
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–ON bond homolysis of alkoxyamines triggered by paramagnetic copper(II) salts†
Gérard
Audran
a,
Elena
Bagryanskaya
b,
Irina
Bagryanskaya
b,
Paul
Brémond
a,
Mariya
Edeleva
b,
Sylvain R. A.
Marque
*ab,
Dmitriy
Parkhomenko
 b,
Evgeny
Tretyakov
b and
Svetlana
Zhivetyeva
b
b,
Evgeny
Tretyakov
b and
Svetlana
Zhivetyeva
b
aAix Marseille Univ, CNRS, ICR, UMR 7273, case 551, Avenue Escadrille Normandie-Niemen, 13397 Marseille Cedex 20, France. E-mail: sylvain.marque@univ-amu.fr
bN. N. Vorozhtsov Novosibirsk Institute of Organic Chemistry SB RAS, Pr. Lavrentjeva 9, 630090 Novosibirsk, Russia
First published on 30th September 2016
Abstract
The metal complexation reactions of bis(hexafluoroacetylacetonato)copper(II) (Cu(hfac)2) with alkoxyamines (diethyl(2,2-dimethyl-1-(tert-butyl-(1-(pyridine-4-yl)ethoxy)amino)propyl)phosphonate and diethyl (2,2-dimethyl-1-(tert-butyl-(1-(pyridine-2-yl)ethoxy)amino)propyl)phosphonate) were studied. According to X-ray analysis, the molecular and crystal structures of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes depend on the configuration of the free alkoxyamines, that is dimeric (RSSR) and chain-polymeric (RR/SS) structures for para-pyridyl-substituted alkoxyamines, and cyclic unimeric (RS/SR) structure for ortho-pyridyl derivative. The complex (2:1 ratio Cu(hfac)2/alkoxyamine) for ortho-pyridyl-substituted alkoxyamine is not resolved. Upon warming, ortho complexes decomposed into free alkoxyamines and only a weak activation was observed. Upon warming, para complexes decomposed into their corresponding unimers, and then, a 21-fold increase in the rate constant of the C–ON bond homolysis was observed compared to the corresponding free alkoxyamines. Tuning of the homolysis rate constant of the C–ON bond via addition of pyridine is also reported.
:1 complexes depend on the configuration of the free alkoxyamines, that is dimeric (RSSR) and chain-polymeric (RR/SS) structures for para-pyridyl-substituted alkoxyamines, and cyclic unimeric (RS/SR) structure for ortho-pyridyl derivative. The complex (2:1 ratio Cu(hfac)2/alkoxyamine) for ortho-pyridyl-substituted alkoxyamine is not resolved. Upon warming, ortho complexes decomposed into free alkoxyamines and only a weak activation was observed. Upon warming, para complexes decomposed into their corresponding unimers, and then, a 21-fold increase in the rate constant of the C–ON bond homolysis was observed compared to the corresponding free alkoxyamines. Tuning of the homolysis rate constant of the C–ON bond via addition of pyridine is also reported.
Introduction
In the last three decades, alkoxyamines have been used as an initiator/controller for Nitroxide Mediated Polymerization (NMP).1,2,3 In recent years, new practical applications arose in materials sciences, e.g., a new system of coding4,5 and self-healing polymers6,7 and led to deeper research into the kinetics of alkoxyamine formation and decomposition. Moreover, recently, the use of alkoxyamines as theranostic8 markers was described,9 that is based on the decomposition of alkoxyamines as a source of nitroxide and alkyl radicals. The nitroxides can be used for diagnostic purposes as spin probes for electron paramagnetic resonance (EPR) tomography or as a DNP agent for magnetic resonance imaging (MRI), whereas alkyl radicals can serve as therapeutic agents due to their oxidant properties.10 The homolysis of the C–ON bond of alkoxyamines is triggered by external events such as a change in pH leading to the protonation of a group of an alkoxyamine or to complexation of an alkoxyamine by a Lewis acid.11,12 Protonation on the alkyl fragment11,12 and deprotonation on the nitroxyl fragment13 substantially promote homolysis of the C–ON bond of alkoxyamines. Complexation at the para position of the pyridine-based alkoxyamine 1 by a Lewis acid such as BH3 increases the rate constant kd of the C–ON bond homolysis by a factor of 10.12 As far as we know, the activation of the C–ON homolysis by the complexation of an alkoxyamine with a metal centre has never been reported. It is reasonable to assume the possible activation of an alkoxyamine by its complexation with a metal centre of a protein in a biological system. This possibility prompted us to investigate for the first time the complexation of a pyridine-based alkoxyamine by metal salts and its influence on the kinetics of alkoxyamine homolysis.It should be noted that recently Hicks and colleagues14 reported that the homolysis of the C–N bond in alkyl verdazyl 3 is increased by the complexation with a ruthenium salt as in 4, according to a decrease in Eaca. 20 kJ mol−1. The difference between the reactions studied by Hicks and colleagues (C–N bond homolysis) and by us (the homolysis of the C–ON bond, Scheme 1) means that different bond energies and influences of different effects are involved. Hence, the above result reinforced our confidence in this approach.
 | ||
| Scheme 1 C–ON bond homolysis in alkoxyamine. | ||
Thus, four complexes RSSR-7, RR/SS-7, (RR/SS)-8 and RS/SR-8 were prepared from alkoxyamines 1 and 2. Their structures were studied by X-ray diffraction (XRD) analysis, and 1H and 31P NMR, and their kd was measured. Tuning of the rate constant kd of the C–ON bond homolysis was investigated using pyridine as a competitor to form the N–Cu bond (Scheme 1).
Experimental section
Synthesis
Infrared (IR) spectra were recorded on a Bruker Vector 22 spectrometer (KBr). Elemental analyses were performed on a Carlo Erba 1106 CHN elemental analyzer. Solvents and reagents were of reagent quality. Alkoxyamines 1 and 2 were prepared as previously reported11,12 as was the monohydrate of bis(hexafluoroacetylacetonato)copper(II) (Cu(hfac)2·H2O) which was sublimated before use.15General procedure for complexes 7 and 8
A solution of Cu(hfac)2·H2O (0.025 g, 0.05 mmol) in hot hexane (2.5 mL) was added dropwise to a solution of alkoxyamine (0.020 g, 0.05 mmol) in hexane (1 mL). The reaction mixture was stirred at room temperature for 1 h and kept in a freezer at −5 °C for 24 h. Characterization of each complex is provided in the ESI.†k d measurements
EPR experiments were performed on an EMX machine equipped with a BVT2000 temperature control unit. The values of kd were measured by recording ESR spectra upon heating of 10−4 M toluene solutions of compounds RSSR-7, RR/SS-7, RS/SR-8, and (RR/SS)-8 in the presence of 3 equivalents of the 2,2,6,6-tetramethylpiperidin-N-oxyl radical (TEMPO) as an alkyl radical scavenger. The solutions were degassed by three cycles of freeze–pump–thaw and sealed under an argon atmosphere prior to measurements to ensure that narrow ESR signals of nitroxide SG1 generated in the course of heating.Profiles of the relative concentration were obtained by integration of the low field EPR line of SG1 and the data were fitted linearly in semi-logarithmic coordinates with eqn (1) as shown for RSSR-7 in Fig. 2. kd values are given in Table 1. Activation energies Ea are given by the Arrhenius equation (eqn (2)) using the value of 2.4 × 1014 s−1 as frequency factor A.16
 | (1) |
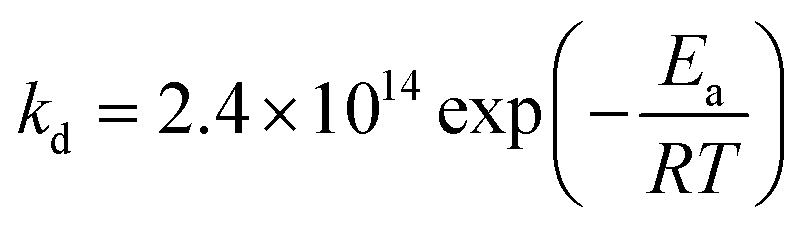 | (2) |
 | ||
| Fig. 1 Structures of copper complexes with alkoxyamines 1 and 2 and the molecules discussed. | ||
 | ||
| Fig. 2 A semi-logarithmic plot for kinetics of SG1 formation upon heating of RSSR-7 in the presence of 0 (■), 1 (○), 6 (▲), and 12 (□) equivalents of pyridine and with TEMPO as an alkyl radical scavenger at 80 °C in toluene. | ||
| Pyridine (eq.) | T (±1 °C) |
k
db (10−3 s−1) |
E
ac (kJ mol−1) |
k
d,120 °Cd (10−3 s−1) |
|
|---|---|---|---|---|---|
| a Complexes 7 and 8 are identified by the species prone to cleave and by the equivalent of pyridine (pn) implied in the reaction. b Given by eqn (1), error is less than 5%. c Given by eqn (2) using data from the 4th column and the frequency factor A = 2.4 × 1014 s−1 as recommended in ref. 16. An error of 1–2 kJ mol−1 is assumed. d Estimated using eqn (2), activation energies Ea given in the 5th column and the frequency factor A = 2.4 × 1014 s−1 (see ref. 16). e See ref. 11. f Averaged values of Ea of the two diastereoisomers. g See ref. 31. h Not available. | |||||
| 1 | 0 | — | — | 123.0f | 10.7 |
| 2 | 0 | — | — | 124.0f | 7.9 |
| 5 | 0 | — | — | 115.5f | 106.7 |
| 6 | 0 | — | — | —h | |
| URSSR-7 | 0 | 80 | 4.6 | 113.0 | 229.4 |
| URSSR-7p1 | 1 | 80 | 3.2 | 114.1 | 168.9 |
| URSSR-7p6 | 6 | 80 | 1.8 | 115.8 | 100.3 |
| URSSR-7p10 | 10 | 80 | 1.2 | 117.0 | 67.4 |
| URSSR-7p60 | 60 | 80 | 0.28 | 121.2 | 18.6 |
| URSSR-7p120 | 120 | 80 | 0.23 | 121.8 | 15.5 |
| URR/SS-7 | 0 | 80 | 3.2 | 114.0 | 168.9 |
| URR/SS-7p1 | 1 | 80 | 2.3 | 115.0 | 124.4 |
| URR/SS-7p4 | 4 | 80 | 1.9 | 115.6 | 103.5 |
| RS/SR-8 | 0 | 80 | 0.46 | 119.7 | 30.0 |
| RS/SR-8p6 | 6 | 80 | 0.42 | 120.0 | 27.3 |
| RS/SR-8p12 | 12 | 80 | 0.42 | 120.0 | 27.3 |
| (RR/SS)-8 | 0 | 100 | 2.0 | 122.0 | 14.8 |
| (RR/SS)-8p2 | 2 | 100 | 2.2 | 121.7 | 16.0 |
| (RR/SS)-8p6 | 6 | 100 | 2.1 | 121.8 | 15.5 |
| (RR/SS)-8p12 | 12 | 100 | 1.7 | 122.5 | 12.5 |
NMR experiments
1H and 31P NMR spectra were recorded for 0.01 M solutions of compounds RSSR-7, RR/SS-7, RS/SR-8, and (RR/SS)-8 (with respect to phosphorus) in deuterated benzene on a conventional NMR spectrometer operating at the resonance frequency of protons 500 MHz. For 1H NMR, the residual protons of the solvent served as a chemical shift reference. For 31P NMR, the signal of H3PO4 was used as an external reference. 1 K to 6 K scans were collected to achieve a good signal to noise ratio in the 31P NMR spectra. For temperature measurement the samples were heated in the cavity of the 500 MHz NMR spectrometer.XRD analysis
Monocrystal X-ray crystallographic analysis of the crystals was carried out on a Bruker Kappa Apex II CCD diffractometer using φ,ω-scans of narrow (0.5°) frames with Mo Kα radiation (λ = 0.71073 Å) and a graphite monochromator. The structures were solved by direct methods and refined by a full matrix least-squares anisotropic–isotropic (for atom H) procedure using the SHELXL97 software suite.17 Absorption corrections were applied using the empirical multiscan method in the SADABS software.18 The hydrogen atom positions were calculated by means of the riding model. In all complexes, some of the CF3 groups are disordered by two positions: occupation ratios have been refined in these cases. The resulting crystal structures were analysed for short contacts between the unbonded atoms using PLATON19,20 and MERCURY software applications.21There were high difference peaks in the structure of RRSS-8. We assumed that this volume is occupied by highly disordered solvent molecules of hexane that could not be modelled as a set of discrete atomic sites. The free solvent accessible volume derived from the PLATON19 routine analysis was found to be 20% (571.0 Å3). We employed the PLATON/SQUEEZE19 procedure to calculate the contribution to the diffraction from the solvent region and thereby produced a set of solvent-free diffraction intensity values.
Crystallographic data on the structures of RSSR-7, RR/SS-7, RS/SR-8 and RRSS-8 are collected in Table S1.†
Results and discussion
Preparation of metal complexes of alkoxyamines 7 and 8
Coordinately unsaturated copper(II) hexafluoroacetylacetonate (Cu(hfac)2) is the most suitable for our purpose due to its solubility in weakly polar media, and because it is a strong Lewis acid and yields well-crystallizing coordination compounds. Nevertheless, when working with Cu(hfac)2, researchers should keep in mind that during its interaction with ligands, there are rather frequent situations (depending on synthesis conditions) when several products can be formed that differ in composition and/or structure.22 Because sometimes different compounds crystallize as a mixture of products, it is necessary to find synthetic conditions where only one product is formed. This situation becomes even more complicated in the case of polydentate and stereochemically non-rigid ligands, such as diastereoisomers RS/SR-1 and RR/SS-1, RS/SR-2 and RR/SS-2. In such cases, it is always advisable to start the selection of conditions with the ones that seem intuitively correct in accordance with the structure and solubility of ligands. In particular, alkoxyamines 1 and 2 contain two strongest donor centres: the N atom of the pyridine ring and the O atom of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety, and compounds 1 and 2 are fairly well soluble in hexane. Accordingly, to obtain the complexes, the choice of the ratio Cu(hfac)2/alkoxyamine 1:1 appears rather reasonable, as does the choice of hexane as the solvent. Then, the reaction of Cu(hfac)2 with RS/SR-1 in the molar ratio 1:1 in hexane afforded the binuclear cyclic complex RSSR-7 with a quantitative yield.
O moiety, and compounds 1 and 2 are fairly well soluble in hexane. Accordingly, to obtain the complexes, the choice of the ratio Cu(hfac)2/alkoxyamine 1:1 appears rather reasonable, as does the choice of hexane as the solvent. Then, the reaction of Cu(hfac)2 with RS/SR-1 in the molar ratio 1:1 in hexane afforded the binuclear cyclic complex RSSR-7 with a quantitative yield.
Under the same conditions, mixing of Cu(hfac)2 with RR/SS-1 yielded the chain polymeric complex RR/SS-7 with the head-to-tail motif because of the coordination of the alkoxyamine via the N atom of the pyridine ring and the O atom of the PO moiety (vide infra). Mixing of Cu(hfac)2 with RS/SR-2 also yielded the complex of 1:1 composition, but this time, the complex RS/SR-8 was formed due to the bidentate-cyclic coordination of the alkoxyamine.‡
In all the aforementioned cases, at the equivalent ratio of reagents, coordination compounds of stereochemistry Cu(hfac)2/alkoxyamine = 1:1 were formed with a high yield. In the case of RR/SS-2, however, only the trinuclear complex RRSS-8 was isolated with a low yield.§ An attempt to change the initial ratio of the reagents to 3:2 led to the formation of the complex (RR/SS)-8 of composition 2:1, and the use of the reagent ratio 2:1 afforded (RR/SS)-8 with 87% yield. The obtained complex outwardly resembled very thin intertwined fibres of light green colour, and all attempts to solve its structure failed.
As SG1-pyridinyl-based alkoxyamines have several sites suitable for complexation, namely, the nitrogen atom on the aromatic ring, the nitrogen and oxygen atoms of the nitroxyl moiety and the oxygen atoms in the diethoxyphosphoryl group, the formation of complexes is not so obvious. Therefore, XRD, 1H and 31P NMR analyses were performed to identify the sites involved in the formation of the complexes as well as the structure of these complexes in solution; this structure can be different from the one in the crystalline state.
Structural analysis of copper complexes 7 and 8
XRD¶ showed the Cu/alkoxyamine ratio of 1:1 for the centrosymmetric dimeric complex RSSR-7, the polymer-chain complex RR/SS-7 and the molecular complex RS/SR-8, as well as the ratio 3:2 for the centrosymmetric trinuclear complex RRSS-8 (Fig. 3).
 | ||
| Fig. 3 X-ray structures of RSSR-7 (a), RR/SS-7 (b), RS/SR-8 (c), and RRSS-8 (d). | ||
Interestingly, the complexes of RSSR-7 and RRSS-8 are composed of the enantiomers of the corresponding alkoxyamines. For the RR/SS diastereoisomer of 2 the less soluble complex of the unknown structure (RR/SS)-8 has the ratio 1:1 (Fig. 1). In all crystals, whatever the alkoxyamine (i.e., ortho or para) or the type of complex, the pyridine moiety is always located as a ligand in the equatorial position and the diethoxyphosphoryl moiety always occupies an axial position as the ligand.|| Typically, the coordination bond lengths vary from 2.26 to 2.37 Å for lCu–OP, from 2.01 to 2.02 Å for lCu–N, from 2.16 to 2.39 Å for lCu–O (axial coordination), and from 1.93 to 1.99 Å for lCu–O (equatorial coordination). The bond lengths, distances and angles in alkoxyamines vary from 1.43 to 1.46 Å for lC–O, from 1.44 to 1.46 Å for lN–O, from 1.83 Å to 1.86 Å for lC–P, from 2.40 to 2.42 Å for dN⋯C and from 111.7° to 112.8° for the <NOC> bond angle; no significant differences are observed with the data11 reported for 1.
Nonetheless, XRD revealed that the coordination bond Cu–OP is the longest one amongst all bonds between copper(II) and the atoms of the first sphere of coordination. Thus, the Cu–OP bond is likely to be the weakest and, consequently, the most prone to be cleaved first in solution.
NMR analyses of copper(II) complexes 7 and 8
Because the X-ray structures are obtained in the solid state, their stability is not ensured in solution and may differ dramatically. Thus to get some clues about the species observed in solution both at room temperature and upon warming, the stability of complexes 7, RS/SR-8 and the complex powder 8 was assessed by 1H and 31P NMR using both temperature and the presence of pyridine to decompose complexes (Fig. 4 and ESI†).** | ||
| Fig. 4 Room temperature (a) and 60 °C (b) data on 31P NMR at 0, 1, 6 and 12 equivalents of pyridine (from bottom to top) added to RSSR-7 (the asterisk denotes free (RR/SS)-1 as impurity). | ||
Indeed, for Cu(II) complexes, pyridine has a valuable property in that it reacts quickly (and often in an irreversible way),23,24 with the Cu(II) atom to displace weakly bonded ligands. Therefore, it is an efficient competitor to investigate the stability of our Cu(II) complexes in solution. For pure RSSR-7 (Fig. 4a), a very broad peak in 31P NMR was observed. In the absence of pyridine no signal was recorded for aromatic protons and line broadening was observed for the nitroxyl fragment signal meaning that all protons are close to the paramagnetic centre, that is the pyridinyl and diethoxyphosphoryl moieties are bonded to Cu(II) atoms as revealed by the X-ray structure (Fig. 3). Moreover, the detection of a broad 31P NMR signal likely denotes a fast equilibrium between the complex and its unimer form (vide infra). Upon addition of pyridine from one to 12 equivalents, the peak in 31P NMR narrowed and shifted to a value close to the one of 1 (free alkoxyamine). The 1H NMR signal of the nitroxyl fragment narrowed significantly meaning that the phosphoryl group is not coordinated to the Cu(II) atom whereas no signal from the aromatic protons of RSSR-7 or from the pyridine was observed.†† A broad signal in the aromatic zone was observed for 12 equivalents and was ascribed to free pyridine in fast exchange with pyridine complexed by the Cu(II) atom. For the amount of 120 equivalents of pyridine, signals of protons of the alkyl fragment are detectable (see the ESI†) meaning that an excess of pyridine greater than 12 equivalents is necessary for efficient competition with the complexation of the Cu(II) atom by the pyridine moiety of the alkoxyamine. The broad peak in 31P NMR denotes a fast exchange between the dimer RSSR-7 and the unimer URSSR-7 whereas the presence of a pyridine signal in 1H NMR (Fig. S1†) only for a large excess of pyridine points to a very fast exchange between the free alkoxyamine and Cu(hfac)2(pyridine)2.
 | ||
| Scheme 2 The kinetic scheme for the C–ON bond homolysis in the absence (route a) and in the presence of pyridine (routes b and c). Starting material (SM) is shown as the NRCu triangle. N, R, and Cu stand for the nitroxyl fragment, alkyl fragment, and copper metal center (Cu(hfac)2). The NRCu triangle is for alkoxyamine complexed to Cu(II) by the pyridyl ring. P is for pyridine and represents a pyridine ligand in NRCuP. Dotted letters are for the radicals issued from the corresponding fragment after the C–ON bond homolysis. | ||
Chemical quenching experiments performed with pyridine showed an efficient quenching meaning that the RSSR-7 complex has been completely decomposed into its unimer URSSR-7p1 carrying one pyridine in the coordination sphere of the Cu(II) centre. The effects of both temperature and pyridine were investigated for RSSR-7 (Fig. 4b) and, because the same trends were observed (Fig. 4b) the comments on the experiments performed at room temperature hold. It is worth mentioning that the experiment, after warming in the absence of pyridine, showed that the equilibrium between complexes RSSR-7 and URSSR-7 is shifted in favour of the latter because the 31P NMR peak is narrower (Fig. 4b) than at room temperature (Fig. 4a).
The 1H and 31P spectra in the presence and in the absence of pyridine as well as at room temperature and 60 °C suggested that the RSSR-7 complex is not stable in solution (Fig. 5), that its decomposition into a unimer as in case (iii) can be disregarded, that its decomposition into the free alkoxyamine 1 as in case (i) requires a large excess of pyridine, and that the unimer URSSR-7p1 as in case (ii) is the species in solution for one equivalent of pyridine and in equilibrium with the RSSR-7 as described in Scheme 2.
 | ||
| Fig. 5 Schematic description of the structures possible for RSSR-7 in solution. On the left side is the solid state and on the right side species expected in solution. Dotted lines are for interaction between the Cu(II) center and alkoxyamine. Full lines are for the cleaved bond. Alkoxamine is schematically described as a yellow square for the nitroxyl fragment and as a blue square for the alkyl fragment. Cu(hfac)2 is schematically described as a green circle. Possibilities of structures in solution are given as (i) to (iii). | ||
The same procedure was applied to the RR/SS-7 complex and to complexes 8 (Fig. S1 and S2†) and afforded the same trends as for the RSSR-7 complex except for broader line widths for complexes 8 than for complexes 7 in 31P NMR (Fig. S2†). Consequently, the comments on complexes RSSR-7 and URSSR-7 hold for RR/SS-7 and URR/SS-7. For complexes 8, broader lines mean that an interaction with the radical centre occurred. Indeed, in the RS/SR-8 complex alkoxyamine plays the role of a chelating agent favouring the complexation of the metal centre and, hence, interactions stronger than in complexes 7. Taking into account the kinetics results (vide infra), the equilibrium for the complex RS/SR-8 is better described as an equilibrium between the complex and the free alkoxyamine ((case i) in Fig. 5). As the structure of the complex (RR/SS)-8 is not resolved and as the same trends are observed by 1H and 31P NMR, no more comments are provided except that complex (RR/SS)-8 is assumed to exhibit the same behaviour as the complex RS/SR-8. These comments hold both at room and high temperatures.
Measurements of kd for complexes 7 and 8
Measurements of kd‡‡ were performed by EPR as reported elsewhere.25 Based on the NMR analysis of complexes 7 and 8, three main kinetic routes (Scheme 2) are proposed to describe the C–ON bond homolysis with pure complexes (k4 for unimers) or in the presence of pyridine (k5 for the unimers carrying a pyridine in the sphere of coordination of Cu(II)): route (a): an equilibrium (k1 and k−1 as rate constants) between RSSR-7, RR/SS-7, RS/SR-8, and (RR/SS)-8 (starting materials SM), and their respective unimer (NRCu); route (b): chemical quenching in the presence of pyridine the equilibrium between the starting materials and unimers is suppressed either by a fast and irreversible quenching of the unimers by pyridine (case (i) in route b, Scheme 2) or fast and irreversible reaction with the starting materials (case (ii) in route b, Scheme 2); route (c): the reaction of the pyridine with complexes is described by an equilibrium. For the sake of simplicity,§§ only case (iii) where pyridine reacts with the unimer species (rate constants k2 and k-2) and case (iv) where the pyridine reacts with the starting materials (rate constants k3 and k−3) are considered in route c (Scheme 2).For RSSR-7 and RS/SR-8, complexes are described, respectively, as cyclic compounds showing two alkoxyamines and two Cu atoms in an alternating fashion (dimer), and a Cu atom bound simultaneously to the alkyl and nitroxyl fragment of the alkoxyamine (cyclic unimer). It has been reported that cyclic alkoxyamines, either because of covalent bonding26 or due to intramolecular hydrogen bonding,27–30 have a stronger C–ON bond than their non-cyclic analogues do. Thus, assuming a fast exchange (vide supra), the homolysis of the C–ON bond in RSSR-7 and RS/SR-8 is disregarded in comparison with the homolysis of the unimeric species URSSR-7 and URS/SR-8 (or the corresponding free alkoxyamine). This comment holds for the kinetics in the presence of pyridine. Complex RR/SS-7 shows a polymeric structure composed of an alternating alkoxyamine and a copper centre in a 1:1 ratio. It has been reported that increasing the electron-withdrawing properties of substituents in the nitroxyl fragment increases kd. Hence, the interaction between copper and the PO moiety in RR/SS-7 is expected to enhance these electron-withdrawing properties, and therefore, to strengthen the C–ON bond. Consequently, the C–ON homolysis in the polymer species is disregarded. Because NMR data indicate the same type of equilibrium for complexes 7 and 8 as for RSSR-7, the aforementioned comments and Scheme 2 still hold.
The rate of generation of nitroxide SG1 (N) released by the homolysis of complexes is given by eqn (3)–(8) depending on the routes (Scheme 2) and assumptions. For route (a), the growth in SG1 is given by eqn (3) and assuming a fast equilibrium with k−1 larger than the C–ON bond homolysis rate constant k4, kd = K1·k4 (K1 = k1/k−1, eqn (4)). For route (b), whatever the cases the chemical quenching conditions afford the growth of SG1 as given by eqn (5), which corresponds to a first-order reaction with k5 as the C–ON bond homolysis rate constant. For route c (Scheme 2), the growth in SG1 is described by the kinetics of the second order as shown in eqn (6) in case (iii) and in eqn (7) and (8) (K2 = k2/k−2), in case (iv).¶¶ Nonetheless, it must be kept in mind that too large an excess of pyridine decomposes the unimer (vide supra) into a free alkoxyamine leading to a decrease in kd.
 | (3) |
 | (4) |
 | (5) |
 | (6) |
 | (7) |
 | (8) |
For the RSSR-7 and RR/SS-7 complexes, the growth in SG1 either in the absence or in the presence of one equivalent of pyridine is described by first-order kinetics. It means that for one equivalent of pyridine the conditions for a chemical quenching are fulfilled, that is there is a fast equilibrium between the starting materials and the corresponding unimers affording a small amount of unimers which is instantaneously and irreversibly scavenged by the pyridine in excess. Consequently, the equilibrium is completely and irreversibly shifted to the formation of unimers. Thus, all other possible routes are disregarded because they provided only second order kinetics for the growth in SG1. Thus, Ea values estimated at 114.1 kJ mol−1 and 115.0 kJ mol−1 in the presence of pyridine for URSSR-7p1 and URR/SS-7p1, respectively, most likely correspond to the true homolysis rate constant kd. Interestingly, the kd values for the homolysis in the absence of pyridine are almost identical to those for one equivalent meaning either K1 = 1 assuming that the C–ON bond homolysis is not sensitive to the presence of pyridine in the coordination sphere of the copper centre or K1 < 1 and k4 for the unimer is significantly greater than k5 for the unimer carrying pyridine.||||
Taking advantage of these results, the tuning of the homolysis of the C–ON bond was studied by varying the amount of pyridine from one (URSSR-7p1) to 120 (URSSR-7p120) equivalents for RSSR-7. As expected, only first-order growth in SG1 was observed with the decreasing kd and with the increasing amount of pyridine (Table 1) meaning that complexes RSSR-7 and URSSR-7 are completely decomposed step-by-step into a free alkoxyamine because Ea is 121.8 kJ mol−1 for 120 equivalents of pyridine (URSSR-7p120): very close to the value of the free alkoxyamine 1 (Ea = 123.0 kJ mol−1).11,12 Thus, these experiments on complexes 7 and pyridine (Table 1) point to the possibility of tuning the C–ON bond homolysis by changing the amount of pyridine.
Whatever the complexes 8 and the conditions are (0 to 12 equivalents of pyridine), Ea values are between 120 and 123 kJ mol−1 (Table 1) and, thus, very close to the values reported for free alkoxyamine 2 (Ea = 124.0 kJ mol−1)31 in sharp contrast to the results reported for complexes 7. Thus, these observations combined with the 31P NMR data (vide supra) support strongly the generation of free alkoxyamine in fast equilibrium with complexes 8. Thus, the kinetics observed are modelled by using eqn (3) and (4) (route a) in Scheme 2) in the absence and in the presence of pyridine as the complexation of the copper(II) occurred after the decomposition of complexes 8 provided N–R–Cu is replaced by N–R. It has to be mentioned that Ea of 6 is not available because the borane adducts decomposed upon heating to release free alkoxyamines 2, the event likely caused by the steric strain at the ortho position of the pyridinyl ring.31 Consequently, because the copper complex is larger than the BH3 molecule, the steric strain is larger and upon warming (whatever the amount of pyridine), complexes 8 mainly decompose into free alkoxyamines 2, accounting for the values of Ea observed. In contrast to BH3 which is a gas, the copper complex stays in solution and can still interact with free alkoxyamines 2 affording Ea values slightly lower than those reported for free 2.
Conclusion
In this work, we demonstrate for the first time that an alkoxyamine's homolysis can be effectively triggered by complexation with a metal. It is shown that for alkoxyamines with several groups suitable for complexation, different types of complexes can be formed such as cyclic, dimeric and linear chain polymeric. The synthesis of five complexes of a pyridine-based alkoxyamine with Cu(hfac)2 was performed and the structure was confirmed for four of five using XRD analysis. The use of pyridine as a competitor for complexation of the metal centre allows us to gain deeper insight into the decomposition kinetics of such complexes.As far as we know, this article is the first report on tuning the C–ON bond homolysis using a nucleophilic reaction based on the exchange of a ligand in a metal center.
The effects due to the size and charge of the metal, and the polarity and the bulkiness of the ligands need to be carefully investigated. Nonetheless, the 21-fold increase in kd observed for the complexation of the pyridyl moiety of 1 at the para-position by a Cu(II) complex as a Lewis acid highlights nicely the potential of Lewis acids based on a metal as activators of the C–ON bond in alkoxyamines. These results may find valuable applicatio in Material Sciences as polymerization initiators3 from the metal surface or as theranostic agents.9
Acknowledgements
SRAM, GA, and PB thank Aix-Marseille Université, CNRS, ANR (grant NAR ANR-14-CE16-0023-01) and the A*MIDEX project (ANR-11-IDEX-0001-02) funded by the “Investissements d'Avenir” French Government program, managed by the French National Research Agency (ANR). SRAM, ME, DP, ET and SZ are grateful to the Russian Science Foundation (grant 15-13-2020) for supporting this work and to the Multi-Access Chemical Service Center SB RAS for spectral and analytical measurements.Notes and references
- D. H. Solomon, E. Rizzardo and P. Cacioli, EP, Appl. 135280, 1985 Search PubMed; D. H. Solomon, E. Rizzardo and P. Cacioli, US Pat, 4581429, 1986 Search PubMed; Chem. Abstr. 1985 102 221335q Search PubMed.
- G. Audran, P. Brémond and S. R. A. Marque, Chem. Commun., 2014, 50(59), 7921–7928 RSC.
- Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science Series, ed. D. Gigmes, RSC Polymer Chemistry Series, 2015 Search PubMed.
- R. K. Roy, A. Meszynska, C. Laure, L. Charles, C. Verchin and J.-F. Lutz, Nat. Commun., 2015, 6, 7237 CrossRef PubMed.
- L. Charles, C. Laure, J.-F. Lutz and R. K. Roy, Macromolecules, 2015, 48, 4319–4328 CrossRef CAS.
- C. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076–5081 CrossRef CAS.
- Z. P. Zhang, M. Z. Rong, M. Q. Zhang and C. Yuan, Polym. Chem., 2013, 4, 4648–4654 RSC.
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670–6683 RSC.
- G. Audran, P. Brémond, J.-M. Franconi, S. R. A. Marque, P. Massot, P. Mellet, E. Parzy and E. Thiaudière, Org. Biomol. Chem., 2014, 12, 719–723 CAS.
- D. Moncelet, P. Voisin, V. Bouchaud, P. Massot, E. Parzy, G. Audran, J.-M. Franconi, S. R. A. Marque, P. Brémond and P. Mellet, Mol. Pharmaceutics, 2014, 11, 2412–2419 CrossRef CAS PubMed.
- P. Brémond and S. R. A. Marque, Chem. Commun., 2011, 47, 4291–4293 RSC.
- P. Brémond, A. Koïta, S. R. A. Marque, V. Pesce, V. Roubaud and D. Siri, Org. Lett., 2012, 14((1), 358–361 CrossRef PubMed.
- M. V. Edeleva, I. A. Kirilyuk, I. F. Zhurko, D. A. Parkhomenko, Y. P. Tsentalovich and E. G. Bagryanskaya, J. Org. Chem., 2011, 76, 5558–5573 CrossRef CAS PubMed.
- C. W. Johnston, T. R. Schwantje, M. J. Ferguson, R. McDonald and R. G. Hicks, Chem. Commun., 2014, 50, 12542–12544 RSC.
- J. A. Bertrand and R. I. Kaplan, Inorg. Chem., 1966, 5, 489–491 CrossRef CAS.
- E. G. Bagryanskaya and S. R. A. Marque, RSC Polymer Chemistry Series, n° = 19, Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Sciences, ed. D. Gigmes, Royal Society of Chemistry, 2016, ch. 2, 45–113 Search PubMed.
- G. M. Sheldrick, SHELX-97, Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, Germany, 1997 Search PubMed.
- SADABS, v. 2008-1, Bruker AXS, Madison, WI, USA, 2008 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool (Version 10 M), Utrecht University, Utrecht, The Netherlands, 2003 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Stree, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- S. Fokin, V. Ovcharenko, G. Romanenko and V. Ikorskii, Inorg. Chem., 2004, 43, 969–977 CrossRef CAS PubMed.
- W. Partenheimer and R. S. Drago, Inorg. Chem., 1970, 9, 47–52 CrossRef CAS.
- J. Pradilla-Sorzano and J. P. Fackler, Inorg. Chem., 1973, 12, 1174–1182 CrossRef CAS.
- S. Marque, C. Le Mercier, P. Tordo and H. Fischer, Macromolecules, 2000, 33(12)), 4403–4410 CrossRef CAS.
- J. Ruehl, N. Ningnuek, T. Thongpaisanwong and R. Braslau, J. Polym. Sci., Part A: Polym. Chem., 2008, 45, 8049–8069 CrossRef.
- S. Harrisson, P. Couvreur and J. Nicolas, Macromol. Rapid Commun., 2012, 33, 805–810 CrossRef CAS PubMed.
- E. G. Bagryanskaya, P. Brémond, T. Butscher, S. R. A. Marque, D. Parkhomenko, V. Roubaud, D. Siri and S. Viel, Macromol. Chem. Phys., 2015, 216(5), 475–488 CrossRef CAS.
- P. Brémond, T. Butscher, V. Roubaud, D. Siri and S. Viel, J. Org. Chem., 2013, 78, 10524–10529 CrossRef PubMed.
- G. Audran, P. Brémond, S. R. A. Marque and T. Yamasaki, J. Org. Chem., 2016, 81, 1981–1988 CrossRef CAS PubMed.
- G. Audran, M. Bim Batsiandzy Ibanou, P. Brémond, S. R. A. Marque, V. Roubaud and D. Siri, Org. Biomol. Chem., 2013, 11, 7738–7750 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation, characterization, and Table S1 with XRD data of complexes 7 and 8. Fig. S1 (1H NMR) and S2 (31P NMR) of complexes 7 and 8. CCDC 1483562–1483565. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00277c |
| ‡ 64% yield was reached for the first purification. Then, after evaporation of the solvent, the dry residue was crystallized from a mixture of hexane/heptane (1:1) to obtain an additional amount (0.012 g) of the product. |
| § As RRSS-8 is a minor compound, no further investigations are performed except X-ray analysis. |
| ¶ For RSSR-7, CCDC 1483562; for RR/SS-7, CCDC 1483563; for RRSS-8, CCDC 1483564; and for RS/SR-8, CCDC 1483565. |
| || The axial positions are determined as the position affording the longest Cu–O bond in 1,1,1,6,6,6-hexafluoropentan-2,4-dione. |
| ** NMR experiments were performed with the same concentration in alkoxyamine for all samples. |
| †† As the peak in 31P NMR is clearly narrower with one equivalent of pyridine than in its absence, it was assumed that the first equivalent of pyridine is mainly used to cleave the PO⋯Cu bond to afford the unimer URSSR-7 (Scheme 2 route b or c). |
| ‡‡ Here, kd is considered as the apparent rate constant of the C–ON bond homolysis. |
| §§ The equilibrium implying the scavenger and pyridine is disregarded. |
| ¶¶ For eqn (6), a first order growth might be observed provided k3·k5·[P] is larger than k−1·(k−3 + k5). |
| |||| Values of K1 cannot be assessed because the concentration of starting materials and unimer cannot be determined accurately and with reliability. |
| This journal is © the Partner Organisations 2016 |