
Configuration-guided reactions: the case of highly decorated spiro[cyclopropane-1,2′(3′H)-pyrrolo[1,2-b]isoxazole] derivatives en route to polyhydroxyindolizidines†‡
F. M.
Cordero
*,
C.
Vurchio
,
C.
Faggi
and
A.
Brandi
*
Department of Chemistry ‘Ugo Schiff’, University of Firenze, Via della Lastruccia 13, 50019 Sesto Fiorentino, FI, Italy. E-mail: franca.cordero@unifi.it; alberto.brandi@unifi.it
First published on 13th September 2016
Abstract
In the course of studies directed toward the synthesis of 7,8-disubstituted 1,2-dihydroxyindolizidines as analogues of the proapoptotic iminosugar lentiginosine, marked reactivity differences of diastereomeric spiro[cyclopropane-1,2′(3H)-pyrrolo[1,2-b]isoxazolidines] precursors were observed. While one minor diastereoisomer gives the Brandi–Guarna rearrangement to indolizidinone very smoothly, and also easily undergoes fluorination of the primary alcohol, the main diastereoisomer behaves in a rather different way. The thermal rearrangement is less efficient and fluorinating agents mainly induce an unexpected cyclization to a highly strained tetracyclic derivative that, in turn, rearranges to form the uncommon 2-oxa-8-azatricyclo[6.2.1.04,9]undecan-5-one system with good yield. In this last process a remarkable macrocyclic dimer has been isolated that sheds light on an unprecedented different aspect of the Brandi–Guarna rearrangement.
Introduction
(+)-Lentiginosine (1), a natural 1,2-dihydroxyindolizidine, is an inhibitor of fungal amyloglucosidases, ATPase, and chaperone activity of Heat shock protein 90 (Hsp 90).1 The non-natural enantiomer (−)-lentiginosine (2) is a potent proapoptotic agent against tumor cells of different origin with low toxicity toward normal cells (Fig. 1).2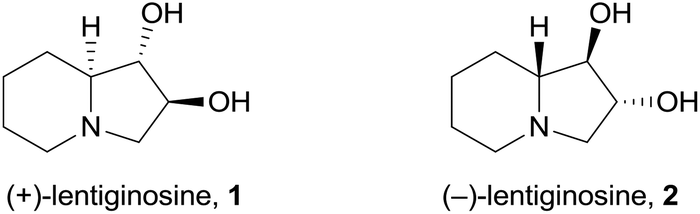 | ||
| Fig. 1 Structure of lentiginosine enantiomers. | ||
In the search for novel lentiginosine analogues endowed with proapoptotic properties, the synthesis of 7,8-disubstituted 1,2-dihydroxyindolizidines 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 was attempted through the convenient domino thermal rearrangement (Brandi–Guarna rearrangement) of suitable 2-spiro[cyclopropane-1,2′(3H)-pyrrolo[1,2-b]isoxazolidines] 4 (Scheme 1).4 Unexpectedly, a marked reactivity difference of diastereomers featuring a 3-hydroxymethyl group on the convex face or on the more crowded concave face of the spiro-fused bicyclic system was found. Herein we describe the synthesis of two new polyhydroxyindolizidine analogues of (−)-lentiginosine and an uncommon 2-oxa-8-azatricyclo[6.2.1.04,9]undecane derivative.
3 was attempted through the convenient domino thermal rearrangement (Brandi–Guarna rearrangement) of suitable 2-spiro[cyclopropane-1,2′(3H)-pyrrolo[1,2-b]isoxazolidines] 4 (Scheme 1).4 Unexpectedly, a marked reactivity difference of diastereomers featuring a 3-hydroxymethyl group on the convex face or on the more crowded concave face of the spiro-fused bicyclic system was found. Herein we describe the synthesis of two new polyhydroxyindolizidine analogues of (−)-lentiginosine and an uncommon 2-oxa-8-azatricyclo[6.2.1.04,9]undecane derivative.
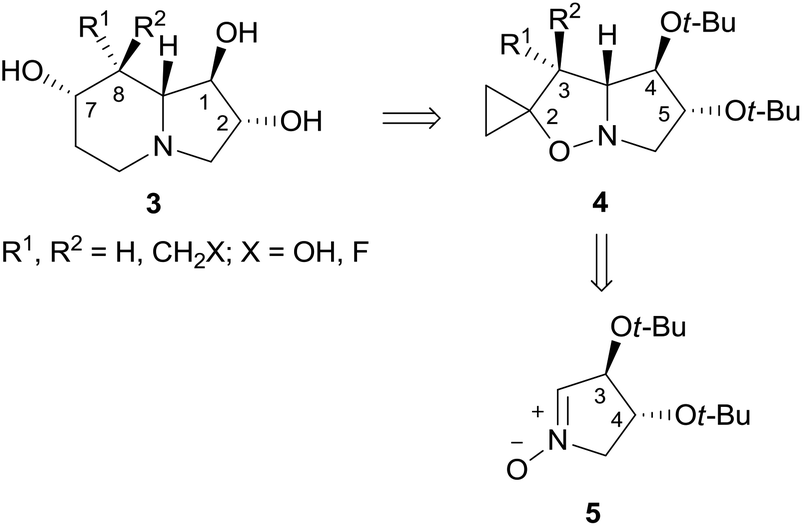 | ||
| Scheme 1 Retrosynthetic pathway to 7,8-disubstituted 1,2-dihydroxyindolizidines 3. | ||
Results and discussion
1,3-Dipolar cycloaddition (1,3-DC) of dihydroxypyrroline N-oxide 5, easily derived from tartaric acid,5 are commonly highly stereo- and regio-selective.6 As nitrone 1,3-DC reactions with alkylidenecyclopropanes exclusively afford thermally stable 4-spirocyclopropane-isoxazolidines, a cyclopropylidene acetate like 6, featuring an electron-withdrawing ester group on the exo-methylene bond, was chosen as a dipolarophile to induce the correct regioselectivity.7,4a Cycloaddition of enantiopure nitrone 5 with ethyl cyclopropylidene acetate 6 in toluene at 30 °C afforded the adducts endo–anti (3-OtBu)-7, exo–anti-8, and endo–syn-9 in a 23:7:1 ratio and 93% overall yield (Scheme 2).8–10 The adduct configuration was assigned by NOE experiments and then validated by the X-ray analysis of the reduced derivatives of 7 and 8 (see below).
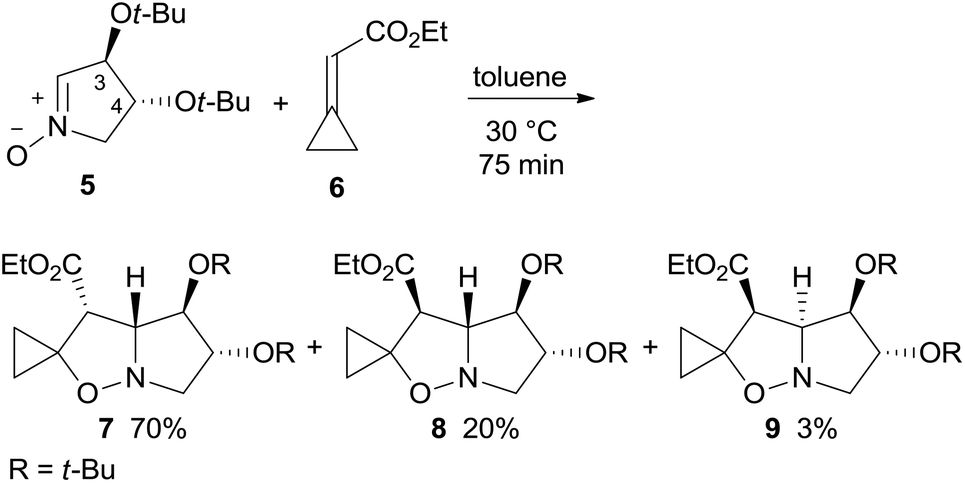 | ||
| Scheme 2 1,3-DC of nitrone 5 and cyclopropylidene acetate 6. | ||
The two major adducts 7 and 8, which have the same relative configuration at the bridgehead carbon atom as lentiginosine, were selectively reduced to the corresponding 3-hydroxymethyl derivatives by diisobutylaluminum hydride (DIBAL) in high yield (Scheme 3). The relative configuration of 10 and 11 was confirmed by a single-crystal X-ray analysis of both the diastereomers (Scheme 3).
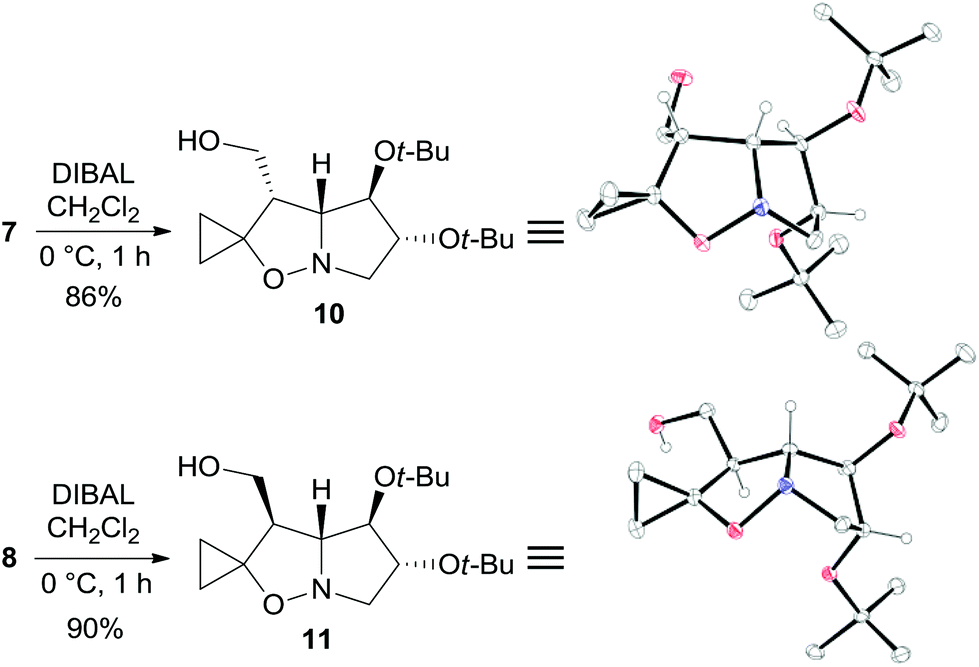 | ||
| Scheme 3 Synthesis and X-ray structures of 10 and 11. Hydrogen atoms of methylene and methyl groups are omitted for clarity. | ||
As fluorinated derivatives play an important role in bioactive compounds,11 it was chosen to introduce the fluorine atom in place of the hydroxyl group in 10 and 11 in advance of carrying out the thermal rearrangement.
Dehydroxyfluorination of the primary alcohol was attempted with (diethylamino)difluorosulfonium tetrafluoroborate (XtalFluorE®)/TEA·3HF.12 Under standard conditions, isoxazolidine 10 featuring the 3-hydroxymethyl group on the concave face of the bicyclic system afforded the unexpected cage compound 12 as the sole product in 73% yield (Scheme 4). When 10 was treated with a less reactive reagent such as perfluoro-1-butanesulfonyl fluoride/1,8-diazabicyclo[5.4.0]undec-7-ene (PBSF/DBU),13 the tetracyclic compound 12 was formed in 38% yield along with the 3-fluoromethyl and 3-methylene derivatives 13 and 14 in 10% and 41% yield, respectively. Epimer 11 afforded the corresponding fluoro derivative 15 in low yield (10%) by treatment with (XtalFluorE®)/TEA·3HF, but with a satisfactory 55% yield with PBSF/DBU (Scheme 4).
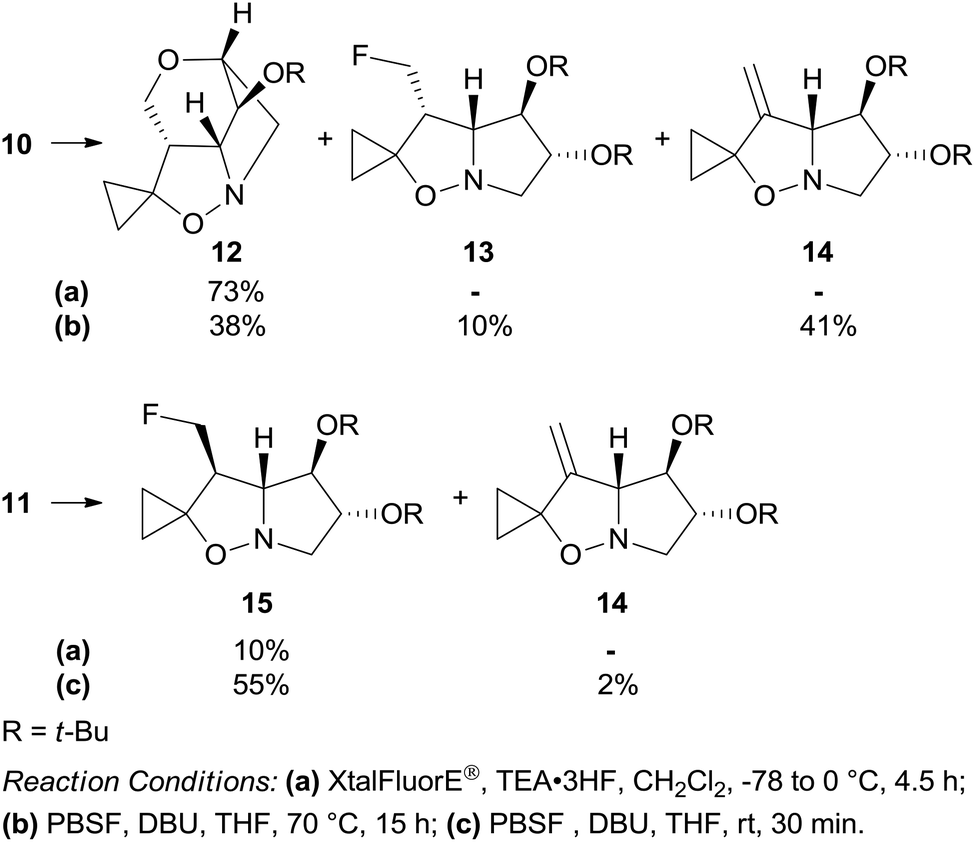 | ||
| Scheme 4 Reaction of diastereomers 10 and 11 with XtalFluorE®/TEA·3HF and PBSF/DBU. | ||
The structure of the unexpected 2,6-dioxa-7-azatricyclo[5.2.1.04,8]decane ring system was confirmed by a single-crystal X-ray analysis of 12 (Scheme 5). Its formation can be rationalized by an intramolecular SN2 reaction where the C-5 tert-butoxy group acts as a nucleophile on the activated primary alcohol in intermediate 16. This process is favored by the close spatial proximity between the two reactive centers as shown by a distance of only 3.4 Å between O-5 and carbinol carbon in the crystal structure of the precursor 10. Analogous fluorinating agent-induced debenzylative cycloetherization is precedented14 but no examples involving a tert-butylether as a nucleophile have been reported up to now.
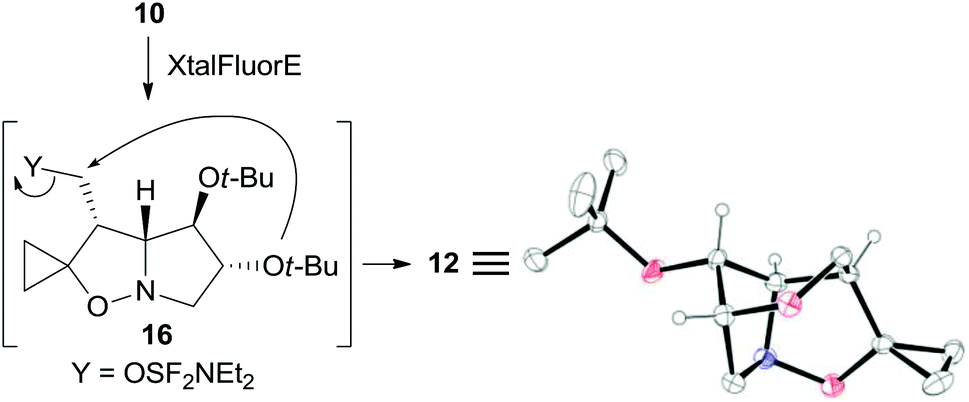 | ||
| Scheme 5 Possible mechanism accounting for the formation of 12 and X-ray structure. Hydrogen atoms of methylene and methyl groups are omitted for clarity. | ||
The next step was the formation of the indolizidine bicyclic system through the Brandi–Guarna thermal rearrangement of 5-spirocyclopropaneisoxazolidines.15 Although this rearrangement reaction has been extensively investigated and applied to the synthesis of various azaheterocycles,4 to date there are no examples of rearrangement of spiro[cyclopropane-1,2′(3H)-pyrrolo[1,2-b]isoxazolidines] monosubstituted on C-3 with a group other than an alkoxycarbonyl moiety.
The rearrangement was carried out in toluene at 170 °C under MW heating (Scheme 6). We were surprised to find a significant difference in rearrangement efficiency between diastereomers 10 and 11. The yield for indolizidinone 17 was significantly lower than that of 18 (25 vs. 71% yield), also highlighting the importance of relative spatial orientation of the substituent on C-3 in this process (Scheme 6).
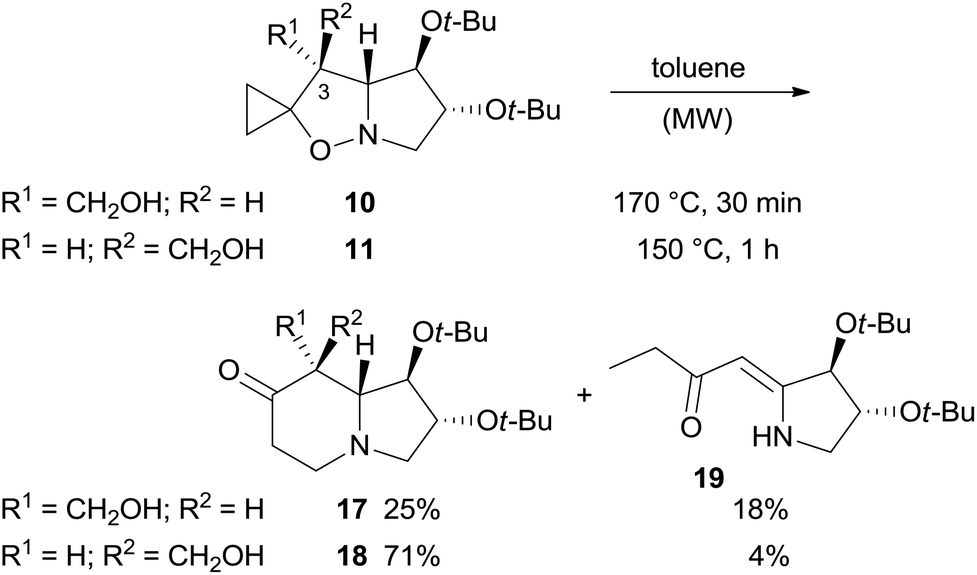 | ||
| Scheme 6 Thermal rearrangement of diastereomers 10 and 11. | ||
Furthermore, enaminone 19 was obtained in place of the expected by-product 22.4 Likely, β-hydroxyketone 21 formed by a 1,5-H shift from diradical intermediate 20, undergoes a retro-aldol reaction with the extrusion of formaldehyde under the reaction conditions (Scheme 7).
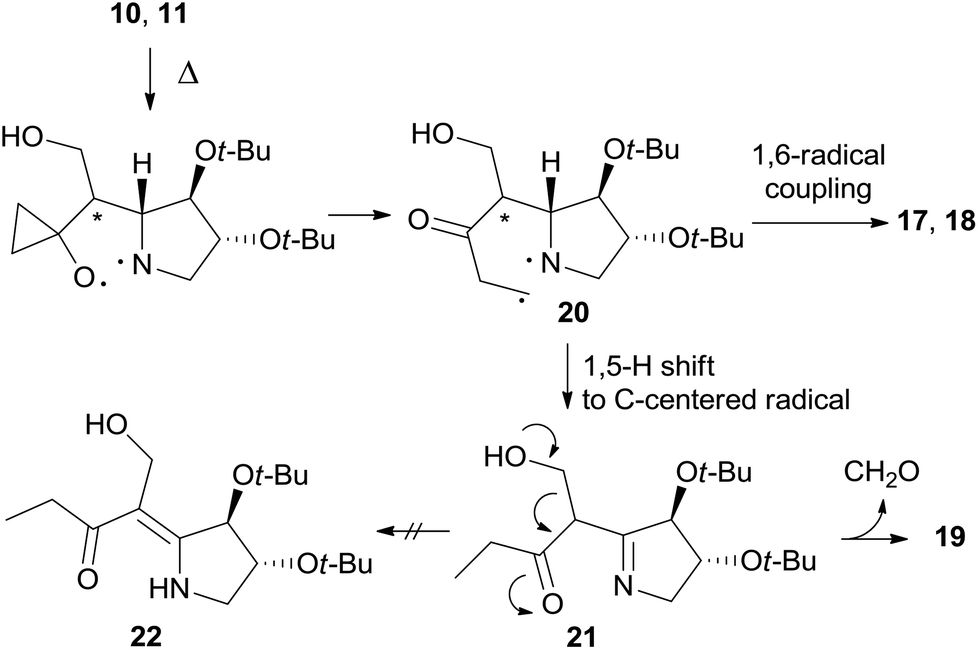 | ||
| Scheme 7 Proposed mechanism for the formation of enaminone 22. | ||
Under the same rearrangement conditions, fluoro derivative 15 affords indolizidinone 23 in 45% yield along with the product of β-elimination 24 whose formation was observed by NMR analysis of the crude reaction mixture (Scheme 8). Likely, ketone 23 is more prone to undergo HF elimination than 15 under the reaction conditions, accordingly, 24 should derive directly from 23 and the overall yield of the thermal rearrangement of fluoro derivative 15 is good (70%) and comparable to that of the corresponding alcohol 11 (71%). With substrate 15 no formation of enaminone by-products was observed. Enone 24 could not be isolated because it undergoes dimerization through hetero-Diels–Alder cycloaddition under high concentration conditions and during chromatography on silica gel. Compound 23 slowly evolves to form the dimer of 24 when stored in a fridge, supporting the hypothesis of the propensity of 23 to eliminate HF (Scheme 8).
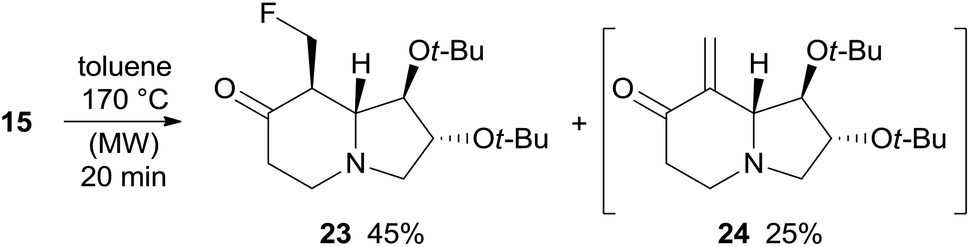 | ||
| Scheme 8 Thermal rearrangement of fluoro derivative 15. | ||
The thermal rearrangement of cage compound 12 was also attempted under standard conditions. The expected 2-oxa-8-azatricyclo[6.2.1.04,9]undecane-5-one 25 was formed in good 55% yield (Scheme 9). Surprisingly, product 26 featuring the double molecular weight of 25 was also isolated in 22% yield. The structures of both 25 and 26 were unambiguously established by single-crystal X-ray analysis (Scheme 9).
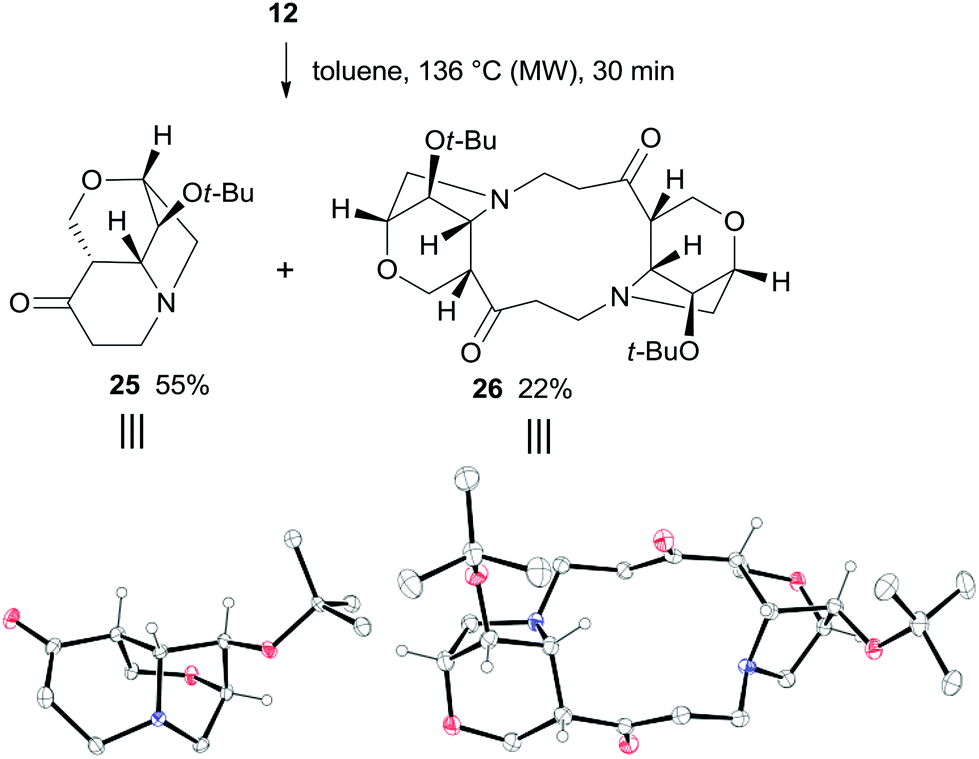 | ||
| Scheme 9 Thermal rearrangement of 12 and X-ray structures of 25 and 26. Hydrogen atoms of methylene and methyl groups are omitted for clarity. | ||
The formation of 26 is an interesting novelty that provides new insights into the Brandi–Guarna rearrangement.4,16 In fact, a macrocyclic dimer such as 26 was never observed in the previous studies of this reaction. We hypothesized that the bicyclic diradical intermediate 27, originated from 12 through the common N–O homolysis of the isoxazolidine ring followed by cyclopropyl opening and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond formation, undergoes a hydrogen-atom abstraction from the carbonyl Cα by the nitrogen-centered radical to give the α,β-unsaturated ketone 29 (Scheme 10). The most common 1,5-H shift occurring in these rearrangements, for example the one leading to compound 21 (Scheme 7), in the rigid intermediate 27 is likely hampered by the lower accessibility of the bridge-head hydrogen α to nitrogen and would lead to a rather strained pyrroline 28. Enone 29, then, can undergo intramolecular Michael addition to pyridinone 2517 or intermolecular head-to-tail cyclization to [1,7]diazacyclododecinedione derivative 26. The presumed intermediate 29, and any open chain dimer, were not observed in the reaction mixture, therefore their formation is only postulated. On the other hand, a double head-to-tail radical coupling of 27, albeit possible, sounds very unlikely due to the short life time of a diradical species such as 27. In addition, a similar behavior has not been observed previously, suggesting a different path of 1,5-H shift for 27.
O double bond formation, undergoes a hydrogen-atom abstraction from the carbonyl Cα by the nitrogen-centered radical to give the α,β-unsaturated ketone 29 (Scheme 10). The most common 1,5-H shift occurring in these rearrangements, for example the one leading to compound 21 (Scheme 7), in the rigid intermediate 27 is likely hampered by the lower accessibility of the bridge-head hydrogen α to nitrogen and would lead to a rather strained pyrroline 28. Enone 29, then, can undergo intramolecular Michael addition to pyridinone 2517 or intermolecular head-to-tail cyclization to [1,7]diazacyclododecinedione derivative 26. The presumed intermediate 29, and any open chain dimer, were not observed in the reaction mixture, therefore their formation is only postulated. On the other hand, a double head-to-tail radical coupling of 27, albeit possible, sounds very unlikely due to the short life time of a diradical species such as 27. In addition, a similar behavior has not been observed previously, suggesting a different path of 1,5-H shift for 27.
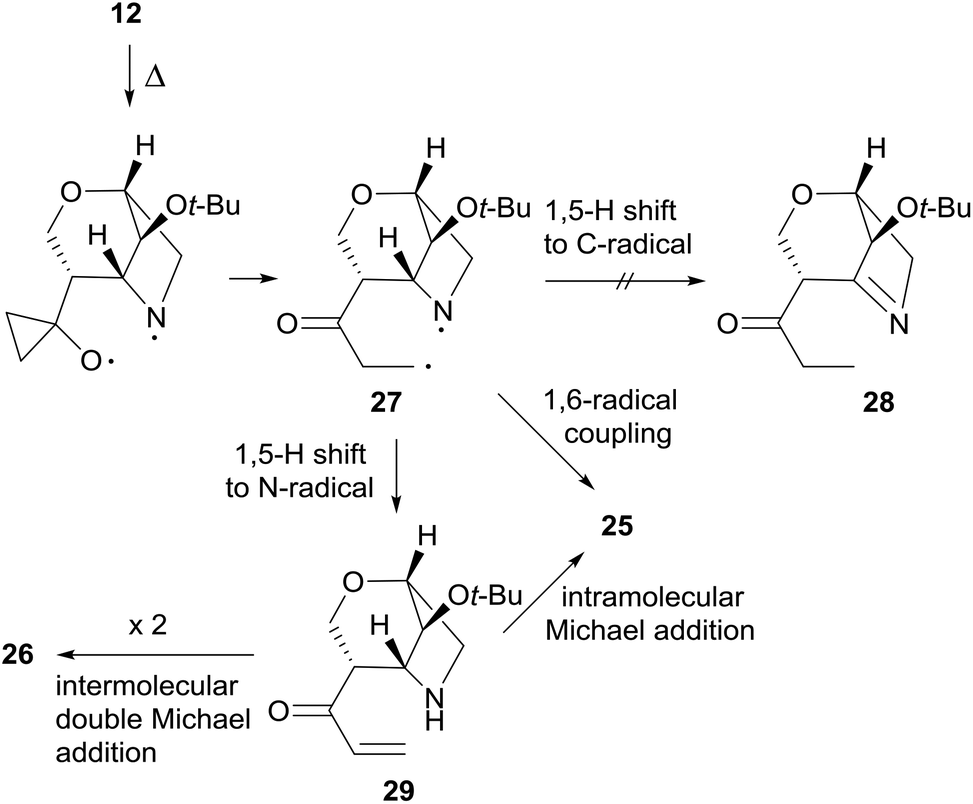 | ||
| Scheme 10 Proposed mechanism of the thermal rearrangement of 12. | ||
Reduction with NaBH4 of cyclic ketones 18, 23, and 25 afforded the corresponding alcohols 30, 32, and 33 in good yield (67–84%) and high diastereoselectivity (Scheme 11). In particular, the attack of the hydride occurred preferentially on the convex face (on the same side of the bridgehead protons) of either the tricyclic or bicyclic systems. Epimeric alcohols were formed in minor amounts (≤10% yield). Deprotection of the tert-butoxy group by treatment with TFA completed the synthesis of lentiginosine analogues 31, 34, and 35 (Scheme 11).
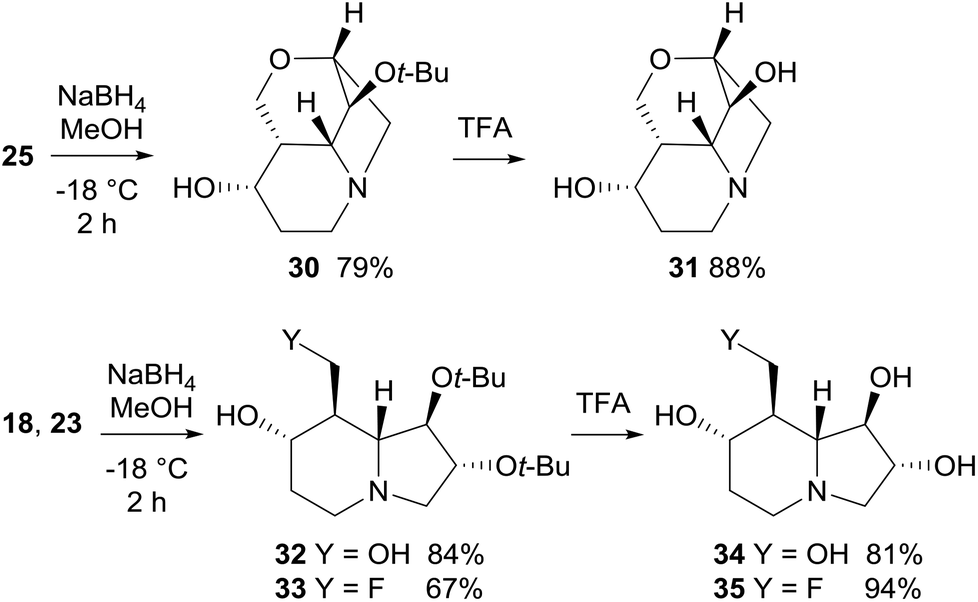 | ||
| Scheme 11 Synthesis of lentiginosine analogues 31, 34, and 35. | ||
Conclusions
The effect on the reactivity of a hydroxymethyl group at C-3 of spiro[cyclopropane-1,2′(3H)-pyrrolo[1,2-b]isoxazolidines] was examined. When this moiety is in the convex face of the spiro-fused bicyclic system it can undergo standard dehydroxyfluorination and thermal rearrangement reactions. In particular, the rearrangement to 8-substituted indolizidinones appears to be favored by the presence of a C-3 alkyl substituent as both the hydroxyl and fluoro derivatives 11 and 15 gave bicyclic rearrangement products in higher yield with respect to C-3 unsubstituted analogues.4 In contrast, a 3-hydroxymethyl group oriented into the more crowded bicyclic concave face partially hampers the thermal rearrangement lowering the indolizidinone yield. Furthermore, activation of the hydroxy group in 10 with fluorinating agents is preferentially followed by the intramolecular attack of the spatially close 6-tert-butoxy group instead of the intermolecular substitution with a fluoride anion. This process allowed the synthesis in good yield of an uncommon hetero-polycyclic system such as 2,6-dioxa-7-azaspiro[tricyclo[5.2.1.04,8]decane-5,1′-cyclopropane] 12 and 2-oxa-8-azatricyclo[6.2.1.04,9]undecane-5-one 25 in turn obtained by the thermal rearrangement of 12 along with a previously never observed macrocyclic byproduct 26. The main indolizidinone derivatives 18, 23, and 25 were then converted into lentiginosine analogues 31, 34, and 35.Biological activities of 31, 34, and 35 and the synthesis of C-8 epimers of 34 and 35 through a different approach are under investigation and will be reported in due course.
Experimental
General
Microwave-assisted reactions were carried out in a CEM Discover™ single-mode microwave reactor with an IR temperature sensor. Rf values refer to TLC on 0.25 mm silica gel plates. Melting points (m.p.) were determined on a Thiele Electrothermal apparatus and on a Bibby–Stuart Scientific apparatus SMP3. Polarimetric measurements were performed on a JASCO DIP-370. NMR spectra were measured on Varian Gemini (1H, 200 MHz,13C, 50 MHz), Varian Mercury (1H, 400 MHz, 13C, 100 MHz) and Varian INOVA (1H, 400 MHz, 13C, 100 MHz) nuclear magnetic resonance spectrometers; CDCl3 was used as the solvent in NMR analyses. NMR data are reported in δ (ppm) from TMS at 25 °C and peak assignments were made on the basis of 1H–1H COSY, HSQC and HMBC experiments. IR spectra were recorded with a Perkin-Elmer Spectrum BX FT-IR System spectrophotometer on CDCl3 solutions. Elemental analyses were performed with a Perkin-Elmer 2400 analyzer and CHN-S Thermo Finnigan Analyzer. MS (ESI) were recorded on a LCQ Fleet Ion Trap Mass Spectrometer with a Surveyor Plus LC System (Thermo Scientific) operating in positive (+ESI) and negative (−ESI) ion mode by direct infusion of a methanolic solution of the sample. Accurate mass spectra were recorded on a LTQ-Orbitrap high-resolution mass spectrometer (Thermo, San Jose, CA, USA), equipped with a conventional ESI source.:1 to Et2O) to afford adducts 7 (1.09 g, 70%), 8 (309 mg, 20%), and 9 (39 mg, 3%) as pale yellow oils.
:1). [α]24D = −135.8 (c = 0.97, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.27 (the A part of an ABX3 system, J = 10.8, 7.2 Hz, 1H, CHHMe), 4.21 (the B part of an ABX3 system, J = 10.8, 7.2 Hz; 1H, CHHMe), 4.17 (dd, J = 6.5, 4.9 Hz, 1H, 4′-H), 4.03 (pseudo dt, J = 10.3, 6.3 Hz, 1H, 5′-H), 3.91 (dd, J = 8.4, 4.9 Hz, 1H, 3a′-H), 3.38 (dd, J = 8.5, 6.1 Hz, 1H, 6′-Ha), 3.30 (dd, J = 10.3, 8.5 Hz, 1H, 6′-Hb), 3.14 (d, J = 8.4 Hz, 1H, 3′-H), 1.31 (t, J = 7.2 Hz, 3H, CH2CH3), 1.16 [s, 9H, C(CH3)3], 1.14 [s, 9H, C(CH3)3], 1.15–1.04 (m, 1H, c-Pr), 1.01–0.89 (m, 2H, c-Pr), 0.64–0.53 (m, 1H, c-Pr) ppm. 13C NMR (CDCl3, 50 MHz): δ = 170.9 (s; CO), 76.7 (d; C-5′), 75.9 (d; C-4′), 73.8 [s; C(CH3)3], 73.5 [s; C(CH3)3], 72.7 (d; C-3a′), 66.0 (s; C-2′), 60.9 (t; CH2CH3), 59.2 (t; C-6′), 53.3 (d; C-3′), 28.6 [q; 3C, C(CH3)3], 28.5 [q; 3C, C(CH3)3], 14.2 (q; CH2CH3), 12.2 (t; c-Pr), 7.9 (t; c-Pr) ppm. IR (CDCl3): v = 2978, 2935, 1732, 1472, 1462, 1365, 1192, 1094 cm−1. MS (ESI) m/z = 356 [M + H]+; 378 [M + Na]+. C19H33NO5 (355.47): calcd C, 64.20; H, 9.36; found C, 64.32; H, 8.97.
:1). [α]24D = +8.29 (c = 0.86, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.18 (the A part of an ABX3 system, J = 10.8, 7.1 Hz, 1H, CHHMe), 4.16 (the B part of an ABX3 system, J = 10.8, 7.1 Hz; 1H, CHHMe), 4.03 (dd, J = 5.6, 3.3 Hz, 1H, 3a′-H), 3.98–3.91 (m, 2H, 5′-H + 4′-H), 3.44 (dd, J = 10.0, 5.5 Hz, 1H, 6′-Ha), 3.26 (d, J = 5.6 Hz, 1H, 3′-H), 3.19–3.12 (m, 1H, 6′-Hb), 1.25 (t, J = 7.1 Hz, 3H, CH2CH3), 1.18 [s, 9H, C(CH3)3], 1.16 [s, 9H, C(CH3)3], 1.24–1.06 (m, 1H, c-Pr), 0.96–0.82 (m, 2H, c-Pr), 0.77–0.66 (m, 1H, c-Pr) ppm. 13C NMR (CDCl3, 50 MHz): δ = 171.5 (s; CO), 80.8 (d; C-4′), 76.2 (d; C-5′), 74.8 (d; C-3a′), 74.03 [s; C(CH3)3], 74.00 [s; C(CH3)3], 64.8 (s; C-2′), 61.0 (t; CH2CH3), 58.8 (t; C-6′), 56.8 (d; C-3′), 28.6 [q; 3C, C(CH3)3], 28.4 [q; 3C, C(CH3)3], 14.5 (t; c-Pr), 14.2 (q; CH2CH3), 4.3 (t; c-Pr) ppm. IR (CDCl3): v = 2978, 2936, 2872, 1734, 1473, 1462, 1366, 1189, 1110 cm−1. MS (ESI) m/z = 356 [M + H]+; 378 [M + Na]+. C19H33NO5 (355.47): calcd C, 64.20; H, 9.36; found C, 63.89; H, 9.40.
:1). [α]25D = −3.22 (c = 0.92, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.55 (pseudo dt, J = 8.7, 7.6 Hz, 1H, 5′-H), 4.27 (pseudo t, J = 8.8 Hz, 1H, 3a′-H), 4.16 (the A part of an ABX3 system, J = 10.9, 7.2 Hz, 1H, CHHMe), 4.12 (the B part of an ABX3 system, J = 10.9, 7.2 Hz; 1H, CHHMe), 4.04 (pseudo t, J = 8.0 Hz, 1H, 4′-H), 3.49 (dd, J = 13.8, 7.4 Hz, 1H, 6′-Ha), 2.96 (d, J = 9.3 Hz, 1H, 3′-H), 2.88 (dd, J = 13.8, 8.7 Hz, 1H, 6′-Hb), 1.28 (t, J = 7.2 Hz, 3H, CH2CH3), 1.15 [s, 9H, C(CH3)3], 1.14 [s, 9H, C(CH3)3], 1.14–1.02 (m, 1H, c-Pr), 0.98–0.80 (m, 2H, c-Pr), 0.63–0.54 (m, 1H, c-Pr) ppm. 13C NMR (CDCl3, 100 MHz): δ = 172.2 (s; CO), 77.2 (d; C-4′), 74.2 [s; 2C, C(CH3)3], 73.5 (d; C-5′), 70.2 (d; C-3a′), 64.6 (s; C-2′), 60.5 (t; CH2CH3), 59.1 (t; C-6′), 53.9 (d; C-3′), 28.5 [q; 3C, C(CH3)3], 28.0 [q; 3C, C(CH3)3], 14.2 (q; CH2CH3), 12.9 (t; c-Pr), 6.7 (t; c-Pr) ppm. IR (CDCl3): v = 2977, 2932, 2872, 1732, 1462, 1365, 1194, 1120 cm−1. MS (ESI) m/z = 356 [M + H]+; 378 [M + Na]+. C19H33NO5 (355.47): calcd C, 64.20; H, 9.36; N, 3.94; found C, 63.83; H, 9.43; N, 3.75.
:1) gave alcohol 10 (745 mg, 86%) as a white solid.
:1). M.p. = 111–113 °C (Et2O). [α]23D = −133.6 (c = 0.84, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.21 (pseudo t, J = 3.2 Hz, 1H, 4′-H), 4.02 (ddd, J = 5.4, 4.4, 3.2 Hz, 1H, 5′-H), 3.89 (dd, J = 9.3, 3.6 Hz, 1H, 3a′-H), 3.83–3.77 (m, 2H, CH2OH), 3.56–3.48 (m, 1H, OH), 3.42 (dd, J = 13.1, 5.4 Hz, 1H, 6′-Ha), 3.21 (dd, J = 13.1, 4.4 Hz, 1H, 6′-Hb), 2.69 (pseudo dt, J = 9.3, 5.0 Hz, 1H, 3′-H), 1.22 [s, 9H, C(CH3)3], 1.20 [s, 9H, C(CH3)3], 1.08–0.98 (m, 1H, c-Pr), 0.93–0.76 (m, 2H, c-Pr), 0.55–0.43 (m, 1H, c-Pr) ppm. 13C NMR (CDCl3, 100 MHz): δ = 79.4 (d; C-5′), 78.6 (d; C-4′), 75.4 (d; C-3a′), 74.8 [s; C(CH3)3], 74.6 [s; C(CH3)3], 63.5 (s; C-2′), 61.1 (t; C-6′), 61.0 (t; CH2-OH), 49.0 (d; C-3), 28.6 [q; 3C, C(CH3)3], 28.3 [q; 3C, C(CH3)3], 10.6 (t; c-Pr), 7.2 (t; c-Pr) ppm. IR (CDCl3): v = 3622, 3365, 2977, 2936,1391, 1366, 1188, 1076 cm−1. MS (ESI) m/z = 314 [M + H]+; 336 [M + Na]+. C17H31NO4 (313.43): calcd C, 65.14; H, 9.97; found C, 64.79; H, 9.60.
:1) gave 11 (200 mg, 90%) as a white solid.
:1). M.p. = 135–137 °C (dec., Et2O). [α]22D = +9.45 (c = 0.51, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 3.98 (pseudo t, J = 5.4 Hz, 1H, 4′-H), 3.89 (pseudo dt, J = 8.4, 5.9 Hz, 1H, 5′-H), 3.75–3.66 (m, 2H, CH2OH), 3.53–3.45 (m, 2H, 3a′-H + 6′-Ha), 3.18 (dd, J = 10.1, 8.8 Hz, 1H, 6′-Hb), 2.51–2.46 (m, 1H, 3′-H), 2.16 (br s, 1H, OH), 1.20 [s, 9H, C(CH3)3], 1.18 [s, 9H, C(CH3)3], 1.10–1.00 (m, 1H, c-Pr), 0.89–0.71 (m, 3H, c-Pr) ppm. 13C NMR (CDCl3, 100 MHz): δ = 80.7 (d; C-4′), 75.7 (d; C-5′), 74.03 [s; C(CH3)3], 73.96 [s; C(CH3)3], 73.4 (d; C-3a′), 64.0 (t; CH2-OH), 63.9 (s; C-2′), 59.3 (t; C-6′), 52.5 (d; C-3′), 29.0 [q; 3C, C(CH3)3], 28.5 [q; 3C, C(CH3)3], 13.4 (t; c-Pr), 1.9 (t; c-Pr) ppm. IR (CDCl3): v = 3626, 2977, 2935, 2878, 1390, 1365, 1191, 1105 cm−1. MS (ESI) m/z = 314 [M + H]+; 336 [M + Na]+. C17H31NO4 (313.43): calcd C, 65.14; H, 9.97; N, 4.47; found C, 64.81; H, 9.99; N, 4.28.
:1) to afford 12 (28.6 mg, 73%) as a white solid.
:1). M.p. = 99–101 °C (i-Pr2O). [α]24D = −14.08 (c = 0.520, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.49–4.46 (m, 1H, 6′-H), 4.06–4.03 (m, 1H, 7′-H), 4.02 (br d, J = 8.1 Hz, 1H, 7a′-H), 3.77 (dd, J = 11.9, 4.2 Hz, 1H, 4′-Ha), 3.56 (dd, J = 14.2, 3.3 Hz, 1H, 8′-Ha), 3.36 (dd, J = 11.9, 1.5 Hz, 1H, 4′-Hb), 3.07 (br d, J = 14.2 Hz, 1H, 8′-Hb), 2.67 (ddd, J = 8.1, 4.2, 1.5 Hz, 1H, 3a′-H), 1.23 [s, 9H, C(CH3)3], 1.04–0.89 (m, 2H, c-Pr), 0.87–0.79 (m, 1H, c-Pr), 0.56–0.50 (m, 1H, c-Pr) ppm. 13C NMR (CDCl3, 100 MHz): δ = 77.5 (d; C-7′), 74.9 [s, C(CH3)3], 71.1 (d, C-6′), 70.8 (d, C-7a′), 67.1 (s, C-3′), 61.7 (t, C-8′), 59.3 (t, C-4′), 48.7 (d, C-3a′), 28.1 [q, 3C, C(CH3)3], 8.9 (t, c-Pr), 8.4 (t, c-Pr) ppm. IR (CDCl3): v = 2977, 2938, 2872, 1365, 1191, 1098 cm−1. MS (ESI) m/z = 240 [M + H]+; 262 [M + Na]+. C13H21NO3 (239.31): calcd C, 65.25; H, 8.84; N, 5.85; found C, 65.31; H, 9.04; N, 5.60.
:1) gave 14 (19 mg, 41%) as a white solid along with 12 (14 mg, 38%), and 13 (5 mg, 10%).
:1). M.p. = 56.7–57.6 °C. [α]25D = −87.24 (c = 0.58, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.93–4.90 (m, 1H, CHH), 4.61–4.58 (m, 1H, CHH), 4.12–4.07 (m, 1H, 3a′-H), 4.03–3.94 (m, 2H, 4′-H + 5′-H), 3.40 (dd, J = 10.1, 6.2 Hz, 1H, 6′-Ha), 2.99 (pseudo t, J = 9.5 Hz, 1H, 6′-Hb), 1.23 [s, 9H, C(CH3)3], 1.16 [s, 9H, C(CH3)3], 1.25–1.09 (m, 2H, c-Pr), 0.91–0.75 (m, 2H, c-Pr) ppm. 13C NMR (CDCl3, 100 MHz): δ = 153.8 (s, C-3′), 100.0 (t, CH2), 81.8 (d; C-4′), 77.6 (d; C-5′), 75.0 (d, C-3a′), 74.3 [s, C(CH3)3], 73.9 [s, C(CH3)3], 63.5 (s, C-2′), 58.7 (t, C-6′), 28.8 [q, 3C, C(CH3)3], 28.5 [q, 3C, C(CH3)3], 17.9 (t, c-Pr), 11.6 (t, c-Pr) ppm. IR (CDCl3): v = 2979, 2935, 2902, 1814, 1793, 1472, 1382, 1190, 1097 cm−1. MS (ESI) m/z = 296 [M + H]+; 318 [M + Na]+. C17H29NO3 (295.42): calcd C, 69.12; H, 9.89; N, 4.74; found C, 69.45; H, 9.50; N, 4.37.
:1). [α]26D = −88.91 (c = 0.550, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.82 (ddd, JF = 46.6; J = 9.4, 7.6 Hz, 1H, CHHF), 4.59 (ddd, JF = 47.2; J = 9.4, 6.7 Hz, 1H, CHHF), 4.08 (pseudo t, J = 3.3 Hz, 1H, 4′-H), 3.78 (pseudo dt, J = 3.6, 5.9 Hz, 1H, 5′-H), 3.91 (dd, J = 8.9, 3.0 Hz, 1H, 3a′-H), 3.44 (dd, J = 11.7, 5.6 Hz, 1H, 6′-Ha), 3.05 (dd, J = 11.7, 6.1 Hz, 1H, 6′-Hb), 2.81 (pseudo ddt, JF = 16.1; J = 9.1, 7.2 Hz, 1H, 3′-H), 1.20 [s, 9H, C(CH3)3], 1.18 [s, 9H, C(CH3)3], 1.00–0.81 (m, 3H, c-Pr), 0.57–0.49 (m, 1H, c-Pr) ppm. 13C NMR (CDCl3, 100 MHz): δ = 83.6 (dt, CH2-F, JF = 166.8), 78.3 (d, C-5′), 77.0 (d, C-4′), 74.5 (dd, C-3a′, JF = 2.8), 74.2 [s, C(CH3)3], 74.0 [s, C(CH3)3], 64.0 (d, C-2′, JF = 8.9), 61.0 (t, C-6′), 46.6 (dd, C-3, JF = 19.4), 28.5 [q, 3C, C(CH3)3], 28.4 [q, 3C, C(CH3)3], 9.3 (t, c-Pr), 7.5 (t, c-Pr) ppm. IR (CDCl3): v = 2978, 2933, 2002, 2872, 1793, 1472, 1382, 1190, 1097, cm−1. MS (ESI) m/z = 316 [M + H]+; 338 [M + Na]+; 631 [2M + Na]+. C17H30FNO3 (315.42): calcd C, 64.73; H, 9.59; N, 4.44; found C, 64.36; H, 9.51; N, 4.08.
:1) gave 15 (28 mg, 55%) and 14 (1 mg, 2%) as white solids.
:1). M.p. = 43–44 °C. [α]24D = +11.59 (c = 0.635, CHCl3) 1H NMR (CDCl3, 400 MHz): δ = 4.51 (ddd, JF = 41.8; J = 9.1, 7.6 Hz, 1H, CHHF), 4.39 (ddd, JF = 40.7; J = 9.1, 6.8 Hz, 1H, CHHF), 3.97 (pseudo t, J = 5.1 Hz, 1H, 4′-H), 3.89 (pseudo dt, J = 8.3, 5.8 Hz, 1H, 5′-H), 3.48 (dd, J = 10.3, 5.9 Hz, 1H, 6′-Ha), 3.36 (pseudo t, J = 4.2 Hz, 1H, 3a′-H), 3.19 (dd, J = 10.3, 8.4 Hz, 1H, 6′-Hb), 2.70 (pseudo ddt, JF = 14.0; J = 3.9, 7.1 Hz, 1H, 3′-H), 1.20 [s, 9H, C(CH3)3], 1.18 [s, 9H, C(CH3)3], 1.08–1.00 (m, 1H, c-Pr), 0.85–0.72 (m, 3H, c-Pr) ppm. 13C NMR (CDCl3, 100 MHz): δ = 84.0 (dt, CH2-F, JF = 171.0), 80.7 (d, C-4′), 75.8 (d, C-5′), 74.1 [s, C(CH3)3], 74.0 [s, C(CH3)3], 73.0 (dd, C-3a′, JF = 5.5), 63.1 (d, C-2′, JF = 6.6), 59.2 (t, C-6′), 51.1 (dd, C-3, JF = 18.9), 28.9 [q, 3C, C(CH3)3], 28.5 [q, 3C, C(CH3)3], 13.1 (t, c-Pr), 1.7 (t, c-Pr) ppm. IR (CDCl3): v = 2977, 2934, 2873, 1391, 1366, 1190, 1099 cm−1. MS (ESI) m/z = 316 [M + H]+; 338 [M + Na]+; 653 [2M + Na]+. C17H30FNO3 (315.42): calcd C, 64.73; H, 9.59; N, 4.44; found C, 64.64; H, 9.24; N, 4.18.
:1) gave 17 (14 mg, 25%) and 19 (8.8 mg, 18%) as pale yellow oils.
:1). [α]26D = −34.27 (c = 0.4, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.21 (dd, J = 11.6, 3.5 Hz, 1H, CHHOH), 4.06 (dm, J = 5.7 Hz, 1H, 1-H), 3.86 (dm, J = 4.7 Hz, 1H, 2-H), 3.83 (ddd, J = 11.6, 3.1, 0.8 Hz, 1H, CHHOH), 3.28–3.21 (m, 1H, 5-Ha), 2.98 (d, J = 9.8 Hz, 1H, 3-Ha), 2.91–2.80 (m, 1H, 6-Ha), 2.59–2.55 (m, 1H, 8-H), 2.51 (dd, J = 5.5, 3.3 Hz, 1H, 8a-H), 2.48–2.35 (m, 3H, 3-Hb + 5-Hb + 6-Hb), 1.21 (s, 9H, CH3 × 3), 1.17 (s, 9H, CH3 × 3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 210.7 (s, C-7), 79.4 (d, C-1), 78.5 (d, C-2), 74.1 (s, CMe3), 74.0 (s, CMe3), 72.6 (d, C-8a), 62.1 (t, CH2OH), 60.0 (t, C-3), 52.4 (d, C-8), 50.1 (t, C-5), 40.7 (t, C-6), 28.9 (q, 3C, Me × 3), 28.7 (q, 3C, Me × 3) ppm. IR (CDCl3): v = 3405, 2978, 2935, 2873, 1707, 1391, 1366, 1191, 1073, 1042 cm−1. MS (ESI) m/z = 314 [M + H]+; 336 [M + Na]+; 346 [M + MeOH + H]+; 649 [2M + Na]+.
:1). 1H NMR (CDCl3, 400 MHz): δ = 9.38 (br s, NH), 5.19 (s, 1H, HC), 4.45 (d, J = 7.3 Hz, 1H, 3-H), 4.08 (pseudo q, J = 7.1 Hz, 1H, 4-H), 3.63 (ddd, J = 10.4, 7.2, 1.1 Hz, 1H, 5-Ha), 3.23 (dd, J = 10.4, 7.0 Hz, 1H, 5-Hb), 2.32 (q, J = 7.6 Hz, 2H, CH2CH3), 1.29 (s, 9H, Me × 3), 1.20 (s, 9H, Me × 3), 1.09 (t, J = 7.6 Hz, 3H, CH2CH3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 200.4 (s, CO), 165.5 (s, C-2), 88.9 (d, CH), 78.1 (d, C-3), 75.7 (d, C-4), 75.3 (s, CMe3), 74.2 (s, CMe3), 50.7 (7, C-5), 35.1 (t, CH2CH3), 29.0 (q, 3C, Me × 3), 28.4 (q, 3C, Me × 3), 9.9 (q, CH2CH3) ppm. IR (CDCl3): v = 3687, 3618, 3019, 2977, 2936, 2876, 1631, 1549, 1367, 1217, 1191, cm−1. MS (ESI) m/z = 306 [M + Na]+; 589 [2M + Na]+.
:1) gave 18 (41.7 mg, 71%) and 19 (2 mg, 4%) as pale yellow oils.
:1). [α]24D = −38.03 (c = 0.69, CHCl3) 1H NMR (CDCl3, 400 MHz): δ = 4.00–3.96 (m, 2H, CHHOH + 2-H), 3.93 (dd, J = 5.2, 2.3 Hz, 1H, 1-H), 3.79 (dd, J = 11.9, 6.6 Hz, 1H, CHHOH), 3.21 (ddd, J = 11.7, 6.5, 1.7 Hz, 1H, 5-Ha), 3.03 (dd, J = 10.1, 2.4 Hz, 1H, 3-Ha), 2.93–2.76 (br s, OH), 2.79 (dd, J = 10.1, 6.6 Hz, 1H, 3-Hb), 2.74–2.54 (m, 3H, 5-Hb + 6-Ha + 8-H), 2.45 (dd, J = 10.6, 5.3 Hz, 1H, 8a-H), 2.26 (dm, J = 14.1 Hz, 1H, 6-Hb), 1.23 (s, 9H, CH3 × 3), 1.19 (s, 9H, CH3 × 3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 211.7 (s, C-7), 84.6 (d, C-1), 78.8 (d, C-2), 74.8 (s, CMe3), 74.0 (s, CMe3), 68.4 (d, C-8a), 59.6 (t, C-3), 59.0 (t, CH2OH), 53.7 (d, C-8), 49.9 (t, C-5), 39.1 (t, C-6), 28.9 (q, 3C, Me × 3), 28.8 (q, 3C, Me × 3)ppm. IR (CDCl3): v = 3567, 2978, 2935, 2874, 2803, 1697, 1392, 1366, 1189, 1070, cm−1. MS (ESI) m/z = 314 [M + H]+; 336 [M + Na]+; 346 [M + MeOH + H]+. C17H31NO4 (313.43): calcd C, 65.14; H, 9.97; N, 4.47; found C, 64.81; H, 9.59; N, 4.06.
:1) gave 23 (21 mg, 45%) and the dimer of 24 (11 mg, 25%) as pale yellow oils.
Further characterization of 23 was not possible as the compound decomposed quickly after isolation. It was immediately reduced to the stable alcohol (see below for product 33).
:1) gave 25 (30 mg, 55%) and 26 (12.1 mg, 22%) as white solids.
:1). M.p. = 101–103 °C (i-Pr2O). [α]27D = −426.73 (c = 0.29, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.13 (br d, J = 3.1 Hz, 1H, 7-H), 3.84 (dd, J = 12.5, 8.3 Hz, 1H, 5-Ha), 3.84 (br s, 1H, 8-H), 3.59–3.48 (m, 3H, 5-Hb + 9-Ha + 2-Ha), 3.42–3.32 (m, 2H, 8a-H + 2-Hb), 2.86 (br d, J = 11.6 Hz, 1H, 9-Hb), 2.84 (dd, J = 7.9, 2.7 Hz, 1H, 4a-H), 2.70 (ddd, J = 15.2, 8.8, 6.5 Hz, 1H, 3-Ha), 2.26 (br pseudo dt, J = 15.2, 5.3 Hz, 1H, 3-Hb), 1.24 [s, 9H, C(CH3)3] ppm. 13C NMR (CDCl3, 100 MHz): δ = 209.4 (s; C-4), 78.4 (d; C-7), 75.4 (d; C-8), 74.9 [s; C(CH3)3], 64.6 (d; C-8), 59.1 (t; C-5), 54.5 (t; C-9), 53.8 (d; C-4a), 49.1 (t; C-2), 35.6 (t; C-3), 28.3 [q; 3C, C(CH3)3] ppm. IR (CDCl3): v = 2977, 2932, 2870, 1703, 1366, 1190, 1091 cm−1. MS (ESI) m/z = 240 [M + H]+; 272 [M + MeOH + H]+. C13H21NO3 (239.31): calcd C, 65.25; H, 8.84; N, 5.85; found C, 64.85; H, 8.43; N, 5.46.
O × 2), 3.65 (dd, J = 11.3, 5.7 Hz, 2H, CHHCHCO × 2), 3.62 (br s, 2H, CHOtBu × 2), 3.54 (br s, 2H, CHN × 2), 3.32–3.25 (m, 2H, OCHCHHN × 2), 3,28 (d, J = 11.7 Hz, 2H, OCHCHHN × 2), 2.86 (dd, J = 11.7, 3.9 Hz, 2H, OCHCHHN × 2), 2.71–2.64 (m, 2H, CH2CHHN × 2), 2.51–2.41 (m, 4H, CHHCO × 2 + CHCO × 2), 2.32 (br dd, J = 18.4, 6.1 Hz, 2H, CHHCO × 2), 1.25 (s, 18H, CH3 × 6) ppm. 13C NMR (CDCl3, 100 MHz): δ = 208.4 (s, 2C, CO), 79.9 (d, 2C, CHOCH2 × 2), 77.2 (d, 2C, CHOtBu × 2), 74.1 (s, 2C, CMe3), 65.9 (d, 2C, CHN × 2), 60.4 (t, 2C, CH2O × 2), 57.7 (t, 2C, CHCH2N × 2), 54.3 (d, 2C, CHCO × 2), 49.2 (t, 2C, CH2CH2N × 2), 42.9 (t, 2C, CH2CO × 2), 28.4 (q, 6C, CH3 × 6) ppm. IR (CDCl3): v = 2976, 2866, 1711, 1390, 1365, 1192, 1090 cm−1. MS (ESI) m/z = 479 [M + H]+; 501[M + Na]+. C26H42N2O6 (478.62): calcd C, 65.25; H, 8.84; N, 5.85; found C, 65.64; H, 8.52; N, 5.41.
:1 to 5:1) afforded 30 (49 mg, 79%) and the C-7 epimer (6 mg, 10%) as white solids.
:1). M.p. = 130–132 °C. [α]27D = −62.66 (c = 0.670, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.07 (d, J = 4.2 Hz, 1H, 2-H), 3.82 (pseudo dt, J = 11.2, 4.7 Hz, 1H, 7-H), 3.78–3.74 (m, 3H, 9-H + 1-H), 3.33–3.20 (m, 2H, 3-Ha + 5-Ha), 3.14–3.04 (m, 1H, 5-Hb), 2.99 (br s, 1H, 8a-H), 2.83 (d, J = 11.9 Hz, 1H, 3-Hb), 2.41–2.33 (m, 1H, 8-H), 1.91 (pseudo ddt, J = 11.6, 5.5, 12.9 Hz, 1H, 6-Ha), 1.46 (dm, J = 13.2 Hz, 1H, 6-Hb), 1.23 (s, 9H, CH3 × 3) ppm. 13C NMR (CDCl3, 100 MHz): δ = 78.8 (d, C-2), 76.9 (d, C-1), 74.4 (s, CMe3), 70.6 (d, C-7), 63.8 (d, C-8a), 58.3 (t, CH2O), 50.8 (t, C-3), 46.7 (t, C-5), 40.0 (d, C-8), 23.2 (q, 3C, CH3 × 3), 23.6 (t, C-6) ppm. IR (CDCl3): v = 3607, 3352, 2977, 2937, 2865, 1392, 1365, 1241, 1193, 1087 cm−1. MS (ESI) m/z = 242 [M + H]+; 264 [M + Na]+; 505 [2M + Na]+. C13H23NO3 (241.33): calcd C, 64.70; H, 9.61; N, 5.80; found C, 64.45; H, 9.26; N, 5.61.
:1), 32 (33 mg, 84%) as a colorless oil and the C-7 epimer (2 mg, 5%).
:1). [α]28D = −22.21 (c = 0.925, CHCl3). 1H NMR (CD3OD, 400 MHz): δ = 4.07 (dd, J = 11.8, 2.0 Hz, 1H, CHHOH), 3.95 (dd, J = 7.6, 2.3 Hz, 1H, 1-H), 3.91–3.87 (m, 1H, 2-H), 3.80 (dd, J = 8.6, 4.0 Hz, 1H, 1-H), 2.91 (dm, J = 10.0 Hz, 1H, 3-Ha), 2.77 (ddd, J = 11.8, 5.9 Hz, 1H, CHHOH), 3.65 (pseudo dt, J = 4.8, 10.3 Hz, 1H, 7-H), 3.04 (d, J = 10.3 Hz, 1H, 3-Ha), 2.99–2.92 (m, 1H, 5-Ha), 2.45–2.35 (m, 1H, 3-Hb), 2.10–1.98 (m, 1H, 5-Hb), 1.96–1.81 (m, 2H, 6-Ha + 8a-H), 1.74 (pseudo ddt, J = 11.1, 4.5, 12.4 Hz, 1H, 6-Hb), 1.68–1.59 (m, 1H, 8-H), 1.28 (s, 9H, CH3 × 3), 1.21 (s, 9H, CH3 × 3) ppm. 13C NMR (CD3OD, 100 MHz): δ = 84.0 (d, C-1), 78.1 (d, C-2), 75.0 (s, CMe3), 74.0 (s, CMe3), 71.6 (d, C-7), 66.6 (d, C-8a), 61.4 (d, CH2OH), 60.7 (t, C-3), 50.3 (t, C-5), 48.1 (d, C-8), 33.0 (t, C-6), 29.2 (s, 3C, Me × 3), 29.0 (s, 3C, Me × 3) ppm. IR (CDCl3): v = 3619, 3412, 2977, 2944, 2798, 1466, 1391, 1368, 1236, 1188, 1071 cm−1. MS (ESI) m/z = 316 [M + H]+; 338 [M + Na]+; 653 [2M + Na]+. C17H33NO4 (315.45): calcd C, 64.73; H, 10.54; N, 4.44; found C, 64.66; H, 10.53; N, 4.09.
:1), 33 (22 mg, 67%) as a colorless oil and only traces of the C-7 epimer.
:1). [α]28D = −27.50 (c = 1.00, CHCl3). 1H NMR (CDCl3, 400 MHz): δ = 4.93 (ddd, JF = 47.4; J = 9.3, 3.0 Hz, 1H, CHHF), 4.70 (ddd, JF = 47.5; J = 9.3, 4.6 Hz, 1H, CHHF), 3.91–3.86 (m, 2H, 1-H + 2-H), 3.71–3.62 (m, 1H, 7-H), 2.98–2.89 (m, 2H, 3-Ha + 5-Ha), 2.55 (dd, J = 10.0, 6.1 Hz, 1H, 3-Hb), 2.18 (pseudo dt, J = 2.8, 12.1 Hz, 1H, 5-Hb), 2.16–2.00 (br s, OH), 2.04 (dd, J = 10.4, 6.5 Hz, 1H, 8a-H), 1.90–1.69 (m, 3H, 6-H + 8-H) ppm. 13C NMR (CDCl3, 100 MHz): δ = 83.8 (d, C-1), 82.8 (dt, JF = 165.1 Hz, CH2F), 78.4 (d, C-2), 74.5 (s, CMe3), 73.9 (s, CMe3), 70.3 (d, C-7), 65.2 (dd, JF = 4.6 Hz, C-8a), 60.0 (t, C-3), 49.0 (t, C-5), 46.4 (dd, JF = 16.5 Hz, C-8), 32.0 (t, C-6), 29.1 (q, 3C, CH3 × 3), 29.0 (q, 3C, CH3 × 3) ppm. IR (CDCl3): v = 3619, 2978, 2870, 2798, 1466, 1391, 1367, 1237, 1190, 1072 cm−1. MS (ESI) m/z = 318 [M + H]+; 340 [M + Na]+. HRMS (ESI): MH+, found 318.24352. C17H33FNO3 (317.44) requires 318.24390.
:1] afforded 31 (30 mg, 88%) as a white waxy solid.
:1]. [α]22D = −60.51 (c = 0.590, MeOH). 1H NMR (CDCl3, 400 MHz): δ = 4.06 (br d, J = 4.3 Hz, 1H, 2-H), 3.85 (br s, 1H, 1-H), 3.81–3.73 (m, 2H, 7-H + CHHO), 3.68 (dd, J = 12.7, 8.7 Hz, 1H, 7-H + CHHO), 3.19 (dd, J = 12.0, 4.4 Hz, 1H, 3-Ha), 3.20–3.12 (m, 1H, 5-Ha), 3.05 (br d, J = 2.9 Hz, 1H, 8a-H), 2.98 (pseudo dt, J = 4.1, 13.9 Hz, 1H, 5-Hb), 2.89 (d, J = 12.0, 1H, 3-Hb), 2.34–2.26 (m, 1H, 8-H), 1.96 (pseudo ddt, J = 11.6, 5.4, 13.2 Hz, 1H, 6-Ha), 1.47–1.39 (m, 1H, 6-Hb) ppm. 13C NMR (CDCl3, 100 MHz): δ = 79.7 (d, C-2), 77.3 (d, C-1), 71.3 (d, C-7), 64.8 (d, C-8a), 59.5 (t, CH2O), 51.1 (t, C-3), 47.2 (t, C-5), 40.8 (t, C-8) ppm. MS (ESI) m/z = 186 [M + H]+. C9H15NO3 (185.22): calcd C, 58.36; H, 8.16; N, 7.56; found C, 58.72; H, 7.79; N, 7.12.
:2] gave 34 (31 mg, 81%) as a white solid.
:2]. M.p. = 140 °C (dec). [α]25D = +24.085 (c = 0.470, MeOH). 1H NMR (CD3OD, 400 MHz): δ = 4.06 (dd, J = 11.3, 4.0 Hz, 1H, CHHOH), 4.01 (ddd, J = 7.1, 3.4, 1.4 Hz, 1H, 2-H), 3.81 (dd, J = 8.0, 3.4 Hz, 1H, 1-H), 3.59 (dd, J = 11.3, 7.8 Hz, 1H, CHHOH), 3.31–3.24 (m, 1H, 7-H), 2.92 (ddd, J = 11.2, 4.4, 2.5 Hz, 1H, 5-Ha), 2.86 (dm, J = 10.5 Hz, 1H, 3-Ha), 2.53 (dd, J = 10.5, 7.1 Hz, 1H, 3-Hb), 2.06 (ddd, J = 12.6, 11.2, 2.7 Hz, 1H, 5-Hb), 1.93–1.86 (m, 1H, 6-Ha), 1.74 (dd, J = 9.9, 8.1 Hz, 1H, 8a-H), 1.70–1.55 (m, 2H, 6-Hb + 8-H) ppm. 13C NMR (CD3OD, 100 MHz): δ = 84.6 (d, C-1), 78.2 (d, C-2), 73.2 (d, C-8a), 70.9 (d, C-7), 62.9 (t, CH2OH), 62.3 (t, C-3), 51.5 (t + d, 2C, C-5 + C-8), 35.0 (t, C-6) ppm. MS (ESI) m/z = 204 [M + H]+; 226 [M + Na]+; 202 [M − H]−. C9H17NO4 (203.24): calcd C, 53.19; H, 8.43; N, 6.89; found C, 53.5; H, 8.27; N, 6.63.
:1] gave 35 (12 mg, 94%) as a white solid.
:1]. M.p. = 121–122 °C (dec). [α]24D = +16.83 (c = 0.890, MeOH). 1H NMR (CD3OD, 400 MHz): δ = 4.80 (ddd, JF = 47.0; J = 9.2, 2.1 Hz, 1H, CHHF), 4.75 (ddd, JF = 47.3; J = 9.2, 3.3 Hz, 1H, CHHF), 3.96 (ddd, J = 6.4, 2.4, 1.0 Hz, 1H, 2-H), 3.82 (dd, J = 7.7, 2.4 Hz, 1H, 1-H), 3.54 (pseudo dt, J = 4.9, 10.7 Hz, 1H, 7-H), 2.94 (ddd, J = 11.3, 4.4, 2.4 Hz, 1H, 5-Ha), 2.84 (d, J = 10.4 Hz, 1H, 3-Ha), 2.52 (dd, J = 10.4, 6.4 Hz, 1H, 3-Hb), 2.09 (dd, J = 11.3, 2.7 Hz, 1H, 5-Hb), 1.98–1.90 (m, 2H, 6-Ha + 8a-H), 1.72–1.47 (m, 2H, 6-Hb + 8-H) ppm. 13C NMR (CD3OD, 100 MHz): δ = 84.8 (d, C-1), 81.4 (dt, JF = 166.1 Hz, CH2F), 79.6 (d, C-2), 70.2 (d, C-8a), 68.7 (dd, JF = 4.9 Hz, C-7), 61.9 (t, C-3), 51.4 (t, C-5), 50.6 (dd, JF = 17.6 Hz, C-8), 35.0 (t, C-6) ppm. MS (ESI) m/z = 206 [M + H]+; 228 [M + Na]+. HRMS (ESI): MH+, found 206.11823. C9H17FNO3 (205.23) requires 206.11870.
Acknowledgements
The authors thank the Italian Ministry of Education, University and Research (MIUR-Rome), for financial support (PRIN20109Z2XRJ).Notes and references
- (a) I. Pastuszak, R. J. Molyneux, L. F. James and A. D. Elbein, Biochemistry, 1990, 29, 1886–1891 CrossRef CAS PubMed; (b) A. Brandi, S. Cicchi, F. M. Cordero, R. Frignoli, A. Goti, S. Picasso and P. Vogel, J. Org. Chem., 1995, 60, 6806–6812 CrossRef CAS; (c) F. Dal Piaz, A. Vassallo, M. G. Chini, F. M. Cordero, F. Cardona, C. Pisano, G. Bifulco, N. De Tommasi and A. Brandi, PLoS One, 2012, 7, e43316 CAS.
- (a) B. Macchi, A. Minutolo, S. Grelli, F. Cardona, F. M. Cordero, A. Mastino and A. Brandi, Glycobiology, 2010, 20, 500–506 CrossRef CAS PubMed; (b) A. Minutolo, S. Grelli, F. Marino-Merlo, F. M. Cordero, A. Brandi, B. Macchi and A. Mastino, Cell Death Dis., 2012, 3, e358 CrossRef CAS PubMed.
- For two recent reviews on lentiginosine and analogues, see: (a) F. Cardona, A. Goti and A. Brandi, Eur. J. Org. Chem., 2007, 1551–1565 CrossRef CAS; (b) F. M. Cordero, D. Giomi and A. Brandi, Curr. Top. Med. Chem., 2014, 1294–1307 CrossRef CAS.
- (a) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213–1269 CrossRef CAS PubMed; (b) F. M. Cordero, F. De Sarlo and A. Brandi, Monatsh. Chem., 2004, 135, 649–669 CrossRef CAS; (c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317–7420 CrossRef CAS PubMed.
- F. M. Cordero, P. Bonanno, B. B. Khairnar, F. Cardona, A. Brandi, B. Macchi, A. Minutolo, S. Grelli and A. Mastino, ChemPlusChem, 2012, 77, 224–233 CrossRef CAS.
- For selection of some recent examples, see: (a) Ref. 5; ; (b) F. M. Cordero, B. B. Khairnar, P. Bonanno and A. Brandi, Eur. J. Org. Chem., 2013, 4879–4886 CrossRef CAS; (c) F. Cardona, D. Lalli, C. Faggi, A. Goti and A. Brandi, J. Org. Chem., 2008, 73, 1999–2002 CrossRef CAS PubMed; (d) J. Jakowiecki, R. Loska and M. Makosza, J. Org. Chem., 2008, 73, 5436–5441 CrossRef CAS PubMed; (e) V. Mannucci, F. M. Cordero, A. Piperno, G. Romeo and A. Brandi, Tetrahedron: Asymmetry, 2008, 19, 1204–1209 CrossRef CAS.
- A. Brandi, F. M. Cordero, F. De Sarlo, R. Gandolfi, A. Rastelli and M. Bagatti, Tetrahedron, 1992, 48, 3323–3334 CrossRef CAS.
- For 1,3-DC reaction of nitrone 5 with methylenecyclopropane and bicyclopropylidene, see: (a) F. M. Cordero, M. Salvati, F. Pisaneschi and A. Brandi, Eur. J. Org. Chem., 2004, 2205–2213 CrossRef CAS; (b) F. M. Cordero, M. Salvati, C. Vurchio, A. De Meijere and A. Brandi, J. Org. Chem., 2009, 74, 4225–4231 CrossRef CAS PubMed.
- For previous 1,3-DC reactions of pyrroline N-oxides with alkoxymethylenecyclopropanes, see: (a) Ref. 7; ; (b) F. M. Cordero, B. Anichini, A. Goti and A. Brandi, Tetrahedron, 1993, 49, 9867–9876 CrossRef CAS; (c) F. Pisaneschi, M. Piacenti, F. M. Cordero and A. Brandi, Tetrahedron: Asymmetry, 2006, 17, 292–296 CrossRef CAS; (d) F. M. Cordero, C. Vurchio, M. Lumini and A. Brandi, Amino Acids, 2013, 44, 769–780 CrossRef CAS PubMed.
- For the use of 6 as a dipolarophile in azomethine ylide 1,3-DC, see: (a) T.-L. Liu, Z.-L. He, H.-Y. Tao, Y.-P. Cai and C.-J. Wang, Chem. Commun., 2011, 47, 2616–2618 RSC; (b) T. Liu, Q. Li, Z. He, J. Zhang and C. Wang, Chin. J. Catal., 2015, 36, 68–77 CrossRef CAS.
- (a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed; (b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- (a) F. Beaulieu, L.-P. Beauregard, G. Courchesne, M. Couturier, F. LaFlamme and A. L'Heureux, Org. Lett., 2009, 11, 5050–5053 CrossRef CAS PubMed; (b) A. L'Heureux, F. Beaulieu, C. Bennett, D. R. Bill, S. Clayton, F. LaFlamme, M. Mirmehrabi, S. Tadayon, D. Tovell and M. Couturier, J. Org. Chem., 2010, 75, 3401–3411 CrossRef PubMed; (c) N. Al-Maharik and D. O'Hagan, Aldrichimica Acta, 2011, 44, 65–74 CAS.
- (a) A. Bennua-Skalmowski and H. Vorbrüggen, Tetrahedron Lett., 1995, 36, 2611–2614 CrossRef; (b) H. Vorbrüggen, Synthesis, 2008, 1165–1174 CrossRef.
- For some recent examples, see: (a) Y.-X. Li, Y. Shimada, K. Sato, A. Kato, W. Zhang, Y.-M. Jia, G. W. J. Fleet, M. Xiao and C.-Y. Yu, Org. Lett., 2015, 17, 716–719 CrossRef CAS PubMed; (b) A. Tikad and S. P. Vincent, Eur. J. Org. Chem., 2013, 7593–7603 CrossRef CAS; (c) D. Waschke, Y. Leshch, J. Thimm, U. Himmelreich and J. Thiem, Eur. J. Org. Chem., 2012, 948–959 CrossRef CAS; (d) T. Tashiro, R. Nakagawa, S. Inoue, M. Shiozaki, H. Watarai, M. Taniguchi and K. Mori, Tetrahedron Lett., 2008, 49, 6827–6830 CrossRef CAS; (e) V. Subramaniam, S. S. Gurcha, G. S. Besra and T. L. Lowary, Tetrahedron: Asymmetry, 2005, 16, 553–567 CrossRef CAS; (f) S.-K. Chung, Y.-U. Kwon, Y.-H. Ahn, T.-H. Jeong and Y.-T. Chang, Bull. Korean Chem. Soc., 2000, 21, 274–276 CAS.
- (a) A. Hassner and I. Namboothiri, Organic Syntheses Based on Name Reactions, Elsevier, 3rd edn, 2012, p. 60 Search PubMed; (b) Z. Wang, Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons, Inc., New York, 2010, pp. 515–520 Search PubMed.
- E. Ochoa, M. Mann, D. Sperling and J. Fabian, Eur. J. Org. Chem., 2001, 4223–4231 CrossRef CAS.
- For a similar intramolecular Michael addition to pyridinones, see: J. Revuelta, S. Cicchi and A. Brandi, J. Org. Chem., 2005, 70, 5636–5642 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Professor Barry M. Trost on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic data of compounds 10, 11, 12, 25 and 26, and NMR spectra of the new compounds. CCDC 1496575–1496579. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00410e |
| This journal is © the Partner Organisations 2016 |