 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemistry at single bimetallic nanoparticles – using nano impacts for sizing and compositional analysis of individual AgAu alloy nanoparticles
En Ning
Saw
a,
Viktoria
Grasmik
b,
Christian
Rurainsky
a,
Matthias
Epple
b and
Kristina
Tschulik
*a
aMicro- & Nano-Electrochemistry and Center for Electrochemical Sciences (CES), Faculty of Chemistry and Biochemistry, Ruhr-University Bochum, D-44780 Bochum, Germany. E-mail: Kristina.Tschulik@rub.de
bInorganic Chemistry and Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitaetsstr. 5-7, D-45117, Essen, Germany
First published on 17th May 2016
Abstract
The increasing interest in producing bimetallic nanoparticles and utilizing them in modern technologies sets the demand for fast and affordable characterization of these materials. To date Scanning Transmission Electron Microscopy (STEM) coupled to energy dispersive X-ray spectroscopy is usually used to determine the size and composition of alloy nanoparticles, which is time-consuming and expensive. Here electrochemical single nanoparticle analysis is presented as an alternative approach to infer the particle size and composition of alloy nanoparticles, directly in a dispersion of these particles. As a proof of concept, 14 nm sized Ag0.73Au0.27 alloy nanoparticles are analyzed using a combination of chronoamperometric single nanoparticle analysis and cyclic voltammetry ensemble studies. It is demonstrated that the size, the alloying and the composition can all be inferred using this approach. Thus, the electrochemical characterization of single bimetallic alloy nanoparticles is suggested here as a powerful and convenient complement or alternative to TEM characterization of alloy nanoparticles.
1. Introduction
Thanks to their many useful properties, nanoparticles are widely employed in a large number of consumer products, ranging from anti-microbial socks and medical implants to additives in sunscreen lotion, food or advanced optical devices.1,2 Key to this success is the ability to obtain high surface to volume ratio and to control the electronic structure of these nanomaterials in contrast to conventional bulk materials, allowing the size of devices to be reduced, their efficiency to be increased, or their optical properties to be changed. While nanotechnology is often considered a recent discovery/development, the ancient Romans had already used a combination of nanometer-sized gold and silver particles to produce dichroic glass.3 Also, the red-coloured ‘activated gold’4 that Michael Faraday produced in the 1850s that led to the discovery of the Faraday–Tyndall effect was a suspension of gold nanoparticles, as we know today. Despite these early studies, only in the past few decades has the use of nanoparticles emerged greatly, thanks to the ability to image, and hence, finally systematically study them by transmission (TEM) or scanning electron microscopy (SEM).5Thus, the research field of nanoscience emerged quickly in the past two decades, and nowadays the combination of different components and functionalities in a single nanoparticle, so called bimetallic and bifunctional nanomaterials, is state-of-the-art. Prominent examples are magnetic-core biocompatible-shell nanoparticles for medical application6,7 or bimetallic alloy nanoparticles with tuneable catalytic activity8,9 and optical properties.10,11
In many cases the synthesis of such bi- or multicomponent nanoparticles is not easy, and their characterization is equally challenging. Their physical properties and chemical reactivity depend not only on their size and shape, but also their spatial composition in terms of alloy vs. core–shell type arrangement of the constituting atoms may play an important role.10,12 The most prominent examples for bimetallic nanoparticles are AgAu nanoparticles due to their many promising applications in medicine, optics and catalysis.9,13,14 Characterization of these particles and their dispersions beyond sizing, however, is difficult. Standard solution-phase nanoparticle characterization techniques like dynamic light scattering (DLS), Nanoparticle Tracking Analysis (NTA) or Centrifugal Disc Sedimentation (DCS) are inherently insensitive to the particle composition. Investigations on dried nanoparticle samples, like nano-diffraction analysis in TEM investigations, are difficult due to the close similarity in crystal structures and lattice parameters of the two elements. Thus, electron dispersive spectroscopy (EDX) coupled to TEM analysis is the state-of-the-art analysis technique to characterize bimetallic nanoparticles.15,16 As this requires expensive equipment, complex sample preparation and the removal of the solvent prior to the analysis, an alternative route to characterize bi- and multi-component nanoparticles is desirable.
It has been demonstrated recently that electrochemical analysis can be used to characterize ensembles of core–shell type nanoparticles in terms of shell quality and shell thickness.17 Furthermore it was demonstrated that electrochemical nano impact experiments enable the sizing of the shell and the core of individual core–shell-type bimetallic nanoparticles.16 In these nano impact experiments either the shell only, or the core plus shell, of individual Au core Ag shell particles were oxidized during their Brownian motion driven collision with an electrode, depending on the potential this electrode was held at.
Here we will demonstrate that the application of single nanoparticle impact experiments can be usefully extended to determine the size and the composition of individual bimetallic alloy nanoparticles. The observed relation between applied potential and measured charge per impact spike for individual alloy nanoparticles will be combined with alloy ensemble studies and compared to the behaviour of monometallic nanoparticles. Thus, electrochemical single nanoparticle analysis will be introduced as a new tool for alloy nanoparticle characterization in terms of the size and composition, using Ag0.73Au0.27 nanoparticles as a proof-of-concept example.
2. Theoretical background
Electrochemical differentiation between alloy- or core–shell-type bimetallic nanoparticles A1−xBx (A having the lower standard potential, that is being less noble than B) can be illustrated based on the Nernst equation, which relates the equilibrium potential for the oxidation reaction to the activity of the component in the particle and the concentration of the ions in the solution| A ⇌ A+ + e− | (1) |
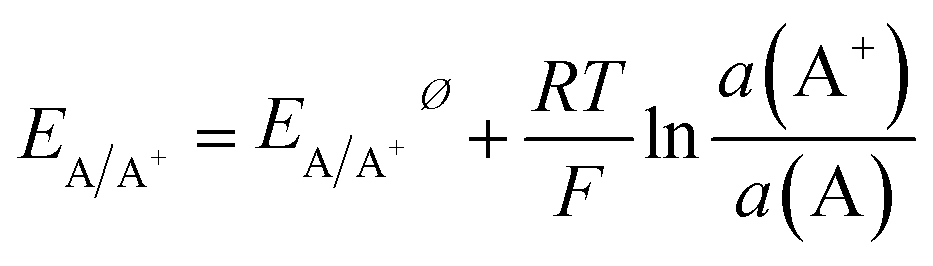 | (2) |
The activity of a pure metal in its standard state is 1, while in an alloy, by assuming the mixture of A and B to be ideal, the activity of A is given by its molar fraction. Thus, for a set activity of A+ in solution, the oxidation potential of component A in an alloy will be shifted to a more positive potential than the pure metal. Fixing the activity of A+ may usefully be achieved by choosing an electrolyte that contains an excess amount of an anion X− that forms a poorly soluble compound AX and thus allows fixing of a(A+) by the solubility product (KL) and the large excess amount of X− in the solution.
| AX(s) ⇌ A+(aq) + X−(aq) | (3) |
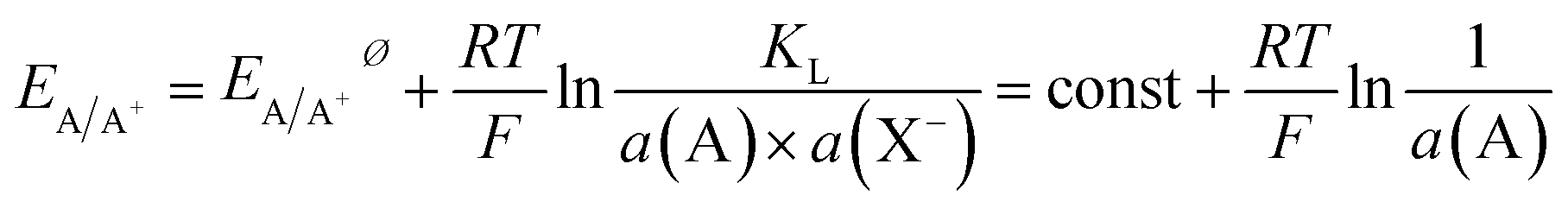 | (4) |
Considering a bimetallic B-core A-shell nanoparticle, the outer shell of the particle exclusively contains A atoms, thus the mole fraction of A at the reaction front will be unity and so oxidation of the shell will occur at the same potential as observed for single monometallic A nanoparticles.17 Once the A shell is oxidized, no faradaic current is seen until a second anodic peak is observed at potentials sufficiently high to facilitate oxidation of the remaining B core. For A-core B-shell nanoparticles, only one anodic peak is observed at the oxidation potential of B, comprising the oxidation of both B and A.
In the case of oxidation of an alloy A1−xBx nanoparticle, leaching of the A component occurs at potentials above the oxidation potential of A and below the B oxidation potential. This so called ‘de-alloying’ increases the B content of the alloy, resulting in a continuous positive shift of the oxidation potentials of the remaining particle until the B oxidation potential is reached and the particle is fully oxidized. Thus, in a cyclic voltammogram a broad oxidation peak above the A oxidation potential and a subsequent B oxidation peak are observed. Depending on the experimental conditions (scan rate, temperature, electrolyte composition, etc.), surface reorganization of the remaining B on the partially leached nanoparticle might additionally occur, causing a further broadening of the oxidation peak.18,19
While electrochemical studies at ensembles of bimetallic nanoparticles enable the characterization of several billions of nanoparticles instantaneously, their individual size cannot be directly determined this way. Hence, electrochemical single entity studies of alloy particles are desirable to derive insights into both the size and the composition of individual bimetallic alloy nanoparticles. Herein, it will be demonstrated that nanoparticle impact experiments may provide a convenient route to gain these additional insights.
In a ‘nano impact’ experiment, the Brownian motion driven sporadic collision of dispersed nanoparticles with a potentiostated electrode is used either to mediate a reaction at the particle surface20–28 or to reduce29–31/oxidize16,32–35 the particle itself and thus, to derive information on a particle-by-particle basis (see Fig. 1). The oxidation of a nanoparticle impacting a suitably polarized electrode results in a burst of faradaic current, observed as a ‘spike’ in a current–time graph. The minimum potential required to drive the faradaic reaction is determined by the oxidation potential, and hence, the composition, of the impacting particle.
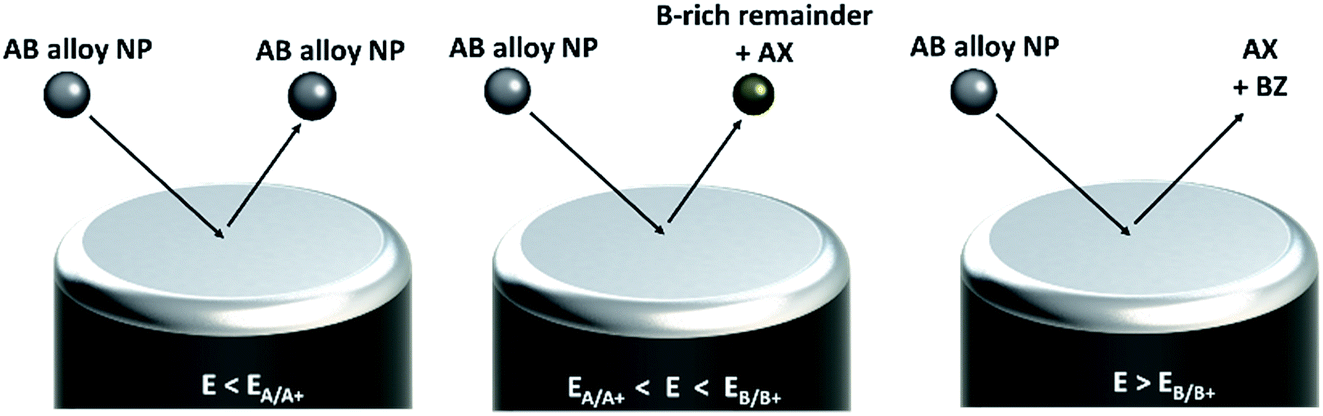 | ||
| Fig. 1 Schematic drawing of nano impact experiments of bimetallic alloy nanoparticles. | ||
The charge Q exchanged during the impact event is determined by the number of atoms contained in the impacting nanoparticle Nnp and the number z of electrons exchanged in the redox reaction:
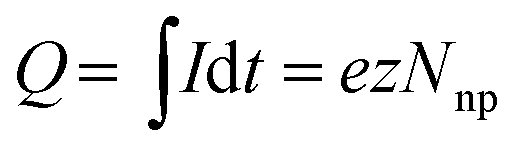 | (5) |
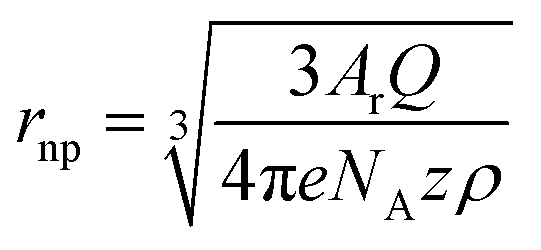 | (6) |
For a AgAu alloy particle, due to the above-mentioned shift of the oxidation potential, a potential significantly higher than the Ag oxidation potential should be required to oxidize the Ag in the alloy particle. The associated Ag leaching further decreases the Ag content in the alloy at the reaction front. Thus, two predictions can be made:
(i) Anodic spikes of AgAu alloy particles should emerge at potentials more positive than the oxidation of pure Ag NPs under otherwise identical conditions (capping agent etc.) and (ii) increasing the potential applied, the charge exchanged per impact should steadily increase until the entire particle is oxidized at potentials sufficient to facilitate Au oxidation. These predictions will be experimentally tested in the following sections.
3. Experimental
3.1. Nanoparticle synthesis
The bimetallic AgAu alloy nanoparticles used in this study were prepared as described in detail elsewhere.15 In brief, the nanoparticles were synthesized by reduction with citrate and tannic acid in aqueous media. All preparations were carried out with double-distilled water. 300 mL of degassed water was heated to 100 °C. Then 0.9 mL of 10 mM HAuCl4 and 2.1 mL of 10 mM AgNO3 were added. A mixture of 3 mL of a solution of 102.6 mg trisodium citrate (116 mM) and 3 mL of a solution of 50.4 mg tannic acid (9.9 mM) was added using a syringe under vigorous stirring. The reaction mixture was then stirred for another 10 min at 100 °C before cooling to room temperature. Further functionalization was carried out by adding 2 mL of a solution of poly(N-vinylpyrrolidone) (PVP; 30 mg mL−1) to the non-purified dispersion. After stirring for at least two more hours, the nanoparticles were separated from the unreacted material by ultracentrifugation (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 30 min) and redispersed in ultrapure water by vortexing. The purification step was repeated twice to remove all synthesis by-products. Monometallic Ag and Au nanoparticles were produced using the same synthesis method to allow a direct comparison of the electrochemical behaviour of the mono- and the bimetallic particles, but 3.0 mL of either 10 mM HAuCl4 or AgNO3 solution were added.
000 rpm, 30 min) and redispersed in ultrapure water by vortexing. The purification step was repeated twice to remove all synthesis by-products. Monometallic Ag and Au nanoparticles were produced using the same synthesis method to allow a direct comparison of the electrochemical behaviour of the mono- and the bimetallic particles, but 3.0 mL of either 10 mM HAuCl4 or AgNO3 solution were added.
3.2. NP characterization
Sizing of the nanoparticles was done using two solution-phase characterization techniques: differential centrifugal sedimentation (DCS) and dynamic light scattering (DLS). A narrow size-distribution and an average nanoparticle diameter of 14 nm of the bimetallic alloy nanoparticles were determined using DCS, as shown in Fig. 2a. DCS measurements were run at 24000 rpm (CPS Instruments Disc Centrifuge DC 24000), and 24 wt% sucrose solution to establish the density gradient. | ||
| Fig. 2 (a) DCS analysis, (b) SEM image, (c) DLS analysis and (d) UV-vis spectrum of the Ag0.73Au0.27 NPs used in this work. | ||
In agreement with this, DLS characterization of the nanoparticles also showed a narrow size distribution with a modal particle size of 16 nm. DLS analysis was performed using a Malvern Instruments Ltd. Zetasizer.
Atomic absorption spectroscopy (AAS) of the alloy nanoparticles dissolved in aqua regia revealed a composition of Ag:Au = 73:27 (Thermo Electron M-Series spectrometer, graphite tube furnace), which is in excellent agreement with the intended composition of ≈Ag0.7Au0.3 previously reported for the synthesis method used.15 The size and compositional information were confirmed using SEM imaging (see Fig. 2b) and SEM-EDX analysis (FEI eSEM Quantax 3D FEG with EDAX Genesis XM2i detector), respectively.
For an extended X-ray diffraction and TEM-EDX analysis of the alloy nanoparticles we refer to Ristig et al.,15,36 who demonstrated that the used synthesis method produces AgAu alloy nanoparticles of adjustable composition, with an increased Au content in the center of the particle. In agreement to their finding, the UV-vis absorption spectrum (Cary 60 UV-vis spectrophotometer by Agilent Technologies Inc.) of the nanoparticle suspension used herein showed a plasmon resonance absorption peak at 447 nm (see Fig. 2d).15
3.3. Electrochemical setup
Electrochemical experiments were run using a three-electrode setup comprising a Ag/AgCl, 3 M KCl reference electrode (potential = 0.197 V vs. SHE) and a Pt counter electrode at constant temperature (24 ± 0.5 °C). The entire cell was placed in a Faraday cage to reduce electronic noise. As a working electrode for nanoparticle ensemble studies a glassy carbon electrode (GCE) was used (Ø = 4 mm) and for nanoparticle impact experiments a carbon fibre (Ø = 7 μm) or Pt micro electrode (Ø = 10 μm) was employed.3.4. Ensemble studies
Cyclic voltammetry (CV) ensemble studies were performed in 0.1 M HCl(aq) in the potential range from −0.2 V to 1.2 V vs. Ag/AgCl at a scan rate of 0.025 V s−1 using a potentiostat (Autolab PG Stat 12). CVs were recorded at blank and nanoparticle-modified GCEs. The modification with bimetallic nanoparticles was achieved by drop casting 2 μL of AgAu nanoparticle stock suspension on the GCE. For comparison pure Au, pure Ag and mixtures of Ag plus Au nanoparticles, respectively, were drop cast on an electrode to study the CV response of the monometallic particles and their mixture. To better resemble the capping agent free Au-rich remainder left by the oxidation of Ag from the bimetallic nanoparticles, PVP-capped Au monometallic nanoparticles were first immobilized on a GCE and cycled from −0.2 V to 1.2 V vs. Ag/AgCl in 0.1 M KNO3 to de-cap the Au nanoparticle surface by oxidation and subsequent reduction of the particle surface.37 Then these particles were analysed in HCl as described above. All CV measurements were repeated three times.3.5. Single nanoparticle experiments
For chronoamperometric nano impact experiments a potentiostat (VA10, npi-electronics, linked to a computer using high speed DA/AD data acquisition cards, plug-in electronics) was used to apply different fixed potentials, ranging from 0 V to 1.2 V. The resulting current response was measured over a time of 50 s, recording 1200 data points per second. To filter the electronic noise without altering the spike charge, a 50 Hz 8 pole Bessel filter was applied. Chronoamperograms were recorded in 0.01 M HCl in the absence or the presence of AgAu nanoparticles at particle concentrations of 10 pM. At each potential at least 5 measurements were performed, scattering the applied potential to omit any possible aging effect. At potentials at which spikes were seen, additional measurements were performed to record a sufficient number of impacts to obtain statistically relevant data. For comparison, Ag nanoparticle impacts were recorded in the same electrolyte.Individual spikes were identified, setting the minimum required peak current to 3 times the noise level, and spike charges were determined using either SignalCounter software (provided by Dr D. Omanovic, Ruder Boscovic Institute Zagreb, Croatia) and Origin 2016 Pro. The mean charge and the error of the mean were determined for each potential applied.
4. Results and discussion
Cyclic voltammograms recorded at a scan rate of 0.025 V s−1 in 0.1 M HCl for the blank GCE and the GCE modified with monometallic Ag and Au nanoparticles are shown in Fig. 3a. An oxidation peak at 0.15 V and reduction peak at 0.05 V are associated with the formation of AgCl on the electrode and the reduction of this sparingly soluble salt.| Ag + Cl− ⇌ AgCl + e− | (7) |
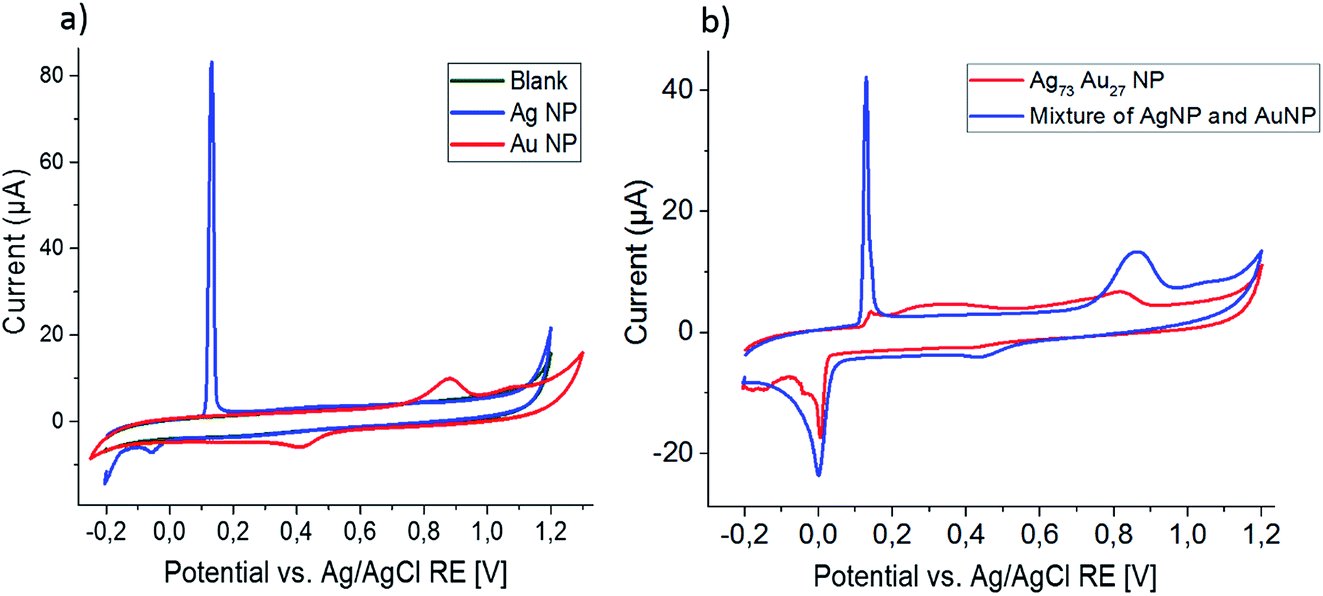 | ||
| Fig. 3 Cyclic voltammogram of (a) a GCE modified with either Ag (blue) or Au (red) nanoparticles and a bare GCE (green); (b) a mixture of these monometallic nanoparticles (blue) and of the bimetallic AgAu alloy nanoparticles (red), recorded in 0.1 M HCl at a scan rate of 0.025 V s−1. | ||
The second cycle (not shown) is identical to the first cycle in shape and position, but has a smaller peak area due to detachment of the AgCl from the electrode or chemical loss of AgCl from the electrode, forming soluble [AgCl1+y]y− species.38
The oxidation of Au under the same experimental conditions occurs under release of a mixture of soluble Au+ and Au3+ species, releasing 1.9 electrons per Au atom on average;16,39 accordingly no peaks are seen in the second cycle of the CV.
| Au + 2Cl− ⇌ [AuCl2]− + e− | (8) |
| Au + 4Cl− ⇌ [AuCl4]− + 3e− | (9) |
CVs recorded using electrodes modified with a mixture of Ag and Au nanoparticles appear as a combination of these two CVs, giving peaks of the same shape and at the same positions as the monometallic nanoparticles (Fig. 3b).
Using the same experimental parameters as above, the bimetallic Ag0.73Au0.27 nanoparticles were analyzed and the recorded CV is plotted in Fig. 3b. While only a small signal is seen at the peak position for Ag oxidation (0.15 V), a very broad peak emerges at potentials slightly higher than that, eventually merging into the Au oxidation peak seen above 0.7 V. The second cycle comprises greatly-increased, sharp oxidation and reduction peaks at the typical position seen for monometallic Ag particles, revealing that the broad peak seen before must be associated to the oxidation of silver from the bimetallic nanoparticles. Similar to the monometallic Ag nanoparticles, this Ag oxidation peak in the second cycle is not suitable for quantitative analysis of the silver released in the first cycle, as it shows a large variation over repeating experiments and is about ten times smaller than expected for Ag0.73Au0.27 nanoparticles. As for the monometallic nanoparticles, no Au oxidation peak is seen in the second scan.
To exclude mass transport limitations as the cause of this peak broadening, repetitive scans were performed starting in smaller potential windows and successively increasing to the full potential range. The obtained set of CVs is shown in Fig. 4. Scanning from −0.2 V to 0.4 V, in the first scan a small Ag oxidation peak at 0.15 V is followed by a broad oxidation peak at more positive potentials, and the increased reduction signal shows that this broad peak is associated with the formation of AgCl. In the second cycle, sharp and increased peaks are seen at 0.15 V and 0 V. Increasing the potential window to 0.7 V in the subsequent third scan, the sharp Ag oxidation peak seen before is followed by a broad oxidation peak starting at the end of the previous potential window (0.4 V) and an increased reduction peak of AgCl, showing that AgCl was formed in this broad oxidation. Continued cycling in the same potential window again confirms the disappearing of the broad silver oxidation peak, and the constant (or slightly decreased) size of the peaks at 0.15 V and 0 V shows that no additional silver is released from the nanoparticle. Further increasing the potential window to 1.2 V, gold oxidation occurs. The charge transferred in the following AgCl reduction was found to be the same as in the previous scan, indicating that almost no silver was left in the leached particle at that stage. Repeated scanning in this potential window shows a slight decrease in the Ag oxidation and AgCl reduction peaks (again due to anticipated loss of material from the electrode by chemical formation of dissolved species), while the absence of a Au oxidation peak confirms the quantitative stripping in the previous (fifth) cycle.
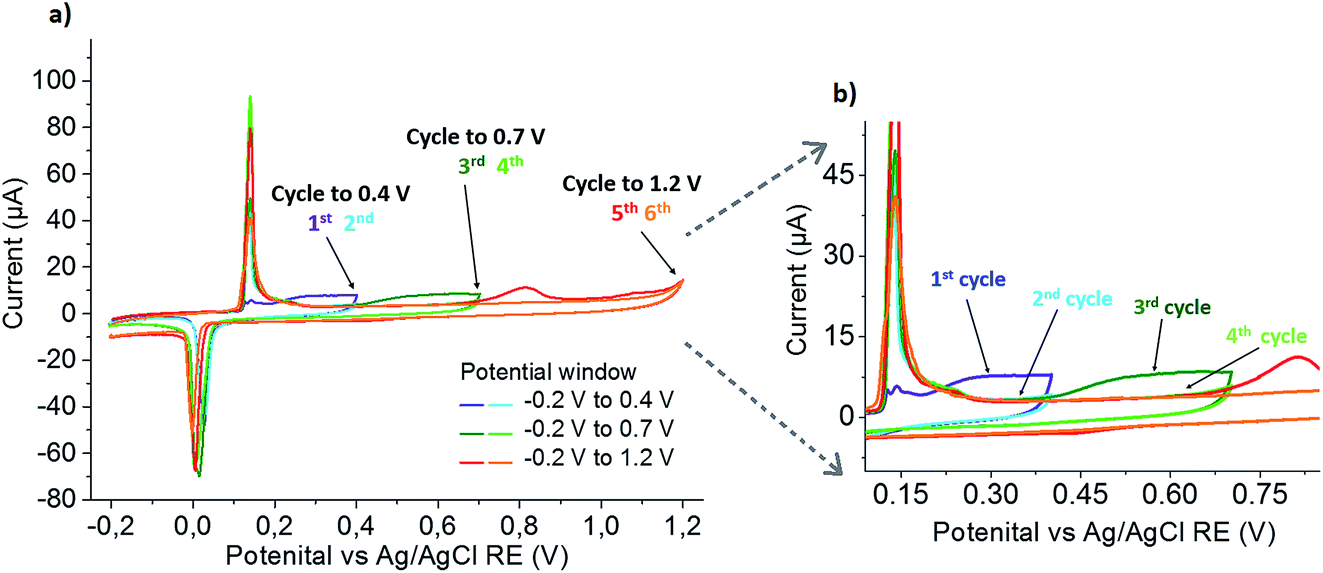 | ||
| Fig. 4 (a) Series of successive CVs of the Au0.73Ag0.27 NPs immobilized on a GCE in 0.1 M HCl, scanning to increasing upper potential limits; (b) an enlarged plot of the region attributed to alloy oxidation. | ||
This clearly proves that the broad Ag oxidation peak at potentials between 0.15 V and 0.7 V is not caused by mass transport limitations, but a consequence of alloying between Au and Ag in the bimetallic particle. Thus, the shift can be attributed to the thermodynamic shift of Ag oxidation to more positive values according to eqn (4), due to the decreased activity of Ag in an alloy Ag1−xAux (a = 1 − x; 0 < x < 1) as compared to its activity in pure Ag (a = 1).18
The oxidative leaching of the Ag from the alloy continuously reduces the Ag content in the alloy, and hence successively increases x and the oxidation potential of Ag to form AgCl. The reduction of AgCl forms monometallic Ag entities on top of the bimetallic sample nanoparticles, as is visible from the increased oxidation peak at 0.15 V in the following cycle.
While it is clear from this ensemble analysis that the bimetallic nanoparticles are alloy particles, the quantitative determination of the composition is not possible due to the broad oxidation peak of the Ag in the first cycle making a quantitative peak integration very difficult and the loss of Ag from the electrode prior to the sharp Ag oxidation in the second cycle. Naturally for the ensemble studies, also no information on the size of the individual nanoparticles can be gained. Thus, the nano impact method will be exploited in the following paragraph to study single bimetallic AgAu particles on a particle-by-particle scale.
Chronoamperometric nano impact experiments were performed using a dispersion of the bimetallic AgAu nanoparticles in 0.01 M HCl(aq) and a particle concentration of ca.10 pM. The current transients recorded at potentials ≥0.6 V showed distinct current spikes of several millisecond duration, while at lower potentials no such features were observed, as shown in Fig. 5a and b. Control experiments in the absence of nanoparticles did not contain any of these features, irrespective of the applied potential, verifying that these spikes originate from charge transfer during nanoparticle impact events (Fig. 5a, ‘blank’).
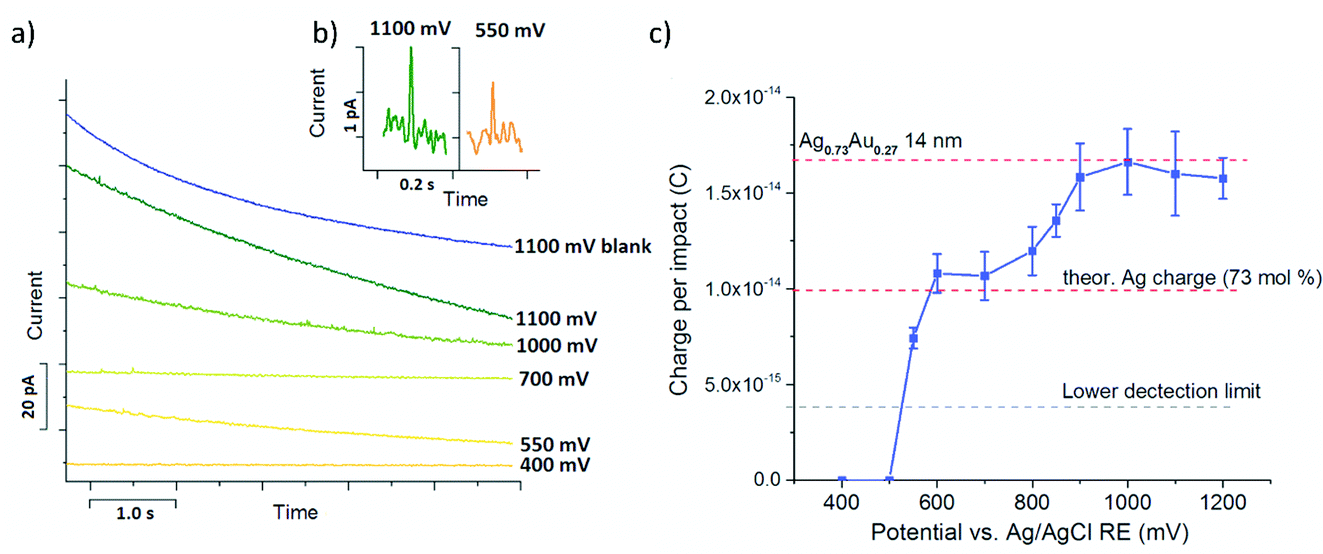 | ||
| Fig. 5 (a) Chronoamperograms recorded in 0.01 M HCl at different potentials in the presence and absence of Au0.73Ag0.27 NPs, shifted along the y-axis for better clarity; (b) typical spike shapes and sizes seen at two different potentials and (c) detected average spike charge as a function of applied potential. | ||
Analysis of the average charge per impact as a function of the potential applied yielded the data summarized in Fig. 5c. Taking eqn (6), the charge expected for quantitative oxidation of a 14 nm sized Ag0.73Au0.27 nanoparticle (7 nm radius), is 1.67 × 10−14 C, while the exclusive oxidation of Ag should result in a charge of 0.98 × 10−14 C (as indicated by the red dashed lines). Also indicated is the detection limit as the grey dashed line.
From this it is evident that spikes recorded at potentials smaller than the Au oxidation potential (≤0.7 V) gave a charge that is in excellent agreement with the charge expected for Ag0.73Au0.27 nanoparticles of 14 nm diameter.
Notably the minimum potentials at which spikes were recorded was significantly above that reported for monometallic citrate-capped Ag nanoparticles in chloride containing solutions (≈0.05 V), and also higher than that seen in the ensemble studies shown above (≈0.1 V). In a previous work employing cetyl trimethylammonium bromide CTAB-capped nanoparticles, a dominating role of the capping agent in the particle oxidation potential was reported.40 Thus, to quantify the stabilizing effect of the PVP capping agent potentially causing this shift, nano impact experiments using monometallic Ag nanoparticles synthesized using the same procedure as for the AgAu particle synthesis were tested in the same electrolyte. Oxidative Ag impacts were already seen at potentials ≥0.2 V (not shown), proving that this increase in the Ag oxidation potential can not be attributed to a mere PVP capping agent-induced effect. Conversely, this demonstrates that alloying of silver in the bimetallic AgAu nanoparticles can be detected on an individual particle scale, due to the associated thermodynamic increase of the Ag oxidation potential. These findings are in excellent agreement with the nanoparticle ensemble studies described above.
The further increase of the spike charge at potentials exceeding 0.7 V reveals the onset of the Au oxidation in the impacting particles. At potentials ≥0.9 V a constant average charge per impact is detected, marking the oxidation of the entire AgAu nanoparticle. As indicated in Fig. 5c, this is in excellent agreement with the charge expected for the quantitative oxidation of the 14 nm sized bimetallic AgAu particles used. This clearly demonstrates that nano impact experiments can provide both size and compositional analysis of bimetallic alloy nanoparticles on an individual particle scale.
Notably the observed relatively shallow increase of the spike charge above this potential is different from the charge-potential dependence reported in previous works for the oxidation of Ag, Au and Au-core Ag-shell nanoparticles. In these cases a steep, step-like increase of the spike charge above a threshold potential was seen in Cl− containing electrolytes.16 It is speculated that this reflects either an increase in the Au oxidation potential in an alloy, due to the fast time scale of the nano impact oxidation as compared to the slower process of de-alloying or a diffusional limitation of Au dissolution due to the quickly-formed porous AgCl network, which hinders the diffusion of Cl− and [AuCl1−y]−. Further investigation using bimetallic alloy particles of different Ag:Au compositions and in different electrolytes are planned to gain further insight into this aspect.
5. Conclusions
In this work the single entity methodology of electrochemical nano impact experiments was used to study 14 nm sized bimetallic AgAu alloy nanoparticles dispersed in an aqueous electrolyte. It was found that both the sizing and compositional analysis of individual nanoparticles is possible by virtue of chronoamperometry at different potentials. The presence of an alloying effect stabilising the silver in the bimetallic particles and thus shifting the onset of silver oxidation to more positive potentials was also demonstrated on this individual particle basis. Cyclic voltammetry ensemble studies confirm the results of these single entity experiments.Thus, the work highlights electrochemistry as a powerful tool to characterise bimetallic alloy nanoparticles and suggests nano impact analysis as a convenient complement or alternative to vacuum characterization techniques used today. It is expected that this methodology can readily be applied to study a variety of different bimetallic and multi-component nanoparticles directly and conveniently in a dispersed state.
Acknowledgements
The authors are grateful to Dr Dario Omanovic (Ruder Boscovic Institute Zagreb, Croatia) for providing the peak analysis software SignalCounter and Sandra Schmidt (RUB) for assistance in the SEM imaging. This work was financially supported by the Ministry of Innovation, Science and Research of the State of North Rhine-Westphalia (NRW Rückkehrerprogramm, K. T.) and the Cluster of Excellence RESOLV (EXC 1069) funded by the German Research Foundation (DFG, K. T. and E. N. S.). M. E. thanks the Deutsche Forschungsgemeinschaft (DFG) for funding of the project Ep 22/44-1.References
- C. N. R. Rao, G. U. Kulkarni, P. J. Thomas and P. P. Edwards, Chem. Soc. Rev., 2000, 29, 27–35 RSC.
- R. Kessler, Environ. Health Perspect., 2011, 119, A120–A125 CrossRef PubMed.
- I. Freestone, N. Meeks, M. Sax and C. Higgitt, Gold Bull., 2007, 40, 270–277 CrossRef CAS.
- M. Faraday, Phil. Trans. R. Soc., 1857, 147, 145–181 CrossRef.
- A. Howie, L. D. Marks and S. J. Pennycook, Ultramicroscopy, 1982, 8, 163–174 CrossRef CAS.
- H. Amiri, L. Bordonali, A. Lascialfari, S. Wan, M. P. Monopoli, I. Lynch, S. Laurent and M. Mahmoudi, Nanoscale, 2013, 5, 8656 RSC.
- P. B. Santhosh and N. P. Ulrih, Cancer Lett., 2013, 336, 8–17 CrossRef CAS PubMed.
- J. R. Croy, S. Mostafa, L. Hickman, H. Heinrich and B. R. Cuenya, Appl. Catal., A, 2008, 350, 207–216 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- P. Mulvaney, M. Giersig and A. Henglein, J. Phys. Chem., 1993, 97, 7061–7064 CrossRef CAS.
- X. López Lozano, C. Mottet and H.-C. Weissker, J. Phys. Chem. C, 2013, 117, 3062–3068 Search PubMed.
- G. Guisbiers, R. Mendoza-Cruz, L. Bazán-Díaz, J. J. Velázquez-Salazar, R. Mendoza-Perez, J. A. Robledo-Torres, J.-L. Rodriguez-Lopez, J. M. Montejano-Carrizales, R. L. Whetten and M. José-Yacamán, ACS Nano, 2016, 10, 188–198 CrossRef CAS PubMed.
- A. J. Logsdail and R. L. Johnston, J. Phys. Chem. C, 2012, 116, 23616–23628 CAS.
- H. Zhang and N. Toshima, Appl. Catal., A, 2012, 447, 81–88 CrossRef.
- S. Ristig, O. Prymak, K. Loza, M. Gocyla, W. Meyer-Zaika, M. Heggen, D. Raabe and M. Epple, J. Mater. Chem. B, 2015, 3, 4654–4662 RSC.
- L. R. Holt, B. J. Plowman, N. P. Young, K. Tschulik and R. G. Compton, Angew. Chem. Int. Ed., 2016, 55, 397–400 CrossRef CAS PubMed.
- K. Tschulik, K. Ngamchuea, C. Ziegler, M. G. Beier, C. Damm, A. Eychmueller and R. G. Compton, Adv. Funct. Mater., 2015, 25, 5149–5158 CrossRef CAS.
- A. L. Pawlowych, D. L. Pile, H. W. Pickering and K. G. Weil, Z. Phys. Chem., 1998, 207, 113–126 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450–453 CrossRef CAS PubMed.
- A. J. Bard, H. Zhou and S. J. Kwon, Isr. J. Chem., 2010, 50, 267–276 CrossRef CAS.
- S. J. Kwon, H. Zhou, F.-R. F. Fan, V. Vorobyev, B. Zhang and A. J. Bard, Phys. Chem. Chem. Phys., 2011, 13, 5394–5402 RSC.
- S. E. F. Kleijn, B. Serrano-Bou, A. I. Yanson and M. T. M. Koper, Langmuir, 2013, 29, 2054–2064 CrossRef CAS PubMed.
- R. Dasari, K. Tai, D. A. Robinson and K. J. Stevenson, ACS Nano, 2014, 8, 4539–4546 CrossRef CAS PubMed.
- D. A. Robinson, J. J. Yoo, A. D. Castañeda, B. Gu, R. Dasari, R. M. Crooks and K. J. Stevenson, ACS Nano, 2015, 9, 7583–7595 CrossRef CAS PubMed.
- X. Li, C. Batchelor-McAuley, S. A. I. Whitby, K. Tschulik, L. Shao and R. G. Compton, Angew. Chem. Int. Ed., 2016, 55, 4296–4299 CrossRef CAS PubMed.
- J. J. Yoo, J. Kim and R. M. Crooks, Chem. Sci., 2015, 6, 6665–6671 RSC.
- M. Kang, D. Perry, Y.-R. Kim, A. W. Colburn, R. A. Lazenby and P. R. Unwin, J. Am. Chem. Soc., 2015, 137, 10902–10905 CrossRef CAS PubMed.
- C.-H. Chen, E. R. Ravenhill, D. Momotenko, Y.-R. Kim, S. C. S. Lai and P. R. Unwin, Langmuir, 2015, 31, 11932–11942 CrossRef CAS PubMed.
- K. Tschulik and R. G. Compton, Phys. Chem. Chem. Phys., 2014, 16, 13909 RSC.
- K. Shimizu, K. Tschulik and R. G. Compton, Chem. Sci., 2016, 7, 1408–1414 RSC.
- W. Cheng and R. G. Compton, Angew. Chem. Int. Ed., 2014, 53, 13928–13930 CrossRef CAS PubMed.
- Y.-G. Zhou, N. V. Rees and R. G. Compton, Angew. Chem. Int. Ed., 2011, 50, 4219–4221 CrossRef CAS PubMed.
- C. S. Lim, S. M. Tan, Z. Sofer and M. Pumera, ACS Nano, 2015, 9, 8474–8483 CrossRef CAS PubMed.
- H. S. Toh, K. Jurkschat and R. G. Compton, Chem.–Eur. J., 2015, 21, 2998–3004 CrossRef CAS PubMed.
- V. Brasiliense, A. N. Patel, A. Martinez-Marrades, J. Shi, Y. Chen, C. Combellas, G. Tessier and F. Kanoufi, J. Am. Chem. Soc., 2016, 138, 3478–3483 CrossRef CAS PubMed.
- S. Ristig, S. Chernousova, W. Meyer-Zaika and M. Epple, Beilstein J. Nanotechnol., 2015, 6, 1212–1220 CrossRef CAS PubMed.
- S. G. D. Shackleford, C. Boxall, S. N. Port and R. J. Taylor, J. Electroanal. Chem., 2002, 538–539, 109–119 CrossRef CAS.
- C. Batchelor-McAuley, A. Martinez-Marrades, K. Tschulik, A. N. Patel, C. Combellas, F. Kanoufi, G. Tessier and R. G. Compton, Chem. Phys. Lett., 2014, 597, 20–25 CrossRef CAS.
- Y.-G. Zhou, N. V. Rees, J. Pillay, R. Tshikhudo, S. Vilakazi and R. G. Compton, Chem. Commun., 2011, 48, 224–226 RSC.
- B. J. Plowman, K. Tschulik, N. P. Young and R. G. Compton, Phys. Chem. Chem. Phys., 2015, 17, 26054–26058 RSC.
| This journal is © The Royal Society of Chemistry 2016 |