
Advancements in the stability of perovskite solar cells: degradation mechanisms and improvement approaches
Abstract
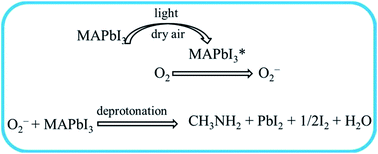
Recently, organic metal halide perovskites have emerged as one of the most promising photoactive materials in the field of photovoltaics. Over the past six years, the efficiency of perovskite solar cells (PSCs) has increased from initially 3.8% to 20.8% through the optimization of perovskite film fabrication and device architecture. However, perovskites suffer inherent instability and degrade rapidly under different exposures (water, oxygen, UV-light irradiation and high temperature), limiting their further application to commercialization. Herein, several studies have been carried out to resolve this problem to some extent. In this paper, possible degradation mechanisms are summarized in order to help us understand the process of perovskite decomposition. Moreover, we also systematically outline and classify the approaches to improve the long-term stability of perovskite devices.
 Please wait while we load your content...
Please wait while we load your content...

