
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chromium complexes bearing amidinato-phosphino ligand: synthesis, characterization, and catalytic properties of ethylene tri-/tetramerization and polymerization†
Rui
Liu
a,
Kongtao
Zhu
a,
Xianghong
Zhong
*b,
Jiancheng
Li
a,
Zhenyu
Liu
b,
Shibing
Chen
b and
Hongping
Zhu
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, National Engineering Laboratory for Green Chemical Productions of Alcohols-Ethers-Esters, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: hpzhu@xmu.edu.cn
bMaoming Branch R&D Institute, SINOPEC, Maoming 525011, China. E-mail: zhongxh.mmsh@sinopec.com
First published on 22nd September 2016
Abstract
An amidinato-phosphino ligand ArN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R)NH(o-Ph2PC6H4) (Ar = 2,4,6-Me3C6H2, R = Ph (1); Ar = 2,6-iPr2C6H3, R = Ph (2); Ar = 2,6-iPr2C6H3, R = tBu (3)) was prepared. The ligand reacted with CrCl3(THF)3 to yield the N,P-chelation complex [ArNHC(R)N(o-Ph2PC6H4)]CrCl3(THF) (4–6), and the ligand’s lithium salt ArNC(R)N(o-Ph2PC6H4)Li reacted with the respective CrCl3(THF)3 and CrCl2(THF)2 to give the N,N,P-chelation complexes [ArNC(R)N(o-Ph2PC6H4)]CrCl2(THF) (7–8) and {[ArNC(R)N(o-Ph2PC6H4)]Cr(μ-Cl)}2 (9–11). Complexes 1–11 were characterized by IR, NMR (for 1–3), EPR (for 4–11) spectroscopy and CHN elemental analysis, of which 3, 5, 8, and 11 were further studied by X-ray crystallography. Upon activation with an organoaluminum cocatalyst, complexes 4–6 were all catalytically active in ethylene tri-/tetramerization along with ethylene polymerization, and complexes 7–11 functioned as well but in ethylene polymerization. The correlation between the structure and the catalytic properties of the catalyst system is discussed.
C(R)NH(o-Ph2PC6H4) (Ar = 2,4,6-Me3C6H2, R = Ph (1); Ar = 2,6-iPr2C6H3, R = Ph (2); Ar = 2,6-iPr2C6H3, R = tBu (3)) was prepared. The ligand reacted with CrCl3(THF)3 to yield the N,P-chelation complex [ArNHC(R)N(o-Ph2PC6H4)]CrCl3(THF) (4–6), and the ligand’s lithium salt ArNC(R)N(o-Ph2PC6H4)Li reacted with the respective CrCl3(THF)3 and CrCl2(THF)2 to give the N,N,P-chelation complexes [ArNC(R)N(o-Ph2PC6H4)]CrCl2(THF) (7–8) and {[ArNC(R)N(o-Ph2PC6H4)]Cr(μ-Cl)}2 (9–11). Complexes 1–11 were characterized by IR, NMR (for 1–3), EPR (for 4–11) spectroscopy and CHN elemental analysis, of which 3, 5, 8, and 11 were further studied by X-ray crystallography. Upon activation with an organoaluminum cocatalyst, complexes 4–6 were all catalytically active in ethylene tri-/tetramerization along with ethylene polymerization, and complexes 7–11 functioned as well but in ethylene polymerization. The correlation between the structure and the catalytic properties of the catalyst system is discussed.
Introduction
Chromium catalysts bearing the diphosphazane ligand were found in the earlier parts of this century, first by BP researchers and later by Sasol researchers, which exhibited high activity and excellent selectivity in ethylene oligomerization.1 The ligand consists of a [PNP] framework with tunable substituents at both the N and P atoms, affording an electronic and steric environment for the Cr center to facilitate ethylene trimerization or tri-/tetramerization through the formation of ultimate CrC6 or CrC6/CrC8 metallocycle(s) as the transient state(s).2 This offers a route for production of 1-hexene and 1-octene capable of challenging the traditional nonselective ethylene oligomerization ways (usually producing Schulz–Flory distributions of oligomers)3 and others such as pyrolysis of petroleum fractions or waxes.2h Both 1-hexene and 1-octene are well known as comonomers for manufacturing linear low-density polyethylene (LLDPE).4 Since then, chromium catalysts bearing various ligands with P–N functionality have received considerable attention. A number of P,N-bidentate ligand-chelation chromium catalysts have been synthesized and employed for this catalytic reaction.5 Besides the above-mentioned selective ethylene oligomerization, nonselective ethylene oligomerization and even ethylene polymerization can also take place by means of these catalysts.A handful of P,N-multidentate ligand-based chromium catalysts have also been developed. The ligands of the A–F types employed in these catalysts are shown in Chart 1. Chromium catalysts incorporated by the A-,6E-,7 or F-type8 ligands were found to be active for ethylene trimerization along with polymerization, producing hexenes and polyethylenes. The B5b coordinated chromium catalyst worked only for polymerization to give polyethylene whereas the C9 coordinated chromium catalyst effected concomitant nonselective oligomerization and polymerization to generate a series of oligomers and polyethylene waxes. The catalytic behavior of the D-type10 ligand chromium catalysts was complex due to change in the variety of either the donor atoms or the substituents on the ligand’s skeleton. Products such as oligomers, polyethylene waxes, and/or high molecular weight polyethylenes were produced.
 | ||
| Chart 1 A summary of N,P-multidentate chelation ligands employed to form chromium catalysts for ethylene oligomerization and/or polymerization. | ||
The nature of the ancillary ligand plays a fundamental role in determining the performance of the chromium catalyst. In the above-mentioned examples the catalytic reaction performances of the state-dependent selective or nonselective ethylene oligomerization or ethylene polymerization were, in fact, affected.5–10 It is worth noting that some combined catalytic performances such as ethylene polymerization plus selective or nonselective ethylene oligomerization were exhibited especially for the same ligand based chromium catalyst system.7–10 The reason for these multiple behaviors may be the formation of multiple active species with chromium in different oxidation states formed under operando conditions.1a,2c,4d,6b,11 The Cr(II) state is suggested to respond to nonselective oligomerization12 whereas the Cr(I)/Cr(III) couple states are for selective oligomerization.13 However, which oxidation state is responsible for polymerization remains unclear.14 Nonetheless, studies on these details appear to have been carried out to a lesser extent.3c,4a Herein, we reported a new type of ligand, a phenyl group-connected amidinato-phosphino ligand with the formula ArNC(R)NH(o-Ph2PC6H4) (Ar, R: aryl or alkyl, Scheme 1). This ligand enabled formation of Cr(III) complexes by either N,P-chelation or N,N,P-chelation prepared under different reaction conditions. Cr(II) complexes with the N,N,P-chelated ligand have also been synthesized. Catalytic tests have shown that in the presence of organoaluminum as the cocatalyst, the N,P-chelation Cr(III) complexes were active in ethylene tri-/tetramerization along with ethylene polymerization whereas the N,N,P-chelation Cr(III) and Cr(II) complexes were only active in ethylene polymerization. These results inspire a discussion of the different catalytic performances which probably occur by forming multiple active species from the same ligand stabilized chromium catalyst system.
 | ||
| Scheme 1 Synthesis of ligands 1–3. | ||
Results and discussion
Synthesis and characterization of the amidinato-phosphino ligands and the chromium complexes
The phenyl group connected amidinato-phosphino ligand has rarely been reported.15 Using o-diphenylphosphinoaniline as the precursor,16 a condensation reaction with imidoyl chloride ArNC(R)Cl was proven to be useful for preparation of the target ligand ArNC(R)NH(o-Ph2PC6H4) (Ar = 2,4,6-Me3C6H2, R = Ph (1); Ar = 2,6-iPr2C6H3, R = Ph (2); Ar = 2,6-iPr2C6H3, R = tBu (3)), with elimination of HCl under reflux conditions in toluene (Scheme 1). Imidoyl chloride can be prepared via initial amination of acylchloride (RC(O)Cl) (R = tBu and Ph) using a primary amine (ArNH2) (Ar = 2,4,6-Me3C6H2 and 2,6-iPr2C6H3), followed by dehydroxylation and subsequent chlorination in the presence of thionyl chloride.17 Then, both the Ar and R groups are easily tuned in the NCN-C6H4-o-PPh2 scaffold.
Ligands 1–3 were characterized by NMR (1H, 13C, and 31P), IR spectroscopy and CHN elemental analysis. The presence of the PPh2 group in 1–3 is indicated by the 31P NMR spectra that show resonances at δ −16.3 ppm for 1, δ −18.2 ppm for 2, and δ −16.1 ppm for 3. These values are a little downfield compared with that of the precursor o-Ph2PC6H4NH2 (δ −21.0 ppm).13 Formation of the amidinato NC–NH functionality is evidenced from the 13C NMR spectra in which carbon resonances at δ 154.0 ppm for 1, δ 153.8 ppm for 2, and δ 153.9 ppm for 3 are exhibited. The IR spectra display the related CN bond vibrations at ν 1619 cm−1 for 1, ν 1620 cm−1 for 2, and ν 1665 cm−1 for 3. Moreover, the resonances at δ 6.20 ppm for 1, δ 6.24 ppm for 2, and δ 6.46 ppm for 3 in the 1H NMR spectra are due to the NH proton. The IR spectra present NH bands for 1–3 in a characteristic area (ν 3336 cm−1 for 1, ν 3335 cm−1 for 2, and ν 3399 cm−1 for 3). To confirm the composition and structure of the ligand in detail, single crystals of 3 were grown and further studied by X-ray diffraction. As expected, the character of the NCN-C6H4-o-PPh2 scaffold in 3 is clearly revealed with the tBu group at the C atom and the 2,6-iPr2C6H3 group at the terminal N atom (Fig. 1). In this motif, the N(1)–C(1) and N(2)–C(1) bond lengths are 1.281(2) and 1.379(2) Å, respectively. The former bond length falls in the range of those observed in the imine compounds (1.24–1.28 Å),18 while the latter is comparably shorter than that of the N–Caryl single bond (1.410(2) and 1.427(2) Å). These data indicate that an electronic conjugation over the NC–N skeleton is formed. It should be mentioned that in the 1H NMR spectra of 1–3, broadening proton resonances were actually observed which implies a slow interchange of the ligand into its resonance form through the NC–N skeleton in solution (Scheme 1). Similar tautomerization has been observed for the N-phosphinoamidine ligand reported by Sydora and coworkers.5b The latter resonance form is also found in the formation of the Cr(III) complexes 4–6 (vide infra).
 | ||
| Fig. 1 X-ray molecular structure of 3 with thermal ellipsoids at 50% probability level. All H atoms, except for that of NH, are omitted for clarity. | ||
The reactions of 1–3 with CrCl3(THF)3 were each carried out at room temperature (in THF for 1 and 2 and in CH2Cl2 for 3), and complexes 4–6 were readily produced with respective isolated yields of 89%, 82%, and 80% (Scheme 2). Further reactions were performed using CrCl2(THF)2 instead. However, no reaction resulted even we treated the reaction at elevated temperatures and even upon reflux. Complexes 4–6 were characterized by IR spectroscopy which showed that the N–H bond remained (the vibration is at ν 3165 cm−1 for 4, ν 3195 cm−1 for 5, and ν 3328 cm−1 for 6). IR bands for the CN bond were found at ν 1595 cm−1 for 4, ν 1588 cm−1 for 5, and ν 1588 cm−1 for 6. Meanwhile, EPR spectral analysis was performed, which gave g values of 4.294 for 4, 3.902 for 5, and 4.646 for 6, indicative of formation of the Cr(III) complexes as these data are close to those of other trivalent chromium complexes (3.75–4.38).19 Furthermore, complex 5 was studied by X-ray crystallography. The structural analysis clearly demonstrated N,P-bidentate chelation of ligand 2 at the Cr(III) center with the NH group away from coordination (Fig. 2). This ligation feature is different from those of the aforementioned P,N,P-chelation chromium complexes having the NH group in the E and F types ligands.7,8 The N(1)–C(7) and N(2)–C(7) bond lengths are 1.322(6) and 1.337(6) Å, respectively, showing great electronic conjugation over the NC–N skeleton. The NH proton facing towards the Cr center then prevents a bond interaction between the Cr and the NNH atom (Cr(1)⋯N(2) 3.312 Å). In this structure, the Cr atom is six-coordinate and adopts a distorted octahedral geometry where P(1), N(1), O(1), Cl(2), and Cr(1) form a basal plane (Δ = 0.0098 Å) and Cl(1) and Cl(3) are located at the top and bottom apex positions (Cl(1)–Cr(1)–Cl(3) 174.3(1)°).
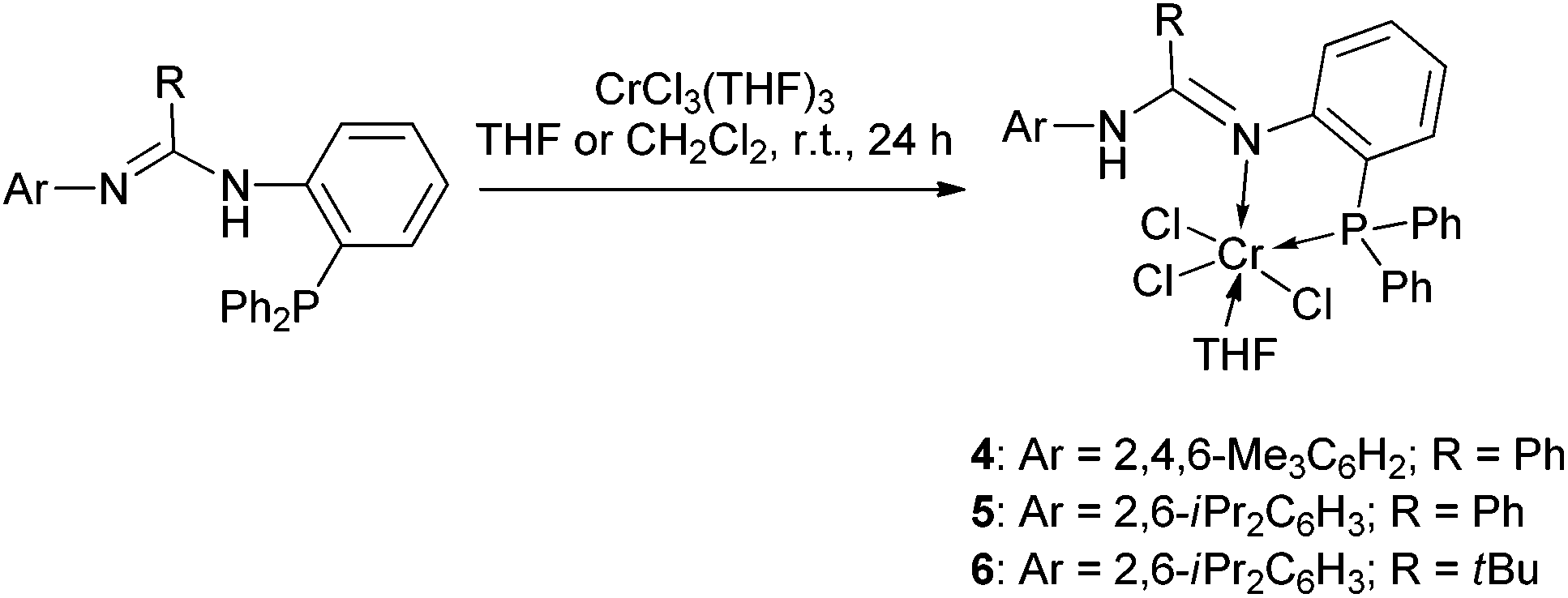 | ||
| Scheme 2 Synthesis of the N,P-chelation Cr(III) complexes 4–6. | ||
 | ||
| Fig. 2 X-ray molecular structure of 5 with thermal ellipsoids at 50% probability level. All H atoms, except for that of NH, are omitted for clarity. | ||
We further employed a metathesis reaction to prepare the chromium complexes. Treatment of 1–3 with nBuLi in THF over the temperature range from −78 to −30 °C produced the related lithium salts that were not isolated but directly used for reaction with CrCl3(THF)3 over the temperature range from −78 °C to room temperature. Complexes 7 and 8 were successfully obtained (Scheme 3), but isolation of a similar complex containing the deprotonated ligand 3 failed, as in the reaction a black suspension was always formed from which it was not possible to obtain the pure target. Still, by this method, complexes 9–11 were readily afforded using CrCl2(THF)2 as the chromium source (Scheme 3).
 | ||
| Scheme 3 Synthesis of the N,N,P-chelation Cr(III) and Cr(II) complexes 7–11. | ||
Complexes 7–11 were characterized by IR spectroscopy which revealed the absence of the N–H bond vibration for each compound. Moreover, the IR band for the CN bond appeared to be red-shifted (ν 1578 cm−1 for 7, ν 1591 cm−1 for 8, ν 1578 cm−1 for 9, ν 1595 cm−1 for 10, and ν 1586 cm−1 for 11) when compared with those of the ligands 1–3. These data are also different from those of complexes 4–6. All of these factors suggest that an N,N,P-coordination mode of the monoanionic ligand at the Cr atom was formed in 7–11, and this mode is confirmed from X-ray crystallographic analysis of the representative complexes 8 and 11. Furthermore, EPR spectral analysis confirmed the Cr(III) state in 7 and 8, which exhibited g values of 3.912 for 7 and 4.349 for 8, both were comparably similar to those of other Cr(III) complexes19 and complexes 4–6. The EPR signals for 9–11 are silent, suggestive of the Cr(II) state of these complexes, as has been commonly discussed in the other divalent chromium compounds.13f Due to the different oxidation states of the Cr atoms present in 8 and 11, differed molecular structures were exhibited (Fig. 3 and 4). Complex 8 is mononuclear with a trivalent chromium center in a distorted octahedral geometry. The ligand’s N, N, and P atoms coordinate at the Cr in a closely meridional arrangement (ΔN(2)C(7)N(1)C(1)C(2)P(1)Cr(1) = 0.0834 Å) and one THF and two Cl− moieties complete the three remaining positions. This structure can be compared to those of the Cr(III) complexes incorporating the neutral PNP (D,10E,7 and F,8Chart 1) and PNN (B,5bChart 1) type ligands. In contrast, complex 11 is dinuclear having two divalent chromium centers. Its formation is probably due to a steric structural compromise of the monomeric form of [2,6-iPr2C6H3NC(tBu)N(o-Ph2PC6H4)]CrCl, as in this form the Cr is four-coordinate and strongly prone to adopting a square-planar geometry.20 In 11, the Cr(1)⋯Cr(1A) separation is 3.410 (1) Å, indicative of almost non-bonding between them. Then, each Cr center adopts a distorted square-pyramidal geometry in which two N and two Cl atoms form the base (ΔN(1)N(2)Cr(1)Cl(1)Cl(1A) = 0.1865 Å) and the P atom stands at the apex.
 | ||
| Fig. 3 X-ray molecular structure of 8 with thermal ellipsoids at 50% probability level. H atoms are omitted for clarity. | ||
 | ||
| Fig. 4 X-ray molecular structure of 11 with thermal ellipsoids at 50% probability level. H atoms are omitted for clarity. Symmetry code for A: 1 − x, y, −z + 1/2. | ||
Selected bond parameters on the NC–N moiety in 3, 5, 8, and 11 are summarized in Table 1. Once they have formed the metal complexes, the parameters for the NC–N skeleton changed dramatically. This indicates a strong bonding interaction between either the Cr(II) or the Cr(III) center with the amidinato-phosphino ligand. The Cr–N bond lengths appear over a close range (1.971(3)–2.114(3) Å). Within the CrCN2 four-membered chelating rings of 8 and 11, the N–Cr–N bond angles are 64.3(1)° and 64.1(1)°, respectively. In addition, the Cr–P bond lengths appear close to those in the Cr(III) complexes (2.464(2) Å in 5 and 2.493(1) Å in 8), but they are significantly shorter than that in the Cr(II) complex 11 (2.772(1) Å) due to the different geometries adopted. The N–Cr–P bond angles within the CrC2NP five-membered chelation ring are 76.3(1)° and 79.2(1)° in the Cr(III) complexes 5 and 8, respectively, and this angle is found to be 71.9(1)° in the Cr(II) complex 11.
| Comp. | 3 | 5 | 8 | 11 |
|---|---|---|---|---|
| N–CNC–N |
1.281(2) | 1.322(6) | 1.313(4) | 1.334(4) |
| 1.379(2) | 1.337(6) | 1.343(4) | 1.344(4) | |
| Cr–N | 2.109(4) | 1.971(3) | 2.054(3) | |
| 2.114(3) | 2.045(3) | |||
| Cr–P | 2.464(2) | 2.493(1) | 2.772(1) | |
| Cr–Cl | 2.298(1) | 2.299(1) | 2.381(1) | |
| 2.323(1) | 2.312(1) | 2.432(1) | ||
| 2.340(2) | ||||
| Cr–OTHF | 2.082(3) | 2.054(2) | ||
| N–C–N | 129.7(2) | 120.3(4) | 110.1(3) | 108.3(3) |
| P–Cr–Nadj. | 76.3(1) | 79.2(1) | 71.9(1) | |
| N–Cr–N | 64.3(1) | 64.1(1) |
Ethylene oligomerization/polymerization test on 4–11
In combination with methylalumoxane (MAO) as the cocatalyst, complex 4 acted as a catalyst to produce oligomers along with polymers when exposed to ethylene at 40 bar and 50 °C in toluene as a solvent. The catalytic activity appeared to change a little when different Al![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cr molar ratios were employed (13.2, 15.1, and 12.5 kg of products per g of Cr per h for the Al:Cr ratios of 800:1, 600:1, and 400:1, entries 3–5 in Table 2). These activities are comparable to those for the Ph2PN(Me)-2-C5H4N/CrCl3(THF)3/MAO system (Al:Cr of 500:1, 30 or 50 bar ethylene, and 60 °C; 8.1–38.9 kg of products per g of Cr per h),5c but lower than those for the [R32PNC(R2)NHR1]CrCl3(THF)/MMAO-3A system (Al:Cr of 400–800, 48 or 60 bar ethylene, and 50–70 °C; 70.1–1051.9 kg of products per g of Cr per h).5b The polymers produced were analyzed to have molecular weights in the range of 1.14–1.97 × 105 g mol−1. Corresponding differential scanning calorimetry (DSC) measurements gave melting points of 135–141 °C, indicating the linear character of these polymers.21 The oligomers were analyzed and found to be composed of 1-C6 and 1-C8 olefins as the dominant parts (66–78%) together with other cyclic C6 components (methylcyclopentane and methylenecyclopentane, 10–19%) and C10+ species (10–14%). These data arise as a result of selective ethylene tri-/tetramerization. This catalytic mode has been extensively studied using Sasol's diphosphazane ligand chromium catalysts and other P,N-chelation chromium catalysts.1–3,5 It was noted that the proportion of C8:C6 changed as well with the Al:Cr ratio (0.48:1 for 800:1, 1.20:1 for 600:1, and 0.50:1 for 400:1). This result, along with the aforementioned activities, implies a non-negligible influence of the Al:Cr molar ratio used in this system. The ratio may relate to the formation of catalytically active site(s) over the course of the interactions between 4 and MAO (vide infra). Nonetheless, in these catalytic reactions an Al:Cr molar ratio of 600:1 is the best ratio to achieve the higher activity as well as the yield of the 1-C8 product.
:Cr molar ratios were employed (13.2, 15.1, and 12.5 kg of products per g of Cr per h for the Al:Cr ratios of 800:1, 600:1, and 400:1, entries 3–5 in Table 2). These activities are comparable to those for the Ph2PN(Me)-2-C5H4N/CrCl3(THF)3/MAO system (Al:Cr of 500:1, 30 or 50 bar ethylene, and 60 °C; 8.1–38.9 kg of products per g of Cr per h),5c but lower than those for the [R32PNC(R2)NHR1]CrCl3(THF)/MMAO-3A system (Al:Cr of 400–800, 48 or 60 bar ethylene, and 50–70 °C; 70.1–1051.9 kg of products per g of Cr per h).5b The polymers produced were analyzed to have molecular weights in the range of 1.14–1.97 × 105 g mol−1. Corresponding differential scanning calorimetry (DSC) measurements gave melting points of 135–141 °C, indicating the linear character of these polymers.21 The oligomers were analyzed and found to be composed of 1-C6 and 1-C8 olefins as the dominant parts (66–78%) together with other cyclic C6 components (methylcyclopentane and methylenecyclopentane, 10–19%) and C10+ species (10–14%). These data arise as a result of selective ethylene tri-/tetramerization. This catalytic mode has been extensively studied using Sasol's diphosphazane ligand chromium catalysts and other P,N-chelation chromium catalysts.1–3,5 It was noted that the proportion of C8:C6 changed as well with the Al:Cr ratio (0.48:1 for 800:1, 1.20:1 for 600:1, and 0.50:1 for 400:1). This result, along with the aforementioned activities, implies a non-negligible influence of the Al:Cr molar ratio used in this system. The ratio may relate to the formation of catalytically active site(s) over the course of the interactions between 4 and MAO (vide infra). Nonetheless, in these catalytic reactions an Al:Cr molar ratio of 600:1 is the best ratio to achieve the higher activity as well as the yield of the 1-C8 product.
| Entry | Cat. | Co-cat. | Al:Cr |
T (°C) | P (bar) | Time (min) | Yield (polym. + oligom., g) | Act.b | Polym. | Oligom. distributionf (wt%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
M
wc (105 g mol−1) |
PDId | MPte | C6(1-C6) | C8(1-C8) | C10+ | |||||||||
|
a General reaction conditions: 70 mL toluene, 10 μmol Cr complex, 30 minutes.
b kg of products per g of Cr per h.
c Determined by GPC relative to polystyrene standards.
d Polydispersity index (PDI) = Mw/Mn.
e Determined by DSC at a heating rate of 10 °C min−1.
f Weight percentage of liquid fraction.
g 10 μmol CrCl3(THF)3.
h 10 μmol CrCl2(THF)2.
i Et2AlCl/Me3Al = 1:1 (mol/mol).
j 20 μmol Cr complex.
|
||||||||||||||
| 1g | CrCl3(THF)3 | MAO | 600 | 50 | 40 | 30 | 0.18 + 0 | 0.7 | nd | nd | nd | — | — | — |
| 2h | CrCl2(THF)2 | MAO | 600 | 50 | 40 | 30 | 0.15 + 0 | 0.6 | nd | nd | nd | — | — | — |
| 3 | 4 | MAO | 600 | 50 | 40 | 30 | 1.94 + 1.98 | 15.1 | 1.97 | 9.2 | 135 | 40(75) | 48(100) | 12 |
| 4 | 4 | MAO | 800 | 50 | 40 | 30 | 1.69 + 1.75 | 13.2 | 1.14 | 2.6 | 140 | 58(65) | 28(100) | 14 |
| 5 | 4 | MAO | 400 | 50 | 40 | 30 | 1.59 + 1.65 | 12.5 | 1.21 | 2.9 | 141 | 60(68) | 30(100) | 10 |
| 6 | 4 | Et2AlCl | 500 | 50 | 40 | 30 | Trace | 0 | — | — | — | — | — | — |
| 7 | 4 | Me3Al | 500 | 50 | 40 | 30 | Trace | 0 | — | — | — | — | — | — |
| 8i | 4 | Et2AlCl/Me3Al | 500 | 50 | 40 | 30 | 0.92 + 0.95 | 7.2 | 1.11 | 73.9 | 137 | 60(69) | 29(100) | 11 |
| 9j | 4 | MAO | 600 | 50 | 40 | 30 | 2.65 + 2.85 | 10.6 | 2.24 | 8.9 | 135 | 41(78) | 47(100) | 12 |
| 10 | 4 | MAO | 600 | 70 | 40 | 30 | 2.44 + 2.48 | 18.9 | 2.07 | 7.9 | 143 | 52(65) | 33(100) | 15 |
| 11 | 4 | MAO | 600 | 30 | 40 | 30 | 1.46 + 1.46 | 11.2 | 5.66 | 13.6 | 145 | 40(62) | 49(100) | 11 |
| 12 | 4 | MAO | 600 | 50 | 30 | 30 | 1.45 + 1.49 | 11.3 | 1.78 | 7.4 | 138 | 43(67) | 46(100) | 11 |
| 13 | 4 | MAO | 600 | 50 | 20 | 30 | 1.31 + 1.35 | 10.2 | 1.51 | 3.6 | 136 | 41(68) | 45(100) | 14 |
| 14 | 4 | MAO | 600 | 50 | 40 | 60 | 4.02 + 3.88 | 15.2 | 1.45 | 4.6 | 133 | 41(78) | 49(100) | 10 |
| 15 | 4 | MAO | 600 | 50 | 40 | 120 | 7.89 + 7.78 | 15.1 | 1.54 | 7.6 | 133 | 42(75) | 47(100) | 11 |
| 16 | 4 | MAO | 600 | 50 | 40 | 180 | 10.3 + 9.25 | 12.5 | 1.87 | 5.9 | 134 | 42(72) | 45(100) | 13 |
| 17 | 4 | MAO | 600 | 50 | 40 | 300 | 12.4 + 8.26 | 7.9 | 1.58 | 6.5 | 134 | 40(76) | 45(100) | 15 |
| 18 | 5 | MAO | 600 | 50 | 40 | 30 | 1.40 + 1.46 | 11.0 | 1.67 | 7.4 | 133 | 58(65) | 29(100) | 13 |
| 19 | 6 | MAO | 600 | 50 | 40 | 30 | 1.33 + 1.38 | 10.4 | 5.91 | 93.5 | 133 | 56(67) | 30(100) | 14 |
| 20 | 7 | MAO | 600 | 50 | 40 | 30 | 3.55 + trace | 13.7 | 1.87 | 5.9 | 134 | — | — | — |
| 21 | 8 | MAO | 600 | 50 | 40 | 30 | 1.70 + trace | 6.5 | 2.45 | 13.3 | 134 | — | — | — |
| 22 | 9 | MAO | 600 | 50 | 40 | 30 | 3.35 + trace | 12.9 | 3.85 | 4.6 | 133 | — | — | — |
| 23 | 10 | MAO | 600 | 50 | 40 | 30 | 2.10 + trace | 8.1 | 2.46 | 10.7 | 133 | — | — | — |
| 24 | 11 | MAO | 600 | 50 | 40 | 30 | 1.75 + trace | 6.7 | 2.58 | 120 | 132 | — | — | — |
Further investigation found that an alteration of the Al cocatalyst had an effect on the reaction result as well. When using either AlMe3 or Et2AlCl instead, there was almost no activity (entries 6 and 7). However, a combination of AlMe3 and Et2AlCl led to a reaction, producing polymers with a significantly broad PDI (73.9) although the distribution of the oligomers generated was comparable to that of entry 5. But, the activity of this reaction decreased remarkably (7.2 kg of products per g of Cr per h, entry 8). Gambarotta, Duchateua and coworkers have reported no activity for the catalyst system of [Ph2PN(Cy)PPh2]CrCl3 activated by AlMe3. But in the additional presence of MAO or other activators the catalyst system worked.22 Similar work has also been reported by the groups of McGuinness, Tooze, and Overett.23 Then, a cooperation between AlMe3 and Et2AlCl may be responsible for getting 4 to work, although the activity appears lower.
On the basis of the conditions used in entry 3, increasing the concentration of the catalyst to double produced more products as expected, but the activity was calculated to decrease a little (10.6 kg of products per g of Cr per h, entry 9), although the results including the Mw, PDI, and DSC data for the polymer and the distribution data for the oligomer are similar to those in entry 3. The higher concentration of the catalyst has a close relation to the formation of the number of real catalytic sites. It has been well indicated that under a steady concentration of the catalyst, catalytic performance often depends on the reaction temperature as well as the ethylene pressure.24 This catalyst system showed decreased reaction activity with decreasing ethylene pressure, but little change was observed for the above-mentioned characters of both the polymers and oligomers produced (11.3 and 10.2 kg of products per g of Cr per h at 30 and 20 bar, entries 12 and 13, reespectively). Keeping the ethylene pressure at 40 bar, and altering the temperature to 30 °C decreased the activity down to 11.2 kg of products per g of Cr per h (entry 11) whereas at 70 °C the activity rose to 18.9 kg of products per g of Cr per h (entry 10). Meanwhile, the C8:C6 ratio of the oligomers changed dramatically with the temperature (1.23:1 at 30 °C and 0.63:1 at 70 °C versus 1.20:1 at 50 °C). Low temperature substantially favors the formation of the CrC8-metallocyclic transient state that results from the insertion of one ethylene molecule into the CrC6-metallocycle intermediate, as has been extensively discussed.1,2,24 To evaluate the effectiveness of the system, an experiment with a longer running time of 4/MAO (Al:Cr of 600:1) at 40 bar ethylene and 50 °C was carried out. In the first 2 h the activity was retained (15.2 and 15.1 kg of products per g of Cr per h within 1 h and 2 h, entries 14 and 15, respectively). By extending the time to 3 h (12.5 kg of products per g of Cr per h, entry 16) and 5 h (7.9 kg of products per g of Cr per h, entry 17) the activity appeared to decrease gradually. However, the features of the oligomer and polymer products produced showed little change.
Complexes 5 and 6 both possess a ligand–metal chelation structure similar to that of 4 and investigation of their catalytic properties was accomplished by controlling the reaction temperature at 50 °C and ethylene pressure at 40 bar in the presence of MAO (Al:Cr of 600:1). As a result, different activities were exhibited (11.0 and 10.4 kg of products per g of Cr per h, entries 18 and 19) when compared with that for 4 under the same conditions (entry 3). Moreover, in the oligomer products the C6 components increased to 58% and 56%, respectively while the C8 part dropped to 29% and 30%. The polymer products showed differences with respect to the Mw, PDI, and MPt values as well. Obviously, variation of the R and Ar substituents on the respective C and N atoms of the ligand backbone has an influence on the catalytic result.
Also under the above conditions, complexes 7–11 were tested. To our surprise, only ethylene polymerization catalysis functioned, all producing polymers without the oligomers (entries 20–24), even though we had further screened the reaction by using greater amounts of the complex and/or MAO as well as by increasing the reaction temperature and/or ethylene pressure. In light of these results, it is worth noting that the same ligand-ligated Cr(III) and Cr(II) complexes 7 and 9 give close activities (13.7 and 12.9 kg of products per g of Cr per h, entries 20 and 22). Moreover, the characteristics of the polymers were similar. The same case is also observed for complexes 8 and 10 (6.5 and 8.1 kg of products per g of Cr per h, entries 21 and 23). But, the results differed between the 7 (or 9)/MAO versus8 (or 10)/MAO systems.
The P,P- or P,N-bidentate chelation chromium catalyst systems have been well known to effect the selective ethylene tri-/tetramerization catalysis that produces the 1-C6 and 1-C8 dominant and C10+ parts. In these systems, ethylene polymerization was sometimes observed, but produced fewer polymers.1b,2,5 Our experiments show that the 4 (5 and 6)/MAO systems produce oligomers and polymers of approximately equal amounts while the 7 (8, 9, 10, and 11)/MAO systems generate only the polymers.25 This suggests that in the 4 (5 and 6)/MAO systems, besides ethylene tri-/tetramerization, the polymerization reaction is not neglected. Structural analysis has indicated the difference between complexes 4–6 and 7–11 where the former complexes feature an N,P-chelation of the ligand at the Cr center while the latter ones feature N,N,P-chelation. Sydora and coworkers have reported that the N-phosphinoamidine N,P-chelation Cr(III) complex/MMAO-3A system initiated selective ethylene tri-/tetramerization while the related N,N,P-chelation Cr(III) complex/MMAO-3A system resulted in polymerization although the polymers were produced in a trace amounts.5b
Gambarotta and coworkers have reported a deprotonation reaction of the ligand’s NH group in the chromium complexes by organoaluminum compounds.10b We have also found a deprotonation reaction from model reactions using 4 with MAO or AlMe3, as indicated by the absence of the NH IR band, by measuring the data of the product upon removal of all volatiles after the reaction, although an isolation of the pure species was not successful. Then, we speculate that the formation of two types of active species in the 4/MAO system is responsible for the multiple catalytic performances. One could be the N,P-chelation chromium complex but with an aluminum-containing moiety attached at the N atom replacing the original H group,11g and the other could be the N,N,P-chelation chromium complex comparable to that resulting from the 7 (or 9)/MAO system. The former can catalyze the ethylene tri-/tetramerization reaction but the latter enables ethylene polymerization. Similar cases are also possible for the same ligand ligated chromium systems of 5/MAO versus8 (or 10)/MAO as well as those of 6/MAO versus11/MAO.
Conclusions
In summary, phenyl group-connected amidinato-phosphino ligands 1–3 have been prepared. Three types of complexes including N,P-chelation Cr(III) (4–6), N,N,P-chelation Cr(III) (7–8) and Cr(II) (9–11) complexes have been subsequently prepared through different synthetic routes. Upon activation with MAO, all of these metal complexes are catalytically active. Complexes 4–6 each catalyze the ethylene tri-/tetramerization reaction along with polymerization while complexes 7–11 enable only polymerization. Complexes 4–6 all contain an additional NH group in the ligand backbone and react with MAO to form two, probably different, active sites for the catalytic function of the respective ethylene tri-/tetramerization and polymerization reactions.Experimental
Materials and methods
All manipulation of air- and moisture-sensitive compounds was carried out using Schlenk techniques or in an argon-filled MBRAUN glove box. Toluene, n-hexane, and tetrahydrofuran were pre-dried over fine sodium wire and then refluxed with sodium/potassium benzophenone under nitrogen prior to use. CDCl3 was degassed and dried over CaH2. 1H (400 MHz), 13C (100 MHz), and 31P (162 MHz) NMR spectra were recorded on a Bruker Avance II 400 MHz spectrometer. IR spectra were measured on a Nicolet 380 spectrometer. All continuous-wave EPR spectra were recorded on an X-band Bruker EMX spectrometer operating at 100 kHz field modulation, 20 mW microwave power, and equipped with a high-sensitivity cavity (4119HS-LC). The g values were determined using a DPPH standard. Melting points of the compounds were measured in a sealed glass tube using a Büchi-540 instrument. Elemental analysis was performed on a Thermo Quest Italia SPA EA 1110 instrument. The molecular weight and polydispersity indices (PDI) of the polymer samples were determined through PL-GPC 220 type high-temperature (150 °C) gel-permeation chromatography. The sample concentration was set in 0.10 mg mL−1, and 1,2,4-trichlorobenzene (TCB) stabilized with 0.0125% butylated hydroxytoluene (BHT) was used as an eluent at a flow rate of 1.00 mL min−1. Differential scanning calorimetry (DSC) measurements were performed on a PerkinElmer DSC 8000 instrument under an N2 atmosphere in which the samples were treated upon heating and then cooling at the same rate of 10 °C min−1. Liquid products were analyzed by GC-FID using a GC-950 with an HP-1 capillary column (60 m × 0.25 mm). Methylaluminoxane (MAO, 10% solution in toluene), AlMe3 (1.0 M solution in hexanes) and Et2AlCl (2.0 M solution in hexanes) were purchased from J & K Chemical Technology Co. Polymerization grade ethylene was obtained from Linde Industrial Gases Co. Compounds CrCl3(THF)3 and CrCl2(THF)2,26 ArNC(R)Cl (Ar = 2,4,6-Me3C6H2, R = Ph; Ar = 2,6-iPr2C6H3, R = Ph; Ar = 2,6-iPr2C6H3, R = tBu),17 and o-Ph2PC6H4NH216 were prepared by following procedures detailed in the literature.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C(Ph)NH(o-Ph2PC6H4) (1).
A mixture of 2,4,6-Me3C6H2NC(Ph)Cl (5.86 g, 22.71 mmol) and o-Ph2PC6H4NH2 (6.30 g, 22.71 mmol) in toluene (200 mL) was heated at reflux for 20 h. After workup, the toluene solvent was removed and the residue was dissolved in ethanol (100 mL). By neutralization using aqueous NH3 (25%, 90 mL), a beige solid of 1 was precipitated and collected by filtration. Recrystallization from toluene (60 mL) at −20 °C for 3 d gave spectroscopically pure 1 (8.94 g, 79%). Mp: 151–152 °C. 1H NMR (400 MHz, CDCl3, 298 K, ppm): δ 7.54–6.50 (m, 21 H, C6H2, C6H4 and C6H5), 6.20 (s, 1 H, NH), 2.25 (s, 3 H, 4-Me), 2.11 (s, 6 H, 2,6-Me). 13C NMR (100 MHz, CDCl3, 298 K, ppm): δ 154.0 (CN), 143.2 (d, JPC = 19.8 Hz), 143.1, 135.2, 134.9 (d, JPC = 9.6 Hz), 133.9 (d, JPC = 20 Hz), 133.0, 131.8, 130.0 (d, JPC = 11.9 Hz), 129.8, 129.2, 129.0, 128.9, 128.8, 128.5, 128.2, 127.7, 124.8, 124.1 (C6H2, C6H4 and C6H5), 18.8 (4-Me), 17.9 (2,6-Me). 31P NMR (162 MHz, CDCl3, 298 K, ppm): δ −16.3. IR (KBr plate, Nujol mull, cm−1): ν 3336 (N–H), 1619 (CN). Anal. Calcd for C34H31N2P (Mr = 498.60): C, 81.90; H, 6.27; N, 5.62. Found: C, 81.76; H, 6.23; N, 5.51.
C(Ph)NH(o-Ph2PC6H4) (2).
A mixture of 2,6-iPr2C6H3NC(Ph)Cl (14.98 g, 50 mmol) and o-Ph2PC6H4NH2 (13.87 g, 50 mmol) in toluene (200 mL) was heated at reflux for 20 h. After workup, the toluene solvent was removed and the residue was dissolved in ethanol (100 mL). By neutralization using aqueous NH3 (25%, 90 mL), an off-white solid was precipitated and collected by filtration to gave spectroscopically pure 2 (23.0 g, 85%). Mp: 156–157 °C. 1H NMR (400 MHz, CDCl3, 298 K, ppm): δ 7.50–6.48 (m, 22 H, C6H3, C6H4 and C6H5), 6.24 (s, 1 H, NH), 3.05 (br, 2 H, CHMe2), 1.14 (br, 12 H, CHMe2). 13C NMR (100 MHz, CDCl3, 298 K, ppm): δ 153.8 (CN), 138.5, 135.6, 134.9 (d, JPC = 9.4 Hz), 133.9 (d, JPC = 19.8 Hz), 133.2, 129.7, 129.2, 128.7 (d, JPC = 7.2 Hz), 128.2, 124.3, 124.0, 123.6, 123.2 (C6H3, C6H4 and C6H5), 28.1 (CHMe2), 23.9 (CHMe2). 31P NMR (162 MHz, CDCl3, 298 K, ppm): δ −18.2. IR (KBr plate, Nujol mull, cm−1): ν 3335 (N–H), 1620 (CN). Anal. Calcd for C37H37N2P (Mr = 540.68): C, 82.19; H, 6.71; N, 5.18. Found: C, 81.88; H, 6.67; N, 5.06.
C(tBu)NH(o-Ph2PC6H4) (3).
A mixture of 2,6-iPr2C6H3NC(tBu)Cl (2.80 g, 10 mmol) and o-Ph2PC6H4NH2 (2.77 g, 10 mmol) in toluene (80 mL) was heated at reflux for 20 h under N2 flow. After workup, the toluene solvent was removed and the residue was extracted with n-hexane (15 mL). The extract was stored in a freezer (−20 °C) for 3 d to give colorless crystals of 3 (3.80 g, 72%). Mp: 121–122 °C. 1H NMR (400 MHz, CDCl3, 298 K, ppm): δ 7.33–6.60 (m, 22 H, C6H3, C6H4 and C6H5), 6.46 (s, 1 H, NH), 3.28 (br, 2 H, CHMe2), 1.29 (br, 12 H, CHMe2), 1.21 (s, 9 H, CMe3). 13C NMR (100 MHz, CDCl3, 298 K, ppm): δ 153.9 (CN), 145.1, 141.6, 135.9, 135.4, 134.2 (d, JPC = 20.1 Hz), 131.9, 129.4, 128.9, 128.2 (d, JPC = 23.9 Hz), 125.5, 124.9, 123.8, 122.0, 121.2 (C6H3, C6H4 and C6H5), 40.0 (CMe3), 29.2 (CMe3), 28.4 (CHMe2), 23.1 (CHMe2). 31P NMR (162 MHz, CDCl3, 298 K, ppm): δ −16.1. IR (KBr plate, Nujol mull, cm−1): ν 3399 (N–H), 1665 (CN). Anal. Calcd for C35H41N2P (Mr = 520.69): C, 80.73; H, 7.94; N, 5.38. Found: C, 80.25; H, 7.56; N, 5.15.
N(o-Ph2PC6H4)]CrCl3(THF)] (4).
A solution of 1 (0.50 g, 1.0 mmol) in THF (15 mL) was added to a suspension of CrCl3(THF)3 (0.37 g, 1.0 mmol) in THF (15 mL) at room temperature. The mixture was stirred for 24 h. Using filtration to remove trace amounts of insoluble solid, the filtrate was evaporated to dryness under reduced pressure. The residue was collected and washed with n-hexane (5 mL) to give complex 4 as a grey green solid (0.65 g, 89%). Mp: 264 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 3165 (N–H), 1595 (CN). EPR (100 kHz field, 90 K): g = 4.294. Anal. Calcd for C38H39Cl3CrN2OP (Mr = 729.06): C, 62.60; H, 5.39; N, 3.84. Found: C, 62.27; H, 5.86; N, 3.49.
N(o-Ph2PC6H4)]CrCl3(THF)] (5).
Complex 5 was prepared in a similar manner to that of 4 but, in the preparation, 2 (0.54 g, 1.0 mmol) was used instead of 1. Complex 5 was obtained as a dark-green solid (0.63 g, 82%). Mp: 265 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 3195 (N–H), ν 1588 (CN). EPR (100 kHz field, 90 K): g = 3.902. Anal. Calcd for C41H45Cl3CrN2OP (Mr = 771.14): C, 63.86; H, 5.88; N, 3.63. Found: C, 64.10; H, 6.01; N, 3.46. X-ray quality single-crystals of 5·0.5THF were obtained from recrystallization in a THF/n-hexane (5v/1v) solution at −20 °C for 3 d.
N(o-Ph2PC6H4)]CrCl3(THF)] (6).
A solution of CrCl3(THF)3 (0.37 g, 1.0 mmol) in CH2Cl2 (15 mL) was added to a solution of 3 (0.50 g, 1.0 mmol) in CH2Cl2 (15 mL) at room temperature, and the mixture was stirred for 24 h. After workup, the CH2Cl2 solvent was removed under reduced pressure and the residue was collected and washed with n-hexane to give complex 6 as a yellow-green solid (0.60 g, 80%). Mp: 236 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 3328 (N–H), ν 1588 (CN). EPR (100 kHz field, 90 K): g = 4.646. Anal. Calcd for C39H49Cl3CrN2OP (Mr = 751.15): C, 62.36; H, 6.58; N, 3.73. Found: C, 62.25; H, 6.43; N, 3.88.
C(Ph)N(o-Ph2PC6H4)]CrCl2(THF) (7).
A solution of nBuLi (2.4 M in n-hexane, 0.42 mL, 1.0 mmol) was added dropwise to a solution of 1 (0.50 g, 1.0 mmol) in THF (30 mL) at −78 °C. The mixture was gently warmed to −30 °C within 2 h and stirred at this temperature for an additional 0.5 h to give the lithium salt of 1. This solution was cooled again to −78 °C and to it a suspension of CrCl3(THF)3 (0.37 g, 1.0 mmol) in THF (15 mL) was added dropwise. The mixture was warmed gently to room temperature within 4 h and then stirred for additional 12 h. After workup, all volatiles were removed under reduced pressure and the residue was extracted with toluene (20 mL). The extract was evaporated to dryness to give complex 7 as a dark-green solid (0.56 g, 81%). Mp: 252 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1578 (CN). EPR (100 kHz field, 90 K): g = 3.912. Anal. Calcd for C38H38Cl2CrN2OP (Mr = 692.60): C, 65.90; H, 5.53; N, 4.04. Found: C, 65.78; H, 5.62; N, 3.99.
C(Ph)N(o-Ph2PC6H4)]CrCl2(THF) (8).
Complex 8 was prepared in a similar manner to that of 7 but, in the preparation, 2 (0.54 g, 1.0 mmol) was used instead of 1. Complex 8 was obtained as a dark-green solid (0.59 g, 86%). Mp: 233 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1591 (CN). EPR (100 kHz field, 90 K): g = 4.439. Anal. Calcd for C41H44Cl2CrN2OP (Mr = 734.68): C, 67.03; H, 6.04; N, 3.81. Found: C, 66.99; H, 6.10; N, 3.79. X-ray quality single-crystals of 8·2.5 toluene were obtained from recrystallization in a toluene/n-hexane (3v/1v) solution at −20 °C for 1 d.
C(Ph)N(o-Ph2PC6H4)]Cr(μ-Cl)}2 (9).
Complex 9 was prepared in a similar manner to that of 7 but, in the preparation, CrCl2(THF)2 (0.27 g, 1.0 mmol) was used instead of CrCl3(THF)3. Complex 9 was obtained as a yellow-green solid (0.48 g, 82%). Mp: 215 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1578 (CN). EPR (100 kHz field, 90 K): silent. Anal. Calcd for C68H60Cl2Cr2N4P2 (Mr = 1170.08): C, 69.80; H, 5.17; N, 4.79. Found: C, 69.65; H, 5.26; N, 4.56.
C(Ph)N(o-Ph2PC6H4)]Cr(μ-Cl)}2 (10).
Complex 10 was prepared in a similar manner to that of 7 but, in the preparation, 2 (0.54 g, 1.0 mmol) and CrCl2(THF)2 (0.27 g, 1.0 mmol) were used instead of the respective 1 and CrCl3(THF)3. Complex 10 was obtained as a green solid (0.54 g, 86%). Mp: 230 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1595 (CN). EPR (100 kHz field, 90 K): silent. Anal. Calcd for C74H72Cl2Cr2N4P2 (Mr = 1254.24): C, 70.86; H, 5.79; N, 4.47. Found: C, 70.68; H, 5.64; N, 4.23.
C(tBu)N(o-Ph2PC6H4)]Cr(μ-Cl)}2 (11).
Complex 11 was prepared in a similar manner to that of 7 but, in the preparation, 3 (0.52 g, 1.0 mmol) and CrCl2(THF)2 (0.27 g, 1.0 mmol) were used instead of the respective 1 and CrCl3(THF)3. Complex 11 was obtained as a dark-green solid (0.50 g, 82%). Mp: 186 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1586 (CN). EPR (100 kHz field, 90 K): silent. Anal. Calcd for C70H80Cl2Cr2N4P2 (Mr = 1214.26): C, 69.24; H, 6.64; N, 4.61. Found: C, 69.45; H, 6.54; N, 4.46. X-ray quality single-crystals of 11·4 toluene were obtained from recrystallization in a toluene/n-hexane (3v/1v) solution at −20 °C for 3 d.
C(Ph)NH(o-Ph2PC6H4) (1).
A mixture of 2,4,6-Me3C6H2NC(Ph)Cl (5.86 g, 22.71 mmol) and o-Ph2PC6H4NH2 (6.30 g, 22.71 mmol) in toluene (200 mL) was heated at reflux for 20 h. After workup, the toluene solvent was removed and the residue was dissolved in ethanol (100 mL). By neutralization using aqueous NH3 (25%, 90 mL), a beige solid of 1 was precipitated and collected by filtration. Recrystallization from toluene (60 mL) at −20 °C for 3 d gave spectroscopically pure 1 (8.94 g, 79%). Mp: 151–152 °C. 1H NMR (400 MHz, CDCl3, 298 K, ppm): δ 7.54–6.50 (m, 21 H, C6H2, C6H4 and C6H5), 6.20 (s, 1 H, NH), 2.25 (s, 3 H, 4-Me), 2.11 (s, 6 H, 2,6-Me). 13C NMR (100 MHz, CDCl3, 298 K, ppm): δ 154.0 (CN), 143.2 (d, JPC = 19.8 Hz), 143.1, 135.2, 134.9 (d, JPC = 9.6 Hz), 133.9 (d, JPC = 20 Hz), 133.0, 131.8, 130.0 (d, JPC = 11.9 Hz), 129.8, 129.2, 129.0, 128.9, 128.8, 128.5, 128.2, 127.7, 124.8, 124.1 (C6H2, C6H4 and C6H5), 18.8 (4-Me), 17.9 (2,6-Me). 31P NMR (162 MHz, CDCl3, 298 K, ppm): δ −16.3. IR (KBr plate, Nujol mull, cm−1): ν 3336 (N–H), 1619 (CN). Anal. Calcd for C34H31N2P (Mr = 498.60): C, 81.90; H, 6.27; N, 5.62. Found: C, 81.76; H, 6.23; N, 5.51.
C(Ph)NH(o-Ph2PC6H4) (2).
A mixture of 2,6-iPr2C6H3NC(Ph)Cl (14.98 g, 50 mmol) and o-Ph2PC6H4NH2 (13.87 g, 50 mmol) in toluene (200 mL) was heated at reflux for 20 h. After workup, the toluene solvent was removed and the residue was dissolved in ethanol (100 mL). By neutralization using aqueous NH3 (25%, 90 mL), an off-white solid was precipitated and collected by filtration to gave spectroscopically pure 2 (23.0 g, 85%). Mp: 156–157 °C. 1H NMR (400 MHz, CDCl3, 298 K, ppm): δ 7.50–6.48 (m, 22 H, C6H3, C6H4 and C6H5), 6.24 (s, 1 H, NH), 3.05 (br, 2 H, CHMe2), 1.14 (br, 12 H, CHMe2). 13C NMR (100 MHz, CDCl3, 298 K, ppm): δ 153.8 (CN), 138.5, 135.6, 134.9 (d, JPC = 9.4 Hz), 133.9 (d, JPC = 19.8 Hz), 133.2, 129.7, 129.2, 128.7 (d, JPC = 7.2 Hz), 128.2, 124.3, 124.0, 123.6, 123.2 (C6H3, C6H4 and C6H5), 28.1 (CHMe2), 23.9 (CHMe2). 31P NMR (162 MHz, CDCl3, 298 K, ppm): δ −18.2. IR (KBr plate, Nujol mull, cm−1): ν 3335 (N–H), 1620 (CN). Anal. Calcd for C37H37N2P (Mr = 540.68): C, 82.19; H, 6.71; N, 5.18. Found: C, 81.88; H, 6.67; N, 5.06.
C(tBu)NH(o-Ph2PC6H4) (3).
A mixture of 2,6-iPr2C6H3NC(tBu)Cl (2.80 g, 10 mmol) and o-Ph2PC6H4NH2 (2.77 g, 10 mmol) in toluene (80 mL) was heated at reflux for 20 h under N2 flow. After workup, the toluene solvent was removed and the residue was extracted with n-hexane (15 mL). The extract was stored in a freezer (−20 °C) for 3 d to give colorless crystals of 3 (3.80 g, 72%). Mp: 121–122 °C. 1H NMR (400 MHz, CDCl3, 298 K, ppm): δ 7.33–6.60 (m, 22 H, C6H3, C6H4 and C6H5), 6.46 (s, 1 H, NH), 3.28 (br, 2 H, CHMe2), 1.29 (br, 12 H, CHMe2), 1.21 (s, 9 H, CMe3). 13C NMR (100 MHz, CDCl3, 298 K, ppm): δ 153.9 (CN), 145.1, 141.6, 135.9, 135.4, 134.2 (d, JPC = 20.1 Hz), 131.9, 129.4, 128.9, 128.2 (d, JPC = 23.9 Hz), 125.5, 124.9, 123.8, 122.0, 121.2 (C6H3, C6H4 and C6H5), 40.0 (CMe3), 29.2 (CMe3), 28.4 (CHMe2), 23.1 (CHMe2). 31P NMR (162 MHz, CDCl3, 298 K, ppm): δ −16.1. IR (KBr plate, Nujol mull, cm−1): ν 3399 (N–H), 1665 (CN). Anal. Calcd for C35H41N2P (Mr = 520.69): C, 80.73; H, 7.94; N, 5.38. Found: C, 80.25; H, 7.56; N, 5.15.
N(o-Ph2PC6H4)]CrCl3(THF)] (4).
A solution of 1 (0.50 g, 1.0 mmol) in THF (15 mL) was added to a suspension of CrCl3(THF)3 (0.37 g, 1.0 mmol) in THF (15 mL) at room temperature. The mixture was stirred for 24 h. Using filtration to remove trace amounts of insoluble solid, the filtrate was evaporated to dryness under reduced pressure. The residue was collected and washed with n-hexane (5 mL) to give complex 4 as a grey green solid (0.65 g, 89%). Mp: 264 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 3165 (N–H), 1595 (CN). EPR (100 kHz field, 90 K): g = 4.294. Anal. Calcd for C38H39Cl3CrN2OP (Mr = 729.06): C, 62.60; H, 5.39; N, 3.84. Found: C, 62.27; H, 5.86; N, 3.49.
N(o-Ph2PC6H4)]CrCl3(THF)] (5).
Complex 5 was prepared in a similar manner to that of 4 but, in the preparation, 2 (0.54 g, 1.0 mmol) was used instead of 1. Complex 5 was obtained as a dark-green solid (0.63 g, 82%). Mp: 265 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 3195 (N–H), ν 1588 (CN). EPR (100 kHz field, 90 K): g = 3.902. Anal. Calcd for C41H45Cl3CrN2OP (Mr = 771.14): C, 63.86; H, 5.88; N, 3.63. Found: C, 64.10; H, 6.01; N, 3.46. X-ray quality single-crystals of 5·0.5THF were obtained from recrystallization in a THF/n-hexane (5v/1v) solution at −20 °C for 3 d.
N(o-Ph2PC6H4)]CrCl3(THF)] (6).
A solution of CrCl3(THF)3 (0.37 g, 1.0 mmol) in CH2Cl2 (15 mL) was added to a solution of 3 (0.50 g, 1.0 mmol) in CH2Cl2 (15 mL) at room temperature, and the mixture was stirred for 24 h. After workup, the CH2Cl2 solvent was removed under reduced pressure and the residue was collected and washed with n-hexane to give complex 6 as a yellow-green solid (0.60 g, 80%). Mp: 236 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 3328 (N–H), ν 1588 (CN). EPR (100 kHz field, 90 K): g = 4.646. Anal. Calcd for C39H49Cl3CrN2OP (Mr = 751.15): C, 62.36; H, 6.58; N, 3.73. Found: C, 62.25; H, 6.43; N, 3.88.
C(Ph)N(o-Ph2PC6H4)]CrCl2(THF) (7).
A solution of nBuLi (2.4 M in n-hexane, 0.42 mL, 1.0 mmol) was added dropwise to a solution of 1 (0.50 g, 1.0 mmol) in THF (30 mL) at −78 °C. The mixture was gently warmed to −30 °C within 2 h and stirred at this temperature for an additional 0.5 h to give the lithium salt of 1. This solution was cooled again to −78 °C and to it a suspension of CrCl3(THF)3 (0.37 g, 1.0 mmol) in THF (15 mL) was added dropwise. The mixture was warmed gently to room temperature within 4 h and then stirred for additional 12 h. After workup, all volatiles were removed under reduced pressure and the residue was extracted with toluene (20 mL). The extract was evaporated to dryness to give complex 7 as a dark-green solid (0.56 g, 81%). Mp: 252 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1578 (CN). EPR (100 kHz field, 90 K): g = 3.912. Anal. Calcd for C38H38Cl2CrN2OP (Mr = 692.60): C, 65.90; H, 5.53; N, 4.04. Found: C, 65.78; H, 5.62; N, 3.99.
C(Ph)N(o-Ph2PC6H4)]CrCl2(THF) (8).
Complex 8 was prepared in a similar manner to that of 7 but, in the preparation, 2 (0.54 g, 1.0 mmol) was used instead of 1. Complex 8 was obtained as a dark-green solid (0.59 g, 86%). Mp: 233 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1591 (CN). EPR (100 kHz field, 90 K): g = 4.439. Anal. Calcd for C41H44Cl2CrN2OP (Mr = 734.68): C, 67.03; H, 6.04; N, 3.81. Found: C, 66.99; H, 6.10; N, 3.79. X-ray quality single-crystals of 8·2.5 toluene were obtained from recrystallization in a toluene/n-hexane (3v/1v) solution at −20 °C for 1 d.
C(Ph)N(o-Ph2PC6H4)]Cr(μ-Cl)}2 (9).
Complex 9 was prepared in a similar manner to that of 7 but, in the preparation, CrCl2(THF)2 (0.27 g, 1.0 mmol) was used instead of CrCl3(THF)3. Complex 9 was obtained as a yellow-green solid (0.48 g, 82%). Mp: 215 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1578 (CN). EPR (100 kHz field, 90 K): silent. Anal. Calcd for C68H60Cl2Cr2N4P2 (Mr = 1170.08): C, 69.80; H, 5.17; N, 4.79. Found: C, 69.65; H, 5.26; N, 4.56.
C(Ph)N(o-Ph2PC6H4)]Cr(μ-Cl)}2 (10).
Complex 10 was prepared in a similar manner to that of 7 but, in the preparation, 2 (0.54 g, 1.0 mmol) and CrCl2(THF)2 (0.27 g, 1.0 mmol) were used instead of the respective 1 and CrCl3(THF)3. Complex 10 was obtained as a green solid (0.54 g, 86%). Mp: 230 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1595 (CN). EPR (100 kHz field, 90 K): silent. Anal. Calcd for C74H72Cl2Cr2N4P2 (Mr = 1254.24): C, 70.86; H, 5.79; N, 4.47. Found: C, 70.68; H, 5.64; N, 4.23.
C(tBu)N(o-Ph2PC6H4)]Cr(μ-Cl)}2 (11).
Complex 11 was prepared in a similar manner to that of 7 but, in the preparation, 3 (0.52 g, 1.0 mmol) and CrCl2(THF)2 (0.27 g, 1.0 mmol) were used instead of the respective 1 and CrCl3(THF)3. Complex 11 was obtained as a dark-green solid (0.50 g, 82%). Mp: 186 °C (decomp.). IR (KBr plate, Nujol mull, cm−1): ν 1586 (CN). EPR (100 kHz field, 90 K): silent. Anal. Calcd for C70H80Cl2Cr2N4P2 (Mr = 1214.26): C, 69.24; H, 6.64; N, 4.61. Found: C, 69.45; H, 6.54; N, 4.46. X-ray quality single-crystals of 11·4 toluene were obtained from recrystallization in a toluene/n-hexane (3v/1v) solution at −20 °C for 3 d.
X-ray crystallographic analysis
The crystals were mounted on a glass fiber using an oil drop method and quickly placed under a low temperature (173 K) atmosphere. Crystallographic data was collected on an Oxford Gemini S Ultra system. During the measurements, graphite-monochromatic Mo-Kα radiation (λ = 0.71073 Å) was used for compounds 3, 5, and 11 while Cu-Kα radiation (λ = 1.54178 Å) was employed for compound 8. Absorption corrections were applied using a spherical harmonics program (multi-scan type). All structures were solved by direct methods (SHELXS-96)27 and refined against F2 using SHELXL-97.28 In general, the non-hydrogen atoms were located by difference Fourier synthesis and refined anisotropically, and hydrogen atoms were included using a riding model with Uiso tied to the Uiso of the parent atoms unless otherwise specified.General procedure for catalytic reaction
The catalytic reaction was carried out in a 300 mL stainless steel autoclave equipped with a mechanical stirrer, temperature controller and internal cooling system. In a typical experiment, the reactor was heated to 100 °C under vacuum for 5 h to maximize the removal of air and moisture inside and then cooled to room temperature prior to use. After flushing with nitrogen gas three times, the catalyst solution (in 20 mL solvent), co-catalyst (MAO), and solvent were injected in turn into the autoclave. The reactor was pressurized with ethylene and then heated to the desired temperature. After the reaction, the ethylene feed was stopped and the reactor was quickly cooled to ∼2 °C through a H2O/glycol low temperature cycling system. By depressurization, the reaction was quenched with a 10% HCl/ethanol solution. The liquid product was dried over anhydrous sodium sulfate and then GC-FID analysis was performed. The solid product was dried overnight in an oven at 50 °C to a constant weight and high-temperature GPC analysis was performed.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21473142), the National Basic Research Program of China (2012CB821704), and the Program for Innovative Research Teams in Chinese Universities (12DZ2260400, 2013B019 and IRT_14R31).Notes and references
- (a) A. Carter, S. A. Cohen, N. A. Cooley, A. Murphy, J. Scutt and D. F. Wass, Chem. Commun., 2002, 858–859 RSC; (b) A. Bollmann, K. Blann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. S. McGuinness, D. H. Morgan, A. Neveling, S. Otto, M. Overett, A. M. Z. Slawin, P. Wasserscheid and S. Kuhlmann, J. Am. Chem. Soc., 2004, 126, 14712–14713 CrossRef CAS PubMed.
- (a) K. Blann, A. Bollmann, J. T. Dixon, F. M. Hess, E. Killian, H. Maumela, D. H. Morgan, A. Neveling, S. Otto and M. J. Overett, Chem. Commun., 2005, 620–621 CAS; (b) M. J. Overett, K. Blann, A. Bollmann, J. T. Dixon, F. Hess, E. Killian, H. Maumela, D. H. Morgan, A. Neveling and S. Otto, Chem. Commun., 2005, 622–624 CAS; (c) P. R. Elowe, C. McCann, P. G. Pringle, S. K. Spitzmesser and J. E. Bercaw, Organometallics, 2006, 25, 5255–5260 CrossRef CAS; (d) K. Blann, A. Bollmann, H. Debod, J. Dixon, E. Killian, P. Nongodlwana, M. Maumela, H. Maumela, A. McConnell and D. Morgan, J. Catal., 2007, 249, 244–249 CrossRef CAS; (e) Z. Weng, S. Teo and T. S. Andy Hor, Dalton Trans., 2007, 3493–3498 RSC; (f) S. Kuhlmann, K. Blann, A. Bollmann, J. Dixon, E. Killian, M. Maumela, H. Maumela, D. Morgan, M. Pretorius, N. Taccardi and P. Wasserscheid, J. Catal., 2007, 245, 279–284 CrossRef CAS; (g) S. Sa, S. M. Lee and S. Y. Kim, J. Mol. Catal. A: Chem., 2013, 378, 17–21 CrossRef CAS; (h) R. Liu, S. Xiao, X. Zhong, Y. Cao, S. Liang, Z. Liu, X. Ye, A. Shen and H. Zhu, Chin. J. Org. Chem., 2015, 35, 1861–1888 CAS.
- (a) C. Bianchini, G. Giambastiani, I. G. Rios, G. Mantovani, A. Meli and A. M. Segarra, Coord. Chem. Rev., 2006, 250, 1391–1418 CrossRef CAS; (b) T. Agapie, Coord. Chem. Rev., 2011, 255, 861–880 CrossRef CAS; (c) D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS PubMed.
- (a) J. T. Dixon, M. J. Green, F. M. Hess and D. H. Morgan, J. Organomet. Chem., 2004, 689, 3641–3668 CrossRef CAS; (b) L. E. Bowen, M. Charernsuk and D. F. Wass, Chem. Commun., 2007, 2835–2837 RSC; (c) P. Crewdson, S. Gambarotta, M.-C. Djoman, I. Korobkove and R. Duchateau, Organometallics, 2005, 25, 5214–5216 CrossRef; (d) W. J. v. Rensburg, C. Grové, J. P. Steynberg, K. B. Star, J. J. Huyser and P. J. Steynberg, Organometallics, 2004, 23, 1207–1222 CrossRef.
- (a) Y. Shaikh, J. Gurnham, K. Albahily, S. Gambarotta and I. Korobkov, Organometallics, 2012, 31, 7427–7433 CrossRef CAS; (b) O. L. Sydora, T. C. Jones, B. L. Small, A. J. Nett, A. A. Fischer and M. J. Carney, ACS Catal., 2012, 2, 2452–2455 CrossRef CAS; (c) Y. Yang, J. Gurnham, B. Liu, R. Duchateau, S. Gambarotta and I. Korobkov, Organometallics, 2014, 33, 5749–5757 CrossRef CAS; (d) A. Alzamly, S. Gambarotta, I. Korobkov, M. Murugesu, J. J. Le Roy and P. H. Budzelaar, Inorg. Chem., 2014, 53, 6073–6081 CrossRef CAS PubMed; (e) J. E. Radcliffe, A. S. Batsanov, D. M. Smith, J. A. Scott, P. W. Dyer and M. J. Hanton, ACS Catal., 2015, 5, 7095–7098 CrossRef CAS.
- (a) D. S. McGuinness, D. B. Brown, R. P. Tooze, F. M. Hess, J. T. Dixon and A. M. Z. Slawin, Organometallics, 2006, 25, 3605–3610 CrossRef CAS; (b) D. S. McGuinness, P. Wasserscheid, W. Keim, C. Hu, U. Englert, J. T. Dixon and C. Grove, Chem. Commun., 2003, 334–335 RSC; (c) D. S. McGuinness, P. Wasserscheid, D. H. Morgan and J. T. Dixon, Organometallics, 2005, 24, 552–556 CrossRef CAS; (d) S. S. Rozenel, W. A. Chomitz and J. Arnold, Organometallics, 2009, 28, 6243–6253 CrossRef CAS.
- M. E. Bluhm, O. Walter and M. Döring, J. Organomet. Chem., 2005, 690, 713–721 CrossRef CAS.
- Q. Liu, R. Gao, J. Hou and W. Sun, Chin. J. Org. Chem., 2013, 33, 808–814 CrossRef CAS.
- S. Liu, R. Pattacini and P. Braunstein, Organometallics, 2011, 30, 3549–3558 CrossRef CAS.
- (a) A. Alzamly, S. Gambarotta and I. Korobkov, Organometallics, 2013, 32, 7204–7212 CrossRef CAS; (b) A. Alzamly, S. Gambarotta and I. Korobkov, Organometallics, 2013, 32, 7107–7115 CrossRef CAS; (c) A. Alzamly, S. Gambarotta and I. Korobkov, Organometallics, 2014, 33, 1602–1607 CrossRef CAS; (d) D. Gong, W. Liu, T. Chen, Z.-R. Chen and K.-W. Huang, J. Mol. Catal. A: Chem., 2014, 395, 100–107 CrossRef CAS.
- (a) R. Emrich, O. Heinemann, P. W. Jolly, C. Kru and G. P. J. Verhovnik, Organometallics, 1997, 16, 1511–1513 CrossRef CAS; (b) H. Mahomed, Appl. Catal., A, 2003, 255, 355–359 CrossRef CAS; (c) D. S. McGuinness, P. Wasserscheid, W. Keim, D. Morgan, J. T. Dixon, A. Bollmann, H. Maumela, F. Hess and U. Englert, J. Am. Chem. Soc., 2003, 125, 5272–5273 CrossRef CAS PubMed; (d) W. J. v. Rensburg, J.-A. v. d. Berg and P. J. Steynberg, Organometallics, 2007, 26, 1000–1013 CrossRef; (e) J. Zhang, P. Braunstein and T. S. A. Hor, Organometallics, 2008, 27, 4277–4279 CrossRef CAS; (f) S. Bhaduri, S. Mukhopadhyay and S. A. Kulkarni, J. Organomet. Chem., 2009, 694, 1297–1307 CrossRef CAS; (g) S. Peitz, N. Peulecke, B. R. Aluri, S. Hansen, B. H. Müller, A. Spannenberg, U. Rosenthal, M. H. Al-Hazmi, F. M. Mosa, A. Wöhl and W. Müller, Eur. J. Inorg. Chem., 2010, 1167–1171 CrossRef CAS; (h) B. R. Aluri, N. Peulecke, S. Peitz, A. Spannenberg, B. H. Muller, S. Schulz, H. J. Drexler, D. Heller, M. H. Al-Hazmi, F. M. Mosa, A. Wohl, W. Muller and U. Rosenthal, Dalton Trans., 2010, 39, 7911–7920 RSC; (i) T. Beweries, C. Fischer, S. Peitz, V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg, D. Heller and U. Rosenthal, J. Am. Chem. Soc., 2009, 131, 4463–4469 CrossRef CAS PubMed.
- (a) J. Skupinska, Chem. Rev., 1991, 91, 613–648 CrossRef CAS; (b) L. A. MacAdams, G. P. Buffone, C. D. Incarvito, J. A. Golen, A. L. Rheingold and K. H. Theopold, Chem. Commun., 2003, 1164–1165 RSC; (c) H. Sugiyama, G. Aharonian, S. Gambarotta, G. P. A. Yap and E. H. M. Budzelaar, J. Am. Chem. Soc., 2002, 124, 12268–12274 CrossRef CAS PubMed; (d) G. Bhandari, Y. Kim, J. M. McFarland, A. L. Rheingold and K. H. Theopold, Organometallics, 1995, 14, 738–745 CrossRef CAS; (e) C. Schulzke, D. Enright, H. Sugiyama, G. LeBlanc, S. Gambarotta and G. P. A. Yap, Organometallics, 2002, 21, 3810–3816 CrossRef CAS.
- (a) K. Albahily, V. Fomitcheva, S. Gambarotta, I. Korobkov, M. Murugesu and S. I. Gorelsky, J. Am. Chem. Soc., 2011, 133, 6380–6387 CrossRef CAS PubMed; (b) K. Albahily, Y. Shaikh, E. Sebastiao, S. Gambarotta, I. Korobkov and S. I. Gorelsky, J. Am. Chem. Soc., 2011, 133, 6388–6395 CrossRef CAS PubMed; (c) S. Licciulli, K. Albahily, V. Fomitcheva, I. Korobkov, S. Gambarotta and R. Duchateau, Angew. Chem., Int. Ed., 2011, 50, 2346–2349 CrossRef CAS PubMed; (d) K. Albahily, V. Fomitcheva, Y. Shaikh, E. Sebastiao, S. I. Gorelsky, S. Gambarotta, I. Korobkov and R. Duchateau, Organometallics, 2011, 30, 4201–4210 CrossRef CAS; (e) A. J. Rucklidge, D. S. McGuinness, R. P. Tooze, A. M. Z. Slawin, J. D. A. Pelletier, M. J. Hanton and P. B. Webb, Organometallics, 2007, 26, 2782–2787 CrossRef CAS. There are some reports also discussed on relating with the Cr(II)/Cr(IV) couple for the tri-/tetramerization catalysis, see: (f) J. Rabeah, M. Bauer, W. Baumann, A. E. C. McConnell, W. F. Gabrielli, P. B. Webb, D. Selent and A. Brückner, ACS Catal., 2013, 3, 95–102 CrossRef CAS.
- (a) T. Xu, Y. Mu, W. Gao, J. Ni, L. Ye and Y. Tao, J. Am. Chem. Soc., 2007, 129, 2236–2237 CrossRef CAS PubMed; (b) K. H. Theopold, Eur. J. Inorg. Chem., 1998, 15–24 CrossRef CAS.
- (a) H. Zhu, K. Zhu, T. Zhu, G. Tan, Y. Li and H. Wan, CN Pat., 102585054, 2012 Search PubMed. More recently, a ligand with the NCN-C6H4-o-P(O)Ph2 scaffold was reported, see: (b) A. O. Tolpygin, T. A. Glukhova, A. V. Cherkasov, G. K. Fukin, D. V. Aleksanyan, D. Cui and A. A. Trifonov, Dalton Trans., 2015, 44, 16465–16474 RSC.
- M. K. Cooper, J. M. Downs, P. A. Duckworth and E. R. T. Tiekink, Aust. J. Chem., 1992, 45, 595–609 CrossRef CAS.
- (a) F.-S. Liu, H.-Y. Gao, K.-M. Song, L. Zhang, F.-M. Zhu and Q. Wu, Polyhedron, 2009, 28, 1386–1392 CrossRef CAS; (b) M. L. Rosenberg, E. Langseth, A. Krivokapic, N. S. Gupta and M. Tilset, New J. Chem., 2011, 35, 2306–2313 RSC.
- (a) B. Gao, X. Luo, W. Gao, L. Huang, S. M. Gao, X. Liu, Q. Wu and Y. Mu, Dalton Trans., 2012, 41, 2755–2763 RSC; (b) G. K. Cantrell, S. J. Geib and T. Y. Meyer, Organometallics, 1999, 18, 4250–4252 CrossRef CAS; (c) C. J. Cooper, M. D. Jones, S. K. Brayshaw, B. Sonnex, M. L. Russell, M. F. Mahon and D. R. Allan, Dalton Trans., 2011, 40, 3677–3682 RSC.
- (a) I. Y. Skobelev, V. N. Panchenko, O. Y. Lyakin, K. P. Bryliakov, V. A. Zakharov and E. P. Talsi, Organometallics, 2010, 29, 2943–2950 CrossRef CAS; (b) A. Brückner, J. K. Jabor, A. E. C. McConnell and P. B. Webb, Organometallics, 2008, 27, 3849–3856 CrossRef.
- (a) J. Wang, G. Tan, D. An, H. Zhu and Y. Yang, Z. Anorg. Allg. Chem., 2011, 637, 1597–1601 CrossRef CAS; (b) J. Wang, D. An and H. Zhu, Chin. J. Struct. Chem., 2010, 29, 933–939 CrossRef CAS.
- (a) Z. Hao, B. Xu, W. Gao, Y. Han, G. Zeng, J. Zhang, G. Li and Y. Mu, Organometallics, 2015, 34, 2783–2790 CrossRef CAS; (b) T. Holtrichter-Rößmann, I. Häger, C.-G. Daniliuc, R. Fröhlich, K. Bergander, C. Troll, B. Rieger, R. S. Rojas and E.-U. Würthwein, Organometallics, 2016, 35, 1906–1915 CrossRef.
- A. Jabri, P. Crewdson, S. Gambarotta, I. Korobkov and R. Duchateau, Organometallics, 2006, 25, 715–718 CrossRef CAS.
- D. S. McGuinness, M. Overett, R. P. Tooze, K. Blann, J. T. Dixon and A. M. Z. Slawin, Organometallics, 2007, 26, 1108–1111 CrossRef CAS.
- (a) R. Walsh, D. H. Morgan, A. Bollmann and J. T. Dixon, Appl. Catal., A, 2006, 306, 184–191 CrossRef CAS; (b) T. Jiang, Y. Ning, B. Zhang, J. Li, G. Wang, J. Yi and Q. Huang, J. Mol. Catal. A: Chem., 2006, 259, 161–165 CrossRef CAS.
- We have performed the catalytic reactions by using either CrCl3(THF)3/MAO or CrCl2(THF)2/MAO under the same conditions as those in entry 3, which gave a little amount of polyethylene (see entries 1 and 2 in the Table 2).
- W. Herwig and H. Zeiss, J. Org. Chem., 1958, 23, 1404–1404 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † CCDC 1471338 (3), 1471337 (5), 1471339 (8) and 1471340 (11). For crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03216h |
| This journal is © The Royal Society of Chemistry 2016 |