be overly dependent on base activation. Silyl esters of dialkyl phosphonate (silyl phosphites), originally introduced by Abramov,11 on the other hand, are free from such limitations and display excellent reactivity.12 Nevertheless, the use of this class of highly nucleophilic silyl phosphites in enantioselective catalysis remained missing until the recent report by List and co-workers.13
In our effort to devise a catalytic enantioselective synthesis of cyclic α-aminophosphonates, we became interested in the addition of silyl phosphites to isoquinolines and their derivatives. Catalytic asymmetric dearomatization has emerged as a remarkably efficient strategy to synthesize a wide range of structurally and functionally important compounds.14 The pioneering works of Reissert have paved the way for decorating nitrogeneous heterocycles through acyl activation followed by nucleophilic addition, commonly known as Reissert-type reactions.15 We realized that the enantioface-selective addition to acyl-activated isoquinolines could be achieved through anion-binding catalysis – a concept, pioneered by Jacobsen et al. (Scheme 1C).16,17 However, despite the emergence of a number of anion-binding catalyst motifs, the asymmetric dearomatization of isoquinolines driven by anion-binding catalysis has so far been restricted mostly to using silyl ketene acetals as the nucleophilic partner.17,18 Rather surprisingly, to date no other silyl nucleophile has been employed in such reactions.
The use of silyl phosphites as a nucleophile in the dearomatization of isoquinolines appears to be tailor-made, in view of the fact that besides being a powerful nucleophile, the silyl group would also act as a sink for the counteranion (XΘ in Scheme 1C) and regenerate the catalyst. This three-component reaction would, in principle, represent a general and enantioselective route to cyclic α-aminophosphonates. The successful implementation of this strategy is presented in this article.
Results and discussion
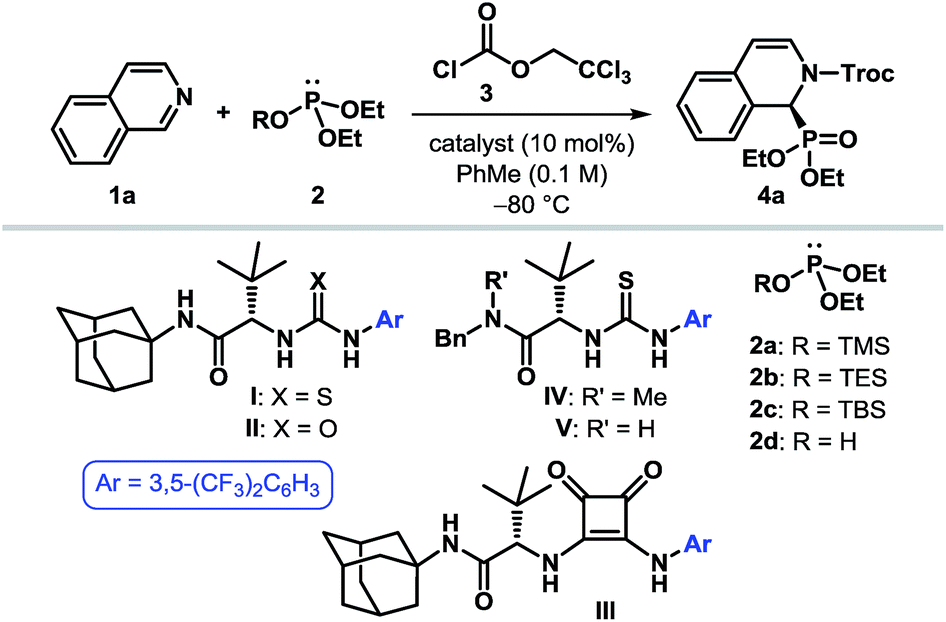
To put our hypothesis into practice, we began our investigation with an equimolar amount of isoquinoline (1a) and diethyl trimethylsilyl phosphite (2a) in the presence of a slight excess of 2,2,2-trichloroethyl chloroformate (TrocCl, 3) (Table 1). The reaction was indeed found to proceed efficiently to generate the desired cyclic N-acyl α-aminophosphonate 4a under the influence of 10 mol% of various hydrogen bond donors. Among the different H-bonding motifs studied (entries 3–5), thiourea proved to be the best anion-binding catalyst.19 Further optimization led to the identification of a simple and easy to prepare tert-leucine-derived thiourea IV,20 which provided 4a with an enantiomeric ratio (er) of 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (entry 6). While the same level of enantioselectivity was observed when diethyl triethylsilyl phosphite (2b) was used as the nucleophile (entry 8), bulkier silyl phosphite 2c furnished the product with a diminished er (entry 9). It must be noted that no conversion to the desired product occurred with diethyl phosphite (2d) as the nucleophile under otherwise identical reaction conditions (entry 10), thereby highlighting the superior nucleophilicity of silyl phosphites. Attempts to optimize the reaction by using different acylating agents or by changing the reaction medium and other parameters failed to provide any improvement in the optical yield.19
:4 (entry 6). While the same level of enantioselectivity was observed when diethyl triethylsilyl phosphite (2b) was used as the nucleophile (entry 8), bulkier silyl phosphite 2c furnished the product with a diminished er (entry 9). It must be noted that no conversion to the desired product occurred with diethyl phosphite (2d) as the nucleophile under otherwise identical reaction conditions (entry 10), thereby highlighting the superior nucleophilicity of silyl phosphites. Attempts to optimize the reaction by using different acylating agents or by changing the reaction medium and other parameters failed to provide any improvement in the optical yield.19
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | 2 | t [h] | Conv.b [%] | erc |
| a Reactions were carried out on a 0.1 mmol scale using 1.0 equiv. of 1a, 1.0 equiv. of 2 and 1.3 equiv. of 3. b Conversion as determined by 1H NMR of crude reaction mixture. c Enantiomeric ratio (er) was determined via HPLC analysis using a stationary phase chiral column. d Reaction at 25 °C. n.d. = not determined. | |||||
| 1d | None | 2a | 1 | >95 | — |
| 2 | None | 2a | 72 | <5 | — |
| 3 | I | 2a | 72 | >95 | 94.5:5.5 |
| 4 | II | 2a | 72 | >95 | 78:22 |
| 5 | III | 2a | 72 | >95 | 60:40 |
| 6 | IV | 2a | 72 | >95 | 96:4 |
| 7 | V | 2a | 72 | >95 | 91.5:8.5 |
| 8 | IV | 2b | 72 | >95 | 96:4 |
| 9 | IV | 2c | 72 | >95 | 94.5:5.5 |
| 10 | IV | 2d | 72 | <5 | n.d. |
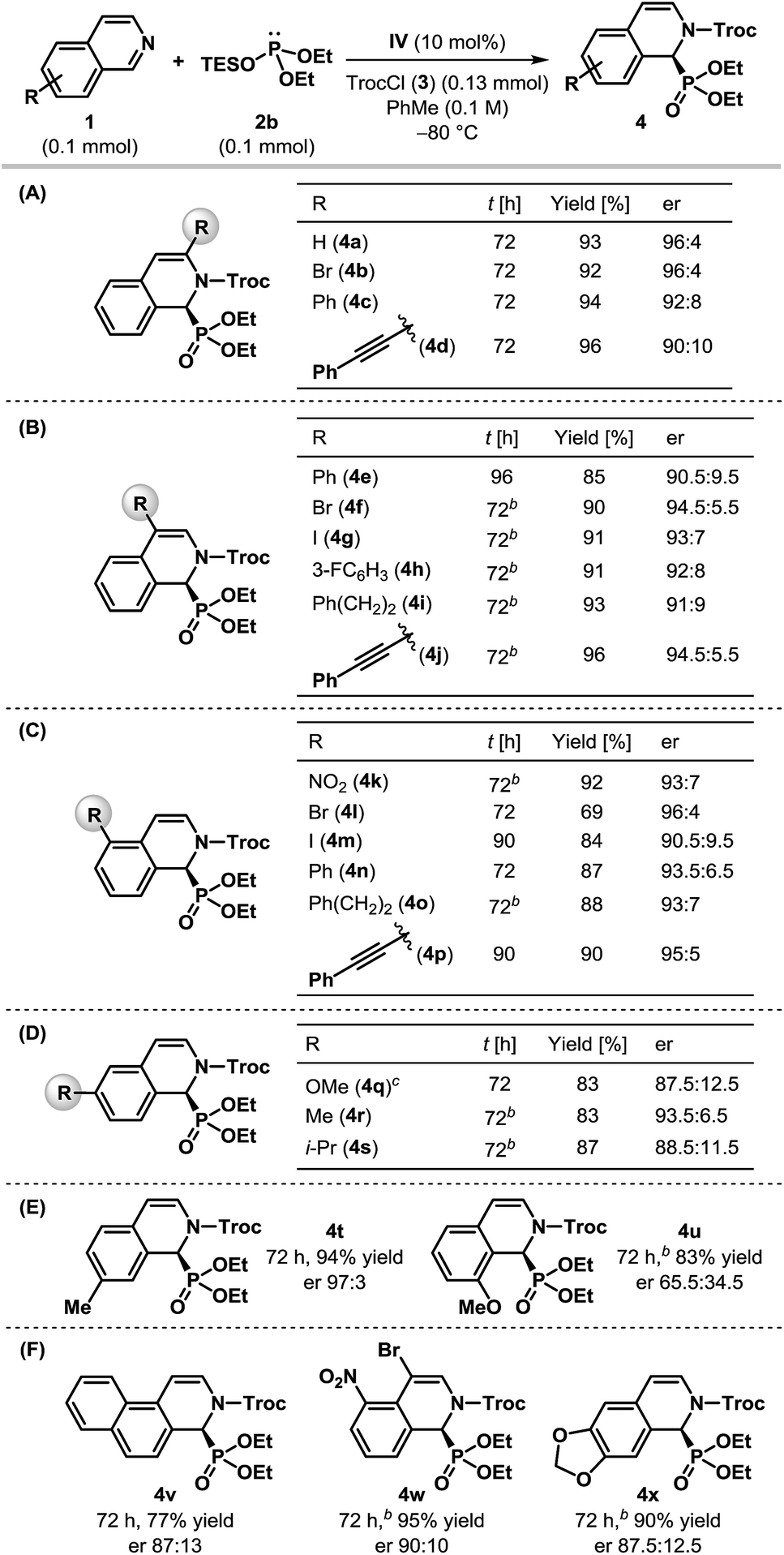
Having optimized the catalyst and the reaction conditions, we sought to test the scope and limitations of this enantioselective dearomatization reaction with respect to other isoquinoline derivatives. Even though identical results were obtained with both silyl phosphites 2a and 2b (Table 1, entries 6 and 8), the enantioselectivity observed for 2a did not appear to be general for other substrates. Therefore, we decided to use diethyl triethylsilyl phosphite 2b for the subsequent studies. The results compiled in Table 2 indicate that our protocol is generally applicable to monosubstituted isoquinolines bearing substituents at nearly every position (Table 2A–E) and even for disubstituted isoquinolines (Table 2F). In all of the cases, the products were obtained in good to excellent yields. Various 3- and 4-substituted isoquinolines were found to be effective substrates and the products were achieved with good to high enantioselectivity (Table 2A and B), although the reaction had to be carried out at −50 °C for most of the 4-substituted isoquinolines. Different substituents at the 5-position of isoquinoline were also tolerated and the desired α-aminophosphonates were generated with good enantioselectivity (Table 2C). Particularly noteworthy is the enantioselectivity observed for 5-nitroisoquinoline (1k), as the nitro group itself is capable of H-bonding with thiourea21 and could potentially upset the anion-binding activation mode of the catalyst. The dearomatization reaction worked well with 6-substituted isoquinolines, although the products were returned with reduced enantioselectivity (Table 2D). While the α-aminophosphonate (4t) derived from 7-methyl isoquinoline was formed with excellent enantioselectivity, the poor er obtained in the case of 8-methoxyisoquinoline 1u is presumably due to the steric influence of the substituent on the reaction center as a result of its proximity (Table 2E). Fused and disubstituted isoquinolines (1v–x) are also competent substrates for this reaction (Table 2F). However, the corresponding products (4v–x) were formed at most with modest enantioselectivities. Our attempts to synthesize cyclic α-aminophosphonates containing a quaternary stereogenic center by using 1-substituted isoquinolines ended in vain as no phosphonylation took place with either 1-methyl or 1-phenyl isoquinolines, even at 25 °C indicating the steric limitation of our protocol.
The absolute configuration of 4w was determined using single crystal X-ray diffraction analysis (Fig. 2).22 The configurations of the other products were assigned as the same, assuming that an analogous catalytic mechanism is operating.
 | ||
| Fig. 2 Absolute configuration of 4w and its X-ray structure. | ||
Besides diethyl triethylsilyl phosphite 2b, trimethylsilyl phosphites bearing different dialkyl substituents were found to be potent nucleophiles for this dearomatization reaction (Scheme 2). While a product (6a) with a reduced er was obtained with dimethyl phosphite (5a), both di(n-propyl) phosphite (5b) and diisopropyl phosphite (5c) rendered the reaction highly enantioselective. In fact, with 5c as the nucleophile, the reaction on a 1.0 mmol scale using only 5 mol% of catalyst (IV) delivered the product (6c) with a comparable yield and er. Notwithstanding the longer reaction time, catalyst recovery was possible with 95% yield.19
 | ||
| Scheme 2 Scope of dearomatization with respect to dialkyl phosphite and the reaction on a larger scale. | ||
The optimized catalyst and reaction conditions were found to be almost equally suitable for dihydroisoquinolines (7) and we were pleased to find that the corresponding α-aminophosphonates (8) were obtained in excellent yields (Table 3). However, the enantioselectivities observed in these reactions were generally found to be lower than those obtained for the corresponding isoquinoline derivatives.

To demonstrate the synthetic utility of the products obtained through the dearomatization of isoquinolines, we undertook the task of removing the protecting groups of cyclic N-Troc α-aminophosphonate 4a (Scheme 3A). The hydrolysis of the phosphonate could be achieved easily using sodium iodide and TMSCl to obtain the N-protected cyclic α-aminophosphonic acid 9 in 65% yield without any erosion in enantioselectivity. However, an attempt to remove Troc by In/NH4Cl23 resulted in rearomatization to the corresponding isoquinoline derivative 10 through aerial oxidation. Such aromatization to isoquinoline derivatives, even under reductive conditions, is documented in the literature24 and can be attributed to the inherent instability of the free secondary amine. The compounds of type 10 are known for their use as corrosion inhibitors.25 Various attempts to remove the Troc group, even from 8a, remained unsuccessful.19 The Fmoc-protected product (11) derived from dihydroisoquinoline, on the other hand, when subjected to piperidine in CH2Cl2, cleanly resulted in cyclic α-aminophosphonate 12 in 91% yield. The reaction of Fmoc-protected dearomatized product 13, under the same conditions, once again produced 10, even though the free amine could be detected initially when the reaction was monitored by 1H-NMR.19
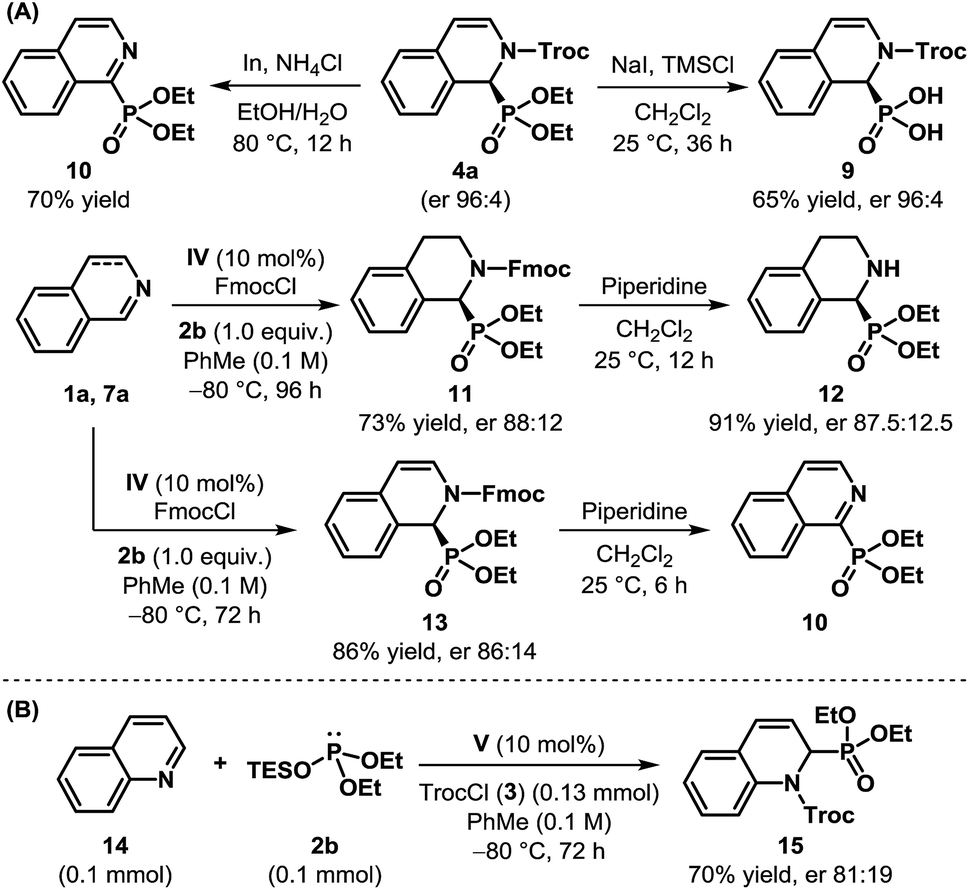 | ||
| Scheme 3 (A) Removal of protecting groups, and (B) the initial results of the catalytic enantioselective dearomatization of quinoline. | ||
After successfully accomplishing the enantioselective dearomatization of isoquinolines, we wondered whether our protocol could be used for quinoline (11).17b While it is more challenging to dearomatize quinoline,26 a preliminary experiment showed that our reaction conditions were indeed suitable for this purpose. Using V as the catalyst, the product 12 was isolated in 70% yield with an er of 81:19 (Scheme 3B). Despite the modest enantioselectivity, these results clearly illustrate the potential of this approach toward the dearomatization of other nitrogenous heteroaromatics.27
A number of pieces of information emerged during the optimization of the reaction conditions which helped to shed light on the mechanism of this catalytic transformation. The reaction medium was found to play a crucial role in the enantioselectivity of this reaction: a homogeneous reaction mixture in CH2Cl2 led to the product with poor er but high enantioselectivity was observed in toluene – a solvent which results in a heterogeneous reaction mixture.19 In addition, the silyl group of silyl phosphite exerts measurable influence on the enantioselectivity of the reaction (Table 1, entry 6 vs. 9). Based on these facts, a tentative mechanism is proposed as depicted in Scheme 4.28 Isoquinoline upon treatment with TrocCl, produces the salt A which exists in equilibrium with its covalent form B. The salt A can readily react with silyl phosphite to furnish Troc-protected α-aminophosphonate 4a. Its covalent form B, on the other hand, is unreactive toward silyl phosphite. The insolubility of A in toluene, especially at −80 °C, accounts for the absence of any background reaction at this temperature (Table 1, entry 2). However, the thiourea catalyst facilitates the ionization of B to generate salt C, in which the chloride ion is bound to thiourea by dual H-bonding. The direct conversion of A to C is also possible in the presence of the thiourea catalyst. The formation of C not only solubilizes the reactive ionic form, but at the same time owing to its proximity to the chiral thiourea-bound chloride anion, suffers an enantioface-selective attack by silyl phosphite. The resulting silylated intermediate D then collapses to release the product (4a) as well as R3SiCl and regenerates the catalyst. The participation of silyl phosphite (as opposed to the thiourea-bound phosphite anion E) in nucleophilic addition accounts for the observed influence of the silyl group on the enantioselectivity of the reaction. The lower er obtained in more polar solvent is most likely due to the competing background reaction which may stem from the superior solubility of A in such solvents.29 Furthermore, the superior solvation of the isoquinolinium cation and the chiral thiourea-bound chloride anion (of C) in more polar solvents possibly prevents the ‘chiral counteranion’ from being in sufficiently close proximity to the reactive center. Such proximity is necessary for the enantioface-selective attack by silyl phosphite.16a
 | ||
| Scheme 4 Proposed mechanism for the dearomatization of isoquinolines. | ||
Conclusions
In conclusion, we have successfully developed an enantioselective dearomatization of diversely substituted isoquinolines through the acyl activation and nucleophilic addition of silyl phosphites. Using a simple and easy to prepare tert-leucine derived thiourea as an anion-binding catalyst, this base-free protocol delivers cyclic α-aminophosphonates in excellent yields with moderate to high enantioselectivities. This is the first example of the use of silyl phosphites as a nucleophile in asymmetric dearomatization reactions driven by anion-binding catalysis. In fact, this is also the first time asymmetric anion-binding catalysis has been applied for the synthesis of α-aminophosphonates. A preliminary experiment with quinoline points toward the potential applicability of this strategy to other nitrogenous heteroaromatics. Efforts in this direction are currently ongoing in our laboratory.Acknowledgements
This work is funded by DAE-BRNS [Grant No. 2013/37C/56/BRNS/2440]. A. R. C. thanks the Council of Scientific and Industrial Research (CSIR), New Delhi for the Shyama Prasad Mukherjee Fellowship. We thank Prodip Howlader (IPC, IISc, Bangalore) for his help with the X-ray structure analysis.Notes and references
- (a) A. Mucha, P. Kafarski and Ł. Berlicki, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed; (b) G. Lavielle, P. Hautefaye, C. Schaeffer, J. A. Boutin, C. A. Cudennec and A. Pierre, J. Med. Chem., 1991, 34, 1998–2003 CrossRef CAS PubMed; (c) M. C. Allen, W. Fuhrer, B. Tuck, R. Wade and J. M. Wood, J. Med. Chem., 1989, 32, 1652–1661 CrossRef CAS PubMed; (d) F. R. Atherton, M. J. Hali, C. H. Hassall, R. W. Lambert and P. S. Ringrose, Antimicrob. Agents Chemother., 1979, 15, 677–683 CrossRef CAS PubMed; (e) J. G. Allen, F. R. Atherton, M. J. Hall, C. H. Hassall, S. W. Holmes, R. W. Lambert, L. J. Nisbet and P. S. Ringrose, Nature, 1978, 272, 56–58 CrossRef CAS PubMed.
- (a) V. Coeffard, I. Beaudet, M. Evain, E. L. Grognec and J.-P. Quintard, Eur. J. Org. Chem., 2008, 3344–3351 CrossRef CAS; (b) M. Drag, M. Pawelczak and P. Kafarski, Chirality, 2003, 15, S104–S107 CrossRef CAS PubMed; (c) A. B. Smith III, K. M. Yager and C. M. Taylor, J. Am. Chem. Soc., 1995, 117, 10879–10888 CrossRef.
- For selected reviews, see: (a) M. Ordóñez, J. L. Viveros-Ceballos, C. Cativiela and F. J. Sayago, Tetrahedron, 2015, 71, 1745–1748 CrossRef; (b) M. Dzięgielewski, J. Pięta, E. Kamińska and Ł. Albrecht, Eur. J. Org. Chem., 2015, 677–702 CrossRef; (c) K. Bera and I. N. N. Namboothiri, Asian J. Org. Chem., 2014, 3, 1234–1260 CrossRef CAS; (d) P. Łyżwa and M. Mikołajczyk, Pure Appl. Chem., 2010, 82, 577–582 CrossRef; (e) K. Moonen, I. Laureyn and C. V. Stevens, Chem. Rev., 2004, 104, 6177–6215 CrossRef CAS PubMed; (f) H. Gröger and B. Hammer, Chem.–Eur. J., 2000, 6, 943–948 CrossRef.
- For selected examples, see: (a) K. Bera and I. N. N. Namboothiri, J. Org. Chem., 2015, 80, 1402–1413 CrossRef CAS PubMed; (b) A. Kumar, V. Sharma, J. Kaur, V. Kumar, S. Mahajan, N. Kumar and S. S. Chimni, Tetrahedron, 2014, 70, 7044–7049 CrossRef CAS; (c) Y.-M. Cao, F.-F. Shen, F.-T. Zhang, J.-L. Zhang and R. Wang, Angew. Chem., Int. Ed., 2014, 53, 1862–1866 CrossRef CAS PubMed; (d) W.-Y. Han, J.-Q. Zhao, Z.-J. Wu, X.-M. Zhang and W.-C. Yuan, J. Org. Chem., 2013, 78, 10541–10547 CrossRef CAS PubMed; (e) P. B. Thorat, S. V. Goswami, R. L. Magar, B. R. Patil and S. R. Bhusare, Eur. J. Org. Chem., 2013, 5509–5516 CrossRef CAS; (f) C. B. Tripathi, S. Kayal and S. Mukherjee, Org. Lett., 2012, 14, 3296–3299 CrossRef CAS PubMed; (g) X. Cheng, R. Goddard, G. Buth and B. List, Angew. Chem., Int. Ed., 2008, 47, 5079–5081 CrossRef CAS PubMed; (h) J. P. Abell and H. Yamamoto, J. Am. Chem. Soc., 2008, 130, 10521–10523 CrossRef CAS PubMed; (i) B. Saito, H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 1978–1986 CrossRef CAS PubMed; (j) D. Pettersen, M. Marcolini, L. Bernardi, F. Fini, R. P. Herrera, V. Sgarzani and A. Ricci, J. Org. Chem., 2006, 71, 6269–6272 CrossRef CAS PubMed; (k) T. Akiyama, H. Morita, J. Itoh and K. Fuchibe, Org. Lett., 2005, 7, 2583–2585 CrossRef CAS PubMed; (l) G. D. Joly and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 4102–4103 CrossRef CAS PubMed; (m) S. Kobayashi, H. Kiyohara, Y. Nakamura and R. Matsubara, J. Am. Chem. Soc., 2004, 126, 6558–6559 CrossRef CAS PubMed; (n) H. Sasai, S. Arai, Y. Tahara and M. Shibasaki, J. Org. Chem., 1995, 60, 6656–6657 CrossRef CAS.
- For the non-enantioselective synthesis of cyclic α-aminophosphonates, see: (a) G. Hu, W. Chen, D. Ma, Y. Zhang, P. Xu, Y. Gao and Y. Zhao, J. Org. Chem., 2016, 81, 1704–1711 CrossRef CAS PubMed; (b) W.-J. Yoo and S. Kobayashi, Green Chem., 2014, 16, 2438–2442 RSC; (c) J. Dhineshkumar, M. Lamani, K. Alagiri and K. R. Prabhu, Org. Lett., 2013, 15, 1092–1095 CrossRef CAS PubMed; (d) J. Xie, H. Li, Q. Xue, Y. Cheng and C. Zhu, Adv. Synth. Catal., 2012, 354, 1646–1650 CrossRef CAS; (e) M. Rueping, S. Zhu and R. M. Koenigs, Chem. Commun., 2011, 47, 8679–8681 RSC.
- (a) Ö. Doğan, H. Babiz, A. G. Gözen and S. Budak, Eur. J. Med. Chem., 2011, 46, 2485–2489 CrossRef PubMed; (b) A. K. Bhattacharya, K. C. Rana, D. S. Raut, V. P. Mhaindarkar and M. I. Khan, Org. Biomol. Chem., 2011, 9, 5407–5413 RSC; (c) B. Boduszek, J. Oleksyszyn, C.-M. Kam, J. Selzler, R. E. Smith and J. C. Powers, J. Med. Chem., 1994, 37, 3969–3976 CrossRef CAS PubMed.
- For selected reviews, see: (a) P. S. Bhaduri and H. Li, Synlett, 2012, 23, 1108–1131 CrossRef; (b) V. A. Alfonsov, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 2637–2644 CrossRef CAS; (c) P. Merino, E. Marqués-López and R. P. Herrera, Adv. Synth. Catal., 2008, 350, 1195–1208 CrossRef CAS.
- (a) I. Schlemminger, Y. Saida, H. Gröger, W. Maison, N. Durot, H. Sasai, M. Shibasaki and J. Martens, J. Org. Chem., 2000, 65, 4818–4825 CrossRef CAS PubMed; (b) H. Gröger, Y. Saida, H. Sasai, K. Yamaguchi, J. Martens and M. Shibasaki, J. Am. Chem. Soc., 1998, 120, 3089–3103 CrossRef; (c) H. Gröger, Y. Saida, S. Arai, J. Martens, H. Sasai and M. Shibasaki, Tetrahedron Lett., 1996, 37, 9291–9292 CrossRef. For a more recent example, see: (d) H. Xie, A. Song, X. Zhang, X. Chen, H. Li, C. Sheng and W. Wang, Chem. Commun., 2013, 49, 928–930 RSC.
- For a cycloaddition approach to cyclic α-aminophosphonates, see: Y. Yamashita, L. C. Nam, M. J. Dutton, S. Yoshimoto and S. Kobayashi, Chem. Commun., 2015, 51, 17064–17067 RSC.
- (a) W. J. Pietro and W. J. Hehre, J. Am. Chem. Soc., 1982, 104, 3594–3595 CrossRef CAS; (b) G. O. Doak and L. D. Freedman, Chem. Rev., 1961, 61, 31–44 CrossRef CAS.
- (a) V. S. Abramov, Dokl. Akad. Nauk SSSR, 1954, 95, 991–992 CAS; (b) V. S. Abramov, Dokl. Akad. Nauk SSSR, 1950, 73, 487–489 CAS.
- For a review, see: (a) L. Woźniak and J. Chojnowski, Tetrahedron, 1989, 45, 2465–2524 CrossRef. For selected examples, see: (b) E. E. Korshin and O. K. Pozdeev, Tetrahedron, 2013, 69, 11109–11115 CrossRef CAS; (c) Z.-H. Cai, G.-F. Du, L. He, C.-Z. Gu and B. Dai, Synthesis, 2011, 2073–2078 CAS; (d) A. D. Blieck, K. G. R. Masschelein, F. Dhaene, E. Rozycka-Sokolowska, B. Marciniak, J. Drabowicz and C. V. Stevens, Chem. Commun., 2010, 46, 258–260 RSC; (e) B. Das, P. Balasubramanyam, M. Krishnaiah, B. Veeranjaneyulu and G. C. Reddy, J. Org. Chem., 2009, 74, 4393–4395 CrossRef CAS PubMed; (f) A. A. Prishchenko, M. V. Livantsov, O. P. Novikova, L. I. Livantsova and V. S. Petrosyan, Heteroat. Chem., 2008, 19, 352–359 CrossRef CAS; (g) D. Liotta, U. Sunay and S. Ginsberg, J. Org. Chem., 1982, 47, 2227–2229 CrossRef CAS.
- J. Guin, Q. Wang, M. van Gemmeren and B. List, Angew. Chem., Int. Ed., 2015, 54, 355–358 CrossRef CAS PubMed.
- (a) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558–2573 CrossRef CAS PubMed; (b) Q. Ding, X. Zhou and R. Fan, Org. Biomol. Chem., 2014, 12, 4807–4815 RSC; (c) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662–12686 CrossRef PubMed.
- (a) A. Reissert, Ber. Dtsch. Chem. Ges., 1905, 38, 1603–1614 CrossRef CAS. For a review, see: (b) M. Ahamed and M. H. Todd, Eur. J. Org. Chem., 2010, 5935–5942 CrossRef CAS.
- For reviews on anion-binding catalysis, see: (a) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed; (b) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed; (c) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef CAS PubMed; (d) Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC. For preliminary propositions on anion-binding by thiourea in catalytic reactions, see: (e) M. Kotke and P. R. Schreiner, Synthesis, 2007, 779–790 CAS; (f) M. Kotke and P. R. Schreiner, Tetrahedron, 2006, 62, 434–439 CrossRef CAS.
- For selected examples, see: (a) O. G. Mancheño, S. Asmus, M. Zurro and T. Fischer, Angew. Chem., Int. Ed., 2015, 54, 8823–8827 CrossRef PubMed; (b) M. Zurro, S. Asmus, S. Beckendorf, C. Mück-Lichtenfeld and O. G. Mancheño, J. Am. Chem. Soc., 2014, 136, 13999–14002 CrossRef CAS PubMed; (c) C. S. Yeung, R. E. Ziegler, J. A. Porco Jr and E. N. Jacobsen, J. Am. Chem. Soc., 2014, 136, 13614–13617 CrossRef CAS PubMed; (d) A. Berkessel, S. Das, D. Pekel and J.-M. Neudörfl, Angew. Chem., Int. Ed., 2014, 53, 11660–11664 CrossRef CAS PubMed; (e) A. G. Schafer, J. M. Wieting, T. J. Fisher and A. E. Mattson, Angew. Chem., Int. Ed., 2013, 52, 11321–11324 CrossRef CAS PubMed; (f) V. Kumar and S. Mukherjee, Chem. Commun., 2013, 49, 11203–11205 RSC; (g) C. K. De, N. Mittal and D. Seidel, J. Am. Chem. Soc., 2011, 133, 16802–16805 CrossRef CAS PubMed; (h) E. G. Klauber, C. K. De, T. K. Shah and D. Seidel, J. Am. Chem. Soc., 2010, 132, 13624–13626 CrossRef CAS PubMed; (i) E. A. Peterson and E. N. Jacobsen, Angew. Chem., Int. Ed., 2009, 48, 6328–6331 CrossRef CAS PubMed; (j) S. E. Reisman, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 7198–7199 CrossRef CAS PubMed; (k) I. T. Raheem, P. S. Thiara, E. A. Peterson and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 13404–13405 CrossRef CAS PubMed; (l) M. S. Taylor, N. Tokunaga and E. N. Jacobsen, Angew. Chem., Int. Ed., 2005, 44, 6700–6704 CrossRef CAS PubMed.
- Seidel et al. has reported an enantioselective dearomatization of isoquinolines through the addition of O-acyled azlactones using a combination of Lewis base and chiral anion-binding catalysis. See ref. 17g.
- See the ESI† for details.
- S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2009, 131, 15358–15374 CrossRef CAS PubMed . Also see: ref. 17g.
- L. S. Aitken, N. R. Arezki, A. Dell’Isola and A. J. A. Cobb, Synthesis, 2013, 45, 2627–2648 CrossRef CAS.
- CCDC 1476699 contains the crystallographic data for 4w.
- M. Valluri, T. Mineno, R. M. Hindupur and M. A. Avery, Tetrahedron Lett., 2001, 42, 7153–7154 CrossRef CAS.
- L. Mengozzi, A. Gualandi and P. G. Cozzi, Chem. Sci., 2014, 4, 3915–3921 RSC.
- (a) D. Redmore, US Pat. 3888627, 1975; (b) D. Redmore, US Pat. 3888626, 1975.
- The aromatic stabilization energy of quinoline and isoquinoline are 198 and 143 kJ mol−1, respectively. See S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, 2004 Search PubMed.
- A single experiment with pyridine as the substrate under the conditions shown in Table 2 revealed no reaction with silyl phosphite 2b, even at 25 °C.
- J. M. Wieting, T. J. Fisher, A. G. Schafer, M. D. Visco, J. C. Gallucci and A. E. Mattson, Eur. J. Org. Chem., 2015, 525–533 CrossRef CAS . Also see ref. 17k.
- The background reaction between 1a, 2a and 3 in CH2Cl2 at −80 °C showed 48% conversion after 48 h.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and analytical data. CCDC 1476699 (4w). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc02466a |
| This journal is © The Royal Society of Chemistry 2016 |