
Macromolecule-based platforms for developing tailor-made formulations for scale inhibition†
Amir
Sheikhi‡
abc,
Na
Li‡
ac,
Theo G. M.
van de Ven
*abc and
Ashok
Kakkar
*ac
aDepartment of Chemistry, McGill University, 801 Sherbrooke St. West, Montreal, Quebec H3A 0B8, Canada. E-mail: ashok.kakkar@mcgill.ca; Fax: +1 514 398 3797; Tel: +1 514 398 6912
bPulp and Paper Research Centre, Department of Chemistry, McGill University, Montreal, Quebec H3A 0B8, Canada. E-mail: theo.vandeven@mcgill.ca
cCentre for Self-Assembled Chemical Structures, McGill University, Montreal, Quebec H3A 0B8, Canada
First published on 8th October 2015
Abstract
Inorganic crystallization, commonly referred to as mineral scale formation, has posed tremendous challenges and is one of the leading assurance problems in water-based industries. A detailed understanding of the mechanism and influencing factors for the initiation and build-up of deposited scale is not only highly relevant for many industries but has also catalyzed academic research in developing efficient antiscalants. Macromolecules that can stop nucleation and inhibit crystallization or interact with forming crystals and modify their morphology to retard further growth have been the focus of intense scientific endeavors. There has been immense activity in developing additives which can regulate unwanted inorganic crystallization and understanding the complexity of how they work in preventing scale deposits. In this review, after a summary of the controlling parameters that define mineral scale growth, we review opportunities generated by using macromolecules as a platform for developing inhibitors for the two most common scale deposits, i.e. calcium salts and silica, with a discussion on their efficiencies in controlling nucleation and changing growing crystal morphology.
 Amir Sheikhi | Amir Sheikhi received his BEng (Hons) and MEng degrees in Chemical Engineering from the University of Tehran (Iran) and subsequently completed his PhD in Chemical Engineering from McGill University (Montreal, Canada) in 2014. He is currently a post-doctoral fellow in the laboratories of Profs. Theo van de Ven and Ashok Kakkar in the Department of Chemistry at McGill University. His research interests focus on a balance of theory and experiments to understand the complex behavior of soft interfaces, cellulose-based advanced nanomaterials, new technologies for environmental remediation, hyperbranched macromolecules, and chemical process design, simulation, and monitoring methods. |
 Na Li | Na Li completed her BSc and MSc studies in Chemical Engineering at Lanzhou University in China. She then went on to obtain a PhD degree in 2014 from University of Bordeaux in France working with Prof. Didier Astruc on a project related to gold nanoparticle sensors. She joined the groups of Professors Ashok Kakkar and Theo van de Ven in the Department of Chemistry at McGill University (Montreal, Canada) as a post-doctoral fellow in October 2014. Her research interests are in the area of designing nanomaterials based on macromolecules as well as metal nanoparticles for a variety of applications. |
 Theo G. M. van de Ven | Theo van de Ven is a Professor in the Department of Chemistry at McGill University in Montreal, Canada, and holds a Senior NSERC/FPInnovations Industrial Research Chair in “Colloid and Papermaking Chemistry”. He is the Scientific Director of a Strategic NSERC Research Network in “Innovative Green Wood Fiber Products”, Chair of FIBRE, a consortium of 8 networks, and the Director of FQRNT Centre for Self-Assembled Chemical Structures. He is an expert in colloid and surface chemistry and has published over 330 papers, among which are a book (Colloidal Hydrodynamics, Academic Press, 1989) and several book chapters. |
 Ashok Kakkar | Ashok Kakkar obtained his training in Chemistry under the directions of Professor Todd B. Marder (PhD University of Waterloo, Waterloo, Ontario, Canada), Professor The Lord Lewis (NSERC Post-doctoral Fellow, University of Cambridge, Cambridge, UK) and Professor Tobin Marks (NSERC Post-doctoral Fellow, Northwestern University, Evanston, USA). He is an Associate Professor in the Department of Chemistry at McGill University, Montreal, Canada. His research interests include developing methodologies to complex architectures such as hyperbranched macromolecules (dendrimers, miktoarm polymers) and metal nanoparticles (gold, iron oxide) for applications in a variety of areas including theranostics, imaging, water treatment etc. |
Water impactMineral scale deposition has plagued water-based manufacturing industries, and the impending economic and environmental costs to address this issue have propelled academics and industrial think tanks worldwide to develop novel antiscalants. Some of the key ingredients that can accelerate growth and aid in developing highly efficient inhibitors include i) a detailed understanding of the factors that contribute to scale build-up and ii) how to incorporate these in the intervention methodologies. This manuscript provides an evaluation of the crystallization mechanism of different inorganic salts and a critical review of the chemical approaches taken to alleviate it. Such an analysis could help in finding a fine balance in scale prevention while minimizing or completely eliminating environmental pollution in water treatment methods. |
Introduction
The growing water demand for manufacturing purposes, energy production, agriculture, and human consumption,1 on the one hand, and the global water shortage, on the other, have necessitated industries to micromanage water consumption for the entire operating process. The average worldwide industrial water usage is about 22% of the total consumption, and for high-income countries it reaches 59%.2 Water demand for manufacturing industries is anticipated to increase 400% by 2050 and is expected to play a key role in the increase of worldwide water demand to 55%.3 In 2008, according to the Water Industry Survey conducted by Environmental Business International Inc. (on behalf of Industry Canada), municipal wastewater treatment plants, oil and gas and food industries, residential usage, mining, power utilities, electronics, and pulp and paper were among the most water-demanding businesses.4 Feed water in these industrial plants, which is obtained from ion-rich natural resources or treated recycled water, often causes typical deposition problems including corrosion products such as metal oxides, microbiological deposits, suspended matter, and mineral scales.5 Industrial processes that utilize high pH/high alkalinity water often suffer from scaling, by which insoluble, strongly adherent deposits are formed on the surface of unit operations. Within 20 years (1980–2001), tailing pond water salinity in the Canada oil sands industry located in Alberta has increased by a significant rate of 75 mg L−1 per year, posing scaling and corrosion problems.6The deposition of sparingly soluble salts from cationic (e.g., Ca2+) and anionic (CO32−) species in industrial water causes serious operating issues. With a salinity of 35 g kg−1, standard sea water contains 10.3 mmol kg−1 (412 mg kg−1) Ca2+ and 1.75 mmol kg−1 (107 mg kg−1) HCO3− at pH ~8 at the sea surface.7 The scaling of oil-field injection water results in pipe blockage, which imposes extensive additional costs to the operating system.8,9 In addition, factors including reduced heat transfer coefficients10 due to the insulating effect of the scale, increased pressure drop, and fluid mechanics problems inside the unit operations due to scale deposition11 are among other important disadvantages of such salt deposits.
The most common scale composition, as for example in reverse osmosis (RO) desalination systems, falls into three main categories: alkaline (e.g., calcium carbonate),12 non-alkaline (calcium sulphate), and silica-based (alumino-silicates).13 A summary of the most common scales and their properties is presented in Table S1 (ESI†). The complex ionic composition of industrial water as well as operational fluctuations and non-uniformity of the streams makes it difficult to establish a screening method to ensure that the water flow will not lead to scaling. Achieving scale-free flow of water in industrial pipes using membranes will be a milestone which has triggered tremendous research in this area.14
The alkaline scale formation is an equilibrium reaction in which, for example, calcium carbonate salt concentration in water is dependent on the carbonic acid species, which are affected by pH, temperature, and ionic strength through the following reactions:15,16
| CO2(g) ⇌ CO2(aq), | (1) |
| CO2(aq) + H2O (l) ⇌ H2CO3(aq), | (2) |
| H2CO3(aq) ⇌ H+(aq) + HCO3−(aq), | (3) |
| HCO3−(aq) ⇌ H+(aq) + CO32−(aq). | (4) |
The resulting CO32− ions react with Ca2+ to form sparingly soluble CaCO3 deposits§:
| CO32− + Ca2+ ⇌ CaCO3(s) ↓ | (5) |
Biomineralization is also related to scaling, and it is a natural process by which a desired element is extracted from the environment and embedded into a structure to acquire a certain functionality in the biosystem.17 Bone development is an example of biomineralization, which is associated with the apatite ordered crystallization in a bed of collagen fibers, leading to crystal {001} and {110} axes along the fiber length and hole zone rows, respectively.18 The matrix of mineralized bone comprises crystals of calcium phosphate, hydroxyapatite, and organic materials.19 Several excellent reviews have been written on the biomineralization process which provide a detailed account of this phenomenon.20,21
According to the Water Quality Association (WQA), the degree of hardness of water is a key indicator of scaling potential.22 Water hardness is commonly regulated by the dissolved di- and trivalent metallic ions such as Ca2+ and Mg2+, which, at high levels (e.g., hard water contains 7–10.5 grains per gallon or 120–180 ppm equivalent of calcium carbonate), cause municipal and industrial water scaling problems.22 The scaling tendency in a system can be characterized by several methods including i) Langelier Saturation Index (LSI is the most common tool based on water pH as compared to the calcium carbonate saturation pH¶); ii) Stiff and Davis Saturation Index (S&DSI)||; iii) supersaturation index (ratio of ion activity product to the scale solubility product), and iv) molar ratio concept.13
Scale inhibition can be achieved through three main alternatives: feed water modification (coagulation, ion-exchange softening, and acidification),23 membrane process optimization (product recovery tuning, feed flow reversal,24 intermediate chemical demineralization,25 and rotation filtration26), and using additives.13 Magnetic pre-treatment is another non-chemical method for decreasing scale formation.27,28
The development of methodologies to overcome scale formation in industrial plants is of paramount importance, which can maximize process efficiency and lower operating costs. Macromolecules have offered a viable approach to develop tailored water treatment technologies and have been widely investigated in this regard. The structural design of macromolecules to optimize their performance requires a good understanding of the stages in crystal development during scale formation. A review of the thermodynamics of crystallization is included below to help understand the scaling process and to initiate a discussion on the mechanistic effects of macromolecules on the scale development. It will be followed by a summary of the scale characterization methods and the most important macromolecular antiscalants, their structure in terms of functional groups and backbone as well as an evaluation of their efficiency.
Thermodynamics of crystallization
Crystallization is a liquid-to-solid phase transition, which is driven by the free energy difference between the physical states. A phase transition is favored when the free energy of a system decreases, and crystallization occurs when the free energy of the product (solution and crystal phases) is lower than that of the starting materials. Free energy can be expressed in terms of solution activities, which is related to the concentration of solutes in an ideal system. For a simple case of one-component (AB) precipitation from two ionic species (A and B),18| aA + bB ⇌ cAB, | (6) |
| Δμ/kBT = ln(a/Ksp), | (7) |
In an unsaturated condition, increase in local ion concentration, for example due to mixing, evaporation, changes in pH, ionic strength, and/or temperature are among some of the factors that can cause local supersaturation and initiate the nucleation process.29 Importantly, a known crystal chemical composition may reflect different supersaturation degrees for its corresponding solid-state species depending on the properties of solid phases, e.g., calcium carbonate can form five crystalline polymorphs and at least one amorphous compound each of which having different solubilities.30
Molecular interactions between the crystal and the solvent (water) introduce several complexities to the thermodynamic interpretations discussed earlier. When the crystallization enthalpy is positive, one way to induce phase transition is to increase the system entropy. This may be possible by solvent molecule release from the crystal structure, bringing the free energy of the final mixture lower than that of the initial solution. One example of such transformation is protein crystallization.31
Gibbs32,33 states that when the free energy of a system, in our case, potentially scaling water, exceeds that of a transformed system, such as scaled water, the phase transition starts with the formation of small-scale regions of the new (solid) phase called nuclei. It should be noted that the nuclei are not necessarily the earliest phase in a crystallization process: the formation of thermodynamically stable solute molecules (e.g., carbonate and phosphate salts of calcium), known as pre-nucleation clusters (PNCs), has been thoroughly investigated.34 The free energy level of the bulk crystal nuclei plays an important role in driving the crystallization forward; however, what happens on the crystal surface is different. Considering that the crystal surface is in direct contact with the solution, it contributes to the solid phase free energy more significantly than the bulk molecules. This plays a key role in the crystallization process.18
Starting with a small nucleus, solute molecules tend to aggregate on top of it, resulting in an increase in the interfacial energy. This is a non-favored state for the crystal growth, competing with the energy contributors such as supersaturation, which can be high enough to bring about crystal dissolution instead of further growth.18,35,36 At this stage, the more the crystal is disordered, e.g., by adsorption of a macromolecule on the crystal surface, the easier is its dissolution. Accordingly, a supersaturated solution may remain in its metastable state without forming a detectable solid phase for a time period called induction time, which is dependent on the supersaturation degree, regulating the growth rate.37
When a crystal is large enough, the interfacial energy gain may not be sufficient to overcome the free energy decrease of the solid phase, and consequently no growth inhibition is possible via the so-called Gibbs–Thomson effect.38 Therefore, the higher the supersaturation degree (i.e., the higher initial phase free energy) and the larger/the more the initial nuclei (e.g., in seeded crystallization),39 the more favored is the crystallization. In the closely related challenge of nanoparticle synthesis, controlling the nucleation rate, e.g., by changing the reactant concentration, can result in a defined particle size.40,41
Once a nucleus is stable (its size is higher than a critical size under which the solid phase dissolves back into the solution), crystal growth rate is dictated by mass and energy transfer between the phases.18 As an important application, by increasing the interfacial energy of the nuclei, it may be possible to retard or inhibit the crystallization process. This molecular aspect of crystallization phase transition together with the properties of the macro phase, such as solute concentration and degree of supersaturation, defines the fate of interacting phases.
When nucleation takes place on a substrate during scaling processes,42 the phase transition free energy change for a hypothetic hemispherical nucleus has a negative contribution from the chemical potential difference between the phases and a positive contribution from the interfacial energy γ of the liquid (l), crystal (c), and substrate (s):18,43
| ΔG = − (0.5V/Vm)Δμ + A(2γcl + γcs − γsl), | (8) |
When the supersaturation degree is low, crystals prefer to have atomically flat surfaces,45 resulting in low surface energy and decelerated growth. By decreasing the surface energy, scale-resistant substrates become achievable.46 This magnifies the important role played by surface defects, heterogeneity, and kinks in attracting an approaching/adsorbed ion, and boosting the growth process. Similarly, impurities may inhibit crystal growth.45 For example, (1) based on the so-called Cabrera–Vermilyea model,47 adsorbed impurity increases the curvature (decreases the velocity) of an advancing step. When the curved steps merge, the impurity is left behind, and the step velocity is restored until the next impurity brings about the same effect. Step growth is completely stopped if the distance between the two impurities is smaller than the critical nucleus diameter, which results in a dead zone; (2) impurities may block the kinks, preventing the adsorption of ions and/or molecules.48 Thus, crystal growth inhibition can be achieved even when as low as one percent of the crystal is covered with a modifier;49 (3) solubility can be modified by incorporating an impurity into the crystal structure;50 and (4) crystal surface energy may be modified by the adsorption of an impurity acting as a surfactant.51
Classical nucleation theory hinging on the gain–loss (Becker–Doring) correlation is among the most common theoretical explanations of crystal growth.52 In an ideal case, crystal growth follows a pathway to adopt the most stable (ordered) structure through which all less stable states are achieved beforehand (Ostwald–Lussac law). Accordingly, disturbing the crystal structure at any low-stability condition may lead to incomplete/unstable crystallization. For calcium carbonate, an increase in structural order results in a phase transition from an amorphous phase to vaterite, aragonite, and eventually calcite;18 however, the coexistence of these phases has been reported in real-life phase transitions.53 A stable phase is expected to have stronger bonding between the ordered molecules, resulting in a higher formation enthalpy and higher entropy change. When a stable phase is formed, crystal growth phase begins.
The time change in crystal size (number of atoms or molecules n) is dictated by the formation rate kf and the elimination rate ke:52
| dn/dt = kf − ke. | (9) |
Assuming a large spherical crystal,
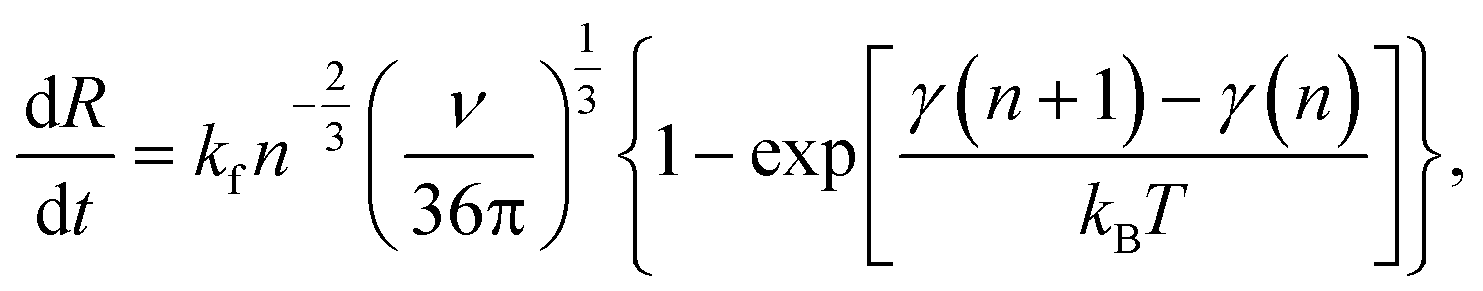 | (10) |
| γ(n + 1) − γ(n) = ΔG(Rc − R). | (11) |
It includes the critical cluster size Rc, which can be neglected in case of large clusters (Rc ≪ R). Note that ΔG is the free energy difference between the bulk solid and the liquid phases. A kinetic model is required to obtain 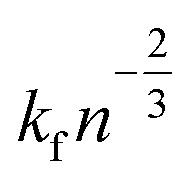 in eqn (10). Hinging on the Fisher–Turnbull (FT) kinetic model,54
in eqn (10). Hinging on the Fisher–Turnbull (FT) kinetic model,54
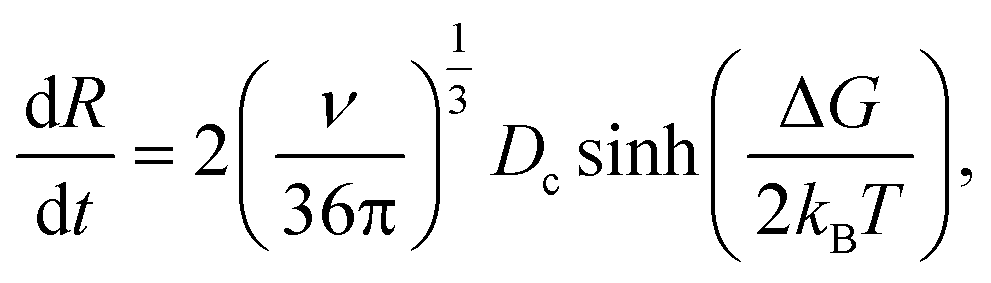 | (12) |
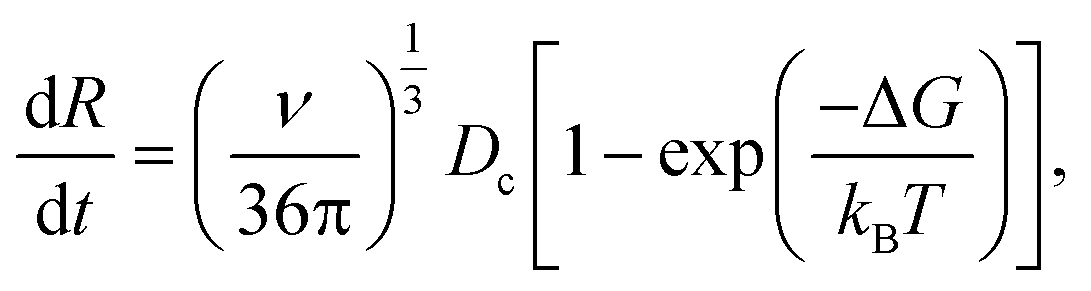 | (13) |
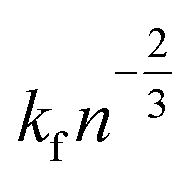 for a cluster of radius Rc. One way of acquiring Dc is to use the Stokes–Einstein equation relating the diffusion to temperature and viscosity.56 During growth, nucleation may still take place;57 however, once the crystal is formed, it is essential to detect and control its growth dynamics to prevent scaling.
for a cluster of radius Rc. One way of acquiring Dc is to use the Stokes–Einstein equation relating the diffusion to temperature and viscosity.56 During growth, nucleation may still take place;57 however, once the crystal is formed, it is essential to detect and control its growth dynamics to prevent scaling.
Scaling process characterization methods
Most common and widely used scale evaluation methods to characterize the effectiveness of scale inhibitors are discussed below. These include static, dynamic, electrochemical, membrane-based, and analytical techniques.58,59Static methods, also called beaker tests, are easy to perform and provide information about the bulk precipitation of the ionic species.58 A bulk property of the solution is monitored upon the addition of precipitating species or changing the pH to induce crystal formation. Some of the important bulk properties include pH,60 conductivity,61 ion concentration,9 and deposited mass.16 To shed light on the kinetics of crystallization, the constant composition method62–64 has been proposed. In this technique, the crystallization is intentionally induced by the addition of crystal seeds to a saturated solution followed by the addition of the same saturated solution to keep the pH constant. The amount of added solution versus time is a good indication of the solution's precipitation tendency. The crystallization can also be induced by increasing the pH, e.g., by degassing CO2, which results in a significant change in the bulk properties such as conductivity or resistivity upon crystallization, providing a reliable detection method called fast controlled precipitation (FCP).65,66 According to NACE (National Association of Corrosion Engineers) standard, static methods provide only lab-scale evaluation, which may be far from the industrial performance of antiscalants.67 To follow the adherence of scales to industrial metal surfaces, a static heat exchanger test68 is utilized in which the ion concentration is monitored while applying a temperature gradient to a saturated solution-filled heat exchanger (e.g., a U-tube). Static methods can be used as tentative screening criteria for antiscalant selection.69
Dynamic scale inhibition tests are designed to monitor the scaling process under flow.58,70 In the so-called dynamic tube-blocking test,71 the tube pressure drop is monitored versus time to understand the dynamics of scale formation. The lower the pressure drop at a desired time, the better is the efficiency of the additive. In closely relevant experiments, the heat flux is kept constant in a heat exchanger72 (e.g., by an electric heater), and the thermal conductivity of the scaling surface is related to the surface temperature and scale layer thickness. A scale layer acts as an insulator, decreasing thermal conductivity.58 To take dynamic techniques one step closer to real industrial conditions, rotating electrodes provide decent mass transfer rate and turbulence. One way of evaluating rotating disc performance is to measure the deposited mass in a given time and compare it to antiscalant-free water.73 Finally, perforated scaling coupons are used to monitor operating industrial systems in action.74 These coupons are placed in a way that induce turbulence and can be replaced without process shutdown.75
Electrochemical methods facilitate the scaling process, providing a solution to characterize the performance of antiscalants in a much shorter time. Through an electrochemical reaction of water and the reduction of dissolved oxygen (at a negative potential, e.g., −1 V/saturated calomel electrode (SCE)), the local concentration of OH− is increased near the cathode, which brings about the precipitation of sparingly soluble salts on the electrode. These methods include chronoamperometry, chronoelectrogravimetry, and electrochemical impedance spectroscopy.75–77
Chronoamperometry, which dates back to 1903,78 is an electrochemical method in which the current is monitored versus time under a constant, controlled potential.76 Scale formation decreases the current until the electrode surface is entirely covered by an insulating layer, blocking ions from reaching the electrode surface, which results in an almost-zero current. An effective antiscalant decreases the slope of current versus time curve, preferably leading to a high constant current, close to the initial current. It is common to use a rotating disc electrode to perform chronoamperometry experiment to improve mass transfer and accelerate the scaling process.79 In a diffusion-controlled process, the square of the current is proportional to the rotation speed.76
In chronoelectrogravimetry, the deposited scale mass (ng cm−2) is monitored using a quartz crystal microbalance (QCM) coupled with an electrochemical cell (EQCM),80 providing similar scaling conditions as for the chronoamperometry. For a film attached to a QCM and with a low dissipation, e.g., a strongly adhered rigid solid, the resonance frequency shift of the quartz crystal is linearly proportional to the deposited mass according to the well-known Sauerbrey equation.81 In electrochemical impedance spectroscopy,82 the frequency of a small-amplitude alternating current is swept and the impedance response of the system is acquired from which the surface coverage and the morphology of the scale layer can be deduced.58,59
Membrane tests are large-scale, expensive methods conducted under various flow directions, such as cross-flow,83 which are designed to simulate real industrial conditions. In this method, the scale formation results in a membrane permeability drop.58 One of the most important phenomena in membrane water treatment processes during scaling, despite the use of non-scaling water, is ion concentration increase in the vicinity of the separating boundary surface.84
To achieve a qualitative understanding of the scaling process, several analytical processes may be used.58 To shed light on the morphology of the precipitated phase, imaging techniques such as scanning electron microscopy (SEM)85 and atomic force microscopy (AFM)86 are commonly used. The polymorphism and composition of the scale can be identified using X-ray diffraction87 and infrared (IR) spectroscopy.88 Wide-angle X-ray scattering (WAXS)89 and small-angle neutron scattering (SANS)90 are also used to acquire the particle structure, size, and morphology evolution of the precipitated solid. Recently, liquid-phase TEM (LP-TEM) has been utilized to monitor the nucleation and growth of calcium carbonate in the presence of an acidic macromolecule (polystyrene sulfonate, PSS). It attested that PSS inhibits crystal formation and results in the formation of amorphous calcium carbonate.91
Macromolecule-based antiscaling platforms
Effect of macromolecules on crystallization and scaling
To change the crystallization pathway, soluble macromolecular additives have been used, which can alter nucleation, growth, and the crystal structure.92 As an interesting example, calcium carbonate crystallization on a glass substrate resulted in a calcite–vaterite crystal mixture in the presence of poly(glutamine acid) bearing carboxyl groups; however, no crystallization was achieved when poly(acrylic acid) or poly(allylamine) with the same functional groups was added to the initial solution, suggesting that the conformation and/or molecular backbone can affect the crystal fate.93 Functional groups such as –COOH, –OH, –NH2, –CH3, –SH, –PO4H2, and –SO3H can direct crystallization.94 Another method to control crystallization is to change the dielectric constant of the medium, which changes supersaturation degree by altering the solvent.30Despite the common coexistence of crystal polymorphs, controlled operating conditions such as temperature, concentration, mixing pattern and residence time can result in pure polymorphs, e.g., anhydrous crystals of CaCO3.95 Further elaboration on these aspects is out of the scope of this review. In Table S2 (ESI†), some examples of the effect of polymeric additives on crystallization processes are presented, and the interested reader is encouraged to follow some of the comprehensive reviews on organic and inorganic crystal modifiers.45,96
In the early stages of developing chemical additives for water treatment, polyphosphates were widely used as industrial antiscalants, and the phosphate functionality was considered as one of the best Ca2+ chelating agents. As the first threshold scale inhibitor, sodium hexametaphosphate (NaPO3)6 (SHMP) was developed in the 1930s to eliminate undesirable Ca2+ scales, the most predominant form of mineral scales.13,97 Due to the weak P–O–P linkage, the polyphosphate species are easily broken down in water at a high temperature, resulting in the formation of orthophosphates; however, such transformation imposes a new crucial problem to water treatment systems, since orthophosphates capture Ca2+ in water and generate new water-insoluble calcium phosphate species.13,98,99 SHMP is commonly used in low-temperature (T < 45 °C) water treatment systems,100,101 whereas most industrial processes, e.g., pulp and paper plants, oil fields, and water recycling heating systems, are operated at high temperatures. This necessitated the development of new thermally stable materials, which are able to efficiently function as antiscalants in a wide temperature range.
Due to their superior ion chelating potential via pendant or backbone-distributed functional groups and excellent dispersion characteristics, macromolecules have been widely explored as antiscalants.102 To date, most of the commercially available compounds for scale inhibition are macromolecule-based formulations.103,104
Polymers as additives
Synthetic polymers offer tremendous advantages by providing their tailorable backbone to host the desired flexibility and functionality, which can potentially adapt to the environmental variation in water treatment processes. It has been demonstrated that macromolecules disperse CaCO3 in water more significantly than an oligomer, since the polymer/copolymer adsorbs more strongly on the calcite crystal facets than the oligomer.105 It is also convenient to tailor the phosphorus content in these macromolecules or completely eliminate it by seeking other chelating entities to reduce phosphorus-induced neutrification/eutrophication in the feed water. Scientists have been very active in this area, and a multitude of polymer-based antiscalants have been developed during the past few decades.102,103,106,107 An evaluation of the progress made in this field is summarized below by categorizing these polymer-based antiscalants according to the build-up of their frameworks or functional groups.The lack of biodegradability of C–C bond based antiscalants is indeed a significant drawback. These compounds cannot be naturally degraded in the environment, and their use in water treatment processes leads to environmental contamination. As an improvement to the C–C bond based polymers, oxygen and/or nitrogen comprising frameworks have recently been incorporated in the polymer synthesis, which brings about degradability and minimizes the environmental footprint. An excellent example of such environmentally benign polymeric scale inhibitors is polyaspartic acid (PASP) (Fig. 1), which contains C–N bonds that are completely biodegradable. It has been demonstrated that PASP performed well in CaCO3, CaSO4, and Ca3(PO4)2 dispersions (Table S3, ESI†).61,108,109 PASP-based scale inhibitors are currently one of the most promising alternatives to conventional non-degradable antiscalants.
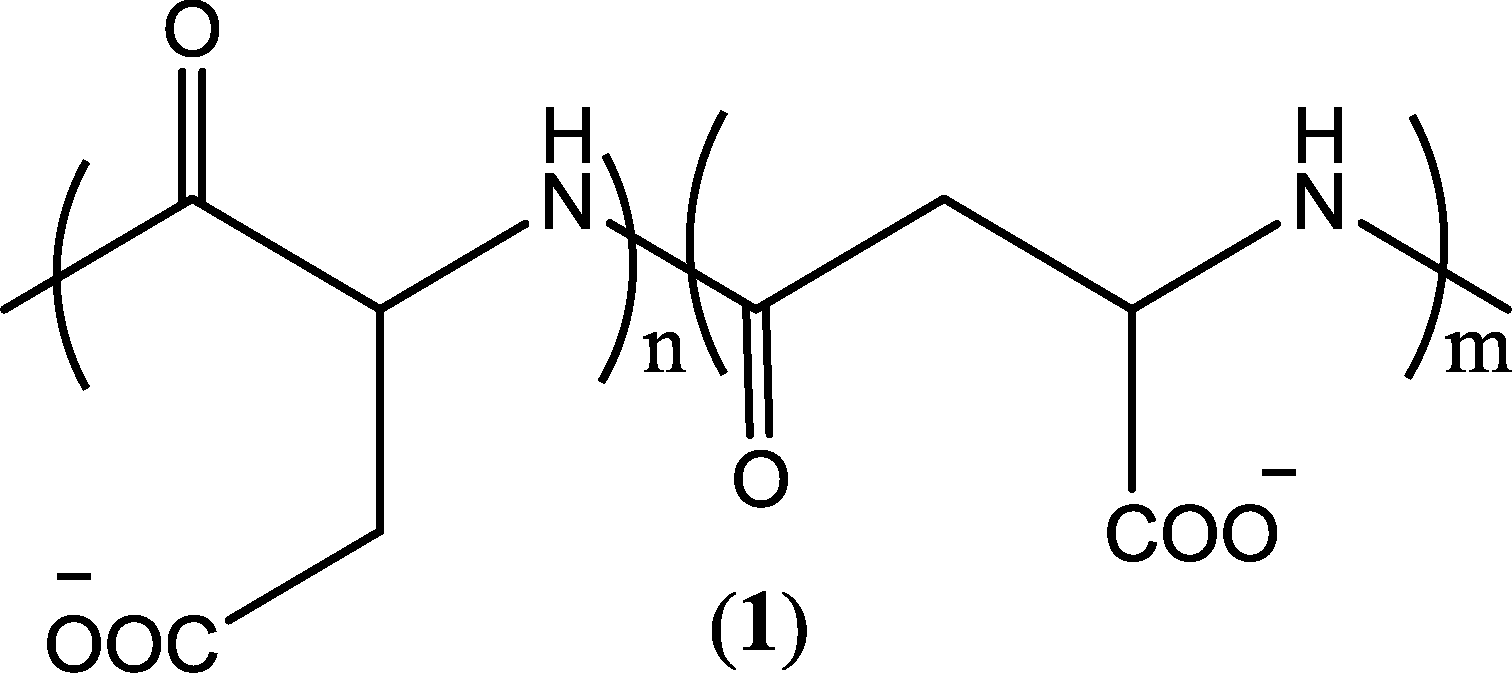 | ||
| Fig. 1 Structure of PASP copolymer.108 | ||
Phosphonate appended polymers based on phosphonic acid and their corresponding salts and esters have been extensively employed as antiscalants in desalination systems.84 Compounds that contain P–C bonds exhibit good thermal stability. Polyphosphonates thus offer better antiscaling performance than polyphosphates at temperatures above 45 °C.112 Accordingly, many attempts have been made to tailor scale inhibitors by incorporating P–C bonds in the backbone. For example, organophosphonic acids with stable P–C bonds have been extensively investigated for preventing calcite scale formation. The most frequently reported organophosphonates include hydroxyethylidene-1,1-diphosphonic acid (HEDP),113 1,2-dihydroxy-1,2-bis(dihydroxyphosphonyl)ethane (DDPE),114 [ethylenediamine-N,N,N′,N′,-tetrakis(methylenephosphonic acid)] (EDTMP),115 nitrilotris-(methylenephosphonic acid) (HNTMP),116 and 1,6-hexylenediamine-N,N,N′,N′-tetrakis-(methylphosphonic acid) (HDATMP).117 The introduction of these active materials to a saturated solution of Ca2+ prolongs the induction time, which consequently retards scale formation. It has been observed that the inhibition rate increases by increasing the antiscalant content; however, an inherent drawback of phosphorus-based inhibitors is that they can serve as nutrients leading to neutrification/eutrophication difficulties;118 therefore, phosphorus content in effluent is strictly regulated in many areas of the world. Accordingly, there has been tremendous effort to develop non-phosphorus-based scale inhibitors for water treatment late in the past century.
Polymers bearing sulfonate moieties (for example, polystyrene sulfonate) have also been employed; however, the efficiency of these macromolecules is not as impressive as that of the carboxylate-based polymers. This has been evidenced by the detailed kinetic studies on the effect of sulfonate polymers on gypsum deposition.111
Compared to phosphonates and sulfonates, carboxylate polymers have been more extensively employed in antiscaling applications. Such polymers benefit from relatively less toxicity, abundant and cost-effective monomers, convenient preparation, superior antiscaling behaviour, and benign nature, as –COOH is a biocompatible unit which inflicts no harm to water systems at low concentrations. Polymers containing carboxylic acid (–COOH) functional groups generally exist as anionic polyelectrolytes in the studied pH ranges. The inhibitory performance of these polymers depends on their molar mass and the number and spatial arrangement of functional groups in the structure.
Poly(acrylic acid) (PAA) is one of the first commercially available polymeric antiscalants tested against CaSO4 and CaCO3 scale build-up. The effect of various characteristics of PAA polymers towards their scale inhibition efficiency has been broadly investigated, which has shown that the molar mass and concentration of PAA are crucial factors. In the early 1960s–1970s, the ppm amount of partially esterified PAA polymers was reported to effectively prevent CaSO4 deposition in sea water evaporation processes.102,119–121 These macromolecules have a promising performance in gypsum crystal modification.119 The suggested molar mass of PAA-based calcium carbonate scale inhibitors for an optimal performance is 1–3 kDa.122
The effect of temperature on the antiscaling performance of PAA was studied by Smith and Sweet.120,121 They synthesized eight partially esterified PAA polymers with molar masses ranging between 1 and 6 kDa and investigated their performance as CaSO4 scale inhibitors at 30 °C and 90 °C. It has been suggested that these polymers were more effective in a low molar mass range and at 30 °C. Their antiscaling efficiency at 90 °C (as a critical temperature) was much lower than at 30 °C, particularly for the higher molar mass additives. The authors explained that a high molar mass made PAA polymers less efficient because each molecule was able to adsorb onto more than one particle in the deposit, producing tightly bound scales.
Importantly, the end-groups of PAA polymers play a vital role in the antiscaling performance. In 2010, Fellow's group123 prepared a series of PAAs (Mw = 1.5–4 kDa) with different end-groups and molar masses, using atom transfer radical polymerization (ATRP), for the inhibition of calcium oxalate deposition. Based on comparative experiments, it was concluded that the inhibition capability of these polymers was strongly dependent on the hydrophobicity and molar mass of end-groups. These results may be explicable in terms of the self-assembly behavior of the polymer or an adsorption–desorption dynamic equilibrium of PAA to crystallite surfaces.
PMA is another widely utilized synthetic homopolymeric antiscalant. Flocon 247, a commercially available PMA-based antiscalant, is reported to be highly efficient in the prevention of CaCO3 and Mg(OH)2 precipitation in seawater desalination.110 It retards the bicarbonate-to-carbonate decomposition rate, which effectively reduces seawater's alkaline scale potential and also disrupts the scale formation stages, resulting in significant inhibition of CaCO3 and Mg(OH)2 scales.110
The effect of heat treatment on the performance of PMA was investigated in a CaCO3 scaling system, which confirmed a weakened antiscaling efficiency at elevated temperatures due to the calcium ion incompatibility originating from thermal stresses.124 On the contrary, PAA did not show such a tendency. The inhibition efficiency of PMA is superior to that of PAA when the temperature is lower than 200 °C.124,125 PAA is more frequently used for CaSO4 scale inhibition, whereas PMA is preferred for CaCO3 precipitation prevention.
It has also been observed that the incorporation of hydrophilic polyethylene glycol (PEG) segments in PAA or PMA causes excellent water solubility and flexibility, providing polymers with more inhibitory effectiveness. Zhou and co-workers126–129 prepared a family of AA–PEG and MA–PEG copolymers, such as acrylic acid–allylpolyethoxy carboxylate (AA–APEC) copolymer,127 acrylic acid–allylpolyethoxy carboxylate copolymer (AA–APEL),128 and polyethylene glycol (degree of polymerization = 8) double-ester of maleic anhydride/acrylic acid (PEG8DMA/AA) (Fig. 2).130 The order of efficiency in both CaCO3 and CaSO4 inhibition was PEG8DMA/AA > AA–APEC > PAA. Specific features of PEG8DMA/AA double-hydrophilic block copolymers allowed an effective encapsulation of calcium ions, resulting in the formation of polyion complex micelles. The outer PEG chain segments stabilize the core of the polyion complex in water, as indicated in the suggested mechanism.130 The maximum inhibition of CaCO3 and CaSO4 were 89% and 99%, respectively, at PEG8DMA/AA dosage levels of 12 and 3 mg L−1. Furthermore, maleic anhydride–allylpolyethoxy carboxylate (MA–APEC)126 polymer provided approximately 99% Ca3(PO4)2 inhibition for industrial recycling water at 6 mg L−1.126
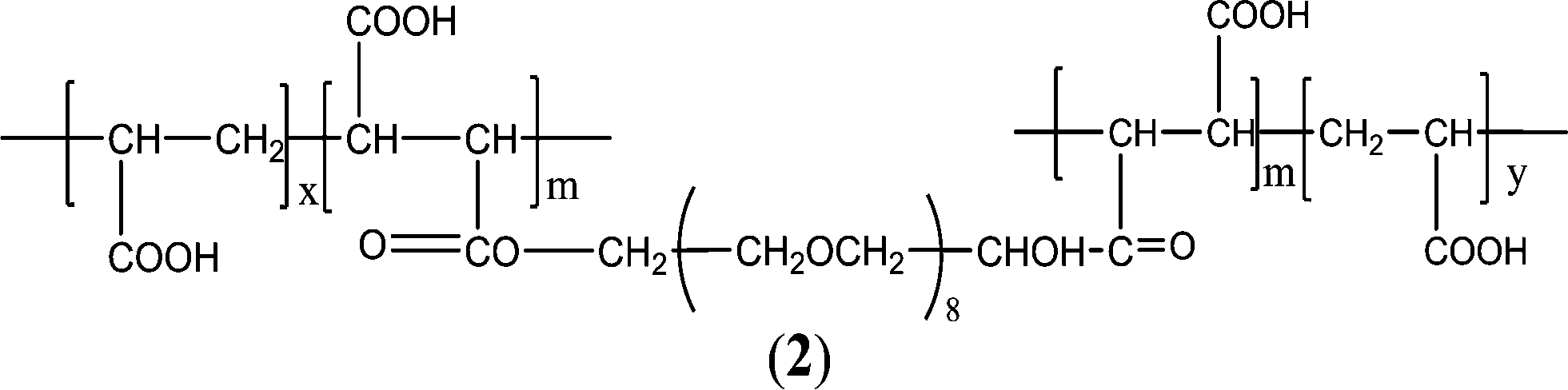 | ||
| Fig. 2 Structure of PEG8DMA/AA.130 | ||
(1) Copolymers with carboxylate groups: in a study of polymers with two or more repeat units (e.g., AA and MA) bearing only –COOH as a pendant group, it was observed that acrylate–maleate copolymer (Mw = 70 kDa) dispersed CaCO3 more significantly than the oligomaleate in water. This was attributed to an enhanced adsorption of acrylate–maleate copolymer to the calcite faces as compared to the weaker performance of oligomaleate.105 The chain segments located in different parts of the polymer adsorb on different calcite faces, resulting in an improved coverage of the crystal faces by the polymer. Interestingly, maleic acid–methacrylic acid (MA–MAA),131 maleic acid–acrylic acid (MA–AA) and maleic acid–ortho-toluidine (MA–OT)132 copolymers were able to inhibit the scale completely, even at low dosage (20–25 ppm) in oilfield desalination.
(2) Polymers containing carboxylate as well as phosphonate and/or sulfonate groups: merging two or more different chelating groups in a polymer is also an effective strategy to improve the efficiency of scale inhibition. Such polymers have demonstrated strong complexation capabilities with inorganic ions due to multiple functionalities and an enhanced dispersion tendency, providing effective nucleation retardation and/or instant prevention of further crystal growth.133 Adsorption of multi-functional polymers onto calcium scale crystal facets has been achieved by the hybrid nature of such polymers, leading to extensive passivation of the crystal surface, which modifies the crystal size and morphology.134–136
A series of phosphorus- and carboxylate-appended polymers bearing either a phosphonate unit inside (such as the commercial polyphosphinocarboxylic acid (PPCA)),135 or on the termini with carboxylic acids and/or sulfonate as pendants have been synthesized.134,136 For instance, a novel AA–APEM–H3PO4 terpolymer (Fig. 3) was prepared through the free radical polymerization reaction of acrylic acid (AA), oxalic acid–allypolyethoxy carboxylate (APEM), and phosphorous acid (H3PO3) in water using a redox system comprising hypophosphorous and ammonium persulfate as initiators.134
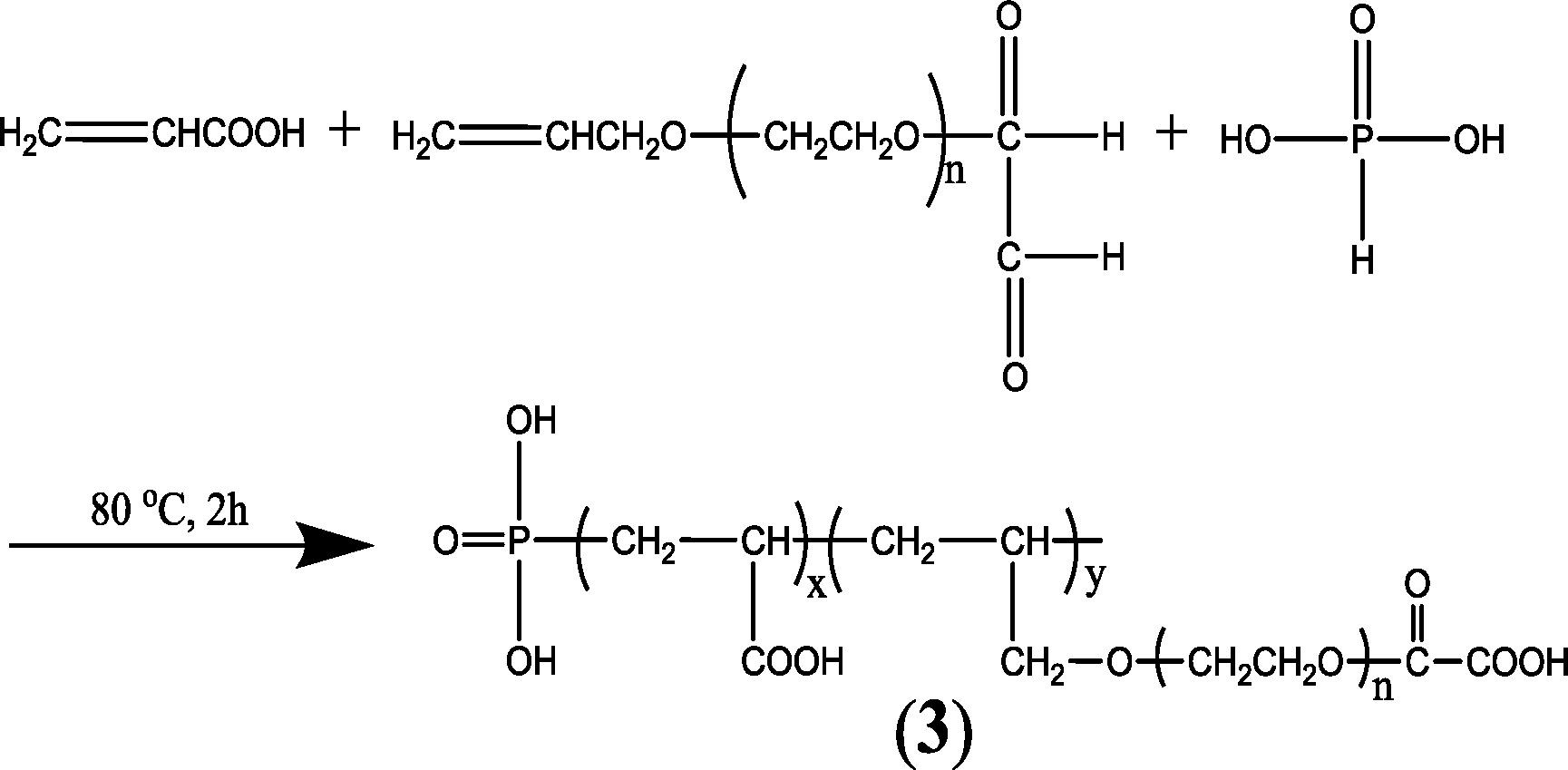 | ||
| Fig. 3 Synthetic scheme for AA–APEM–H3PO4.134 | ||
In addition, a phosphonate–maleate–sodium p-styrenesulfonate copolymer (MA-SS) has been synthesized in water using a redox combination of hypophosphorous and hydrogen peroxide as initiators. This polymer was able to effectively inhibit CaCO3 (ref. 137 and 138) and CaSO4 (ref. 139) deposition in cooling water systems, as evidenced by static scale inhibition tests.
A well-defined block copolymer comprising acrylic acid (AA) and 2-acrylamido-2-methyl-propane sulfonate (AMPS) units (PAA–PAMPS) was obtained in a one-step reaction by reversible addition–fragmentation chain transfer RAFT copolymerization (Fig. 4). The antiscaling behavior of the copolymer was investigated during different stages of CaCO3 formation, which proved that the copolymers not only retarded nucleation but they also stabilized nanoscale CaCO3 particles in the post-nucleation stage (growth phase).133
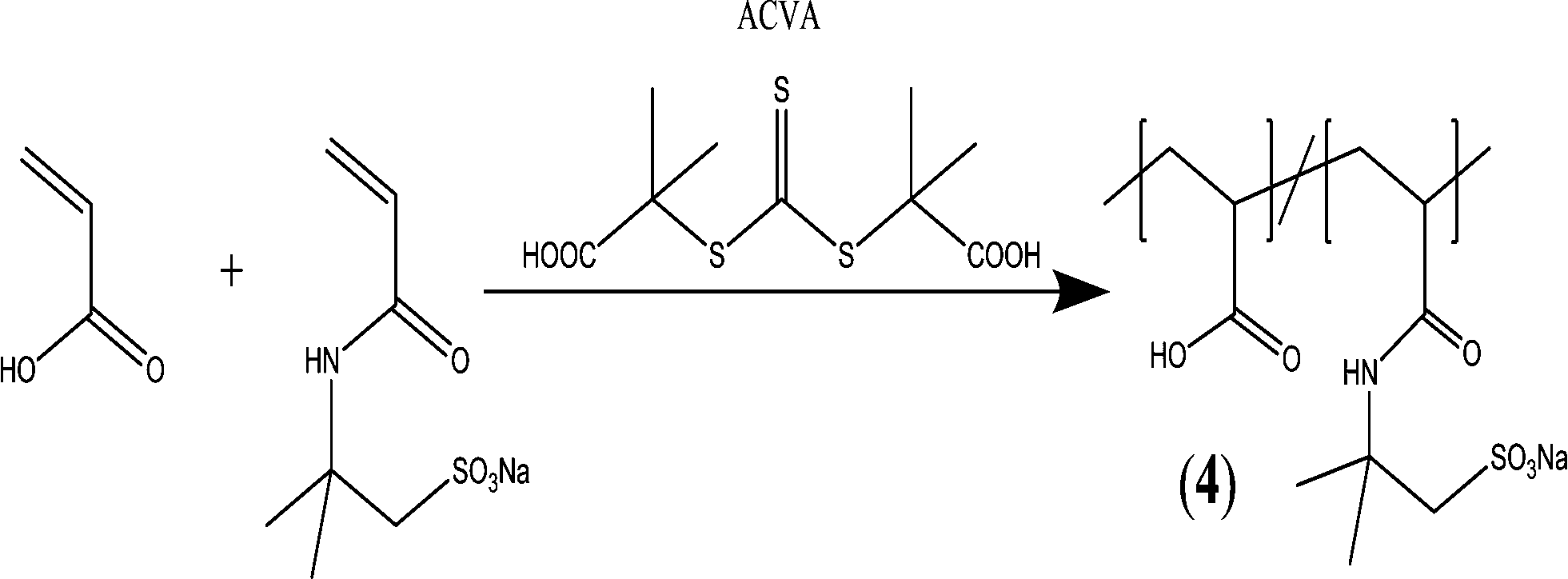 | ||
| Fig. 4 Synthesis of PAA–PAMPS block copolymer via RAFT copolymerization.133 | ||
A range of diallylammonium-based cyclo-polymers and cyclo-copolymers has been successively developed by Ali and co-workers using Butler's cyclopolymerization140 protocol. It was revealed that 10–20 ppm diallylammonium-based dianionic polyelectrolytes (DAPEs) bearing phosphopropyl pendants were highly effective in inhibiting the formation of CaSO4 scale at 50 °C.141,142 Furthermore, 10–20 ppm pH-responsive polyzwitterion acid (PZA) polymers bearing both phosphorus and sulfopropyl pendant groups in the same repeat unit were found to be effective antiscalants for the inhibition of CaSO4 at 40 °C in reverse osmosis desalination.143–145
(3) Polymers containing –COOH and other groups such as –NH2/CN: new functional moieties (such as –NH2 and –CN) have been added to carboxylate-containing polymers. Vasudevan's group146 reported that the inhibition of CaSO4 and CaCO3 deposition can be achieved by using acrylonitrile–acrylic acid (AN-AA) and acrylonitrile–methacrylic acid (AN-MAA) copolymers over a temperate range of 50–80 °C at pH ranging from 7.0 to 8.5. The efficiency of the copolymers in scale inhibition was assessed by various techniques such as the chemical screening test, impedance measurements, and constant potential electrolysis. A comprehensive summary of the antiscaling performance of such macromolecules is presented in Table S3 (ESI†).
A hydrolyzed pectin-based graft copolymer has been synthesized and utilized as a green antiscalant, and its CaSO4 antiscaling performance was found to be independent of the temperature and pH variations. The pectinH-g-poly(AAm-co-amine) exhibited up to 100% and 97.7% scale inhibition in a basic solution of Ca2+ and SO42−, respectively. In addition, the observed homogeneous crystallization induction time was more than 25 days in the presence of the pectinH-g-poly(AAm-co-amine) (Fig. 5).147
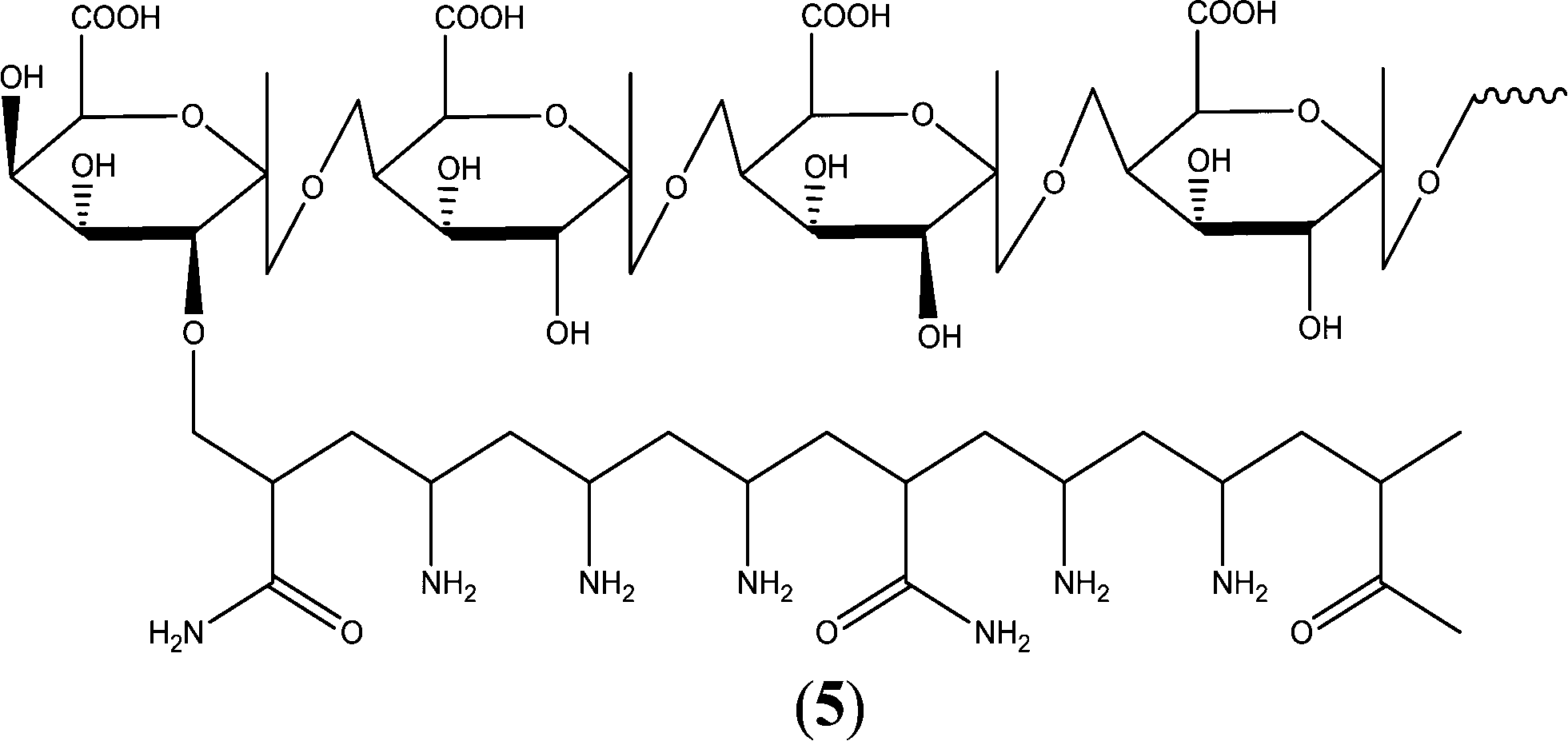 | ||
| Fig. 5 Proposed structure of pectinH-g-poly(AAm-co-amine) polymer.147 | ||
Hyperbranched macromolecule (dendrimers/dendrons)-based antiscalants
Another interesting venue for the development of antiscalants is to incorporate hyperbranched 3-D structures, such as dendrimers or dendritic polymers, into the antiscaling agents. Dendritic macromolecules have been demonstrated to be effective additives for controlling scale formation. Dendrimers offer intriguing properties due to their multivalent, globular, hyperbranched architecture, which is assembled in a layer-by-layer fashion leading to monodisperse macromolecules.148,149 In addition, the number, spatial location, and polarity of the functional groups can be precisely controlled.150Hyperbranched polyglycerols have been synthesized starting from pentaerythritol and glycidol by anionic ring-opening polymerization and were modified to sulfate- or carboxylate-based polyglycerols using SO3–Py complex and sodium chloroacetate, respectively (Fig. 6).151 These modified polyglycerols have been used as additives for the biomimetic crystallization of CaCO3. The morphology of calcite crystals was controlled by adjusting the functionality and molar mass of polyglycerol, and a structural transition from a single crystal to a meso- to polycrystalline state has been observed.151
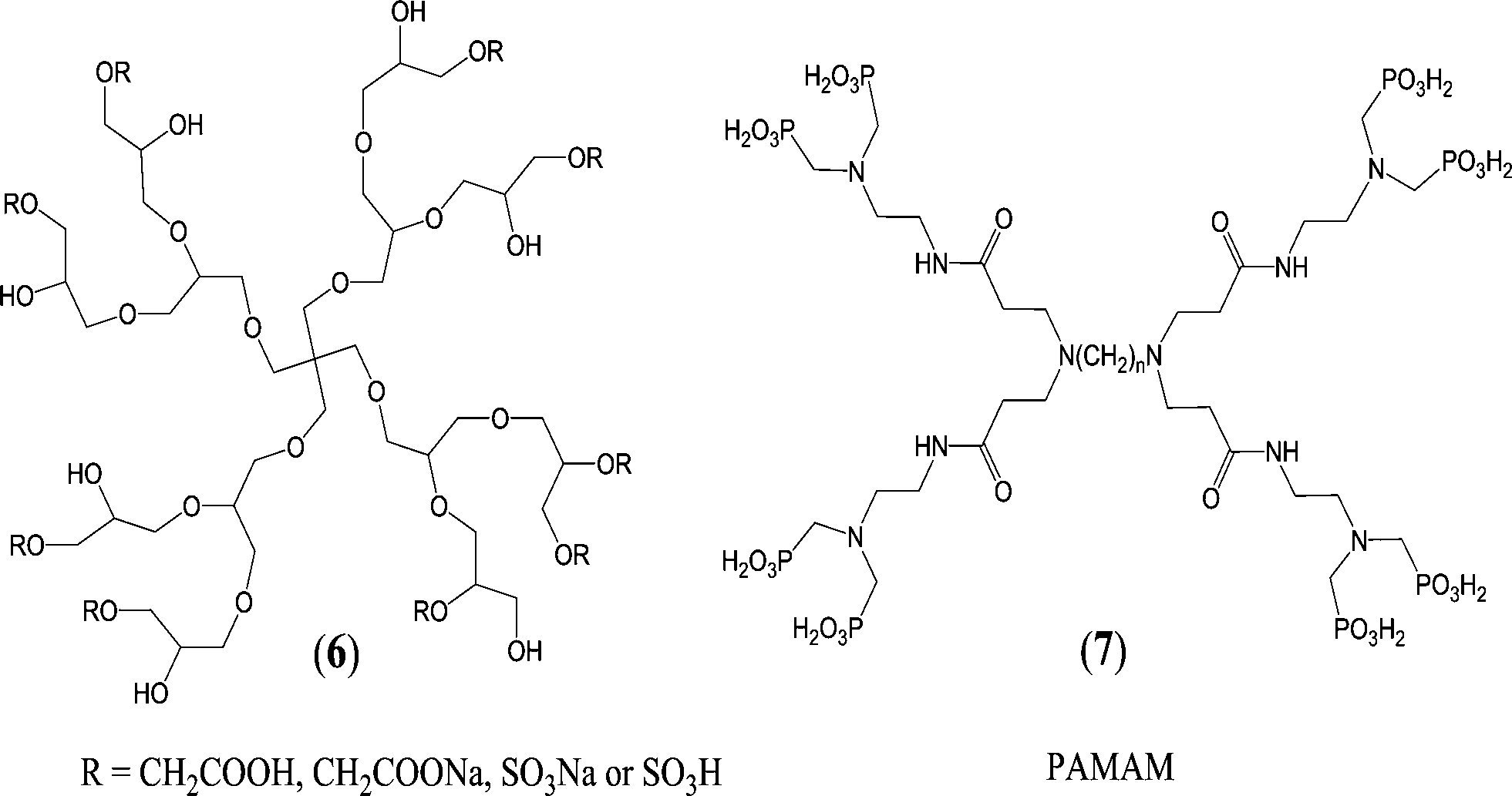
The octamethylenephosphonic acid terminated polyamidoamine (PAMAM) dendrimer (Fig. 6) has been introduced as a suitable antiscalant for high Ca2+ concentrations in industrial water treatments in boilers, cooling systems, desalination, and oil production.152 When the antiscalant concentration was 14 mg L−1, the inhibition rate of CaCO3 approached 100%. This PO3H2-terminated dendrimer was found to be superior to most of the classical commercially available PO3H2-containing small molecules, such as amino trimethylene phosphonic acid (ATMP) and ethylene diamine tetra(methylene phosphonic acid) (EDTMP).152
Natural antiscalants
Some naturally occurring organic molecules, in particular plant extracts, have been used as new “green” antiscalants recently. These “environmentally friendly” inhibitors are advantageous in various domains where using synthetic macromolecules is limited by the environmental regulations and/or associated process difficulties in drinking water supply and the food industry. Comprehensive reviews on “natural” antiscalants have been published recently to which the reader is directed to for further study.153,154Polymers and dendrimers for the inhibition of silica/silicate deposition
Silica (SiO2) scale formation and deposition is a major problem in high-silica-containing cooling water,155,156 where the concentration of dissolved silica exceeds the solubility equilibrium limit of amorphous silica at the pipeline or reservoir operating temperature and pH.157 Tremendous efforts and expenditures have been invested on the dissolution and removal of colloidal SiO2; however, the efficiency of commonly employed silica dissolution additives is generally low and dosage dependent.158–160 Therefore, scale prevention rather than removal is highly desired to facilitate the operation of RO systems. Aiming at the prevention of silica/silicate precipitation, polymers and dendrimers have now been widely investigated. These macromolecules, which are effective in inhibiting silicate-based scales, generally have one or more functional groups such as –COOH, –NH2, –CONH2 or –NH3+, which possess strong chelation properties.Polyamine/polyammonium cationic macromolecules have also demonstrated inhibition capability against colloidal silica particle growth. Polyethyleneimine (PEI) (a highly branched cationic polymer bearing primary, secondary, and tertiary amines, Fig. 7) and polyallylamine hydrochloride (PALAM) have been shown to possess high inhibition efficiency.162 Environmentally friendly polyammonium cationic macromolecules including poly(acrylamide-co-diallyldimethylammonium chloride) (PAMALAM),162,163 and cationic inulin (CATIN) polymers163 were also shown to be powerful silica deposition inhibitors. The optimum dosages were 10 ppm for PEI, 20 ppm for PALAM, and 80–100 ppm for PAMALAM, and the inhibition was 55%, 65%, and 60% within 24 h, respectively. A significant dependence of the silica inhibition efficiency on additive dosage and polymer backbone cationic charge density has been widely reported.163
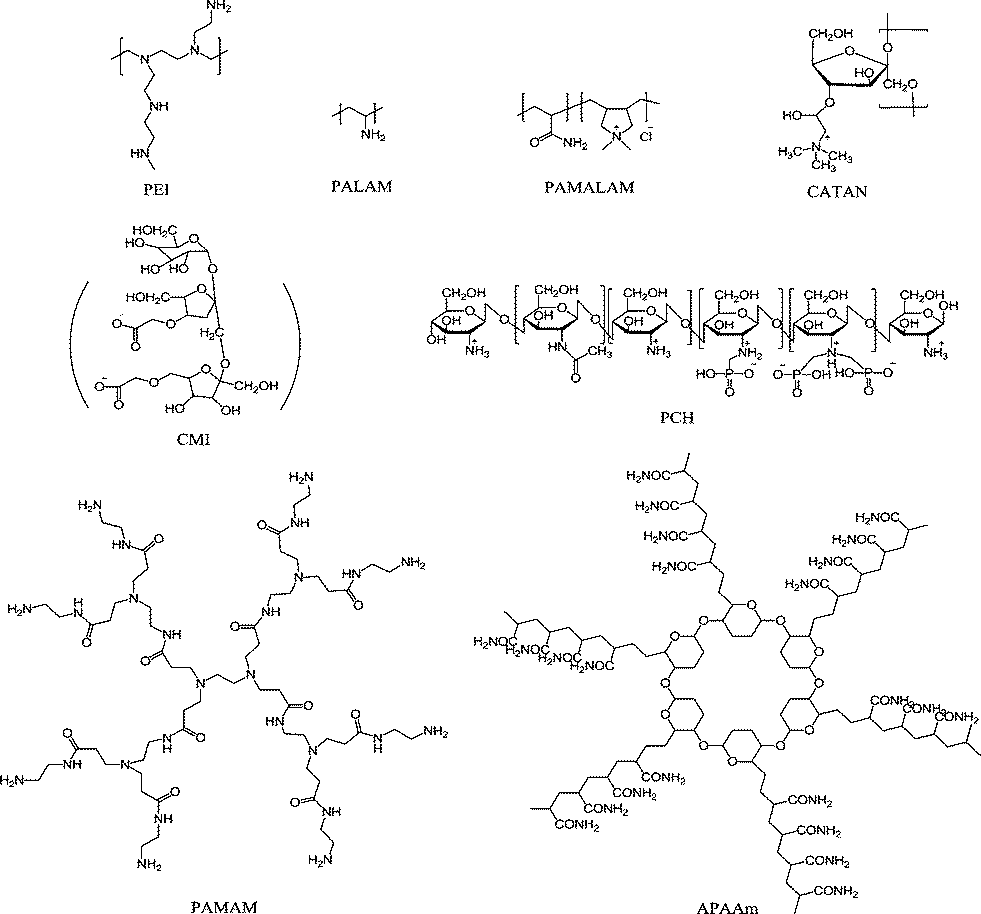 | ||
| Fig. 7 Examples of efficient SiO2 scale inhibitors including PEI,162 PALAM,162 PAMALAM,166 CATAN,163 CMI164,165 and PCH polymers,166 as well as PAMAM160 and APAAm dendrimers.171 | ||
The use of other “green” additives in SiO2 scale inhibition has also been reported. For instance, carboxymethylinulin (CMI), a derivative of a biopolymer extracted from the roots of the chicory plant, had antiscaling behavior;164,165 a zwitterionic polymer phosphonomethylated chitosan (PCH) was able to retard silicic acid condensation at circumneutral pH in aqueous supersaturated solutions.166 Moreover, PEG (Mw = 20 kDa) presented a good inhibition performance.166 Interestingly, positive synergistic effects of two or three of the above-mentioned additives for SiO2 scale inhibition have been demonstrated.164–166
Demadis and co-workers168–170 have investigated the influence of structural features of PAMAM dendrimers including the generation number and nature of end groups in inhibiting silica/silicate deposition. PAMAM dendrimers of different generations (0.5, 1.5, 2.5 with –COOH termini, and 1 and 2 with –NH2 termini) were tested for the prevention of SiO2 deposition. Dendrimers with –NH2 termini were found to be superior to those with –COOH for the inhibition of SiO2. More than 80% of silica deposition was avoided using PAMAM generation 1.0 (with either 8-NH2 termini or 8-COOH termini) at 40 ppm dosage level employed for 12 h. PAMAM dendrimers are not only effective for SiO2 scale inhibition but they are also used as silica nucleators, forming SiO2–PAMAM composites.
Novel “green” star-shaped dendrimers with algae core and poly(acrylamide)–poly(acrylic acid) arms (APAAm) (see structure in Fig. 7) were recently synthesized and applied as silica polymerization inhibitors.171 The inhibition achieved by this APAAm dendrimer was over 90% within 12 h, and the induction time was delayed to 10 days, indicating a better inhibition performance than PAMAM-G0 (generation zero of PAMAM dendrimer) inhibitor
Removal of macromolecular scale inhibitors from effluents
Post-treatment, especially in municipal wastewater treatment plants (MWWTPs), is of utmost importance to reduce the ecological footprints of antiscaling additives. The presence of excessive amounts of phosphorus and nitrogen (two main constituents of scale-inhibiting macromolecules) in water effluents results in an ecological catastrophe called eutrophication.172 Owing to current advances in pre- and post-treatment, early elimination of organic micropollutants has become feasible. Upgrades such as post-ozonation and sand filtration to conventional municipal wastewater plants have facilitated the elimination of a wide range of organic materials.173 Other methods include hydrogen peroxide and ultraviolet (UV) treatments, both of which provide efficient oxidation of macromolecules using radicals.174 Several comprehensive reviews have discussed various aspects of organic material elimination from wastewater for reclamation, recycling, and reusing purposes.175–181Conclusions
Mineral scale deposition is a major industrial problem, and much effort has been devoted in developing additives that could prevent scale build-up during water processing. Macromolecules that control or completely inhibit inorganic crystal growth have offered tremendous potential in water treatment and are the most promising inhibitors currently employed in industry. The conventional intervention for scale inhibition in the past has been with polymers that exploit their basic property, backbones that cannot be broken down easily and with natural means. The strong drive towards balancing environmental impact and efficiency has mobilized the scientific community to design “green” and benign antiscalants that can readily biodegrade. Another important parameter that has to be taken into consideration is the cost-effectiveness since industries are profit driven. New strategies in preventing scale build-up will have to include these factors and design antiscalants that will be used at low dosage for maximum efficacy. Sequestering inorganic ions effectively may also require more than one type of environment in a single platform. Dendrimers and hyperbranched polymers with tailored layer-by-layer construction and multivalent nature provide this opportunity. Initial reports investigating their antiscaling efficiency are very encouraging, and we expect to see much more research exploration in this area. The focus of the current review has been on the macromolecular additives for taming inorganic scales, however, it is essential that the scientific community consider their side effects on the municipal water quality. Fortunately, micropollutant elimination science has been flourishing to provide reliable removal of C-, N-, and P-containing macromolecules through advance methods, including ozonation and active filtration, in municipal waste water treatment plants.Acknowledgements
We would like to thank Natural Sciences and Engineering Research Council (Canada), Kemira Chemicals, Fonds de Recherche du Québec-Nature et technologies (FRQNT, Quebec, Canada), and Centre for Self-assembled Chemical Structures (CSACS) for financial support.Notes and references
- S. Renzetti, Land Econ., 1992, 68, 396–404 CrossRef
.
- UNESCO, United Nations World Water Dev. Rep., 2003, pp. 1–36 Search PubMed.
- UNESCO, United Nations World Water Dev. Rep., 2014, pp. 1–230 Search PubMed.
- Industry Canada Report, Environmental Business Journal, Environmental Business International Inc., 2008, pp. 1–14, http://uvpure.com/docs/CanadianWaterIndustry-EBJReport.pdf Search PubMed
- Z. Amjad, R. T. Landgraf and J. L. Penn, Int. J. Corros. Scale Inhib., 2014, 3, 35–47 CrossRef
- E. W. Allen, J. Environ. Eng. Sci., 2008, 7, 499–524 CrossRef CAS
- K. Al-Anezi and N. Hilal, Sep. Purif. Rev., 2006, 35, 223–247 CrossRef CAS
- W. N. Al Nasser and F. H. Al Salhi, Chem. Eng. Sci., 2013, 86, 70–77 CrossRef CAS
- Y. Zhang, H. Shaw, R. Farquhar and R. Dawe, J. Pet. Sci. Eng., 2001, 29, 85–95 CrossRef CAS
- F. Detrick, Ind. Eng. Chem. Process Des. Dev., 1964, 3, 345–348 Search PubMed
- J. D. Doyle and S. A. Parsons, Water Res., 2002, 36, 3925–3940 CrossRef CAS PubMed
- A. A. Al-Hamzah, C. P. East, W. O. S. Doherty and C. M. Fellows, Desalination, 2014, 338, 93–105 CrossRef CAS
- A. Antony, J. H. Low, S. Gray, A. E. Childress, P. Le-Clech and G. Leslie, J. Membr. Sci., 2011, 383, 1–16 CrossRef CAS
- A. S. Al-Amoudi, Desalination, 2010, 259, 1–10 CrossRef CAS
- A. A. Al-Hamzah and C. M. Fellows, Desalination, 2014, 332, 33–43 CrossRef CAS
- Y. Wang, J. Davidson and L. Francis, J. Sol. Energy Eng., 2005, 127, 3–14 CrossRef CAS
-
S. Mann, Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry, Oxford University Press, 2001 Search PubMed
- J. J. DeYoreo and P. G. Vekilov, Rev. Mineral. Geochem., 2003, 54, 57–93 CrossRef CAS
- H. Ozawa, K. Hoshi and N. Amizuka, J. Oral Biosci., 2008, 50, 1–14 CrossRef
- S. Weiner and L. Addadi, Annu. Rev. Mater. Res., 2011, 21–40 CrossRef CAS
- S. Mann, J. Mater. Chem., 1995, 5, 935–946 RSC
- Water Quality Association webpage, 2015.
-
Mineral Scales and Deposits: Scientific and Technological Approaches, ed. Zahid Amjad and K. D. Demadis, Elsevier B.V., Amsterdam, 2015 Search PubMed
- H. Gu, A. R. Bartman, M. Uchymiak, P. D. Christofides and Y. Cohen, Desalination, 2013, 308, 63–72 CrossRef CAS
- C. J. Gabelich, M. D. Williams, A. Rahardianto, J. C. Franklin and Y. Cohen, J. Membr. Sci., 2007, 301, 131–141 CrossRef CAS
- S. Lee and R. M. Lueptow, Desalination, 2003, 155, 131–139 CrossRef CAS
- J. S. Baker and S. J. Judd, Water Res., 1996, 30, 247–260 CrossRef CAS
- A. Alabi, M. Chiesa, C. Garlisi and G. Palmisano, Environ. Sci.: Water Res. Technol., 2015, 1, 408–425 CAS
-
J. W. Mullin, Crystallization, Butterworth-Heinemann, Oxford, 2001 Search PubMed
- F. C. Meldrum and H. Cölfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed
- Z. S. Derewenda and P. G. Vekilov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 116–124 CrossRef PubMed
- J. W. Gibbs, Trans. Conn. Acad. Arts Sci., 1876, 3, 108–248 Search PubMed
- J. W. Gibbs, Trans. Conn. Acad. Arts Sci., 1878, 16, 343–524 Search PubMed
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergström and H. Cölfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC
- D. Kashchiev, J. Chem. Phys., 2003, 118, 1837–1851 CrossRef CAS
- S. Auer and D. Frenkel, Nature, 2001, 413, 711–713 CrossRef CAS PubMed
- D. Verdoes, D. Kashchiev and G. M. van Rosmalen, J. Cryst. Growth, 1992, 118, 401–413 CrossRef CAS
- M. Perez, Scr. Mater., 2005, 52, 709–712 CrossRef CAS
- M. M. Reddy and W. D. Gaillard, J. Colloid Interface Sci., 1981, 80, 171–178 CrossRef CAS
- G. Frens, Int. J. Phys. Sci., 1973, 241, 20–22 CAS
- T. K. Sau and A. L. Rogach, Adv. Mater., 2010, 22, 1781–1804 CrossRef CAS PubMed
- M. Sluyters-Rehbach, J. H. O. J. Wijenberg, E. Bosco and J. H. Sluyters, J. Electroanal. Chem. Interfacial Electrochem., 1987, 236, 1–20 CrossRef CAS
-
D. Kashchiev, Nucleation: Basic Theory with Applications, Butterworths, Heinemann, Oxford, 1999 Search PubMed
- N. H. Fletcher, J. Chem. Phys., 1958, 29, 572–576 CrossRef CAS
- A. G. Shtukenberg, S. S. Lee, B. Kahr and M. D. Ward, Annu. Rev. Chem. Biomol. Eng., 2014, 5, 77–96 CrossRef CAS PubMed
- G. Azimi, Y. Cui, A. Sabanska and K. K. Varanasi, Appl. Surf. Sci., 2014, 313, 591–599 CrossRef CAS
- N. Cabrerra and D. A. Vermilea, in Proceedings of the International Conference, Cooperstown, NY, 1958, pp. 393–410 Search PubMed.
-
G. Bliznakov, Adsorpt. Croissance Cristal. Ed. du Cent. Natl. la Rech. Sci. Paris, 1965, pp. 291–301 Search PubMed
- H. Fleisch, Kidney Int., 1978, 13, 361–371 CrossRef CAS PubMed
- H. E. Buckley, Faraday Discuss., 1946, 37, 243–254 Search PubMed
- M. D. Sikirić and H. Füredi-Milhofer, Adv. Colloid Interface Sci., 2006, 128–130, 135–158 CrossRef PubMed
- M. C. Weinberg, W. H. Poisl and L. Granasy, C. R. Chim., 2002, 5, 765–771 CrossRef CAS
- G. Nehrke, H. Poigner, D. Wilhelms-Dick, T. Brey and D. Abele, Geochem., Geophys., Geosyst., 2012, 13, 1–8 CrossRef
- D. Turnbull and J. C. Fisher, J. Chem. Phys., 1949, 17, 71–73 CrossRef CAS
- B. Sheva, J. Non-Cryst. Solids, 1985, 74, 85–95 CrossRef
-
A. Einstein, Investigations on the theory of the Brownian movement, Dover Publications, New York, 1956 Search PubMed
- D. B. Warren, H. Benameur, C. J. H. Porter and C. W. Pouton, J. Drug Targeting, 2010, 18, 704–731 CrossRef CAS PubMed
-
Mineral Scales in Biological and Industrial Systems, ed. Z. Amjad, CRC Press, Taylor & Francis Group, 2014 Search PubMed
- F. Hui and J. Lédion, Eur. J. Water Qual., 2002, 1, 1–27 Search PubMed
- H. Feitler, Mater. Prot. Perform., 1972, 11, 31–35 CAS
- S. A. Ali, I. W. Kazi and F. Rahman, Desalination, 2015, 357, 36–44 CrossRef CAS
- M. B. Tomson and G. H. Nancollas, Science, 1978, 200, 1059–1060 CAS
- T.
F. Kazmierczak, M. B. Tomson and G. H. Nancollas, J. Phys. Chem., 1982, 86, 103–107 CrossRef CAS
- R. Beck, M. Seiersten and J. P. Andreassen, J. Cryst. Growth, 2013, 380, 187–196 CrossRef CAS
- G. Gauthier, Y. Chao, O. Horner, O. Alos-Ramos, F. Hui, J. Lédion and H. Perrot, Desalination, 2012, 299, 89–95 CrossRef CAS
- D. Peronno, H. Cheap-Charpentier, O. Horner and H. Perrot, Journal of Water Process Engineering, 2015, 7, 11–20 CrossRef
- NACE Standard Test Method. Laboratory screening tests to determine the ability of scale inhibitors to prevent the precipitation of calcium sulfate and calcium carbonate from solution (for oil and gas production systems) TM0374, 2007.
- D. H. Troup and J. A. Richardson, Chem. Eng. Commun., 1978, 2, 167–180 CrossRef CAS
- C. Tzotzi, T. Pahiadaki, S. G. Yiantsios, A. J. Karabelas and N. Andritsos, J. Membr. Sci., 2007, 296, 171–184 CrossRef CAS
-
N. I. T. Group, Dynamic Scale Inhibitor Evaluation Apparatus and Procedures in Oil and Gas Production, 2005 Search PubMed
- S. J. Dyer and G. M. Graham, J. Pet. Sci. Eng., 2002, 35, 95–107 CrossRef CAS
- D. Hasson, M. Avriel, W. Resnick, T. Rozenman and S. Windreich, Ind. Eng. Chem. Res., 1968, 7, 59–65 Search PubMed
- A. Neville, Energy Fuels, 2012, 26, 4158–4166 CrossRef CAS
- R. Sharp, E. Vadiveloo, R. Fergen, M. Moncholi, P. Pitt, D. Wankmuller and R. Latimer, Water Environ. Res., 2013, 85, 675–686 CrossRef CAS PubMed
-
Z. Amjad, Mineral scale formation and inhibition, Springer Science + Business Media, LLC, New York, 1995 Search PubMed
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2001 Search PubMed
- C. Gabrielli, M. Keddam, H. Perrot, A. Khalil, R. Rosset and M. Zidoune, J. Appl. Electrochem., 1996, 26, 1125–1132 CrossRef CAS
- F. G. Cottrell, Z. Phys. Chem., 1903, 42, 385–431 Search PubMed
- R. G. Compton, M. E. Laing, D. Mason, R. J. Northing and P. R. Unwin, Proc. R. Soc. London, 2014, 418, 113–154 CrossRef
- C. Gabrielli, J. Electrochem. Soc., 1998, 145, 2386–2396 CrossRef CAS
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed
- J. Macdonald, Ann. Biomed. Eng., 1992, 20, 289–305 CrossRef CAS PubMed
- C. Tzotzi, T. Pahiadaki, S. Yiantsios, A. Karabelas and N. Andritsos, J. Membr. Sci., 2007, 296, 171–184 CrossRef CAS
- E. G. Darton, Desalination, 1997, 113, 227–229 CrossRef CAS
- Y. Tang, W. Yang, X. Yin, Y. Liu, P. Yin and J. Wang, Desalination, 2008, 228, 55–60 CrossRef CAS
- S. L. S. Stipp, C. M. Eggleston and B. S. Nielsen, Geochim. Cosmochim. Acta, 1994, 58, 3023–3033 CrossRef
- S. Xu, J. Electrochem. Soc., 1999, 146, 3315–3323 CrossRef CAS
- E. Dalas, J. Cryst. Growth, 2001, 222, 287–292 CrossRef CAS
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed
- H. Endo, D. Schwahn and H. Cölfen, J. Chem. Phys., 2004, 120, 9410–9423 CrossRef CAS PubMed
- P. J. M. Smeets, K. R. Cho, R. G. E. Kempen, N. A. J. M. Sommerdijk and J. J. De Yoreo, Nat. Mater., 2015, 14, 394–399 CrossRef CAS PubMed
- N. Blagden, M. de Matas, P. T. Gavan and P. York, Adv. Drug Delivery Rev., 2007, 59, 617–630 CrossRef CAS PubMed
- T. Kato, T. Suzuki, T. Amamiya, T. Irie, M. Komiyama and H. Yui, Supramol. Sci., 1998, 5, 411–415 CrossRef CAS
- H. Deng, X. C. Shen, X. M. Wang and C. Du, Front. Mater. Sci., 2013, 7, 62–68 CrossRef
- H. Nebel and M. Epple, Z. Anorg. Allg. Chem., 2008, 634, 1439–1443 CrossRef CAS
- F. Jones and M. I. Ogden, CrystEngComm, 2010, 12, 1016–1023 RSC
- E. G. Darton, Desalination, 2000, 132, 121–131 CrossRef CAS
- S. Patel, in Corrosion-National Association of Corrosion Engineers Annual Conference, NACE International, Houston, TX, 1998, pp. 56–60 Search PubMed.
- S. Ghani and N. S. Al-Deffeeri, Desalination, 2010, 250, 463–472 CrossRef CAS
- D. E. Abd-El-Khalek and B. A. Abd-El-Nabey, Desalination, 2013, 311, 227–233 CrossRef CAS
- F. H. Butt, F. Rahman and U. Baduruthamal, Desalination, 1997, 109(3), 323–332 CrossRef CAS
- D. H. Solomon and P. F. Rolfe, Desalination, 1966, 1, 260–266 CrossRef CAS
- J. Macadam and S. A. Parsons, Rev. Environ. Sci. Bio/Technol., 2004, 3(2), 159–169 CrossRef CAS
- Z. Amjad, in CORROSION 96, The NACE International Annual Conference and Exposition, 1996 Search PubMed.
- E. Hädicke, J. Rieger, I. Ursula Rau and D. Boeckh, Phys. Chem. Chem. Phys., 1999, 1, 3891–3898 RSC
- X. Li, B. Gao, Q. Yue, D. Ma, H. Rong, P. Zhao and P. Teng, J. Environ. Sci., 2015, 29, 124–130 CrossRef CAS PubMed
-
R. W. Zuhl and Z. Amjad, in The Science and Technology of Industrial Water Treatment, ed. Z. Amjad, CRC Press, Taylor & Francis Group, 2010, pp. 81–103 Search PubMed
- K. Burns, Y. T. Wu and C. S. Grant, Langmuir, 2003, 19, 5669–5679 CrossRef CAS
- J. Chen, L. Xu, J. Han, M. Su and Q. Wu, Desalination, 2015, 358, 42–48 CrossRef CAS
- S. W. Walinsky and B. J. Morton, Desalination, 1979, 31, 289–298 CrossRef CAS
- Z. Amjad, Desalination, 1985, 54, 263–276 CrossRef CAS
- R. Ketrane, B. Saidani, O. Gil, L. Leleyter and F. Baraud, Desalination, 2009, 249, 1397–1404 CrossRef CAS
- R. E. Cooper, K. G. Hanlon, L. G. Smart and G. M. Talbot, Desalination, 1979, 31, 257–266 CrossRef
- P. G. Koutsoukos and C. G. Kontoyannis, J. Cryst. Growth, 1984, 69, 367–376 CrossRef CAS
- N. Abdel-Aal and K. Sawada, J. Cryst. Growth, 2003, 256, 188–200 CrossRef CAS
- A. T. Kan, G. Fu and M. B. Tomson, J. Colloid Interface Sci., 2005, 281, 275–284 CrossRef CAS PubMed
- F. Zeng, J. Zhang, S. Gong and X. Wang, Chin. J. Chem., 2010, 28, 337–343 CrossRef
- S. Lattemann and T. Höpner, Desalination, 2008, 220, 1–15 CrossRef CAS
- P. F. Rolfe, Desalination, 1966, 1, 359–366 CrossRef CAS
- F. Sweet, B. R. Smith, P. Casamento and R. N. Taylor, Desalination, 1970, 8, 167–175 CrossRef
- B. R. Smith, Desalination, 1967, 3, 263–268 CrossRef CAS
- Z. Amjad and J. Hooley, Tenside, Surfactants, Deterg., 1994, 1, 12–17 Search PubMed
- C. M. Wallace, A. D. Al-Hamzah, A. East, C. P. Doherty and W. O. S. Fellows, J. Appl. Polym. Sci., 2010, 116, 1165–1171 Search PubMed
-
R. W. Amjad and Z. Zuhl, in Heat Treatment of Synthetic Polymers as CaCO3 Inhibitors-Part II, 2008, pp. 50–55 Search PubMed
- R. W. Goeldner, Desalination, 1983, 41, 25–35 CrossRef
- C. Fu, Y. Zhou, H. Xie, W. Sun and W. Wu, Ind. Eng. Chem. Res., 2010, 49, 8920–8926 CrossRef CAS
- C. Fu, Y. Zhou, G. Liu, J. Huang, W. Sun and W. Wu, Ind. Eng. Chem. Res., 2011, 50, 10393–10399 CrossRef CAS
- G. Liu, M. Xue, J. Huang, H. Wang, Y. Zhou, Q. Yao, L. Ling, K. Cao, Y. Liu, Y. Bu, Y. Chen, W. Wu and W. Sun, Front. Environ. Sci. Eng., 2014, 9, 545–553 CrossRef
- K. Cao, Y. Zhou, G. Liu, H. Wang and W. Sun, J. Appl. Polym. Sci., 2014, 131, 40193(1)–40193(9) Search PubMed
- Y. Liu, Y. Zhou, Q. Yao, J. Huang, G. Liu, H. Wang, K. Cao, Y. Chen, Y. Bu, W. Wu and W. Sun, J. Appl. Polym. Sci., 2014, 131, 39792(1)–39792(12) Search PubMed
- B. Senthilmurugan, B. Ghosh, S. Kundu and B. Kameshwari, Pet. Sci. Technol., 2011, 29, 2077–2085 CrossRef CAS
- B. Senthilmurugan, B. Ghosh and S. Sanker, J. Ind. Eng. Chem., 2011, 17, 415–420 CrossRef CAS
- M. Dietzsch, M. Barz, T. Schüler, S. Klassen, M. Schreiber, M. Susewind, N. Loges, M. Lang, N. Hellmann, M. Fritz, K. Fischer, P. Theato, A. Kühnle, M. Schmidt, R. Zentel and W. Tremel, Langmuir, 2013, 29, 3080–3088 CrossRef CAS PubMed
- Y. Chen, Y. Zhou, Q. Yao, Y. Bu, H. Wang, W. Wu and W. Sun, Desalin. Water Treat., 2014, 1–11 Search PubMed
- T. Chen, A. Neville, K. Sorbie and Z. Zhong, Chem. Eng. Sci., 2009, 64, 912–918 CrossRef CAS
- Z. Liu, S. Wang, L. Zhang and Z. Liu, Desalination, 2015, 362, 26–33 CrossRef CAS
- X. Wang, C. Zhu and D. Wang, Polym. Polym. Compos., 2013, 21, 449–456 Search PubMed
- C. Wang, S. P. Li and T. D. Li, Desalination, 2009, 249, 1–4 CrossRef CAS
- C. Wang, T. Shen, S. Li and X. Wang, Desalination, 2014, 348, 89–93 CrossRef CAS
- G. B. Butler, Acc. Chem. Res., 1982, 15, 370–378 CrossRef CAS
- I. W. Kazi, F. Rahman and S. A. Ali, Polym. Eng. Sci., 2014, 54, 166–174 CAS
- S. A. Ali, I. W. Kazi and F. Rahman, Polym. Int., 2014, 63, 616–625 CrossRef CAS
- S. A. Ali, S. A. Haladu and H. A. Al-Muallem, J. Appl. Polym. Sci., 2014, 131, 40615(1)–40615(11) CrossRef
- S. A. Ali and S. A. Haladu, Polym. Int., 2014, 63, 1682–1690 CrossRef CAS
- S. A. Haladu and S. A. Ali, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 5130–5142 CrossRef CAS
- P. Shakkthivel, R. Sathiyamoorthi and T. Vasudevan, Desalination, 2004, 164, 111–123 CrossRef CAS
- K. Chauhan, R. Kumar, M. Kumar, P. Sharma and G. S. Chauhan, Desalination, 2012, 305, 31–37 CrossRef CAS
- A. W. Bosman, H. M. Janssen and E. W. Meijer, Chem. Rev., 1999, 99, 1665–1688 CrossRef CAS PubMed
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS PubMed
- M. K. Jensen and M. A. Kelland, J. Pet. Sci. Eng., 2012, 94–95, 66–72 CrossRef CAS
- G. Wang, L. Li, J. Lan, L. Chen and J. You, J. Mater. Chem., 2008, 18, 2789 RSC
-
F. Li, B. Zhang and F. Chen, US Pat. 2014/0319067 A1, 2014 Search PubMed
- M. Chaussemier, E. Pourmohtasham, D. Gelus, N. Pécoul, H. Perrot, J. Lédion, H. Cheap-Charpentier and O. Horner, Desalination, 2015, 356, 47–55 CrossRef CAS
- D. Hasson, H. Shemer and A. Sher, Ind. Eng. Chem. Res., 2011, 50, 7601–7607 CrossRef CAS
- B. W. H. Goodman, M. R. Godfrey and T. M. Miller, in International Water Conference, San Antonio, Texas, 2010, pp. 1–8 Search PubMed.
- A. S. Behrman and H. Gustafson, Ind. Eng. Chem. Res., 1940, 32, 468–472 CrossRef CAS
- R. Y. Ning, Desalination, 2003, 151, 67–73 CrossRef CAS
- K. D. Demadis, E. Mavredaki and M. Somara, Ind. Eng. Chem. Res., 2011, 50, 12587–12595 CrossRef CAS
- K. D. Demadis and E. Mavredaki, Environ. Chem. Lett., 2005, 3, 127–131 CrossRef CAS
- D. L. Gallup and E. Barcelon, Geothermics, 2005, 34, 756–771 CrossRef CAS
- J. S. Gill, Colloids Surf., A, 1993, 74, 101–106 CrossRef CAS
- K. D. Demadis and A. Stathoulopoulou, Ind. Eng. Chem. Res., 2006, 45, 4436–4440 CrossRef CAS
- A. Ketsetzi, A. Stathoulopoulou and K. D. Demadis, Desalination, 2008, 223, 487–493 CrossRef CAS
- S. D. Demadis, K. D. Neofotistou, E. Mavredaki, E. Tsiknakis, M. Sarigiannidou and E. M. Katarachia, Development, 2005, 125, 2457–2467 Search PubMed
- E. Mavredaki, A. Stathoulopoulou, E. Neofotistou and K. D. Demadis, Desalination, 2007, 210, 257–265 CrossRef CAS
- K. D. Demadis and M. Preari, Desalin. Water Treat., 2014, 55, 1–7 Search PubMed
- R. M. Crooks, M. Q. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS PubMed
- E. Neofotistou and K. D. Demadis, Desalination, 2004, 167, 257–272 CrossRef CAS
- E. Neofotistou and K. D. Demadis, Colloids Surf., A, 2004, 242, 213–216 CrossRef CAS
- K. D. Demadis, J. Chem. Technol. Biotechnol., 2005, 80, 630–640 CrossRef CAS
- K. Chauhan, P. Patiyal, G. S. Chauhan and P. Sharma, Water Res., 2014, 56, 225–233 CrossRef CAS PubMed
- D. L. Correll, J. Environ. Qual., 1998, 27, 261–266 CrossRef CAS
- J. Hollender, S. G. Zimmermann, S. Koepke, M. Krauss, C. S. Mcardell, C. Ort, H. Singer, U. Von Gunten and H. Siegrist, Environ. Sci. Technol., 2009, 43, 7862–7869 CrossRef CAS PubMed
- W. H. Glaze, J.-W. Kang and D. H. Chapin, Ozone: Sci. Eng., 1987, 9, 335–352 CrossRef CAS
- V. Camel and A. Bermond, Water Res., 1998, 32, 3208–3222 CrossRef CAS
- M. S. Siddiqui, G. L. Amy and B. D. Murphy, Water Res., 1997, 31, 3098–3106 CrossRef CAS
- G. K. Morse, S. W. Brett, J. A. Guy and J. N. Lester, Sci. Total Environ., 1998, 212, 69–81 CrossRef CAS
- L. E. de-Bashan and Y. Bashan, Water Res., 2004, 38, 4222–4246 CrossRef CAS PubMed
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed
- A. Matilainen, M. Vepsäläinen and M. Sillanpää, Adv. Colloid Interface Sci., 2010, 159, 189–197 CrossRef CAS PubMed
- O. Legrini, E. Oliveros and A. M. Braun, Chem. Rev., 1993, 93, 671–698 CrossRef CAS
- W. F. Langelier, D. H. Caldwell, W. B. Lawrence and C. H. Spaulding, Ind. Eng. Chem., 1950, 42, 126–130 CrossRef CAS
- R. Dooly and J. Glater, Desalination, 1972, 11, 1–16 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ew00158g |
| ‡ These authors contributed equally to this work. |
| § The reaction between the species is usually more complex than what is shown here; as an example, in thermal desalination plants, the alkaline scale results from the competitive bimolecular182 and unimolecular183 decomposition reactions of bicarbonate ion.15 |
¶ LSI = pH – pHs, where pHs (CaCO3) = (9.3 + 0.1[log10(TDS) −1] − 13.12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10[T] + 34.55) − (log10[Ca2+] − 0.4 + log10[alkalinity]). log10[T] + 34.55) − (log10[Ca2+] − 0.4 + log10[alkalinity]). |
| || S&DSI = pH – pHs, where pHs = pCa + pAlk + K, and K is an indicator of ionic strength and temperature. |
| This journal is © The Royal Society of Chemistry 2016 |