DOI:
10.1039/C6QO00360E
(Research Article)
Org. Chem. Front., 2016,
3, 1661-1667
3,3-Dibromo-2-trifluoromethyl acrylic acid ethyl ester: a versatile platform for the stereoselective preparation of functionalized-α-trifluoromethyl α,β-unsaturated lactones and trifluoromethyl pyrazolinones †
Received
14th July 2016
, Accepted 14th September 2016
First published on 27th September 2016
Abstract
We herein describe a method for synthetic routes to multi-functionalized-α-trifluoromethyl α,β-unsaturated lactones and trifluoromethyl pyrazolinones. This involves a tandem stereoselective functionalization of 3,3-dibromo-2-trifluoromethyl acrylic acid ethyl ester and intramolecular cyclization reaction to afford precursors for a Suzuki–Miyaura cross-coupling reaction with arylboronic acids.
Introduction
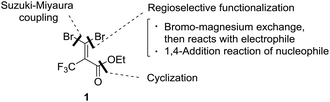
Trifluoromethyl heterocycles have been an important motif of pharmaceutical drugs and agrochemicals because the presence of a CF3 group can cause improved metabolic stability, lipophilicity and bioavailability.1 Nowadays, numerous CF3 substituted heterocycle-containing pharmaceuticals on the market can be witnessed,2 with examples such as trifluridine,3 efavirenz,4 celecoxib5 and mefloquin (Fig. 1).6 Over the past decades, there has been increasing interest in the development of methods for the efficient synthesis of such fluorinated heterocyclic molecules as potential biological targets.7 Nevertheless, synthetic methods for accessing an array of CF3-containing heterocycles remain underdeveloped, in particular for nonaromatic heterocycles.8 Considering the difficulty of introducing the CF3 moiety in nonaromatic ring systems,9 divergent synthesis using a simple and readily available CF3-containing precursor to convert into the diverse set of trifluoromethyl heterocyclic compounds may be one of the versatile and straightforward strategies for drug discovery.10 In the divergent synthesis, the design of building blocks to improve compound quality and accelerate drug discovery must be chosen carefully at an early stage. In a previous paper, we disclosed an efficient synthesis of α-hydroxy-α-trifluoromethyl γ-lactams using ethyl trifluoropyruvate and enamines via a tandem aldol condensation and cyclization reaction.11 In addition, we developed an efficient synthesis of a series of bicycle trifluoromethyl pyrazolinone compounds using reactions of 2-aryl-3-chloro-3-trifluoromethyl acrylate.12 Meanwhile, gem-dibromoalkenes have been widely used as important building blocks for organic synthesis because they act as not only coupling partners in transition-metal catalysis but also precursors of metal carbenoid intermediates formed by monometalation with organo-lithium and magnesium.13 Consequently, we envisaged that trifluoropyruvate derived ethyl 3,3-dibromo-2-trifluoromethylacrylate (1) bearing multi-reaction sites toward regioselective functionalization (bromine–magnesium exchange and 1,4-addition reaction), cyclization and a Pd-catalyzed cross-coupling reaction might serve as a suitable precursor for the diverse synthesis of trifluoromethyl heterocycles (Fig. 2).
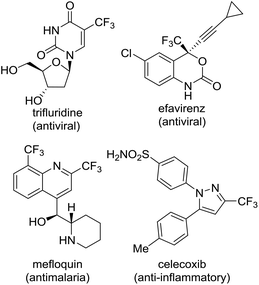 |
| | Fig. 1 Examples of trifluoromethyl heterocycle-containing drugs. | |
 |
| | Fig. 2 Our approach for synthesis of trifluoromethyl heterocycles using a novel building block, gem-dibromoalkene 1. | |
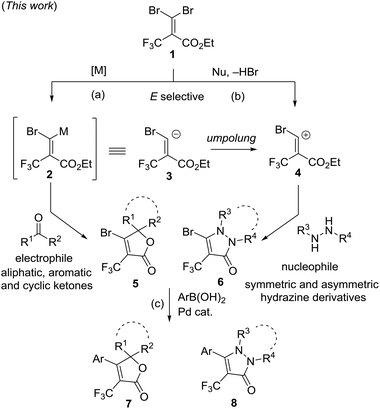
Herein, we would like to report our efforts using two new approaches involving an umpolung strategy, with gem-dibromoalkene 1 as a single precursor, leading to the multi-functionalized-α-trifluoromethyl α,β-unsaturated lactones and trifluoromethyl pyrazolinones. The synthetic routes are summarized in Fig. 3(a), a regioselective magnesium–bromine exchange reaction of 1 with organometals generates the magnesium carbenoid intermediate 2. The intermediate 2 subsequently undergoes a tandem C–C bond formation and cyclization reaction with electrophiles such as aliphatic, aromatic and cyclic ketones, affording α-trifluoromethyl α,β-unsaturated lactones 5. The intermediate 2 can be proposed to be an equivalent to carbanion 3. Another way is a 1,4-addition reaction of hydrazine derivatives to gem-dibromoalkene 1 as a Michael acceptor followed by the elimination of HBr and intramolecular cyclization to provide trifluoromethyl pyrazolinone derivatives 6 (Fig. 3(b)). The precursor 1 is a carbocation of synthon 4 which is also in a relationship with umpolung of 3. (c) Further functionalization by using a Pd-catalyzed arylation of 5 and 6 affords the corresponding products 7 and 8, respectively.
 |
| | Fig. 3 Approach for the diverse synthesis of multi-functionalized trifluoromethyl heterocycles. | |
Results and discussion
Generation and reaction of magnesium carbenoids
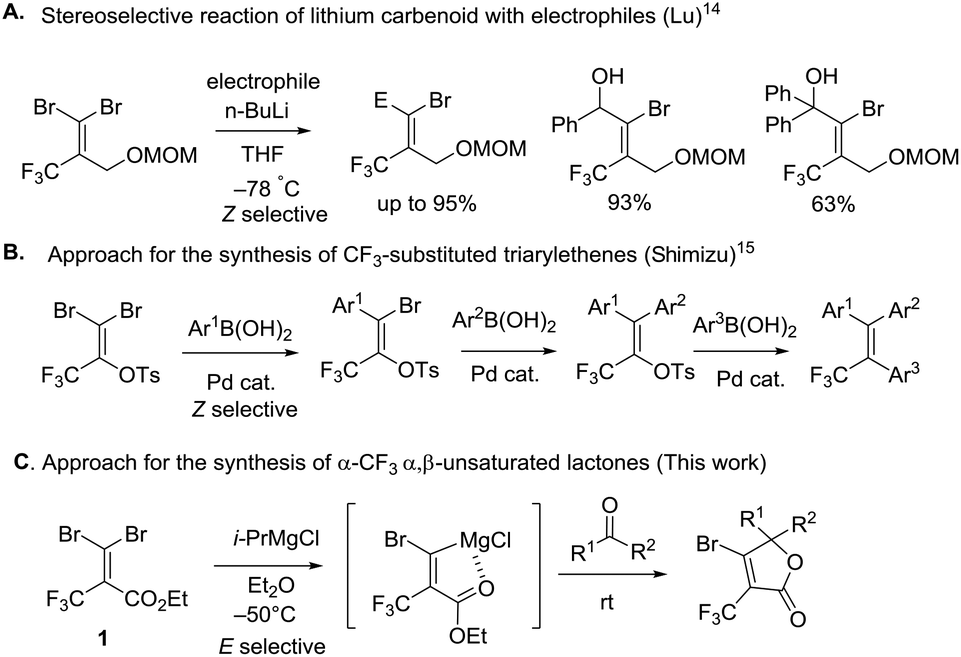
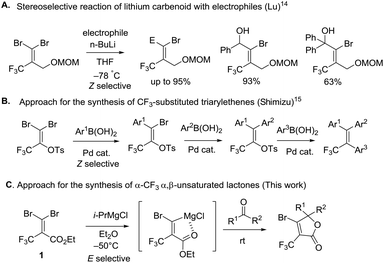
To the best of our knowledge, there are few reports about the selective functionalization of fluorine-containing gem-dibromoalkenes. In 2004, Lu et al. reported that the Z-selective bromine–lithium exchange reaction of O-protected 2-trifluoromethyl-3,3-dibromoallylic alcohol proceeded in THF under thermodynamically controlled conditions to give the corresponding carbenoid intermediate which could react with electrophiles including benzaldehyde and acetophenone, finally furnishing geometrically pure products in high yield (Fig. 4A).14 The stereoselectivity may be controlled by the chelation of a lithium atom with a fluorine atom. Besides, Shimizu and co-workers have engineered Pd-catalyzed stereoselective threefold cross-coupling reactions of 1,1-dibromo-3,3,3-trifluoro-2-tosyloxy-propene, in which the Pd⋯F interaction was proposed to account for the Z selectivity for the first cross-coupling process (Fig. 4B).15 Based on this knowledge, as reactions of fluorine-containing gem-dibromoalkenes usually provide E-products predominantly due to the fluorine–metal interaction, the selective reaction of bromine trans to the trifluoromethyl group is still challenging. In our synthetic plans, the presence of the ester of 1 was needed to aid E selective metal–bromine exchange due to the electrostatic interaction between the cationic magnesium and the partially negatively charged carbonyl oxygen (Fig. 4C). The carbenoid intermediate undergoes a nucleophilic addition to a variety of ketones, followed by intramolecular cyclization accessing β-bromo-α-trifluoromethyl α,β-unsaturated lactones.
 |
| | Fig. 4 Examples of novel building blocks for regioselective functionalization. | |
To begin our study, gem-dibromoalkene 1 was prepared from the reaction of 2,2,2-trifluoromethylpyruvate with carbon tetrabromide and triphenyl phosphine according to a previous report.16 Next, some experiments were performed in order to examine the stereoselectivity and reactivity of the bromine–metal exchange reaction of 1. When i-PrMgCl (1.0 equiv., 2 M in Et2O) was added to a THF solution of 1 at −80 °C, after quenching in methanol a mixture of 11 and 12 was obtained in a ratio of 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (entry 1, Table 1). The ratio of isomers was carefully determined using 1H and 19F NMR of the crude mixtures because it was difficult to isolate products due to their volatility. The stereochemistry for 11 is determined to be in (E)-configuration based on the H–F coupling constant.14,17 Next, we further investigated the impact of temperature, solvent and organometals concerning the reactivity and selectivity. The reaction with i-PrMgCl in THF was performed at −50 °C, and gave several unidentified by-products along with isomers 11 and 12 (E/Z = 58:42, entry 2). When the reaction was performed at 0 °C, the decomposition of 1 was observed. It should be noted that the carbenoid intermediates 9 and 10 were quite unstable. Interestingly, the use of Et2O solvent caused the opposite selectivity and improved conversion, (Z)-product 12 was obtained predominantly in moderate selectivity and yield (E/Z = 27:73, 60% yield). When the reaction was carried out at −50 °C, the selectively was slightly increased to E/Z = 20:80 (entries 3 and 4). When the reaction was conducted in hexane at −50 °C instead of Et2O, both the E/Z ratio and yield were decreased to 2:3 and 20%, respectively (entry 5). In addition, the treatment of 1 with MeMgCl and n-BuLi at −80 °C in Et2O resulted in no conversion (entries 6 and 7). Therefore, these results reveal that the combination of i-PrMgCl and Et2O are a good choice for the generation of carbenoid 10.
:1 (entry 1, Table 1). The ratio of isomers was carefully determined using 1H and 19F NMR of the crude mixtures because it was difficult to isolate products due to their volatility. The stereochemistry for 11 is determined to be in (E)-configuration based on the H–F coupling constant.14,17 Next, we further investigated the impact of temperature, solvent and organometals concerning the reactivity and selectivity. The reaction with i-PrMgCl in THF was performed at −50 °C, and gave several unidentified by-products along with isomers 11 and 12 (E/Z = 58:42, entry 2). When the reaction was performed at 0 °C, the decomposition of 1 was observed. It should be noted that the carbenoid intermediates 9 and 10 were quite unstable. Interestingly, the use of Et2O solvent caused the opposite selectivity and improved conversion, (Z)-product 12 was obtained predominantly in moderate selectivity and yield (E/Z = 27:73, 60% yield). When the reaction was carried out at −50 °C, the selectively was slightly increased to E/Z = 20:80 (entries 3 and 4). When the reaction was conducted in hexane at −50 °C instead of Et2O, both the E/Z ratio and yield were decreased to 2:3 and 20%, respectively (entry 5). In addition, the treatment of 1 with MeMgCl and n-BuLi at −80 °C in Et2O resulted in no conversion (entries 6 and 7). Therefore, these results reveal that the combination of i-PrMgCl and Et2O are a good choice for the generation of carbenoid 10.
Table 1 Bromine–magnesium exchange reaction of 1
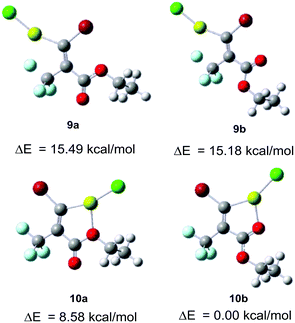
To elucidate this reaction process, we calculated the four initial geometries of the carbenoid intermediates and estimated the stability of 9vs.10 (Fig. 5).18 The relative energies show that the geometry of the 5-membered metallacycle 10b formed by the interaction between magnesium and the carbonyl oxygen is more stable than the other geometries 9a, b and 10a. Relevant reports about the chelation of O with lithium or magnesium to control the selectivity of monometalation of dibromoalkene would support these calculated results.19 In contract to 10b, their relative energies reveal that 9a, b are substantially less stable by ca. 15 kcal mol−1. Therefore, the transformation of 1 to carbenoid intermediate 10b would be favorable, while the transformation to 9a, b would be unfavorable. Nevertheless, the experimentally observed ratio was moderate (E/Z = 1:4, entry 4, Table 1). We deduced from the large differential relative energies between 9 and 10 that (E)-11 was the kinetic product, and no isomerisation occurred. In summary, the presence of a carbonyl group of the ester was crucial for (E)-selectivity by the coordination with magnesium. Furthermore, we assumed that the bromine–magnesium exchange reaction in Et2O of 1 at low temperature (−80 or −50 °C) gave the thermodynamic product 12 as the major isomer and the minor isomer 11 spontaneously. The solvent effect in detail is currently under investigation.
 |
| | Fig. 5 Geometries and relative energies of carbenoid intermediates. | |
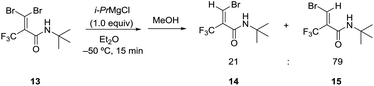
We further investigated the bromine–magnesium exchange reaction of 3,3-dibromo-N-(tert-butyl)-2-(trifluoromethyl)acrylamide (13). As shown in Scheme 1, the treatment of 13 with 1.0 equiv. of i-PrMgCl at −50 °C in Et2O by adding methanol after 15 min gave isomers 14 and 15 with moderate selectivity (E/Z = 21:79). The result was similar to those using 1 (entry 4, Table 1). Thus, the amide group for 13 had a minor influence on the E/Z ratio. However, we could ascertain that the chelation between the carbonyl oxygen and magnesium was supposed to be the significant factor to give the regioselectivity in the bromine–magnesium exchange reaction.
 |
| | Scheme 1 | |
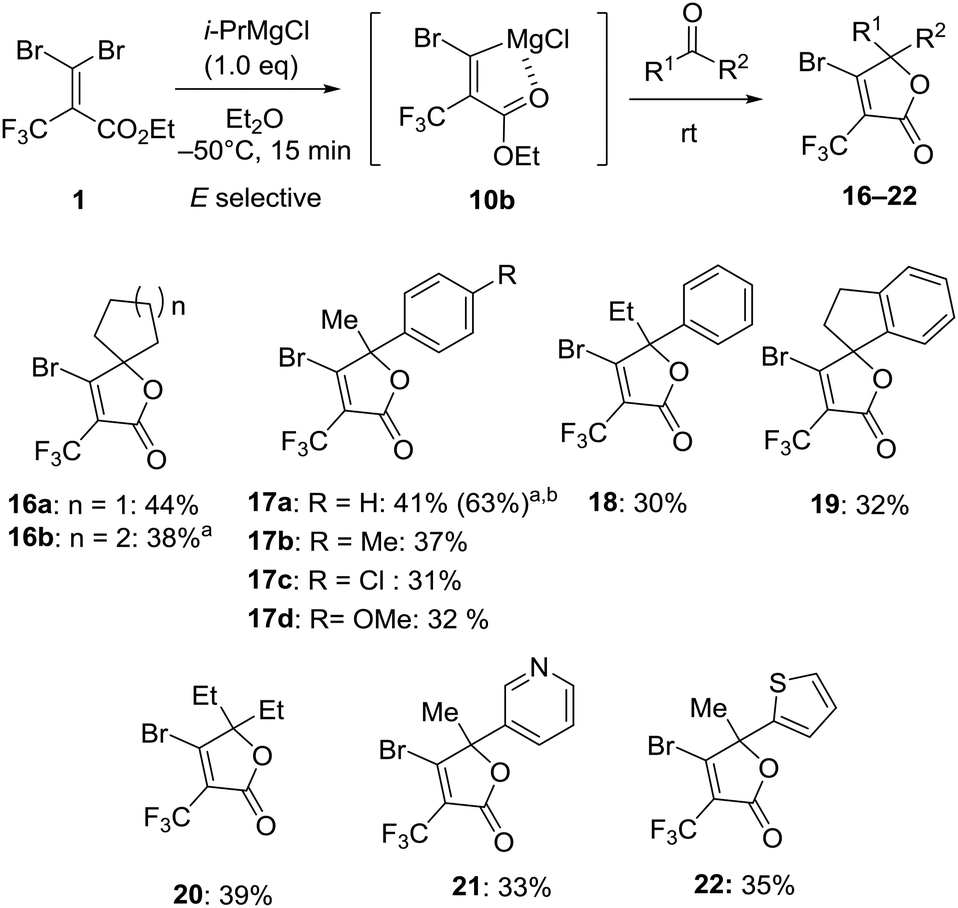
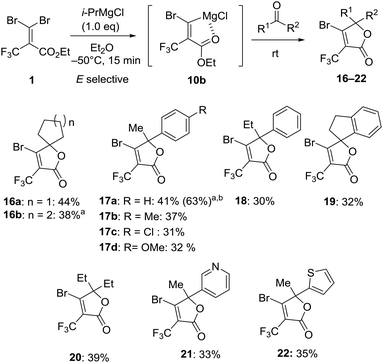
With the optimum conditions in hand, we explored the reactivity of intermediate 10b generated by the treatment of 1 with i-PrMgCl in Et2O at −50 °C for 15 minutes to carbonyl substrates (Fig. 6). In the case of benzaldehyde, the reaction gave benzylalcohol by the reduction by i-PrMgCl. The reaction with acetophenone provided β-bromo-α-trifluoromethyl α,β-unsaturated lactone 16a in 44% isolated yield without any of the (Z)-bromine exchange product, presumably due to concerns over the decomposition of carbenoid intermediate 9 prior to the reaction with the electrophile. Therefore, we demonstrated that the carbenoid 10b could be trapped by a series of ketones like cyclic ketone, acetophenone, 1-indanone and 3-pentanone affording the corresponding lactones 16–20 in 30–44% yield. The transformation showed that some para-substituted groups of acetophenone were tolerated, including methyl, chloro and methoxy groups furnishing the corresponding products 17a–d in 31–41% yield. By using 2.0 equiv. of 1 and i-PrMgCl to acetophenone, the isolated yield for 17a was increased to 63% based on the carbonyl substrate. Otherwise, reactions with 3-acetylpyridine and 2-acetylthiophene also gave trifluoromethyl α,β-unsaturated lactones 21 and 22 in 33% and 35% yield, respectively.
 |
| | Fig. 6 Reaction of magnesium carbenoid 10b with ketones. aReaction was performed at −80 °C. b2.0 equiv. for 1 and i-PrMgCl to ketone was used. | |
Tandem 1,4-addition–cyclization reaction of 1 with hydrazine derivatives
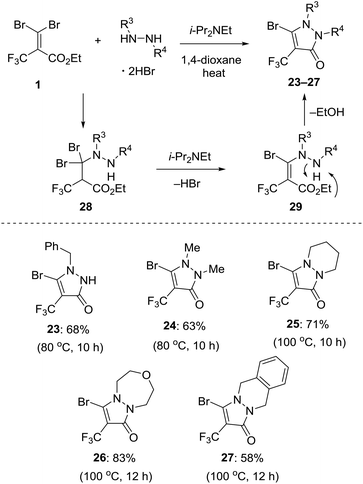
Previously, we disclosed that reaction of gem-dihaloalkene with cyclic hydrazine derivatives in 1,4-dioxane in the presence of an organic base at raised temperature could provide a variety of bicyclic pyrazolinone compounds.12 According to the procedure, we demonstrated the further utility of 1 as a novel building block for highly regioselective synthesis of 5-bromo-4-trifluoromethyl pyrazolinone derivatives. A plausible mechanism of the transformation to pyrazolinone is shown in Fig. 7. Initially, addition of either of two nitrogen atoms in the hydrazine derivatives to the Michael acceptor 1 at the β position forms 1,4-adduct 28, which undergoes regioselective elimination of HBr in the presence of i-Pr2NEt, giving the corresponding acrylate intermediate 29. Finally, the intramolecular cyclization of 29 allows for the formation of the pyrazolinone ring. This tandem 1,4-addition-cyclization reaction with some hydrazine derivative hydrobromide was amenable to give 5-bromo-4-trifluoromethyl pyrazolinones 23–27 in moderate yield with high regioselectivity. The reaction of 1 using monobenzyl hydrazine furnished the single isomer 23 in 68% isolated yield. The reaction of N,N-dimethyl hydrazine allowed for the formation of product 24 in 63% yield. This approach allowed us to use cyclic pyrazolinone derivatives such as hexahydropyridazine and [1,3,5]oxadiazepane. Actually, these reactions successfully gave bicycle products 25 and 26 in 71% and 83% yield, respectively. Otherwise the reaction of 1,2,3,4-tetrahydrophthalazine provided tricyclic compound 27 in 58% yield.
 |
| | Fig. 7 Synthesis of trifluoromethyl pyrazolinones. | |
Pd-catalyzed cross-coupling reactions of bromolactones and bromopyrazolinones
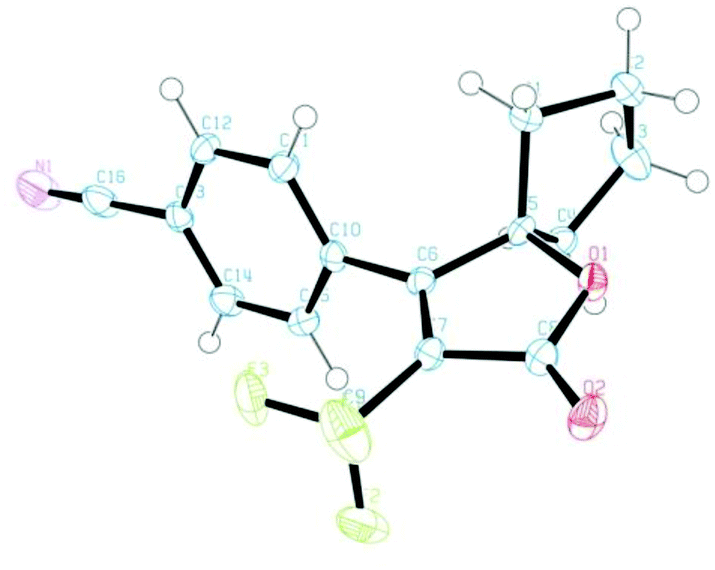
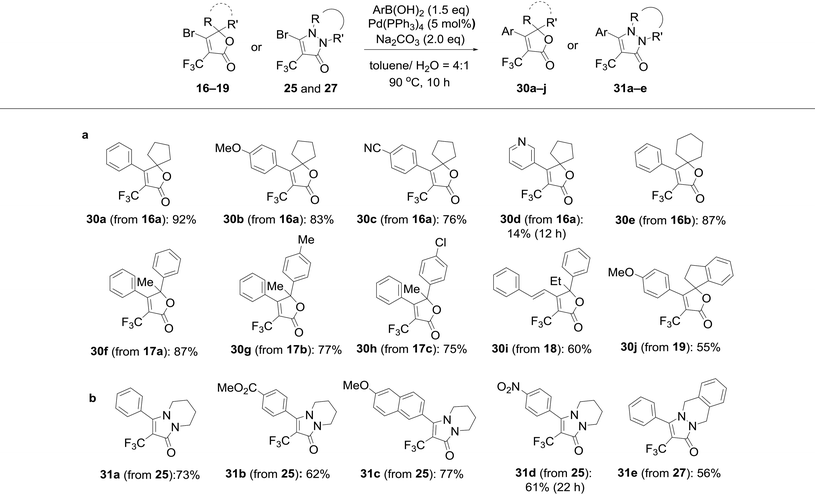
To this end, we executed further transformation of bromolactones 16–19 and bromopyrazolinone 25 and 27 by using a Suzuki–Miyaura cross-coupling reaction with various arylboronic acids (Table 2).13a,20 Bromolactone 16a was chosen as a model substrate for the Pd-catalyzed cross-coupling reaction. Indeed, the reaction of 16a with phenylboronic acid in the presence of palladium(0) tetrakis(triphenylphosphine) (5 mol%) and Na2CO3 (2.0 eq.) at 90 °C for 10 hours in a mixture of toluene and H2O furnished the arylated product 30a in 92% yield. Without optimization of the reaction conditions, other substrates were subjected to Suzuki coupling reactions under the same conditions. As illustrated in Table 2, a wide range of functional groups is tolerated, including methoxy, cyano, ester and nitro groups. The cross-coupling reactions with such arylboronic acids involving (E)-strylboronic acid were successful, and obtained the corresponding arylated products 30b,c,e–j (55–87% yield), but 3-pyridine boronic acid resulted in poor conversion to 30d (14% yield). Additionally, we also performed single-crystal X-ray diffraction analysis for 30c (Fig. 8). The structure of 30c in the solid-state shows that the geometry of the 4-CN phenyl group is unequivocally cis to the CF3 group. In extensive experiments, we explored the reactions of bromopyrazolinones 25 and 27 with the same conditions. Four examples of arylboronic acid were performed and arylated pyrazolinones 31a–e were obtained in up to 77% yield. Use of 27 also was successful, and the reaction of 27 with PhB(OH)2 proceeded smoothly to furnish the polycyclic compound 31e in 56% yield. Consequently, we were pleased to find both bromolactones 16–19 and bromopyrazolinones 25 and 27 to be facile coupling partners for the Pd-catalyzed cross coupling reaction with various arylboronic acids.
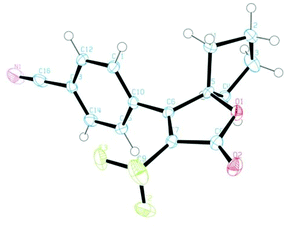 |
| | Fig. 8 ORTEP drawing of the X-ray structure of 30c. Hydrogen atoms are omitted for clarity and ellipsoids displayed at 50% probability. | |
Table 2 Substrate scope for cross-coupling reaction with a variety of arylboronic acids
|
a, Introduction of an aromatic group to bromolactones 16–19. b, Pd-catalyzed cross-coupling reactions of bromopyrazolinones 25 and 27 with arylboronic acids. |
|
Conclusions
In summary, we have developed a novel synthetic method accessing various multi-functionalized-α-trifluoromethyl α,β-unsaturated lactones and trifluoromethyl pyrazolinones from 3,3-dibromo-2-trifluoromethyl-acrylic acid ethyl ester 1 as a single precursor. The significant feature of our method involves two approaches: one is that a carbenoid intermediate is in situ-generated through the bromine–magnesium exchange reaction of 1, another is a condensation of 1 with hydrazine derivatives with high stereoselectivity. Further modification of bromo-lactones and pyrazolinones by using Suzuki-coupling reactions with arylboronic acids led to a series of arylated trifluoromethyl compounds. Since α,β-unsaturated lactones and pyrazoline compounds are an important part of the structure of natural bioactive compounds,21,22 the potential importance of these synthetic entries has stimulated our interest to develop new drugs with medicinal and agricultural applicability. The further application to evaluate their biological activity is currently underway. The detailed outcome will be reported soon.
Acknowledgements
This study was supported financially by Grants-in-Aid for Scientific Research (no. 26713041) from the Japan Society for Promotion of Science (JSPS). We thank Mrs Junko Nagaoka, of the Faculty of Engineering of Nagasaki University, for providing X-ray diffractometer measurements and analysis of the structure.
Notes and references
-
(a)
Biomedical Frontiers of Fluorine Chemistry, ACS Symposium Series 639, ed. I. Ojima, J. R. McCarthy and J. T. Welch, American Chemical Society, Washington, DC, 1996 Search PubMed;
(b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed;
(c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
-
Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Blackwell, United Kingdom, 2009 Search PubMed.
- A. A. Carmine, R. N. Brogden, R. C. Heel, T. M. Speight and G. S. Avery, Drugs, 1982, 23, 329–353 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- M. J. Brodie, C. N. Hensby, A. Parke and D. Gordon, Life Sci., 1980, 27, 603–608 CrossRef CAS PubMed.
- M. Müller, C. M. Orben, N. Schützenmeister, M. Schmidt, A. Leonov, U. M. Reinscheid, B. Dittrich and C. Griesinger, Angew. Chem., Int. Ed., 2013, 52, 6047–6049 CrossRef PubMed.
-
(a) P. Lin and J. Jiang, Tetrahedron, 2000, 56, 3635–3671 CrossRef CAS;
(b) Y. Kamitori, J. Heterocycl. Chem., 2001, 38, 773–775 CrossRef CAS;
(c) Z.-X. Jiang and F.-L. Qing, Tetrahedron Lett., 2001, 42, 9051–9053 CrossRef CAS;
(d) V. D. Matteis, F. L. van Delft, H. Jakobi, S. Lindell, J. Tiebes and F. P. J. T. Rutjes, J. Org. Chem., 2006, 71, 7527–7532 CrossRef PubMed;
(e) K. Uneyama, T. Katagiri and H. Amii, Acc. Chem. Res., 2008, 41, 817–829 CrossRef CAS PubMed;
(f) M. A. Honey, R. Pasceri, W. Lewis and C. J. Moody, J. Org. Chem., 2012, 77, 1396–1405 CrossRef CAS PubMed;
(g) W. Zhu, J. Wang, S. Wang, Z. Gu, J. L. Aceña, K. Izawa, H. Liu and V. A. Soloshonok, J. Fluorine Chem., 2014, 167, 37–54 CrossRef CAS;
(h) F. Meyer, Chem. Commun., 2016, 52, 3077–3094 RSC;
(i) Z.-M. Zhang, B. Xu, S. Xu, H.-H. Wu and J. Zhag, Angew. Chem., Int. Ed., 2016, 55, 6324–6328 CrossRef CAS PubMed.
- For selected examples of methods for introducing trifluoromethyl groups to heteroarenes, see:
(a) D. A. Nagib and W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed;
(b) Y. Ji, T. Brueckl, R. D. Baxter, Y. Fijiwara, I. B. Seiple, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411–14415 CrossRef CAS PubMed;
(c) E. J. Cho and S. L. Buchwald, Org. Lett., 2011, 13, 6552–6555 CrossRef CAS PubMed;
(d) Y. Fujiwara, J. A. Dixon, F. O'Hara, E. Daa Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95–100 CrossRef CAS PubMed;
(e) E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679–1681 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
-
(a) S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed;
(b) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 42, 46–58 CrossRef PubMed;
(c) F. W. Goldberg, J. G. Kettle, T. Kogej, M. W. D. Perry and N. P. Tomkinson, Drug Discovery Today, 2015, 20, 11–17 CrossRef PubMed.
- N. Shibata, H. Fujimoto, S. Mizuta, S. Ogawa, Y. Ishiuchi, S. Nakamura and T. Toru, Synlett, 2006, 3484–3488 CrossRef CAS.
-
O. Kobayashi, N. Niikura, T. Inoue, S. Mizuta, R. Takatsuna, K. Hirai, K. Shirouzu and M. Obata, PCT Int. Appl, WO2014142308A1, 2014 Search PubMed.
- For reviews:
(a)
Metal-catalyzed Cross-Coupling Reactions, ed. F. Diederich and P. J. Stang, Wiley-VCH, Weiheim, 1998 Search PubMed;
(b)
Topics in Current Chemistry, ed. N. Miyaura, Springer-Verlag, Berlin, 2002, vol. 219 Search PubMed.
- Y. Li and X. Zhao, Org. Lett., 2004, 6, 4467–4470 CrossRef CAS PubMed.
- Y. Takeda, M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2007, 46, 8659–8661 CrossRef CAS PubMed.
- Y.-H. Li, X.-M. Zhao and L. Lu, J. Fluorine Chem., 2004, 125, 1821–1824 CrossRef CAS.
- For details, see the ESI.†.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta Jr., F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
-
(a) H. Mahler and M. Braun, Tetrahedron Lett., 1987, 28, 5145–5148 CrossRef CAS;
(b) H. Mahler and M. Braun, Chem. Ber., 1991, 124, 1379–1395 CrossRef CAS;
(c) T. Harada, T. Katsuhira and A. Oku, J. Org. Chem., 1992, 57, 5805–5807 CrossRef CAS;
(d) T. Harada, T. Katsuhira, K. Hattori and A. Oku, Tetrahedron, 1994, 50, 7987–8002 CrossRef CAS;
(e) V. A. Vu, I. Marek and P. Knochel, Synthesis, 2003, 1797–1802 CAS.
- Review on Suzuki–Miyaura coupling, see:
(a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS;
(b) N. Miyaura, Top. Curr. Chem., 2002, 219, 1–59 CrossRef.
-
(a) F. Q. Alali, X.-X. Liu and J. L. McLaughlin, J. Nat. Prod., 1999, 62, 504–540 CrossRef CAS PubMed;
(b) G. R. Flematti, E. L. Ghisalberti, K. W. Dixon and R. D. Trengove, Science, 2004, 305, 977 CrossRef CAS PubMed;
(c) Y. Aoyagi, A. Yamazaki, C. Nakatsugawa, H. Furuya, K. Takeya, S. Kawauchi and H. Izumi, Org. Lett., 2008, 10, 4429–4432 CrossRef CAS PubMed;
(d) R. A. Pilli, G. B. Rosso and M. C. F. de Oliveira, Nat. Prod. Rep., 2010, 27, 1908–1937 RSC.
-
(a) M. J. Kornet and R. J. Garrett, J. Pharm. Sci., 1979, 68, 377–388 CrossRef CAS PubMed;
(b) T.-S. Jeong, K. S. Kim, S.-J. An, K.-H. Cho, S. Lee and W. S. Lee, Bioorg. Med. Chem. Lett., 2004, 14, 2751–2717 Search PubMed;
(c) M. P. Clark, S. K. Laughlin, M. J. Laufersweiler, R. G. Bookland, T. A. Brugel, A. Golebiowski, M. P. Sabat, J. A. Townes, J. C. VanRens, J. F. Djung, M. G. Natchus, B. De, L. C. Hsieh, S. C. Xu, R. L. Walter, M. J. Mekel, S. A. Heitmeyer, K. K. Brown, K. Juergens, Y. O. Taiwo and M. J. Janusz, J. Med. Chem., 2004, 47, 2724–2727 CrossRef CAS PubMed;
(d) Z. Özdemir, H. B. Kandilci, B. Gümüşel, Ü. Çaliş and A. A. Bilgin, Eur. J. Med. Chem., 2007, 42, 373–379 CrossRef PubMed;
(e) K. Tran, P. J. Lombardi and J. L. Leighton, Org. Lett., 2008, 10, 3165–3167 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all novel compounds, additional crystallographic information and NMR spectra. CCDC 1491776. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00360e |
|
| This journal is © the Partner Organisations 2016 |