 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward fully quantum modelling of ultrafast photodissociation imaging experiments. Treating tunnelling in the ab initio multiple cloning approach
Dmitry V.
Makhov
a,
Todd J.
Martinez
b and
Dmitrii V.
Shalashilin
a
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: D.Makhov@leeds.ac.uk; D.Shalashilin@leeds.ac.uk
bDepartment of Chemistry, Stanford University, Stanford, CA 94305, USA
First published on 2nd May 2016
Abstract
We present an account of our recent effort to improve simulation of the photodissociation of small heteroaromatic molecules using the Ab Initio Multiple Cloning (AIMC) algorithm. The ultimate goal is to create a quantitative and converged technique for fully quantum simulations which treats both electrons and nuclei on a fully quantum level. We calculate and analyse the total kinetic energy release (TKER) spectra and Velocity Map Images (VMI), and compare the results directly with experimental measurements. In this work, we perform new extensive calculations using an improved AIMC algorithm that now takes into account the tunnelling of hydrogen atoms. This can play an extremely important role in photodissociation dynamics.
I. Introduction
Quantum non-adiabatic molecular dynamics is a powerful tool for understanding the details of the mechanisms of important photo-induced processes, such as the photodissociation of pyrrole and other heteroaromatic molecules. In these processes, quantum effects such as electronically non-adiabatic transitions and tunnelling are important, and an approach that goes beyond surface hopping, such as multiconfigurational time dependent Hartree (MCTDH),1 for example, is often required. MCTDH can be very accurate, and was recently used to simulate the dissociation of pyrrole.2 However it needs a parameterized potential energy surface as a starting point, which significantly restricts its practicality. A good alternative is represented by a variety of methods3–11 based on trajectory-guided Gaussian basis functions (TBF). Despite the fact that such approaches use classical trajectories, they are still fully quantum mechanical because these trajectories are employed only for propagating the basis, while the evolution of their amplitudes and, thus, of the total nuclear wave-function is determined by the time-dependent Schrödinger equation. An important advantage of trajectory-guided quantum dynamics methods is that they are fully compatible with direct or ab initio molecular dynamics where excited state energies, gradients, and non-adiabatic coupling terms are evaluated on the fly simultaneously with the nuclear propagation. The disadvantage is that trajectory based direct dynamics is very expensive due to the high cost of electronic structure calculations and typically can afford only a limited number of trajectories, which can be an obstacle to full convergence.Recently, we introduced the ab initio multiple cloning (AIMC)10 method, where TBFs are moving along Ehrenfest trajectories, as in the multiconfigurational Ehrenfest (MCE)8,9 approach, with bifurcation of the wave-functions taken into account via basis function cloning. While leading to the growth of the number of trajectories, the use of cloning helps to adopt the basis set to quantum dynamics significantly better than in the classical MCE approach. AIMC also uses a number of tricks to efficiently sample the trajectory basis and to use the information obtained on the fly: (1) similar to the previously developed trajectory based methods AIMC relies on importance sampling of initial conditions. (2) AIMC uses the so called time displaced or train basis sets,10,12,13 which increase the basis set size almost without any additional extra cost by reusing the ab initio data which has already been obtained. (3) The method calculates quantum amplitudes in a “post-processing technique” after the trajectories of the basis set functions have been found. As a result, the trajectories can be calculated one by one in parallel and good statistics can be accumulated.
In this work, we present a new implementation of the AIMC approach that is improved to take into account the tunnelling of hydrogen atoms by identifying possible tunnelling points and placing additional TBFs of the other side of the barrier. We use this new implementation to simulate the dynamics of the photodissociation of pyrrole, a process where tunnelling can play a very important role. We calculate the TKER spectrum and velocity map image (VMI), and directly compare the results of our calculations with experimental observations.14
The paper is organized as follows. In Section II we describe the proposed implementation of the AIMC approach. Section III contains the computational details of our simulations. In Section IV, we present and discuss the results. Conclusions are given in Section V.
II. Theory
II.1 Working equations
The AIMC method10 is based on the same ansatz as the multiconfigurational Ehrenfest (MCE) approach,8,9 in which the total wave-function |Ψ(t)〉 is represented in a trajectory-guided basis |ψn(t)〉: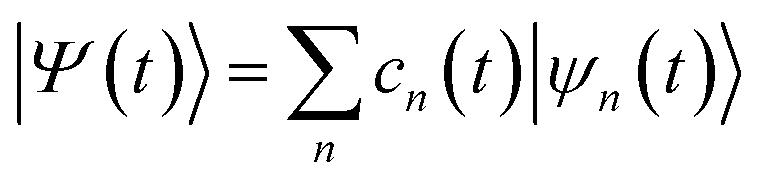 | (1) |
The basis functions |ψn(t)〉 are composed of nuclear and electronic parts:
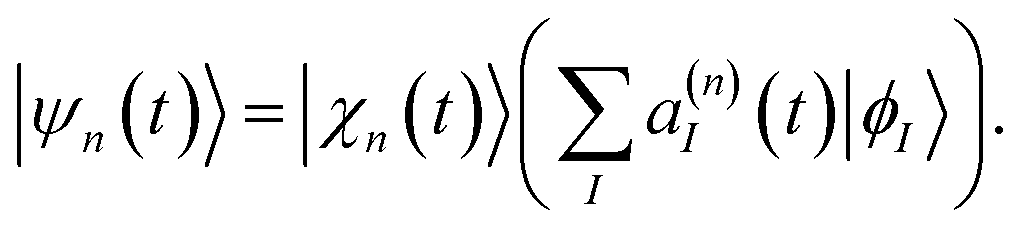 | (2) |
The nuclear part |χn(t)〉 is a Gaussian coherent state moving along an Ehrenfest trajectory:
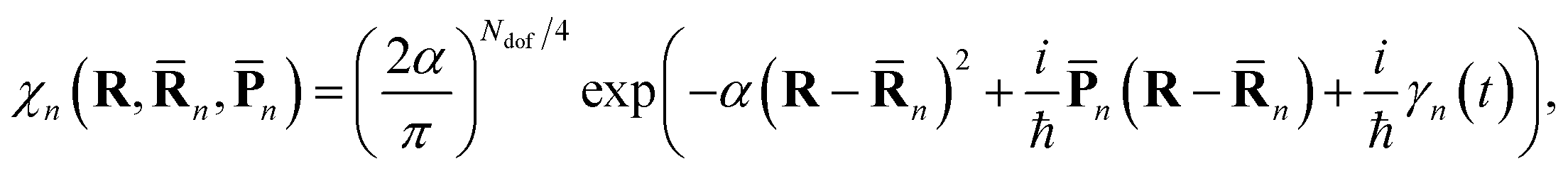 | (3) |
![[R with combining macron]](https://www.rsc.org/images/entities/b_char_0052_0304.gif) n(t) and
n(t) and ![[P with combining macron]](https://www.rsc.org/images/entities/b_char_0050_0304.gif) n(t) are the phase space coordinate and momentum vectors of the basis function centre, γn(t) is a phase, and the parameter α determines the width of the Gaussians. The electronic part of basis functions |ψn(t)〉 is represented as a superposition of several adiabatic eigenstates |ϕI〉 with quantum amplitudes a(n)I.
n(t) are the phase space coordinate and momentum vectors of the basis function centre, γn(t) is a phase, and the parameter α determines the width of the Gaussians. The electronic part of basis functions |ψn(t)〉 is represented as a superposition of several adiabatic eigenstates |ϕI〉 with quantum amplitudes a(n)I.
The time dependence of the Ehrenfest amplitudes a(n)I is given by the equations
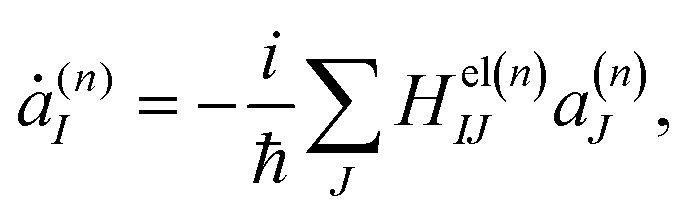 | (4) |
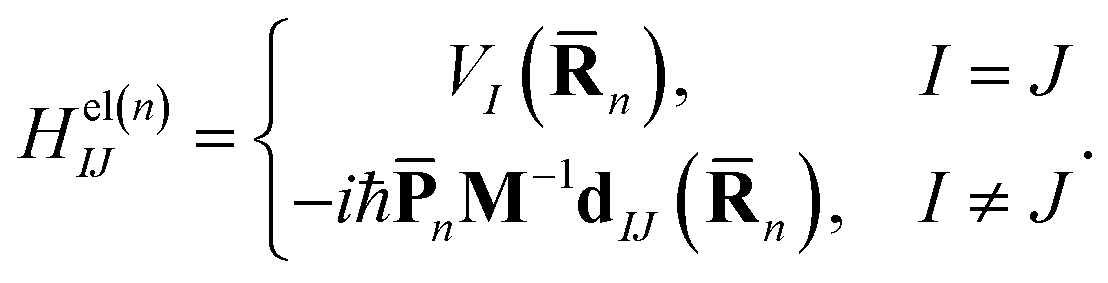 | (5) |
n) is the Ith potential energy surface and dIJ(n) = 〈ϕI|∇R|ϕJ〉 is the non-adiabatic coupling matrix element (NACME).
The motion of the centres of the Gaussians follows the standard Newton's equations:
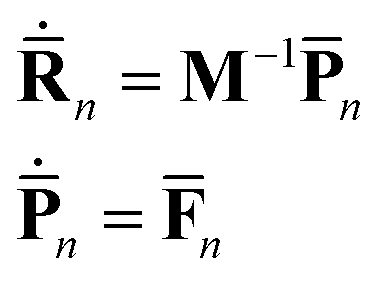 | (6) |
![[F with combining macron]](https://www.rsc.org/images/entities/b_char_0046_0304.gif) n is an Ehrenfest force that includes both the usual gradient term and the additional term related to the change of quantum amplitudes as a result of non-adiabatic coupling:
n is an Ehrenfest force that includes both the usual gradient term and the additional term related to the change of quantum amplitudes as a result of non-adiabatic coupling: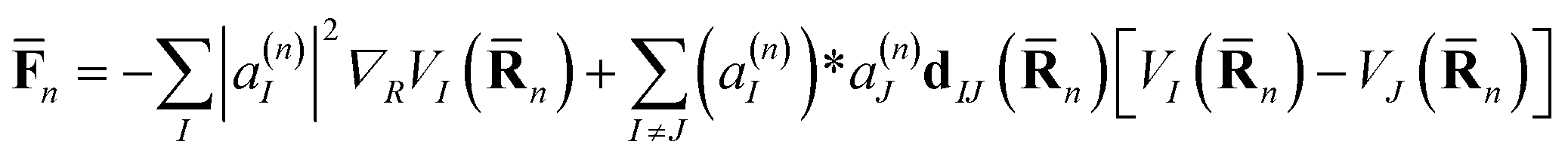 | (7) |
Finally, the phase γn evolves as:
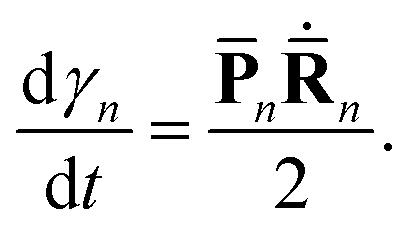 | (8) |
Eqn (3)–(8) form a complete set, determining the basis and its time evolution.
The evolution of the total wave-function |Ψ(t)〉 (eqn (1)) is defined by both the evolution of the basis functions |ψn(t)〉 and the evolution of the relevant amplitudes cn(t). The time dependence of the amplitudes cn(t) is given by the equation
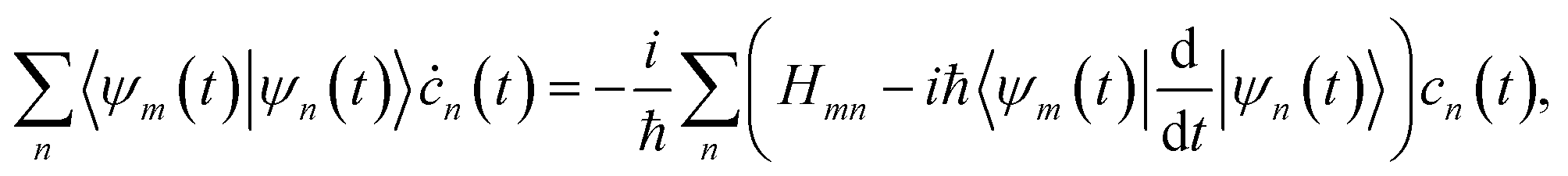 | (9) |
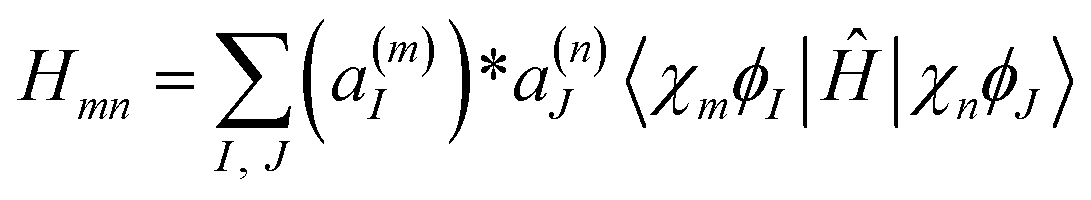 | (10) |
Assuming that the second derivative of the electronic wave-function |ϕI〉 with respect to R can be disregarded, we get:
 | (11) |
The matrix elements of the kinetic energy operator 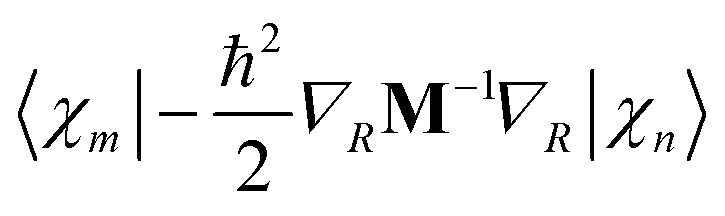 can be calculated analytically. For potential energy and non-adiabatic coupling matrix elements, we use a simple approximation:10
can be calculated analytically. For potential energy and non-adiabatic coupling matrix elements, we use a simple approximation:10
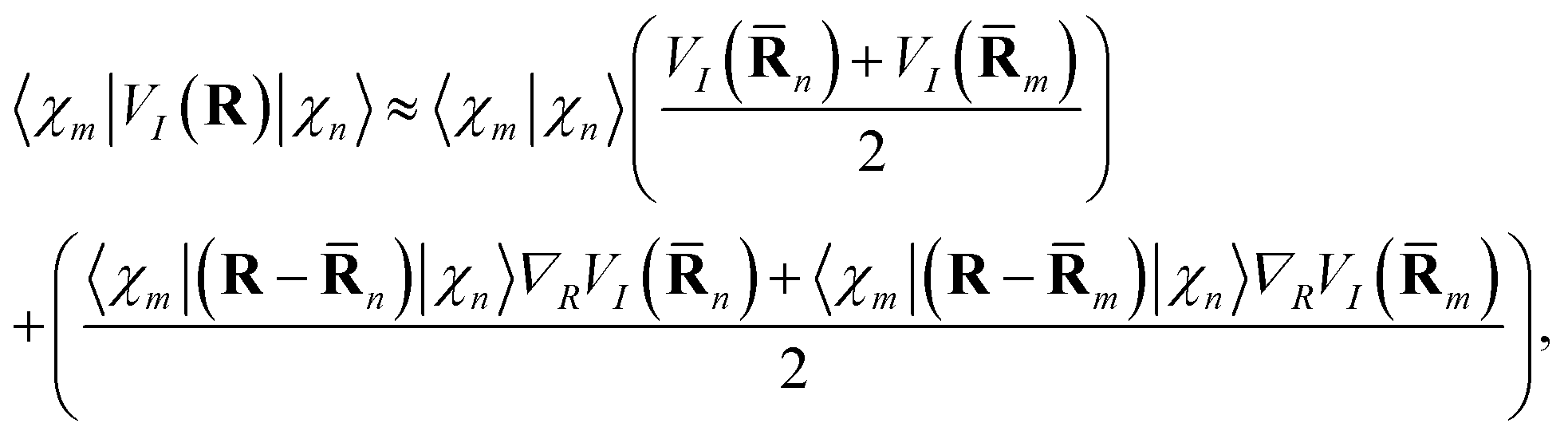 | (12) |
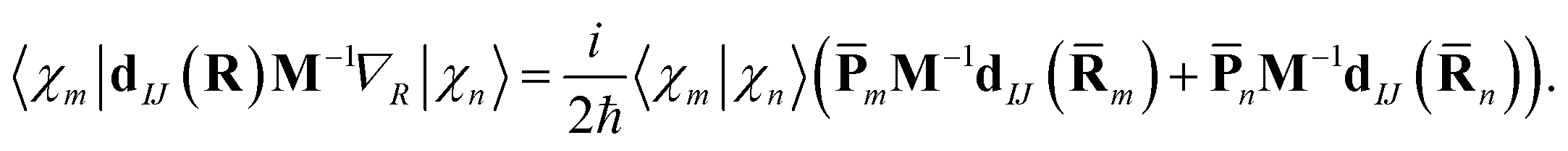 | (13) |
The approximation (12) represents a linear interpolation of the potential energy between the two points and can be improved further at the cost of calculating higher derivatives of the potential energy along the trajectories. It has been tested previously,10 and no visible change of the results was found when this approximation was applied compared to the saddle point approximation which expands around a distinct centroid for each pair of TBFs.4
The term 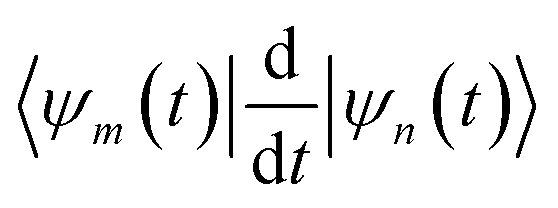 in eqn (9), which originates from the time dependence of the basis, can be expressed as:
in eqn (9), which originates from the time dependence of the basis, can be expressed as:
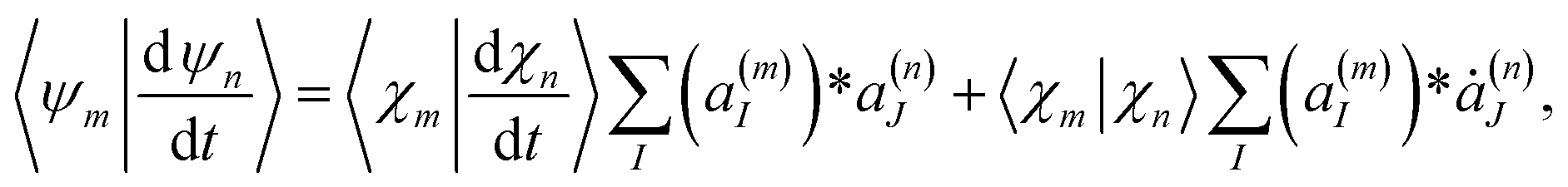 | (14) |
 | (15) |
Notice that in the AIMC approach, all off-diagonal matrix elements entering eqn (9) are calculated from the electronic structure data at the TBF centres, which is needed for the propagation of the basis. Thus, quantum coupling between the configurations comes at almost no extra cost. Moreover, eqn (9) can be solved after the trajectories have been calculated, provided the appropriate electronic structure information has been saved.
The detailed derivation of MCE equations together with the expressions for relevant matrix elements can be found in our previous works.10,11
II.2 Basis set sampling and cloning
The Ehrenfest basis set is guided by an average potential, which can be advantageous when quantum transitions are frequent. However, it becomes unphysical in regions of low non-adiabatic coupling when two or more electronic states have significant amplitudes: in this case, the difference of the shapes of potential energy surfaces for different electronic states should lead to branching of the wavepacket.In order to reproduce the bifurcation of the wave-function after leaving the non-adiabatic coupling region, AIMC methods adopt the cloning procedure,10 where the appropriate basis function is replaced by two basis functions, each guided (mostly) by a single potential energy surface. After the cloning event, an Ehrenfest configuration 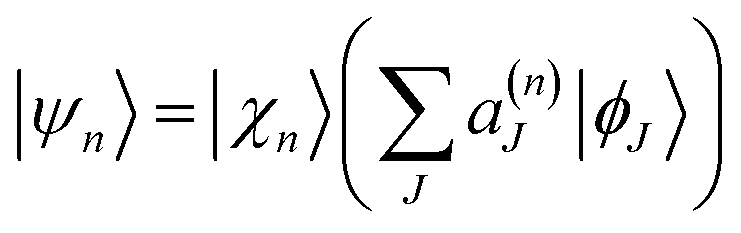 yields two configurations:
yields two configurations:
 | (16) |
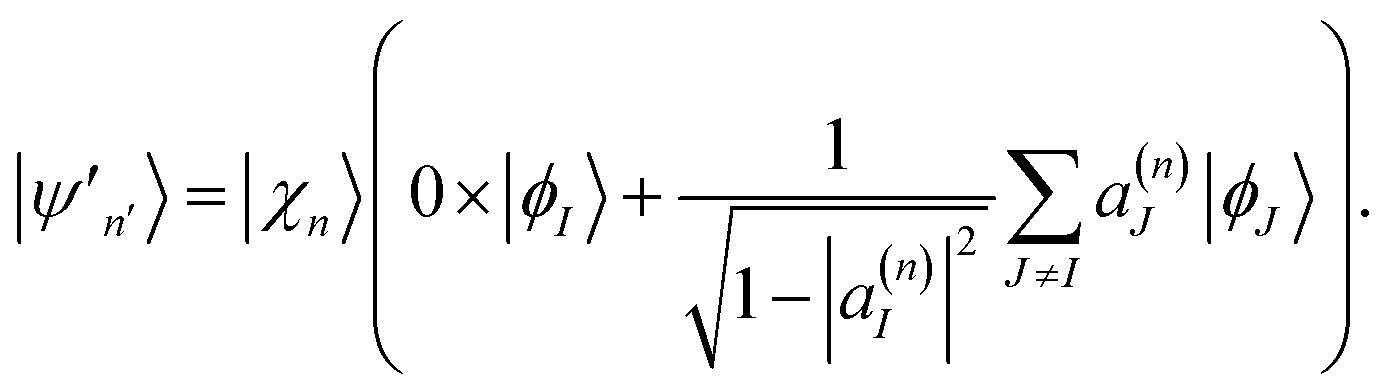 | (17) |
The first clone configuration has non-zero amplitudes for only one electronic state, and the second clone contains contributions of all other electronic states. The amplitudes of the two new configurations become:
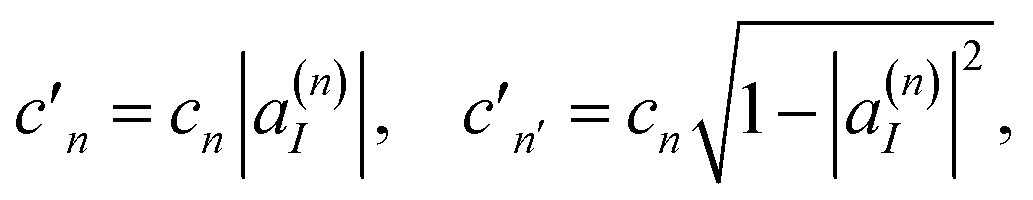 | (18) |
 | (19) |
We apply the cloning procedure shortly after a trajectory passes near a conical intersection, when the non-adiabatic coupling is lower than a threshold, and, at the same time, the so-called breaking force
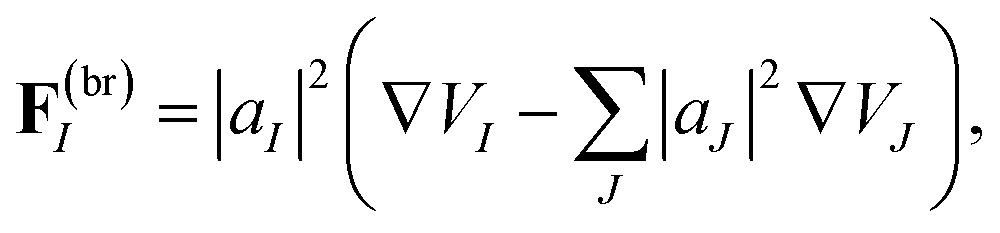 | (20) |
The cloning procedure is very much in spirit of the spawning, used in the Ab Initio Multiple Spawning approach (AIMS). Cloning does not require any back-propagation of spawned/cloned basis functions, unlike many4 (but not all15,16) implementations of spawning.
As has been described in our previous work,7 we rely on importance sampling when generating the initial conditions. Using the linearity of the Schrödinger equation, we first represent the initial wave-function as a superposition of Gaussians and then propagate each of them independently, “bit-by-bit”.7 We use a time-displaced basis set (coherent state trains), where several Gaussian basis functions are moving along the same trajectory but with a time-shift Δt, allowing us to reuse the same electronic structure data for each of the basis functions in the “train.” Fig. 1 shows a time displaced basis guided by a trajectory and its bifurcation via cloning. The best possible result with AIMC can be achieved when a swarm of trains is used to propagate each “bit” of the initial wave-function.
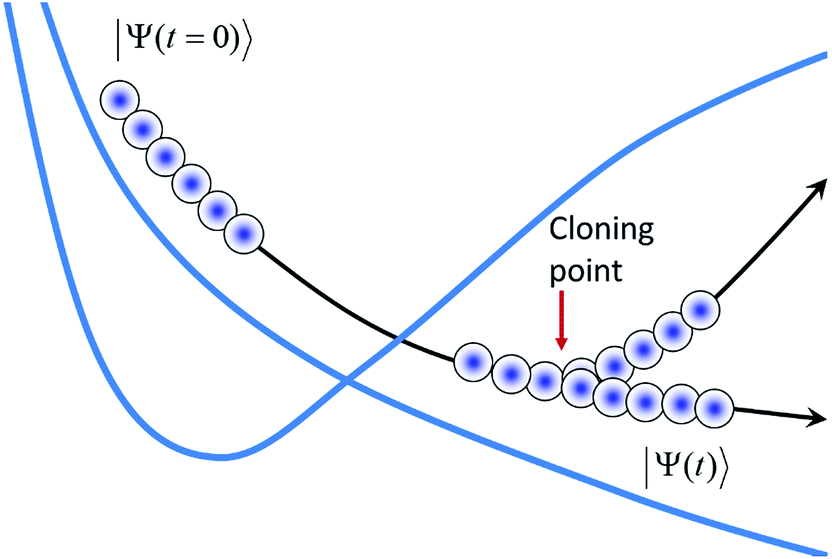 | ||
| Fig. 1 A sketch of the AIMC propagation scheme. The wave-function is represented as a superposition of Gaussian coherent states, which form a train moving along the trajectory. After passing the intersection, the train branches in the process of cloning. The figure shows a single train with cloning. In the most detailed AIMC calculation, a basis of several cloning trains interacting with each other is used. | ||
II.3 Tunnelling
The tunnelling of hydrogen atoms can play an important role in photodissociation processes. As mentioned above, MCE, AIMC and AIMS are fully quantum methods because classical trajectories are used only to propagate the basis, while the amplitudes cn(t) are found by solving the time dependent Schrödinger equation. When Gaussian basis functions are present on two sides of the potential barrier, the interaction between them can provide quantum tunnelling through the barrier. However, in the case of direct ab initio dynamics, the basis is usually very small, far from being complete. As a result, no basis functions normally would be present on the other side, and they must be placed there by hand in order to take tunnelling into account.In this paper we adopt the ideas17,18 previously used in the AIMS method to describe tunnelling for use with the AIMC technique. Fig. 2 illustrates the algorithm that we apply. First, we calculate the usual AIMC trajectories and find turning points, where the distance between the hydrogen atom and the radical reaches a local maximum. Then, for each of these turning points, we calculate the shape of the potential barrier: we increase manually the length of the N–H bond keeping all other degrees of freedom frozen, calculate potential energies, and find the point on the other side of the barrier with the same energy as in the turning point. If this point lies further than a set threshold from the turning point, we assume that tunnelling is not possible here, as the potential barrier is too wide. Otherwise, we use it as a starting point for an additional AIMC trajectory. The new trajectory is calculated both forward and backward in time, and the initial momenta are taken as the same as in the turning point, ensuring that new trajectories have the same total classical energies as their parent trajectories. This is exactly the procedure used in the multiple spawning approach, thus our method combines cloning for non-adiabatic events and spawning for tunnelling events. The forward propagation of new trajectories often involves branching as a result of cloning; backward propagation is performed without cloning and for a sufficiently short time, until new and parent trajectories separate in phase space.
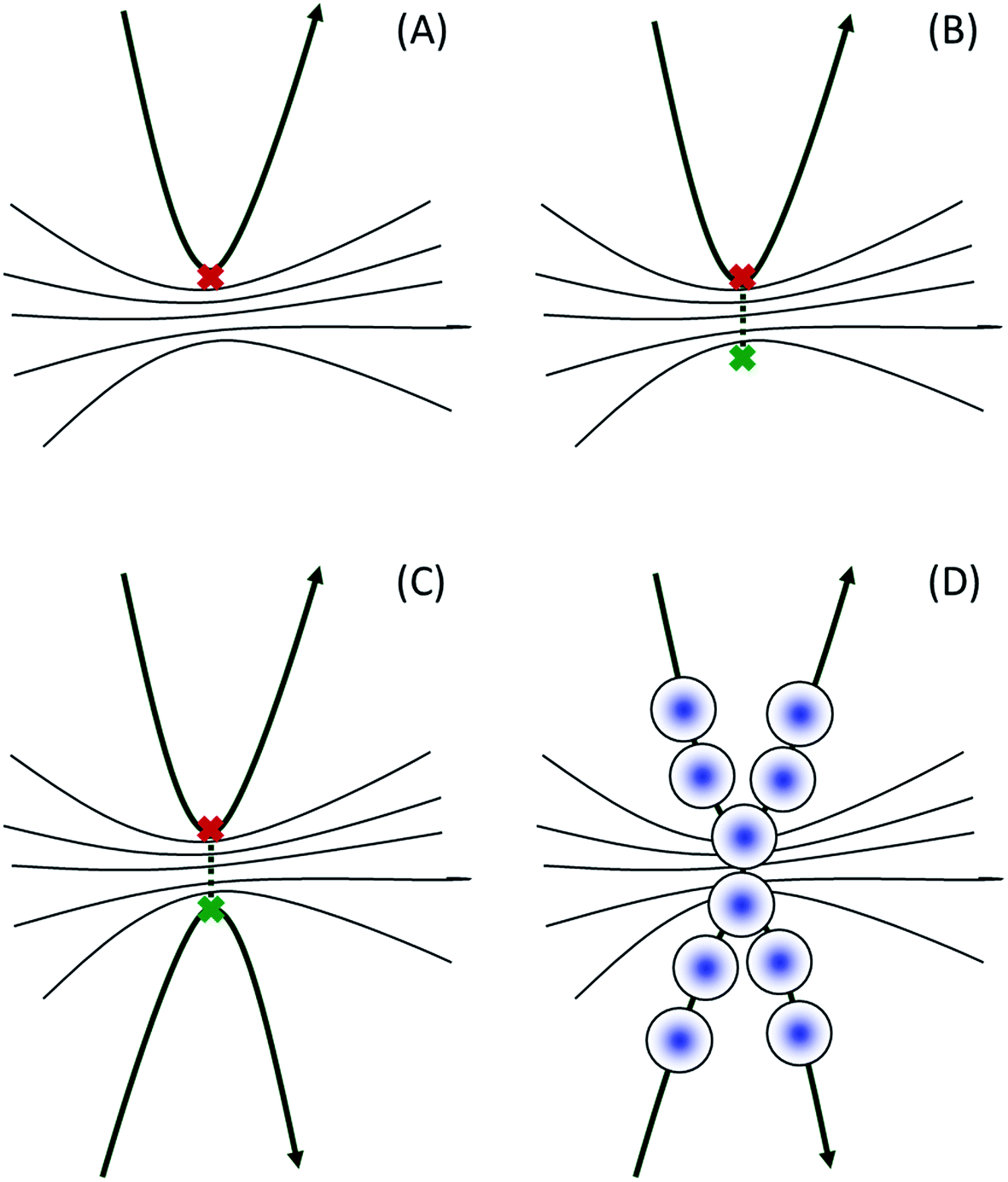 | ||
| Fig. 2 Illustration of the algorithm used to treat tunnelling in our approach. (A) Identify turning point; (B) find a point with the same potential energy on the opposite side of the barrier; (C) run an additional trajectory through this point; (D) solve time-dependent Schrödinger equation in the basis of a coherent state trains10 moving along the trajectories on both sides of the barrier. | ||
When all the trajectories are calculated, we solve eqn (9) for quantum amplitudes cn(t) in a time-displaced basis set (coherent state trains). This is similar to our previous approach10,11 but with the difference that now the basis is better adapted to treat tunnelling. The train basis on the new trajectory is placed in such way that it reaches the tunnelling point at the same time as the train basis on the parent trajectory. Because the new trajectory differs from its parent by only one coordinate at a tunnelling point, namely by the length of the N–H bond, there is a significant overlap between Gaussian basis functions belonging to these two trajectories. This interaction is retained for a significant time while the coherent state trains are passing the tunnelling point, ensuring the transfer of quantum amplitude across the barrier.
III. Computational details
Using our AIMC approach, we have simulated the dynamics of pyrrole following excitation to the first excited state. Trajectories were calculated using the AIMS-MOLPRO19 computational package, which has been modified to incorporate Ehrenfest dynamics. Electronic structure calculations were performed with the complete active space self-consistent field (CASSCF) method using the cc-pVDZ basis set. As in our previous works,9,11 we used an active space of eight electrons in seven orbitals (three ring π orbitals and two corresponding π* orbitals, one σ orbital and a corresponding σ* orbital). State averaging was performed over four singlet states using equal weights, i.e. the electronic wave-function is SA4-CAS(8,7)/cc-pVDZ. The width of Gaussian functions α was taken as 4.7 bohr−2 for hydrogen, 22.7 bohr−2 for carbon, and 19.0 bohr−2 for nitrogen atoms, as suggested in ref. 20. Three electronic states were taken into consideration during the dynamics – the ground state and the two lowest singlet excited states.The initial positions and momenta were randomly sampled from the ground state vibrational Wigner distribution in the harmonic approximation using vibrational frequencies and normal modes were calculated at the same CASSCF level of theory. We approximate the photoexcitation by simply lifting the ground state wavepacket to the excited state, as would be appropriate for an instantaneous excitation pulse within the Condon approximation. Of course, the fine details of the initial photoexcited wavepacket are lost in this approximation, however, we do not expect these details to have much effect on the observables shown in this paper.
We have run 900 initial Ehrenfest trajectories, each propagated with a time-step of ∼0.06 fs (2.5 a.u.) for 200 fs or until the dissociation occurred, defined as an N–H distance exceeding 4.0 Å. For a small number of trajectories, simulations exhibiting N–H dissociation were carried out to the full 200 fs in order to investigate the dynamics of the radical. Cloning was applied to TBFs when the breaking acceleration of eqn (20) exceeded a threshold of 5 × 10−6 a.u. and the norm of the non-adiabatic coupling vector was simultaneously less than 2 × 10−3 a.u. For all initial trajectories, as well as for their branches resulting from cloning, we identified turning points for the N–H bond length and calculated the width of the potential barrier. Additional trajectories on the other side of the barrier were placed if the width of the barrier did not exceed 0.5 bohr, which corresponds to an overlap of ∼0.3 between Gaussian basis functions. The new trajectories were propagated backward for 20 fs to accommodate the train basis set, and forward until dissociation or until the trajectory time exceeds 200 fs.
For each initial trajectory with all its branches and tunnelling sub-trajectories, we solved eqn (9) using a train basis set of N = 21 Gaussians per branch, separated by 10 time steps, which corresponds to an average overlap of ∼0.6 between the nearest Gaussians in the train. The total size of the basis is constantly changing because of the inclusion of new branches. The final amplitudes cn give statistical weights for each of the branches, which are used in the analysis that follows.
IV. Results
As a result of cloning, 900 initial configurations give rise to 1131 trajectory branches. This corresponds to an average of ∼0.25 cloning events per initial trajectory. For these branches, we have found 7702 local maxima of N–H bond length, of which 2376 have been identified as possible tunnelling points. For all these points, we run sub-trajectories, which finally gives 3203 additional branches, 4334 branches in total. The majority of these branches undergo N–H dissociation within our computational time of 200 fs: the total statistical weight of dissociative trajectories is 92%, of which 53% is the contribution of tunnelling sub-trajectories.The kinetic energy distribution of the ejected hydrogen atom is presented in Fig. 3 together with the experimental TKER spectrum.14 Both distributions clearly exhibit two contributions: a large peak at higher energies, and a small contribution at lower energies. It is important to note that adopting the basis set to tunnelling shifts the high-energy peak of TKER spectrum toward the lower energies by about ∼1000 cm−1 and makes the low-energy peak slightly more pronounced. While the calculated energies are still on average about 1.5 times higher than experimental values, this difference can be ascribed to the lack of dynamic electron correlation in the CASSCF potential energy surfaces. We previously showed11 that a more accurate MS-CASPT2 PES would lead to a shift in the kinetic energy peak of approximately ∼1800–1900 cm−1 towards lower energies, significantly improving the agreement with experiment.
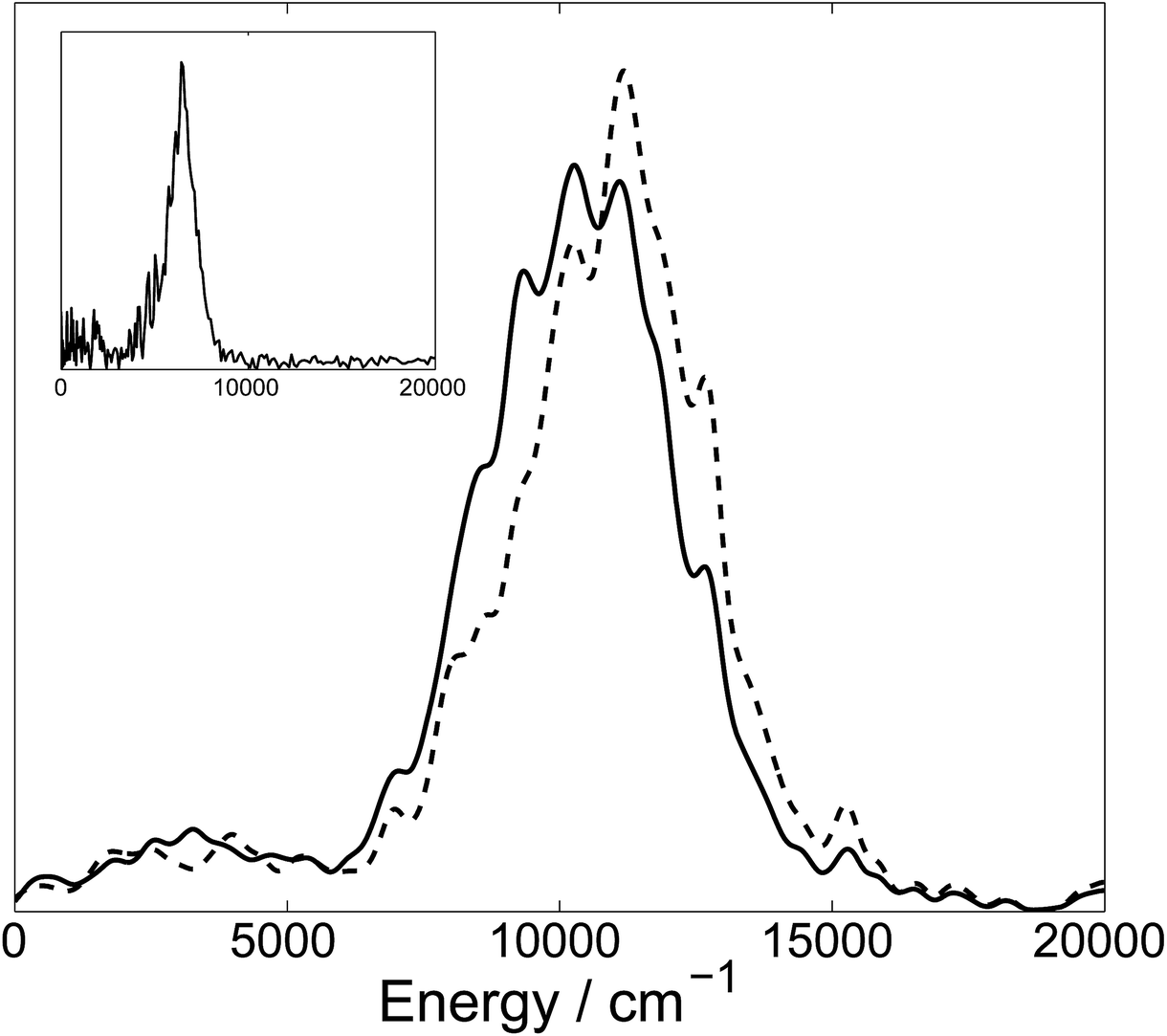 | ||
| Fig. 3 Total kinetic energy release (TKER) spectrum of hydrogen atoms after dissociation calculated with (solid) and without (dash) taking tunnelling into account. Both spectra are averaged over the same ensemble of initial configuration. The curves are smoothed by replacing delta-functions with Gaussian functions (σ = 200 cm−1). The inset shows the experimentally measured spectrum.14 | ||
Analysis of the electronic state amplitudes in the Ehrenfest configurations (eqn (2)) shows that the bifurcation of the wave-function while passing through a conical intersection plays an important role in the formation of a two-peak spectrum: the high kinetic energy product is predominantly in the ground state, while the low energy peak is formed by mostly low-weight branches with substantial contribution from excited electronic states. Fig. 4 presents an example of such a bifurcating trajectory. At about 55 fs after photoexcitation, this trajectory reaches an intersection for the first time. After passing the intersection, the ground and first excited states of the original TBF are approximately equally populated, so the cloning procedure is applied creating instead two TBF, one in the ground state and one in the excited state. At this point, the potential energy surfaces for the ground and excited states have opposite gradients. This leads to the acceleration of the hydrogen atom for the TBF associated with the ground state and, at the same time, slows it down for the exited state TBF. As a result, although both branches are leading to dissociation, the kinetic energies of the ejected atoms are significantly different: the ground state branch contributes to the high energy peak of the distribution in Fig. 3, while the excited state branch contributes to the low energy peak. For the ground state branch, the remaining vibrational energy of the radical is low, so it remains in the ground state for the rest of the run and does not reach the intersection again. For the excited state branch, the energy taken away by the hydrogen atom is lower, leaving the pyrrolyl radical with sufficient energy to pass through numerous intersections with population transfer between the ground and both excited states. Naturally, quenching to the ground state will happen eventually for this branch but the time scale of this process is much longer than that for the dissociation, while the TKER spectrum is only affected by the radical dynamics until the H atom is lost.
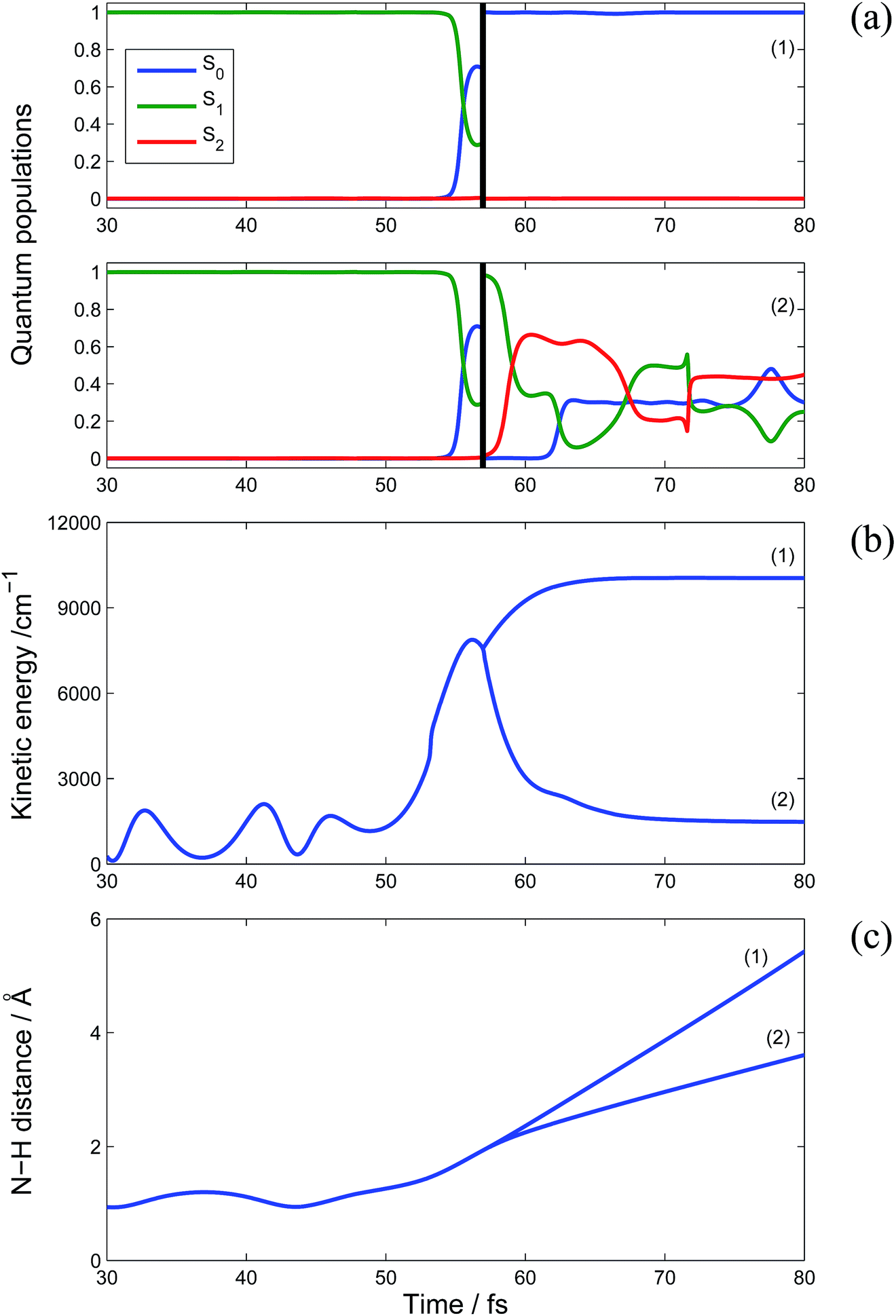 | ||
| Fig. 4 An example of trajectory bifurcation on conical intersection. Electronic state populations (a), the kinetic energy of the H atom (b) and the N–H distance (c) as a function of time. Fast and slow branches are referred as (1) and (2) respectively. The black vertical line indicates the moment when cloning was applied. | ||
In order to calculate the velocity map image with respect to the laser pulse polarization, we must average the velocity distribution of hydrogen atoms relative to the axes of the molecule, given by calculations, over all possible orientations of the molecule:
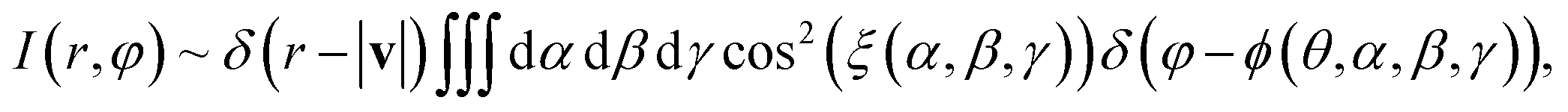 | (21) |
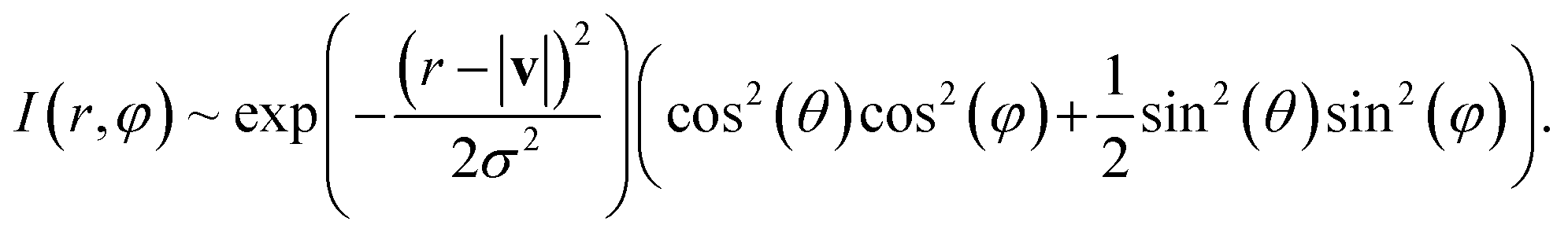 | (22) |
Fig. 5 shows the simulated velocity map with respect to the laser pulse polarization assuming that the transition dipole is normal to the molecular plane. The simulations reproduce well the main feature of the velocity map image, which is the anisotropy of the intense high energy part. Our results are also consistent with experiment14 in the low energy region showing an isotropic distribution, although admittedly the statistics of both experiment and simulation are poorer in the region of low energy.
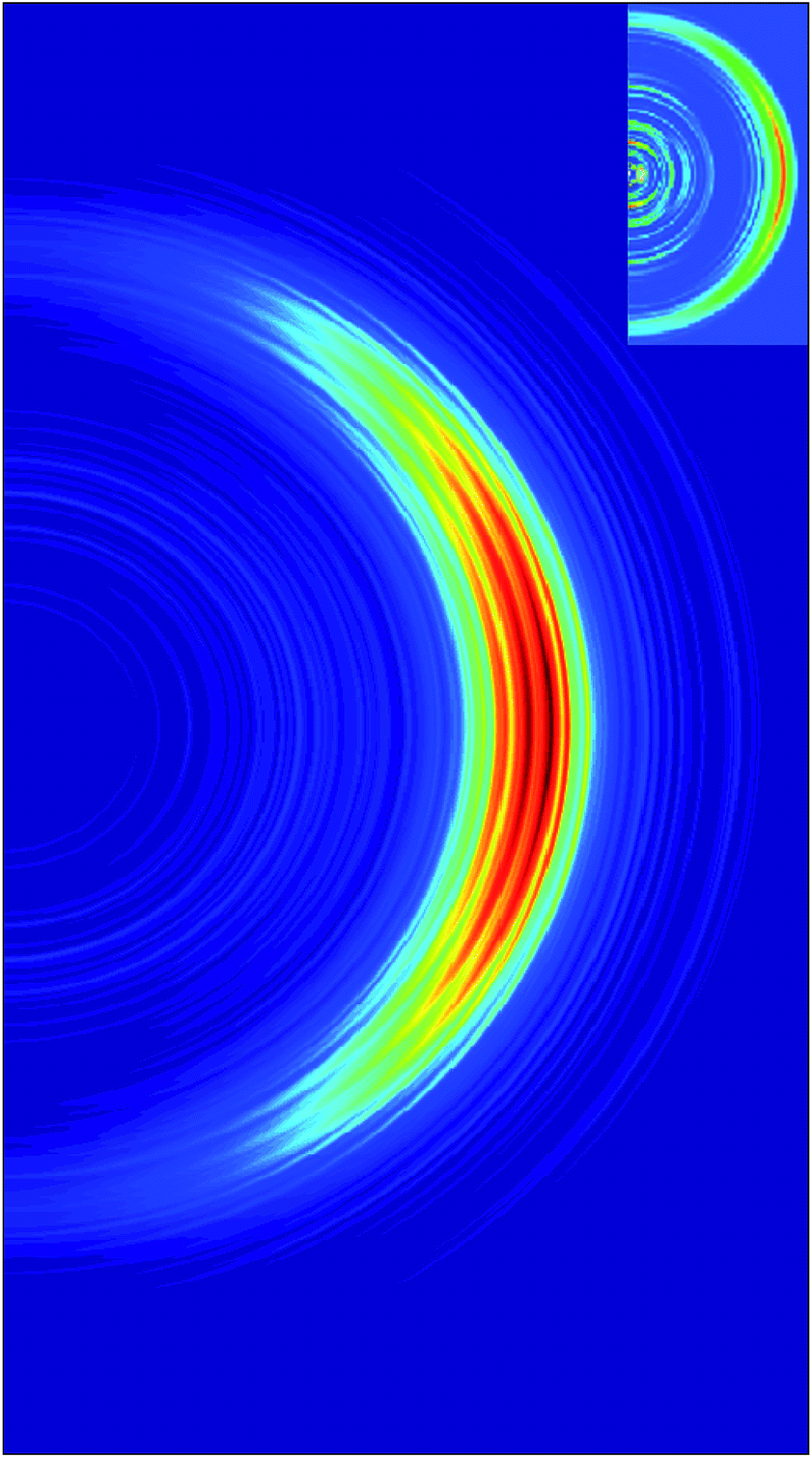 | ||
| Fig. 5 Simulated velocity map image with respect to the laser pulse polarization assuming that the transition dipole moment is normal to the molecule plane. The experimental VMI14 is shown in the inset. | ||
V. Conclusion
We simulated the photodissociation dynamics of pyrrole excited to the lowest singlet excited state (1A1 → 1A2) using a new implementation of the AIMC approach, which now is modified to take into account the tunnelling of hydrogen atoms more accurately. AIMC is a fully quantum technique but its computational cost in our implementation is compatible with classical “on the fly” molecular dynamics, which allows the accumulation of sufficient statistics to clarify the details of photo-induced processes in pyrrole. The treatment of tunnelling in our implementation provides a promising starting point for the further development of fully quantum methods for non-adiabatic dynamics and tunnelling with the ultimate goal of reaching well converged quantitative results. The current version of AIMC is already accurate enough to reproduce features of the experimentally observed TKER spectrum and velocity map images.Acknowledgements
DM and DS acknowledge the support from EPSRC through grants EP/J001481/1 and EP/N007549/1.References
- G. A. Worth, H.-D. Meyer, H. Köppel, L. S. Cederbaum and I. Burghardt, Using the MCTDH wavepacket propagation method to describe multimode non-diabatic dynamics, Int. Rev. Phys. Chem., 2008, 27, 569–606 CrossRef CAS
.
- G. Wu, S. P. Neville, O. Schalk, T. Sekikawa, M. N. R. Ashfold, G. A. Worth and A. Stolow, Excited state non-adiabatic dynamics of pyrrole: a time-resolved photoelectron spectroscopy and quantum dynamics study, J. Chem. Phys., 2015, 142, 074302 CrossRef PubMed
- T. J. Martinez, M. Ben-Nun and G. Ashkenazi, Classical/quantal method for multistate dynamics: a computational study, J. Chem. Phys., 1996, 104, 2847 CrossRef CAS
- M. Ben-Nun and T. J. Martínez, Ab Initio Quantum Molecular Dynamics, Adv. Chem. Phys., 2002, 121, 439 CrossRef CAS
- D. V. Shalashilin, Quantum mechanics with the basis set guided by Ehrenfest trajectories: theory and application to spin-boson model, J. Chem. Phys., 2009, 130(24), 244101 CrossRef PubMed
- S. L. Fiedler and J. Eloranta, Nonadiabatic dynamics by mean-field and surface hopping approaches: energy conservation considerations, Mol. Phys., 2010, 108(11), 1471–1479 CrossRef CAS
- D. V. Shalashilin, Nonadiabatic dynamics with the help of multiconfigurational Ehrenfest method: improved theory and fully quantum 24D simulation of pyrazine, J. Chem. Phys., 2010, 132(24), 244111 CrossRef PubMed
- D. V. Shalashilin, Multiconfigurational Ehrenfest approach to quantum coherent dynamics in large molecular systems, Faraday Disc., 2011, 153, 105 RSC
- K. Saita and D. V. Shalashilin, On-the-fly ab initio molecular dynamics with multiconfigurational Ehrenfest method, J. Chem. Phys., 2012, 137, 8 CrossRef PubMed
- D. V. Makhov, W. J. Glover, T. J. Martinez and D. V. Shalashilin,
Ab initio multiple cloning algorithm for quantum nonadiabatic molecular dynamics, J. Chem. Phys., 2014, 141(5), 054110 CrossRef PubMed
- D. V. Makhov, K. Saita, T. J. Martinez and D. V. Shalashilin,
Ab initio multiple cloning simulations of pyrrole photodissociation: TKER spectra and velocity map imaging, Phys. Chem. Chem. Phys., 2015, 17, 3316 RSC
- D. V. Shalashilin and M. S. Child, Basis set sampling in the method of coupled coherent states: coherent state swarms, trains and pancakes, J. Chem. Phys., 2008, 128, 054102 CrossRef PubMed
- M. Ben-Nun and T. J. Martinez, Exploiting Temporal Non-Locality to Remove Scaling Bottlenecks in Nonadiabatic Quantum Dynamics, J. Chem. Phys., 1999, 110, 4134–4140 CrossRef CAS
- G. M. Roberts, C. A. Williams, H. Yu, A. S. Chatterley, J. D. Young, S. Ullrich and V. G. Stavros, Probing ultrafast dynamics in photoexcited pyrrole: timescales for (1)pi sigma* mediated H-atom elimination, Faraday Discuss., 2013, 163, 95–116 RSC
- M. Ben-Nun and T. J. Martínez, A Continuous Spawning Method for Nonadiabatic Dynamics and Validation for the Zero-Temperature Spin-Boson Problem, Isr. J. Chem., 2007, 47, 75–88 CrossRef CAS
- S. Yang, J. D. Coe, B. Kaduk and T. J. Martínez, An “Optimal” Spawning Algorithm for Adaptive Basis Set Expansion in Nonadiabatic Dynamics, J. Chem. Phys., 2009, 130, 134113 CrossRef PubMed
- M. Ben-Nun and T. J. Martinez, Semiclassical tunneling rates from ab initio molecular dynamics, J. Phys. Chem. A, 1999, 103(31), 6055–6059 CrossRef CAS
- M. Ben-Nun and T. J. Martínez, A Multiple Spawning Approach to Tunneling Dynamics, J. Chem. Phys., 2000, 112, 6113–6121 CrossRef CAS
- B. G. Levine, J. D. Coe, A. M. Virshup and T. J. Martinez, Implementation of ab initio multiple spawning in the Molpro quantum chemistry package, Chem. Phys., 2008, 347(1), 3–16 CrossRef CAS
- A. L. Thompson, C. Punwong and T. J. Martinez, Optimization of width parameters for quantum dynamics with frozen Gaussian basis sets, Chem. Phys., 2010, 370, 70–77 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2016 |