
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The half-sandwich 18- and 16-electron arene ruthenium iminophosphonamide complexes†
Tat'yana A.
Peganova
a,
Iana S.
Sinopalnikova
a,
Alexander S.
Peregudov
a,
Ivan V.
Fedyanin
a,
Albert
Demonceau
b,
Nikolai A.
Ustynyuk
a and
Alexander M.
Kalsin
*a
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilov str., 119991 Moscow, Russia. E-mail: kalsin@ineos.ac.ru
bLaboratory of Macromolecular Chemistry and Organic Catalysis, Department of Chemistry, University of Liège, Sart-Tilman (B6a), 4000 Liège, Belgium
First published on 16th September 2016
Abstract
Novel half-sandwich 18ē and 16ē arene ruthenium iminophosphonamide complexes [(η6-C6Me6)RuCl{(R′N)2PR2}] (3a–c) and [(η6-C6Me6)Ru{(R′N)2PR2}]+(X−) (4a–c) (a, R = Ph, R′ = p-Tol; b, R = Et, R′ = p-Tol; c, R = Ph, R′ = Me. X = BF4, PF6 or BArF4) were synthesized. The elongated Ru–Cl bond in the 18ē complexes is shown to dissociate even in apolar solvents to form the corresponding 16ē cations, which can be readily isolated as salts with non-coordinating anions. The coordinatively unsaturated 16ē complexes are stable species due to efficient π-electron donation from the nitrogen atoms of the zwitterionic NPN-ligand. The ruthenium iminophosphonamides are moderately active in the ROMP polymerization of norbornene; the 16ē complexes 4a,b yield high molecular weight polymers (Mn ∼ 300 × 103) with a narrow distribution Mw/Mn ∼ 1.6, while the 18ē complexes 3a,b give polymers of lower molecular weight (Mn < 50 × 103) with a wider polydispersity index Mw/Mn ∼ 2.5.
Introduction
Among transition metal complexes with κ2-N,N-heteroallylic ligands, the iminophosphonamides bearing a coordinated R2P(NR′)2− anion (NPN) are studied fragmentarily; there have been less than a hundred of molecular structures of NPN complexes established to date, that is in sharp contrast to more than a thousand of transition metal amidinate structures, according to the Cambridge Structural Database (CSD). The IV group metals,1–6 chromium,7–10 nickel11–15 and copper16–22 iminophosphonamides are the most studied, which is due to their catalytic application in cyclopropanation,16,21,23 olefin oligomerization7–9 and polymerization.2,6,12,15,24,25 A few platinum metal group iminophosphonamides have been reported for palladium24 and ruthenium26 before 2009, when we started systematic studies of these complexes. We have demonstrated experimentally from the precision X-ray data by determining the deformational electron density for the palladium complex [Pd{(p-iPrC6H4N)2PPh2}2] that the iminophosphonamide ligand is zwitterionic N−–P+–N− having single P–N bonds and bearing full negative charges at the nitrogen atoms.27 This result shows a big difference between the electronic properties of iminophosphonamide and amidinate complexes, which previously have been considered as having a similar heteroallylic delocalized π-electronic system. The HOMO orbital of the zwitterionic NPN ligand may have either C2v or Cs symmetry,28 of which the latter can efficiently donate the π-electron density from the nitrogen atoms to the dxz-orbital of the metal located in the plane of the ligand, similarly to the β-diketiminate complexes (Chart 1A and B). In contrast, the C2v symmetry of the HOMO orbital in the amidinate ligand allows π-donation only by lateral coordination of the amidinate ligand resulting in a strong folding of the four-membered metallacycle (Chart 1C).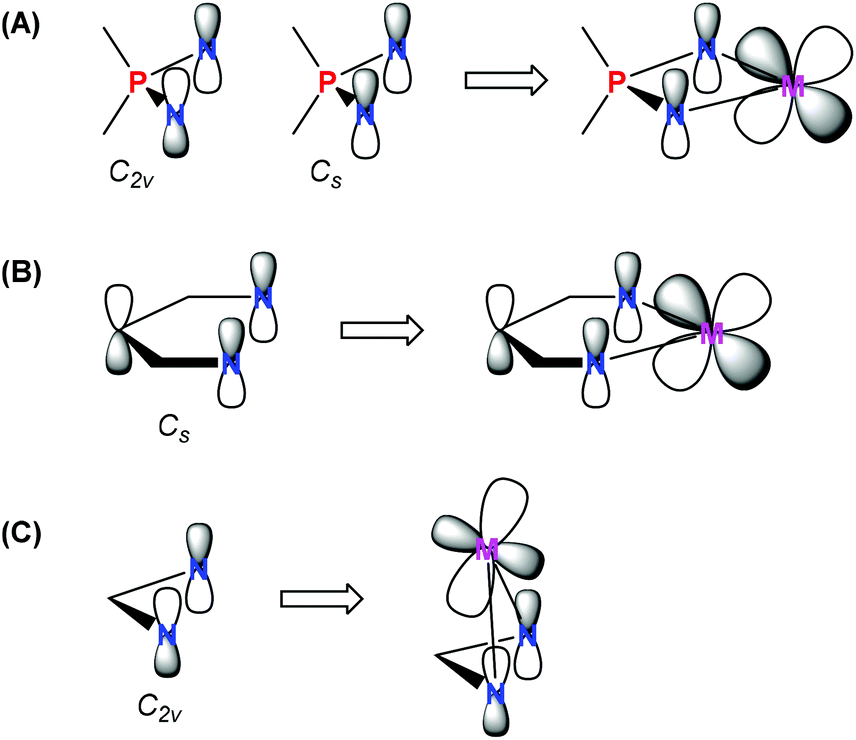 | ||
| Chart 1 Schematic drawing of the HOMO orbitals of (A) iminophosphonamide, (B) β-diketiminate, and (C) amidinate ligands (on the left) and possible π-bonding with the d-orbital of the metal (on the right). | ||
Indeed, the electron deficient ruthenium complexes can be stabilized by intramolecular π-coordination of the amidinate ligand, which leads to strong puckering of the Ru–N–C–N metallacycle in the 16ē complexes [(Cp*)Ru{(tBuN)2C(Mes)}]29 and [(C6Me6)Ru{(iPrN)2CMe}]30 to 39.9° and 31.5°, respectively. The lateral coordination of the amidinate ligand stabilizes these 16ē complexes inefficiently since it weakens the M–N σ-bonds; such species are very reactive and can readily coordinate 2ē donors30–32 or other organometallic moieties to form dinuclear μ2-amidinate complexes.33 At the same time, the solely reported ruthenium iminophosphonamide, the stable 16ē [(p-cymene)Ru{(iPrN)2PPh(NHiPr)}](BPh4), did not react either with [Et3NH]Cl, [PPh4]Cl or with triphenylphosphine and triphenyl phosphite to form 18ē adducts; only carbon monoxide or cyanide could coordinate, however the corresponding 18ē complexes were not isolated.26 For this complex the authors proposed a possibility of pseudo π-allyl donation to ruthenium based on a slight puckering of the Ru–N–P–N plane by 13.9°. Since that time the chemistry of ruthenium iminophosphonamides has not progressed, although two structures of 18ē [(p-cymene)RuBr{(p-TolN)2PMe(p-TolNH)}] and 17ē [(Cp*)RuCl{(p-TolN)2PMe(p-TolNH)}] complexes were deposited in CSD in 2004–2005 but their synthesis and properties were not published.
Recently, an interesting arene ruthenium bis(phosphinimino)methanide complex [(p-cymene)Ru(L)Cl] (L = PhN(PPh2)CH(PPh2)NPh) has been shown to exist as a cationic 18ē complex with a tridentate κ3-C,N,N ligand L.34 Although this ligand closely relates to iminophosphonamides, the stabilization of the 16ē species occurs by intramolecular coordination of the methanide group but not by the unpaired electron density from the nitrogen atoms. Indeed the displacement of the methanide group by Cl− or MeCN is highly unfavorable with the 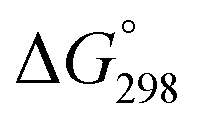 calculated for the latter reaction to be +19 kcal mol−1. This complex fails to react with CO under ambient conditions, which has also been attributed to the high nucleophilicity of the methanide group and the weak π-basicity of the ruthenium center.35
calculated for the latter reaction to be +19 kcal mol−1. This complex fails to react with CO under ambient conditions, which has also been attributed to the high nucleophilicity of the methanide group and the weak π-basicity of the ruthenium center.35
Here we report the synthesis of a series of new 18ē and 16ē half-sandwich arene ruthenium complexes with various iminophosphonamide ligands distinguished by the electronic properties of their N- and P-substituents, their physico-chemical and structural data in comparison to other κ2-N,N-heteroallylic arene ruthenium complexes, and preliminary catalytic data for the ring-opening metathesis polymerization (ROMP) of norbornene.
Results and discussion
Synthesis and characterization of the complexes 3–4
The synthesis of arene ruthenium NPN-complexes (3–4) from the diaminophosphonium salts 1a–c is summarized in Scheme 1.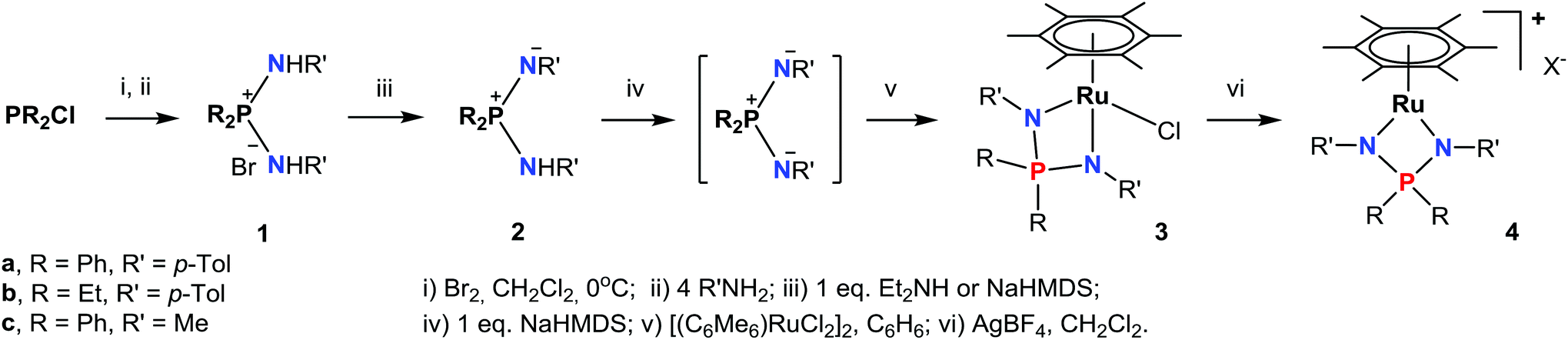 | ||
| Scheme 1 Synthesis of complexes 3–4. | ||
The diaminophosphonium salts [R2P(NHR′)2]Br (1a–c) were prepared in high yields, according to the earlier developed procedure.36 The salts 1a–c can be monodeprotonated with strong bases like NaHMDS or n-BuLi to give the corresponding iminophosphonamines 2a–c, while the more acidic 1a is deprotonated easily with an equimolar amount of Et2NH to yield 2a quantitatively. At the same time, NaHMDS is preferred for the synthesis of 2b and 2c; when n-BuLi is employed, the isolation of these iminophosphonamines is very laborious due to their complexation with lithium salts. The new compounds 1b and 2a,b were fully characterized spectroscopically and by elemental analysis, while we were not able to obtain satisfactory elemental analysis for 2c due to its high moisture sensitivity. In 31P NMR the phosphorus signals of 2a–c are shifted by 27–32 ppm to less positive values compared to those of 1a–c. In 1H NMR, the NH hydrogen of 1a (δ 9.26) is more acidic than the NH groups of 1b (δ 8.77) and 1c (δ 6.61) thus reflecting the electron-releasing effect of the N- and P-substituents on the electron density at the nitrogen atoms. Expectedly, the basic NH resonances of the iminophosphonamines 2a (δ 5.55) and 2b (δ 3.85) are shifted to less positive values. The signals from chemically inequivalent substituents at the nitrogen atoms in iminophosphonamines 2a,b are averaged tentatively due to intermolecular N–H⋯N proton exchange;37 perhaps such exchange is responsible for not observing the NH signal for 2c.
Further deprotonation of 2a–c with 1 equiv. of NaHMDS results in the formation of sodium iminophosphonamides Na[R2P(NR′)2], as indicated by the new signals in 31P NMR at more positive values δ 7.0 (R = Ph, R′ = p-Tol), δ 29.0 (R = Et, R′ = p-Tol) and δ 28.5 (R = Ph, R′ = Me) with respect to the corresponding iminophosphonamines 2a–c;14,27,38 however they were not isolated due to extremely high moisture sensitivity. The iminophosphonamides Na[R2P(NR′)2] generated in situ react with the dimeric ruthenium complex [(η6-C6Me6)RuCl2]2 to give the corresponding 18ē arene ruthenium(II) complexes 3a–c with the chelating bidentate NPN-ligand in moderate-to-high isolated yields (62–86%). The chloride ligand in 3a–c was easily replaced with the non-coordinating anions (PF6−, BF4−, BArF4) by treating them with the corresponding silver or sodium salts in dichloromethane to afford the new 16ē cationic complexes [(η6-C6Me6)Ru{(p-TolN)2PPh2}](PF6) (4a), [(η6-C6Me6)Ru{(p-TolN)2PEt2}](BF4) (4b) and [(η6-C6Me6)Ru{(MeN)2PPh2}](BArF4) (4c) in nearly quantitative yields as deep-violet solids. All the complexes obtained were fully characterized by NMR spectroscopy and elemental analysis and their molecular structures were confirmed by single crystal X-ray diffraction studies. The selected structural parameters of 3a–c and 4a–c are given in Table 1 and their projections are shown in Fig. 1–6.
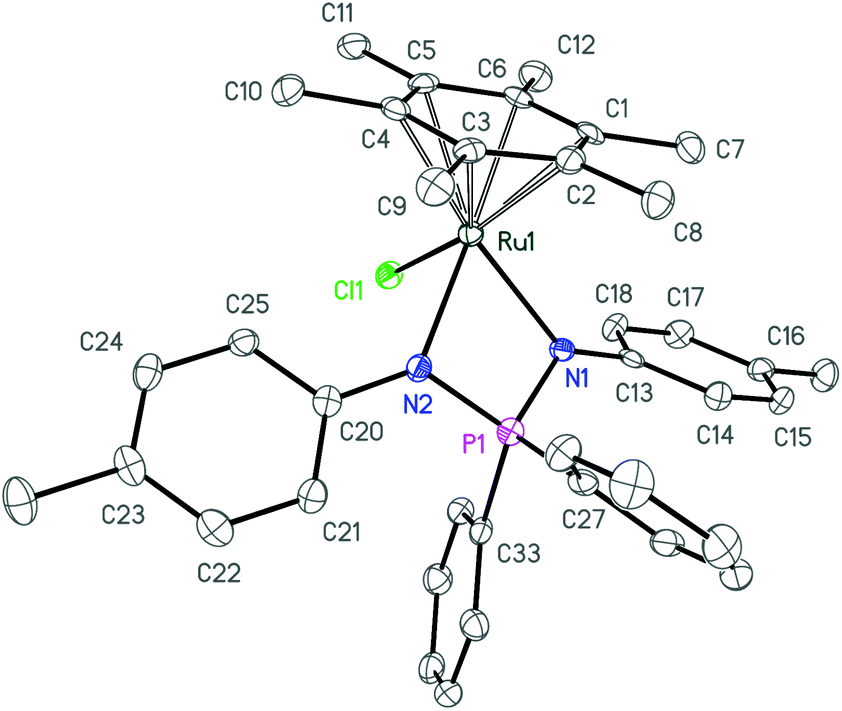 | ||
| Fig. 1 ORTEP diagram of complex 3a. Ellipsoids are shown at 50% probability, hydrogen atoms are omitted for clarity. | ||
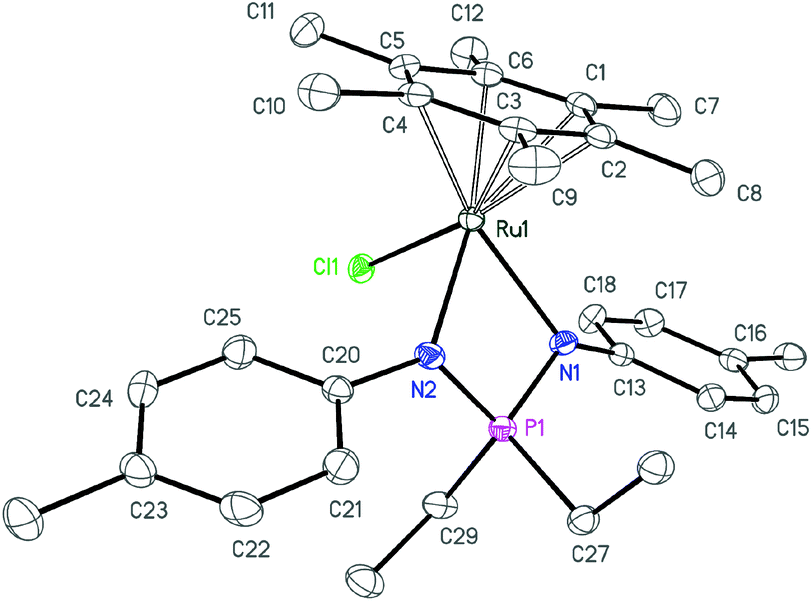 | ||
| Fig. 2 ORTEP diagram of complex 3b. Ellipsoids are shown at 50% probability, hydrogen atoms are omitted for clarity. | ||
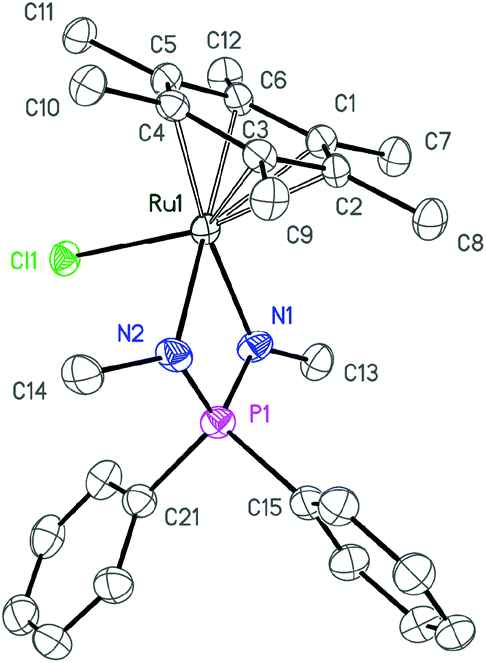 | ||
| Fig. 3 ORTEP diagram of complex 3c. Ellipsoids are shown at 50% probability, hydrogen atoms are omitted for clarity. | ||
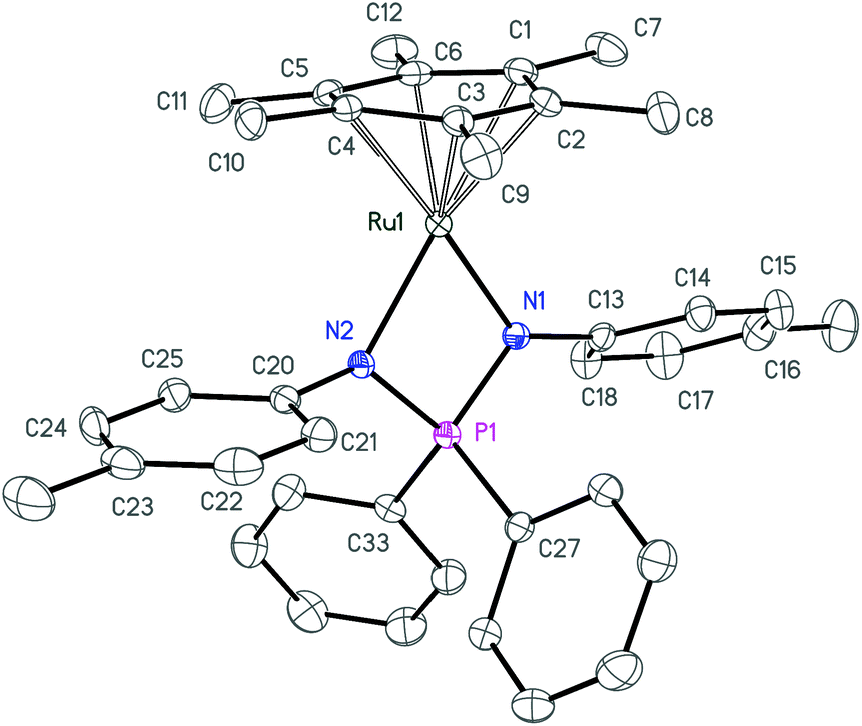 | ||
| Fig. 4 ORTEP diagram of cation 4a. Ellipsoids are shown at 50% probability, hydrogen atoms and the anion are omitted for clarity. | ||
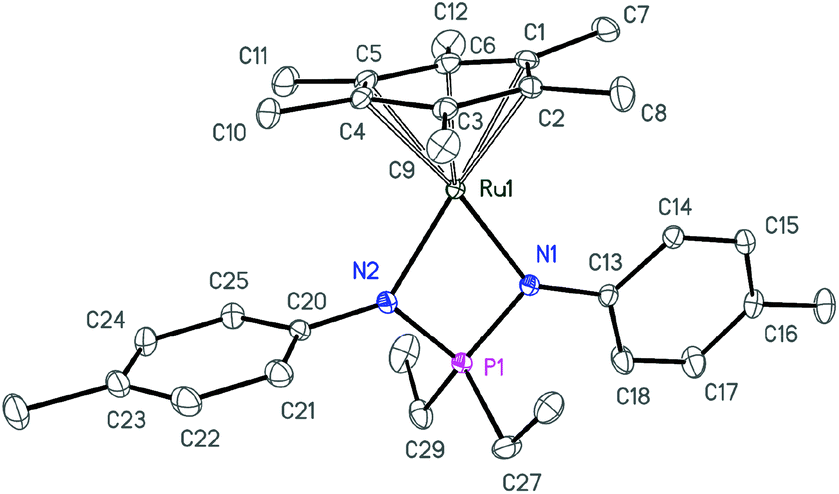 | ||
| Fig. 5 ORTEP diagram of cation 4b. Ellipsoids are shown at 50% probability, hydrogens atoms and the anion are omitted for clarity. | ||
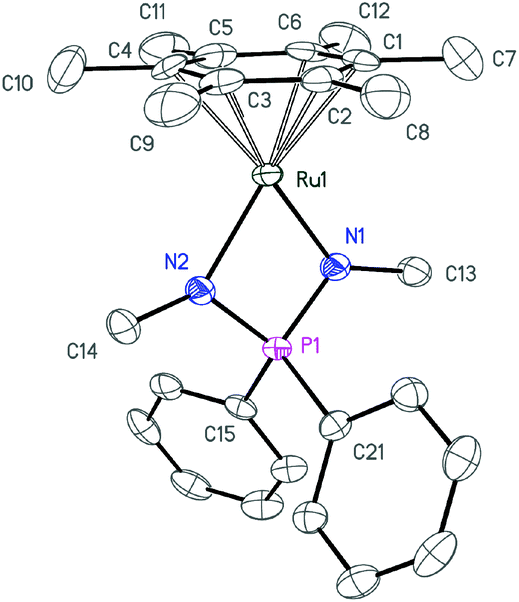 | ||
| Fig. 6 ORTEP diagram of the cation 4c. Ellipsoids are shown at 50% probability, hydrogen atoms and the anion are omitted for clarity. | ||
| 3a | 3b | 3c | 4a | 4b | 4c | |
|---|---|---|---|---|---|---|
| a The sum of bond angles at the corresponding nitrogen atom. | ||||||
| Ru⋯C6Me6(centroid) | 1.675(3) | 1.666(2) | 1.662(4) | 1.662(2) | 1.675(1) | 1.652(4) |
| Ru–C(arene) | 2.175–2.233(3) | 2.171–2.217(2) | 2.159–2.218(4) | 2.149–2.252(2) | 2.143–2.275(1) | 2.142–2.208(4) |
| Ru–N1 | 2.137(2) | 2.151(2) | 2.137(4) | 2.017(2) | 2.0119(11) | 2.011(4) |
| Ru–N2 | 2.171(2) | 2.161(2) | 2.159(4) | 2.036(2) | 2.0733(10) | 2.020(3) |
| P–N1 | 1.600(2) | 1.603(2) | 1.605(4) | 1.623(2) | 1.6352(10) | 1.615(4) |
| P–N2 | 1.613(3) | 1.615(2) | 1.602(4) | 1.624(2) | 1.6233(10) | 1.619(4) |
| Ru–Cl | 2.438(3) | 2.437(2) | 2.445(4) | |||
| N1–Ru–N2 | 68.08(9) | 68.08(7) | 69.39(14) | 72.33(9) | 72.17(4) | 72.93(14) |
| N1–P–N2 | 97.30(12) | 97.22(10) | 99.36(19) | 94.87(12) | 95.22(5) | 95.57(18) |
| Ru–N1–P | 97.98(11) | 96.81(9) | 95.94(18) | 96.54(11) | 94.82(5) | 95.50(17) |
| Ru–N2–P | 96.25(11) | 96.07(9) | 95.15(17) | 95.81(11) | 97.56(5) | 95.01(17) |
| Ru–N(1)–N(2)–P | 173.69(15) | 166.25(12) | 175.9(3) | 173.23(15) | 175.02(6) | 169.7(2) |
| C6Me6(centroid)–Ru–P | 142.2 | 148.7 | 148.2 | 176.0 | 174.1 | 175.6 |
| Ru–C(1)–C(4)/Ru–N(1)–N(2) | 6.3 | 5.7 | 26.1 | |||
| ∑(N1)a | 359.2(5) | 354.5(4) | 357.6(8) | 360.0(5) | 359.8(2) | 359.0(8) |
| ∑(N2)a | 358.6(5) | 354.6(4) | 344.4(8) | 354.9(5) | 360.0(2) | 356.9(8) |
The 18ē complexes 3a–c exhibit a three-legged piano stool geometry with a pseudo-octahedral configuration of the ligands around the ruthenium atom. The Ru–C6Me6 (centroid) distance is in the range of 1.675(3)–1.662(4) Å, which is typical of neutral half-sandwich arene ruthenium complexes. Similarly to the β-diketiminate arene ruthenium complex,39 in both 3a,b the C–C bond lengths in the η6-coordinated arene noticeably alternate: the bonds C(2)–C(3), C(4)–C(5), and C(1)–C(6), which are trans to the N(1), N(2) and Cl(1), are shorter (1.412–1.421 Å for 3a and 1.417–1.423 Å for 3b) than the bonds C(1)–C(2), C(3)–C(4), and C(5)–C(6) (1.436–1.442 Å for 3a and 1.442–1.447 Å for 3b). In contrast to this, in 3c the coordinated arene gives a nearly eclipsed conformation with the chloride and the NPN-ligands; only N(1) significantly deviates from that (the torsion angle N(1)–Ru–C6Me6(centroid)–C(1) is 18.5°), which results in shortening of the C(4)–C(5) bond trans to N(1) (1.417 Å), while the other C–C bonds are slightly longer (1.428–1.441 Å).
In iminophosphonamide complexes 3a–c the Ru–N bonds (2.137–2.171 Å) and the Ru–Cl bonds (2.437–2.445 Å) are considerably longer than those in analogous 18ē arene ruthenium amidinate (2.078–2.139 Å and 2.400–2.434 Å, respectively)40–44 or triazenide (2.104–2.133 Å and 2.386–2.397 Å)45 complexes, perhaps due to the high negative charge located at the nitrogen atoms of the zwitterionic NPN-ligand. Indeed, the Ru–Cl bond is elongated in arene ruthenium complexes with highly efficient σ,π-donating β-diketiminate (2.461–2.521 Å)39,46,47 or dianionic bis(imidazolin-2-iminate) (2.4853(4) Å) ligands,48 up to full dissociation in the latter example.
The chlorine atom has intramolecular close CH⋯Cl contacts with one ortho-hydrogen (H18A) of the N-tolyl substituents. The H18A⋯Cl distances in 3a (2.762 Å) and 3b (2.813 Å) fall below the sum of the van der Waals radii of 2.95 Å (ref. 49 and 50) and the corresponding angles Cl⋯H–C of 140.4° (3a) and 140.8° (3b) are typical of such a type of non-directed interaction. The chlorine atom is almost coplanar to the plane of the tolyl ring involved in the H⋯Cl contact and the corresponding torsion angle Cl–H(18A)–C(18)–C(13) is 7.6° and 8.4° for 3a and 3b, respectively. A few other intra- and intermolecular close contacts H⋯Cl are observed in 3a,b with the hydrogens of the C6Me6 ligand (the Cl⋯H11C, Cl⋯H11B distances are 2.772, 2.749 Å in 3a and the Cl⋯H11C, Cl⋯H8A are 2.899, 2.871 Å in 3b) and of the P-substituent (the Cl⋯H34A is 2.868 Å in 3a and the Cl⋯H29A is 2.812 Å in 3b). Similar intra- and intermolecular close contacts can be seen in 3c between the chlorine atom and the hydrogen atoms of the methyl group at N(2) (Cl⋯H14B is 2.872 Å) and C6Me6 (Cl⋯H10C is 2.840 Å), respectively.
The chelate angle N(1)–Ru–N(2) in 3a–c (68.1–69.4°) is significantly larger than that in the analogous 18ē ruthenium amidinate complexes (61–62°),40–43,51 since the P–N bonds are longer than the corresponding C–N bonds in the amidinates. It is worth noting that the torsion angle Ru–N(1)–N(2)–P is close to 180° showing small puckering of the Ru–N(1)–P–N(2) metallacycle from the planarity (6.3° for 3a, 13.7° for 3b and 4.1° for 3c). The pyramidalization of the nitrogen atoms is rather small in 3a,b, while it is strong for one of the nitrogens in 3c, for which the sum of the angles at the N(2) atom ∑(N) is 344.4°. Recently, a much wider range of puckering angles (up to 23.4°) and relatively strong pyramidalization of one of the nitrogen atoms (348–351°) were observed in the square-planar palladium iminophosphonamide complexes, presumably as a result of steric repulsion between the N-cumyl substituents of the NPN- and co-ligands.27 However this seems to be not the case of 3c bearing sterically small N-methyl groups; more probably the pyramidalization of N(2) is due to the high unpaired electron density located fully at the nitrogen atom and being not able to delocalize on any other electron system like aromatic N-tolyl groups in 3a,b.
The 16ē cationic complexes 4a–c expectedly exhibit a two-legged piano-stool geometry with the chelating NPN-ligand positioned nearly perpendicular to the C6Me6 ring; the C6Me6 (centroid)–Ru–P angle is 174–176°. The Ru–C6Me6 (centroid) distance is 1.652–1.675 Å and is similar to that in 3a–c. The Ru–N bond lengths in 4a–c (2.011–2.073 Å) are close to those in [(p-cymene)Ru{(iPrN)2PPh(NHiPr)}](BPh4) (5) (2.011, 2.017 Å)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and are significantly shorter than in 3a–c due to stronger binding with the positively charged ruthenium atom. Slightly shorter Ru–N distances were reported for 16ē cationic β-diketiminates (1.994–1.997 Å)39 and bis(imidazolin-2-iminates) (1.977–2.003 Å),48 while in the analogous ruthenium amidinate complexes these bonds are longer (2.058–2.065 Å),30,32 showing the intermediate donating ability of the NPN-ligand. The C6Me6 ring is not planar but is significantly distorted towards the boat conformation making four Ru–C(arene) bonds shorter (2.149–2.178(2) Å in 4a and 2.143–2.199(1) Å in 4b) and two bonds, namely Ru–C(1) and Ru–C(4) (2.238(2), 2.252(2) Å for 4a and 2.228(1), 2.275(1) Å for 4b), longer. Unlike in the 18ē complexes 3a,b, the C–C bonds in C6Me6 in 4a,b do not alternate, instead the bonds C(2)–C(3) and C(5)–C(6) (1.440–1.444 Å) are slightly longer than the other four C–C bonds (1.418–1.428 Å) as a result of the stronger bonding of these carbons with the ruthenium atom. Importantly, in 4a,b the longer Ru–C(1) and Ru–C(4) bonds are always trans to the Ru–N bonds (the dihedral angle between the planes Ru–C(1)–C(4) and Ru–N(1)–N(2) is 5.7–6.3°) with the longest Ru–C(4) bond trans to the shortest Ru–N(2) bond. A similar distortion of the coordinated arene planarity and the Ru–C(arene) bond distribution was earlier observed for 526 and dicationic arene ruthenium complexes with the bis(imidazolin-2-imine) ligand.48 It should be noted that in 4c the arene and the NPN-ligand are in a staggered conformation (the dihedral angle between the planes Ru–C(2)–C(5) and Ru–N(1)–N(2) is 86.1°), which leads to slight shortening of the Ru–C(2) and Ru–C(5) (2.142(4), 2.163(4) Å) compared to the other four Ru–C(arene) bonds (2.177–2.208 Å) resulting in a flipped boat conformation of the arene.
26 and are significantly shorter than in 3a–c due to stronger binding with the positively charged ruthenium atom. Slightly shorter Ru–N distances were reported for 16ē cationic β-diketiminates (1.994–1.997 Å)39 and bis(imidazolin-2-iminates) (1.977–2.003 Å),48 while in the analogous ruthenium amidinate complexes these bonds are longer (2.058–2.065 Å),30,32 showing the intermediate donating ability of the NPN-ligand. The C6Me6 ring is not planar but is significantly distorted towards the boat conformation making four Ru–C(arene) bonds shorter (2.149–2.178(2) Å in 4a and 2.143–2.199(1) Å in 4b) and two bonds, namely Ru–C(1) and Ru–C(4) (2.238(2), 2.252(2) Å for 4a and 2.228(1), 2.275(1) Å for 4b), longer. Unlike in the 18ē complexes 3a,b, the C–C bonds in C6Me6 in 4a,b do not alternate, instead the bonds C(2)–C(3) and C(5)–C(6) (1.440–1.444 Å) are slightly longer than the other four C–C bonds (1.418–1.428 Å) as a result of the stronger bonding of these carbons with the ruthenium atom. Importantly, in 4a,b the longer Ru–C(1) and Ru–C(4) bonds are always trans to the Ru–N bonds (the dihedral angle between the planes Ru–C(1)–C(4) and Ru–N(1)–N(2) is 5.7–6.3°) with the longest Ru–C(4) bond trans to the shortest Ru–N(2) bond. A similar distortion of the coordinated arene planarity and the Ru–C(arene) bond distribution was earlier observed for 526 and dicationic arene ruthenium complexes with the bis(imidazolin-2-imine) ligand.48 It should be noted that in 4c the arene and the NPN-ligand are in a staggered conformation (the dihedral angle between the planes Ru–C(2)–C(5) and Ru–N(1)–N(2) is 86.1°), which leads to slight shortening of the Ru–C(2) and Ru–C(5) (2.142(4), 2.163(4) Å) compared to the other four Ru–C(arene) bonds (2.177–2.208 Å) resulting in a flipped boat conformation of the arene.
The chelate angle N(1)–Ru–N(2) in 4a–c (72.1–72.9°) is almost equal to that in 5 (71.8°)26 and larger by ca. 4° than in 3a–c. The pyramidalization of the nitrogen atoms (∑(N) is 355–360°) and the puckering of the plane Ru(1)N(1)P(1)N(2) (5.0–10.3°) are small. This is in sharp contrast to the strong puckering (31.4°) of the chelate metallacycle M–N–C–N in the analogous amidinate 16ē arene ruthenium complex [(η6-C6Me6)Ru{(NiPr)2CMe}](PF6), required for additional stabilization of the coordinatively unsaturated species by a π-heteroallyl system.30
The structural peculiarities of the coordinated arene, the flattened RuNPN metallacycle and the short Ru–N bonds in 4a–c are indicative of the strong σ,π-bonding of the iminophosphonamide ligand26via nitrogen atoms, similarly to β-diketiminates and bis(imidazolin-2-iminates), and in contrast to allylic π-stabilization in metal amidinates. The elongated Ru–Cl bonds in 3a–c and the pyramidalization of the nitrogen atom in the most electron-rich 3c are also in agreement with the zwitterionic structure of the NPN ligand bearing enhanced negative charges at the nitrogen atoms.
In the 31P NMR spectra of 3a,b the phosphorus resonance (δ 43.9 for 3a and δ 72.4 for 3b) is shifted by ca. 47–49 ppm to more positive values compared to the precursors 2a and 2b. Interestingly, in the 1H NMR spectra of 3a and 3b in CDCl3 the two chemically inequivalent substituents at the phosphorus atom give only one set of signals for phenyl and ethyl groups, respectively. However, in the 1H NMR spectra recorded in apolar C6D6, the P-substituents of 3a and 3b give rise to two sets of the corresponding resonances. Similarly, in the 13C NMR spectra of 3a and 3b in CDCl3 the resonances for only one type of phenyl and ethyl groups are observed. It is noteworthy that in 13C NMR the characteristic doublets for ipso-carbons of phenyls are not found, perhaps due to their strong broadening. Apparently, in polar solvents fast exchange between the two P-substituents takes place. Indeed, heating a solution of 3a and 3b in apolar toluene-d8 (T = 273–353 K) leads to broadening of the inequivalent phenyl and ethyl resonances, respectively for 3a and 3b, in 1H NMR, though their coalescence is not achieved (see the ESI†). In a more polar CD2Cl2 solution of 3b at 298 K the signals for only one ethyl substituent are observed, while decreasing the temperature to 193 K gives two separate resonances of methyl groups (Fig. 7). At the coalescence temperature of Tc = 238 K in dichloromethane, the estimated exchange rate constant is 1370 s−1 and the free energy of activation ΔG≠ calculated from the Eyring equation is about 10.4 kcal mol−1.52
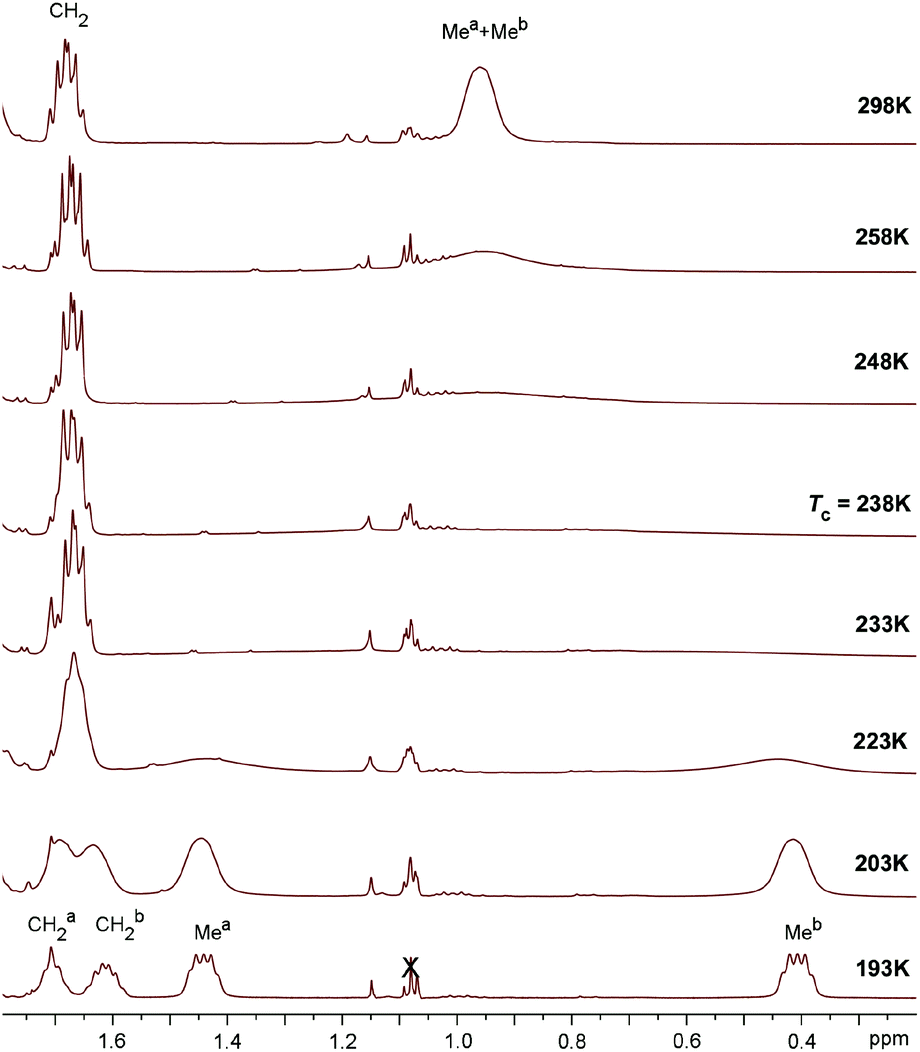 | ||
| Fig. 7 VT NMR spectra of complex 3b recorded in CD2Cl2 at T = 193–298 K (x – stands for grease). | ||
The exchange between the P-substituents Ra and Rb seems to proceed via a C2v-symmetric intermediate or a transition state with two equivalent R groups, tentatively the cationic complex [(η6-C6Me6)Ru{R2P(N-p-Tol)2}]+Cl− (vide infra), formed from 3a,b by dissociation of the chloride anion (Scheme 2).
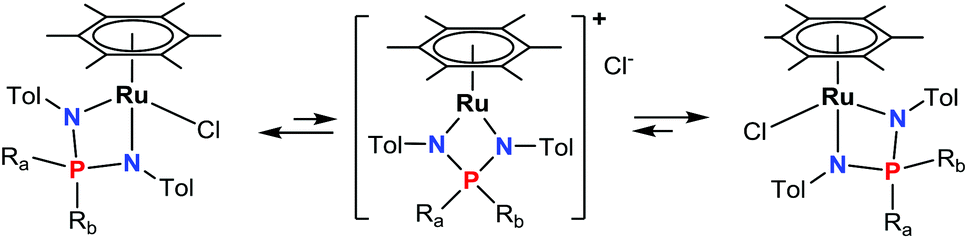 | ||
| Scheme 2 The exchange between Ra and Rb substituents via putative dissociation–association of the chloride anion.53 | ||
Although the dissociation of the chloride anion in 18ē complexes 3a,b is facile in CDCl3 or CD2Cl2, the equilibrium concentration of the dissociated 16ē form is negligible, as far as their signals in 31P NMR in CDCl3 (δ 43.9 for 3a, δ 72.4 for 3b) and C6D6 (δ 43.3 for 3a, δ 71.1 for 3b) remain virtually unchanged. The phosphorus resonance of the 16ē cationic complexes 4a (δ 71.9) and 4b (δ 102.3) is strongly shifted by ∼30 ppm to more positive values compared to the neutral complexes 3a,b. In the 1H and 13C NMR spectra of 4a,b in CD2Cl2 the P-substituents are chemically equivalent, independent of the temperature (193–298 K) as it is expected for C2v-symmetric complexes.
In apolar C6D6 complex 3c also gives rise to two inequivalent phenyl groups in the 1H and 13C NMR spectra, which is consistent with the non-dissociated 18ē chloride complex, though the signals are broadened. Whereas in more polar CDCl3 complex 3c has violet color and its 31P resonance (δ 76.9) is strongly shifted to more positive values than in C6D6 (δ 59.8) and becomes close to the signal of the cationic complex 4c (δ 80.8). The 1H NMR spectra of 3c and 4c in CDCl3 are nearly identical and in line with the cationic C2v-symmetric complex. Hence, in sharp contrast to 3a,b, in CDCl3 solution 3c undergoes facile dissociation to give the cationic complex [(η6-C6Me6)Ru{Ph2P(NMe)2}]+Cl−, which is prevalent in the equilibrium mixture. Apparently strongly electron-releasing N-methyl substituents enhance the π-donating ability of the NPN-ligand compared to that of the complexes 3a,b bearing weaker N-tolyl donors.
The arene ruthenium complexes having monoanionic β-diketiminate39 and dianionic bis(imidazolin-2-iminate)48 ligands have also been reported earlier to undergo facile chloride dissociation, whereas in the amidinate ruthenium complexes the counter-ion dissociation has been observed only for the weakly coordinating triflate ligand but not the chloride.51 Hence the capability of the iminophosphonamide ligand to donate electrons and to stabilize the electron-deficient states is much higher than that of the amidinate ligand and comparable to the β-diketiminate and zwitterionic bis(imidazolin-2-iminate) ligands.
In the UV-vis spectra the complexes 4a–c have a broad medium intensity band at λmax 540–550 nm shifted to lower energies compared to the corresponding complexes 3a,b (in CH2Cl2) and 3c (in C6H6) having a band at λmax 410–450 nm. Similar bands at λmax 520–530 nm have also been observed for 16ē pentamethylcyclopentadienyl amidinate,29 β-diketiminate47 and bis(imidazolin-2-iminate)48 ruthenium complexes, which has been attributed to d–d centered transitions.47
The 16ē complexes 4a–c are remarkably stable in solution to air and moisture in sharp contrast to the 18ē complexes 3a–c, which hydrolyze to produce the corresponding phosphinoxides R2P(O)(NHR′). Apparently the nitrogen atoms in 3a–c are highly basic due to their free electron pairs and thus are prone to the attack by water molecules, whereas in 4a–c these electron pairs participate in π-bonding to the ruthenium atom and are much less basic. Indeed, the susceptibility to hydrolysis is higher for the complex bearing more electron-releasing N-substituents; thus 3c decomposes within minutes in wet CD2Cl2 while 3a,b are stable for hours. Under similar conditions 4a–c do not hydrolyze and do not form the 18ē aqua NPN-complexes; their NMR spectra in dried and wet solvents are the same and remain unchanged for days. However, when 10 equivalents of MeCN were added to a solution of 4a in CD2Cl2 the colour immediately changed from violet to red and the phosphorus resonance shifted from δ 72.2 to δ 63.8 indicating the formation of the new complex, presumably the cationic adduct [(η6-C6Me6)Ru(MeCN){Ph2P(N-p-Tol)2}](PF6). The attempt to isolate it by removing the excess of acetonitrile in vacuo returned the starting complex 4a (violet solution in CD2Cl2 with the phosphorus resonance at δ 71.0, no acetonitrile signal in 1H NMR). Bubbling carbon monoxide into a solution of 4a in CH2Cl2 leads to the formation of a yellow CO adduct (νCO = 1984 cm−1), however the CO ligand is labile and easily decoordinates upon removing the solvent under reduced pressure to give back 4a. A similar 16ē ruthenium iminophosphonamide 5 has also been recently reported to coordinate reversibly CO to form the unstable CO adduct (νCO = 1993 cm−1) and to not react with Cl−, PPh3, and P(OPh)3.26 In sharp contrast, the more electron-deficient arene ruthenium amidinate [(η6-C6H6)Ru{PhC(NtBu)2}](BArF4) readily forms the stable carbonyl complex [(η6-C6H6)Ru(CO){PhC(NtBu)2}](BArF4), in which the CO band is observed at a much higher frequency (νCO = 2050 cm−1).30 On the other hand, the carbonyl band in the arene ruthenium complex with the dianionic dithiolate ligand [(η6-C6Me6)Ru(CO)(SXyl)2] (νCO = 1965 cm−1)54 is close to that in the CO adduct of the iminophosphonamide 4a and hence indirectly evidences for the zwitterionic nature of the NPN-ligand. Apparently in cationic iminophosphonamides 4a–c the positive charge is predominantly located on the phosphorus atom rather than on the ruthenium atom and thus their electronic properties are more like those of neutral arene ruthenium complexes with dianionic ligands.
ROMP polymerization of norbornene
Half-sandwich arene ruthenium complexes with sterically bulky phosphanes have been shown earlier to be readily accessible precatalysts for ring-opening metathesis polymerization of both strained (norbornene) and low-strain (cyclooctene) cycloolefins and their functionalized derivatives.55,56 Typically, the polymers obtained from norbornene had a number-average molecular weight Mn of 60–80 × 103 and a molecular weight distribution Mw/Mn of 1.6. We have demonstrated that under the same catalytic conditions the complexes 3a–c and 4a,b activated by trimethylsilyldiazomethane (TMSD) catalyze the polymerization of norbornene, except for almost inactive 4c (Table 2).|
|
|||||
|---|---|---|---|---|---|
| Entry | Complex | Monomer conv. (%) | Polymer yield (%) | M n (kg mol−1) | M w/Mn |
| a Norbornene (7.5 mmol); catalyst (0.03 mmol); TMSD (0.1 mmol); chlorobenzene (30 mL); 2 h at 60 °C under argon. | |||||
| 1 | 3a | 87 | 76 | 50 | 2.7 |
| 2 | 3b | 93 | 82 | 36 | 2.5 |
| 3 | 3c | 59 | 51 | 40 | 6.9 |
| 4 | 4a | 91 | 70 | 283 | 1.65 |
| 5 | 4b | 98 | 79 | 341 | 1.55 |
| 6 | 4c | 10 | 6 | — | — |
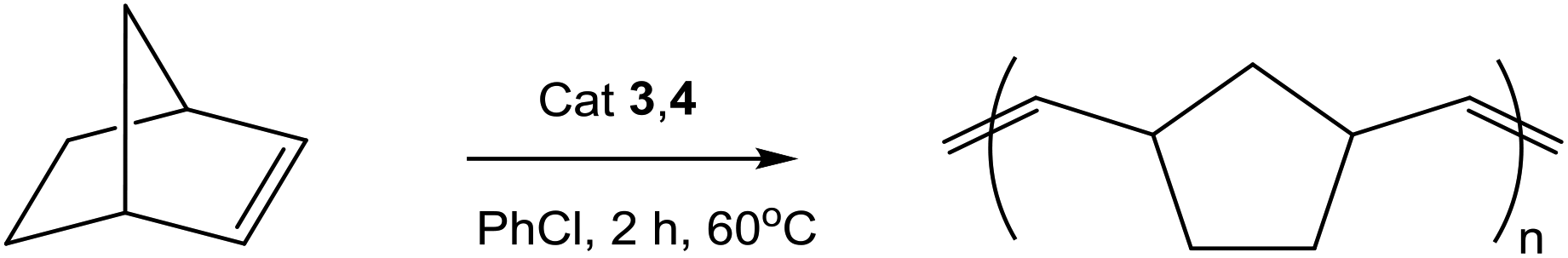
The 18ē complexes 3a–c produced polymers of low molecular weight Mn = 36–50 × 103, which is close to the expected Mn = 23.5 × 103 calculated from the catalyst/substrate ratio (1/250), meaning that all ruthenium centers are involved in catalysis. The molecular weight distribution is relatively wide: Mw/Mn is 2.5–2.7 for 3a,b and is 6.9 for 3c. The polydispersity of the polymer can be significantly improved when 16ē complexes 4a,b are employed. They yield polymers of a much higher molecular weight Mn = 283–341 × 103, which is supposedly due to their limited solubility in chlorobenzene and hence only a minor fraction (about 10%) of these cationic complexes can participate in the catalytic cycle. This assumption would mean that the actual activity of 4a,b is about ten times higher than that of 3a,b. Both types of complexes give a considerable fraction (11–20%) of low-weight oligomers soluble in methanol.
In the absence of TMSD the ruthenium iminophosphonamides studied are inactive. The actual catalytically active species in the ROMP of olefins are carbene complexes, which are typically generated in situ from the precatalysts and TMSD. Recently, the 16ē cationic ruthenium amidinate complexes have been reported to react with TMSD to result in migration of the SiMe3 group to the ruthenium atom and formation of the amidinato-carbene complexes, in which the carbene moiety is inserted into the RuNCN metallacycle.57 These amidinate complexes provided low activity in the ROMP polymerization of norbornene; higher activity was mentioned for polymerization of high-strain norbornadiene catalyzed by the 18ē complex [(C6H6)RuBr{MeC(NiPr)2}] although the detailed results were not published.32 Perhaps, a similar carbene insertion into the Ru–N bond occurs in the NPN-precatalysts.58 Not surprisingly, the more electron rich iminophosphonamide arene ruthenium complexes 3a,b and 4a,b excel the corresponding ruthenium amidinates in the ROMP polymerization of norbornene. However, our attempts to involve them in the ROMP of low-strain cyclooctene were unsuccessful.
It is noteworthy that strongly electron-releasing N-methyl substituents drastically decrease the activity of the complexes 3c and 4c. The negligible activity of the 16ē complex 4c could be a result of very efficient stabilization of the unsaturated ruthenium center to make it insusceptible to the reaction with the generated in situ carbene CHSiMe3 rather than of the solubility issues. The same can be attributed to the reduced activity of 3c, which should partially dissociate in polar chlorobenzene to form stable 16ē cationic species (vide supra).
Experimental
General procedures
All manipulations were carried out using standard Schlenk techniques under an atmosphere of dry argon. Solvents were purified by standard methods and distilled prior to use. Ph2PCl and Et2PCl were distilled under high vacuum or at ambient pressure, respectively, prior to use; other commercially available compounds were used as received. 1H, 31P and 13C NMR spectra were obtained on a Bruker AMX-400 spectrometer and referenced to the residual signals of deuterated solvent (1H and 13C), and to 85% H3PO4 (31P, external standard). Elemental analyses were performed on a Carlo Erba 1106 CHN analyzer. The following compounds were prepared according to described procedures: [Ph2P(NH-p-Tol)2]Br (1a),36 [(η6-C6Me6)RuCl2]2,59 Na[B(3,5-C6H3(CF3)2)4] (NaBArF4).60:v). Finally, it was redissolved in benzene; the hazy solution was filtered to remove the traces of the ammonium salts and then evaporated to give off-white powder. Yield 1.62 g (97%). Anal. calcd for C26H25N2P: C, 78.77; H, 6.36; N, 7.06%. Found: C, 78.94; H, 6.49; N, 6.95%. 31P NMR (CDCl3): δ −3.5. 1H NMR (CDCl3): δ 7.93 (ddd, 3JHP = 12.4, 3J = 8.1, 4J = 1.8, 4H, o-HPh), 7.39–7.51 (m, 6H, m,p-HPh), 6.96 (d, 3J = 7.5, 4H, C6H4), 6.92 (d, 3J = 7.5, 4H, C6H4), 5.55 (br. s, 1H, NH), 2.21 (s, 6H, CH3). 13C NMR (C6D6): δ 132.7 (d, 1J = 130.6, i-CPh), 132.2 (d, 2J = 9.1, o-CPh), 131.3 (s, p-CPh), 129.8 (s, β-CTol), 128.9 (br. s, i-CTol(Me)), 128.5 (d, 3J = 12.9, m-CPh), 121.3 (br. d, 3J = 11.8, α-CTol), 20.5 (s, MeTol).
Analogously, from 2b (0.51 g, 1.70 mmol) in THF (50 mL), NaHMDS (2.0 M, 0.90 ml, 1.80 mmol) and [(η6-C6Me6)RuCl2]2 (0.56 g, 0.85 mmol), complex 3b was obtained. The product was purified by precipitation from a benzene solution (3–5 mL) with hexane (10–15 mL) and further recrystallized from Et2O. Yield 0.74 g (73%). Anal. calcd for C30H42ClN2PRu: C, 60.24; H, 7.08%. Found: C, 60.37; H, 7.11%. 31P NMR (CDCl3): δ 72.4. 1H NMR (CDCl3): δ 7.04 (d, 3J = 8.0, 4H, C6H4), 6.89 (d, 3J = 8.0, 4H, C6H4), 2.23 (s, 6H, MeTol), 1.89 (s, 18H, C6Me6), 1.77 (dq, 2JHP = 11.7, 3J = 7.6, 4H, CH2CH3), 1.05 (dt, 3JHP = 16.2, 3J = 7.6, 6H, CH2CH3). 1H NMR (C6D6): δ 7.48 (d, 3J = 7.7, 4H, C6H4), 7.11 (d, 3J = 7.7, 4H, C6H4), 2.33 (s, 6H, MeTol), 2.17 (br. dq, 2JHP = 14.4, 3J = 7.8, 2H, CH2′CH3), 1.86 (s, 18H, C6Me6), 1.47 (br. dt, 3JHP = 15.6, 3J = 7.2, 3H, CH2CH3), 1.31 (br. dq, 2JHP = 14.4, 3J = 7.2, 2H, CH2CH3), 0.54 (br. dt, 3JHP = 15.6, 3J = 7.8, 3H, CH2CH3′). 13C NMR (CDCl3): δ 145.1 (d, 2JCP = 4.0, i-CTol(N)), 128.9 (s, i-CTol(Me)), 128.8 (s, β-CTol), 126.1 (d, 3JCP = 6.7, α-CTol), 88.7 (s, C6Me6), 25.7 (d, 1JCP = 54.8, CH2Me), 20.6 (s, MeTol), 15.9 (s, C6Me6), 6.5 (d, 2JCP = 4.8, CH2Me). UV-vis (CH2Cl2; λmax, nm; ε, M−1 cm−1): 450 (940).
Analogously, from 3b (0.45 g, 0.75 mmol) and AgBF4 (0.15 g, 0.77 mmol), complex 4b was obtained (0.46 g, 93%). Anal. calcd for C30H42BF4N2PRu: C, 55.48; H, 6.52%. Found: C, 55.61; H, 6.44%. 31P NMR (CDCl3): δ 102.3. 1H NMR (CDCl3): δ 7.14 (d, 3J = 6.0, 4H, C6H4), 6.91 (d, 3J = 6.0, 4H, C6H4), 2.31 (s, 6H, MeTol), 2.02 (s, 18H, C6Me6), 1.59 (br. dq, 2JHP = 10, 3J = 8, 4H, CH2CH3), 0.99 (br. dt, 3JHP = 16, 3J = 8, 6H, CH2CH3). 13C NMR (CDCl3): δ 141.4 (d, 2JCP = 4.0, i-CTol(N)), 134.9 (d, 5JCP = 2.0, i-CTol(Me)), 130.3 (s, β-CTol), 125.7 (d, 3JCP = 5.2, α-CTol), 89.2 (s, C6Me6), 20.6 (s, MeTol), 19.8 (d, 1JCP = 53.6, CH2Me), 16.3 (s, C6Me6), 5.2 (d, 2JCP = 5.4, CH2Me). UV-vis (CH2Cl2; λmax, nm; ε, M−1 cm−1): 540 (1410).
Analogously, from 3c (0.14 g, 0.26 mmol) and NaBArF4 (0.23 g, 0.27 mmol), complex 4c was obtained (0.34 g, 96%). Anal. calcd for C58H46BF24N2PRu: C, 50.86; H, 3.38%. Found: C, 50.85; H, 3.34%. 31P NMR (CDCl3): δ 80.8. 1H NMR (CDCl3): δ 7.71 (br. s., 8H, o-HBARF), 7.60 (ttd, 3J = 7.5, 4J = 1.5, 5JHP = 1.4, 2H, p-HPh), 7.50 (br. s., 4H, p-HBARF), 7.46 (td, 3J = 7.5, 4JHP = 3.0, 4H, m-HPh), 7.25 (dd, 3JHP = 12.9, 3J = 7.2, 4H, o-HPh), 2.74 (d, 3JHP = 18.3, 6H, NMe), 2.27 (s, 18H, C6Me6). 13C NMR (CDCl3): δ 161.7 (q, 1JCB = 49.8, i-CB(BArF)), 128.7 (qq, 2JCF = 31.2, 3JCB = 2.4, m-C(BArF)), 117.4 (m, o-C(BArF)), 134.7 (br. s, p-C(BArF)), 124.5 (q, 1JCF = 272.6, CF3), 133.8 (d, 4JCP = 2.4, p-CPh), 131.1 (d, 2JCP = 9.9, o-CPh), 129.3 (d, 3JCP = 11.6, m-CPh), 124.9 (d, 1J = 87.5, i-CPh), 89.3 (s, C6Me6), 33.2 (s, NMe), 16.4 (s, C6Me6). UV-vis (CH2Cl2; λmax, nm; ε, M−1 cm−1): 550 (1300).
Typical procedure for the ROMP of norbornene
A 50 mL round-bottom flask equipped with a magnetic stirring bar and capped with a three-way stopcock was charged with the ruthenium complexes 3–4 (0.03 mmol) and degassed chlorobenzene (20 mL) was added under an argon atmosphere. The solution was stirred for a few minutes at room temperature and then in an oil bath thermostated at 60 °C. Norbornene (1.5 M in chlorobenzene, 5 mL, 7.5 mmol) and eventually trimethylsilyldiazomethane, TMSD (0.1 M in a hexanes–chlorobenzene mixture, 1 mL, 0.1 mmol) were added with a syringe, and the reaction mixture was stirred for 2 h at 60 °C. The conversion was monitored by gas chromatography using norbornane as an internal standard. The resulting gel was diluted with CHCl3 (20 mL) and slowly poured into MeOH (500 mL) under vigorous stirring. The precipitated polymer was filtered, dried under dynamic vacuum, and characterized by NMR spectroscopy and GPC in THF using a polystyrene calibration.X-ray crystal structure determination
Single crystals of 3 and 4 were obtained by slow diffusion of Et2O to a solution of a complex (3a, 4a–c) in CH2Cl2, or by diffusion of hexane to a solution of 3b and 3c in benzene. Data collection for all samples was performed on a Bruker SMART APEX II diffractometer (MoKα radiation, λ = 0.71073 Å) equipped with an Apex II CCD detector. Frames were integrated using the Bruker SAINT software package61 by a narrow-frame algorithm. A semiempirical absorption correction was applied with the SADABS62 program using the intensity data of equivalent reflections. The structures were solved with direct methods and refined by the full-matrix least-squares technique against F2hkl in anisotropic approximation with the SHELX63 software package. The positions of hydrogen atoms were calculated, and all hydrogen atoms were refined in a riding model with 1.5Ueq(Cm) and 1.2Ueq(Ci), where Ueq(Cm) and 1.2Ueq(Ci) are respectively the equivalent thermal parameters of the methyl and all other carbon atoms to which the corresponding H atoms are bonded. Detailed crystallographic information is given in Table 3. Crystallographic data have been deposited to the Cambridge Crystallographic Data Centre, CCDC numbers 1475876–1475879, 1494098 and 1494099.| 3a | 3b | 3c | 4a | 4b | 4c | |
|---|---|---|---|---|---|---|
| Formula | C38H42ClN2PRu | C30H42ClN2PRu | C26H34ClN2PRu | C38H42F6N2P2Ru | C30H42BF4N2PRu | C58H46BF24N2PRu |
| Formula weight | 694.22 | 598.14 | 542.04 | 803.74 | 649.50 | 1369.82 |
| T, K | 100 | 100 | 100 | 100 | 100 | 120 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | C2/c | P21/c |
| Z/Z′ | 4/1 | 4/1 | 2/1 | 4/1 | 8/1 | 4/1 |
| a, Å | 8.6451(9) | 9.890(3) | 8.8288(13) | 9.7804(6) | 25.9645(3) | 12.2605(8) |
| b, Å | 20.932(2) | 36.718(9) | 8.8550(13) | 32.185(2) | 9.96760(10) | 28.9349(15) |
| c, Å | 17.7244(19) | 8.509(2) | 16.886(3) | 12.0350(8) | 23.1951(3) | 17.1266(10) |
| α, ° | 87.525(3) | |||||
| β, ° | 99.594(2) | 113.639(7) | 79.413(2) | 107.2960(10) | 96.5526(6) | 108.197(2) |
| γ, ° | 68.626(2) | |||||
| V, Å3 | 3162.6(6) | 2830.4(13) | 1208.0(3) | 3617.1(4) | 5963.76(12) | 5771.9(6) |
| d calc., g cm−3 | 1.458 | 1.404 | 1.490 | 1.476 | 1.447 | 1.576 |
| μ, cm−1 | 6.62 | 7.26 | 8.42 | 5.83 | 6.27 | 4.18 |
| 2θmax, ° | 54 | 60 | 54 | 60 | 60 | 56 |
| Refls. collected/independent | 32206/6902 |
26013/8183 |
16106/5286 |
46296/10527 |
74134/8695 |
41189/13856 |
| Observed reflections [I > 2σ(I)] | 4730 | 6453 | 4315 | 7940 | 8146 | 7865 |
| R 1 | 0.0388 | 0.0385 | 0.0506 | 0.0461 | 0.0218 | 0.0653 |
| wR2 | 0.0670 | 0.0835 | 0.1262 | 0.1043 | 0.0641 | 0.1570 |
| GOF | 1.007 | 1.040 | 1.036 | 1.087 | 1.043 | 1.060 |
| Residual density, e Å−3 (dmax/dmin) | 0.798/−0.735 | 0.761/−0.664 | 2.120/−0.945 | 1.526/−0.927 | 0.568/−0.509 | 1.277/−0.863 |
Conclusions
A series of new 18ē and 16ē hexamethylbenzene ruthenium complexes with the iminophosphonamide ligand have been synthesized and fully characterized. The elongated Ru–Cl bonds and an easy dissociation of the chloride anion in the 18ē complexes 3a–c as well as short Ru–N bonds in the 16ē complexes 4a–c and small puckering of the Ru–N–P–N metallacycle indeed suggest the zwitterionic nature and strong σ,π-donor character of the iminophosphonamide ligand being able to donate 4ē or 6ē. Due to charge separation between the phosphorus and the nitrogen atoms, the electronic properties of the NPN-ligand are similar to those of dianionic ligands and therefore it can efficiently stabilize 16ē electron deficient complexes. Among other arene ruthenium complexes with κ2-N,N chelating ligands, the properties of 16ē iminophosphonamide complexes resemble those of strongly donating monoanionic β-diketiminates and dianionic bis(imidazolin-2-iminates), and render them air stable unlike the analogous air-sensitive 16ē amidinate complexes. Thus arene ruthenium complexes 3a,b and 4a,b bearing the strongly donating NPN-ligand perform better than the analogous ruthenium amidinates in the ROMP of strained norbornene. To properly address the unusually low activity of complexes 3c, 4c with the most electron-rich NPN-ligand, which seems to block the activation of the ruthenium center by carbene, a deeper mechanistic investigation is required. The detailed kinetic and thermodynamic studies on the dissociation of the chlorides 3a–c and the coordination of various ligands to their 16ē counterparts 4a–c as well as the ROMP mechanistic study and application in the transfer hydrogenation of ketones are in progress and to be reported soon.Acknowledgements
The authors thank the Russian Foundation for Basic Research (grant no. 14-03-00345) for financial support.Notes and references
- R. Vollmerhaus, R. Tomaszewski, P. Shao, N. J. Taylor, K. J. Wiacek, S. P. Lewis, A. Al-Humydi and S. Collins, Organometallics, 2005, 24, 494–507 CrossRef CAS.
- R. Vollmerhaus, P. Shao, N. J. Taylor and S. Collins, Organometallics, 1999, 18, 2731–2733 CrossRef CAS.
- M. Witt, M. Noltemeyer, H.-G. Schmidt, T. Lübben and H. W. Roesky, J. Organomet. Chem., 1999, 591, 138–147 CrossRef CAS.
- E. Müller, J. Müller, F. Olbrich, W. Brüser, W. Knapp, D. Abeln and F. T. Edelmann, Eur. J. Inorg. Chem., 1998, 1998, 87–91 CrossRef.
- C. Qi and S. Zhang, Appl. Organomet. Chem., 2006, 20, 70–73 CrossRef CAS.
- R. Tomaszewski, R. Vollmerhaus, A. Al-Humydi, Q. Wang, N. J. Taylor and S. Collins, Can. J. Chem., 2006, 84, 214–224 CrossRef CAS.
- K. Albahily, V. Fomitcheva, S. Gambarotta, I. Korobkov, M. Murugesu and S. I. Gorelsky, J. Am. Chem. Soc., 2011, 133, 6380–6387 CrossRef CAS PubMed.
- K. Albahily, V. Fomitcheva, Y. Shaikh, E. Sebastiao, S. I. Gorelsky, S. Gambarotta, I. Korobkov and R. Duchateau, Organometallics, 2011, 30, 4201–4210 CrossRef CAS.
- K. Albahily, S. Licciulli, S. Gambarotta, I. Korobkov, R. Chevalier, K. Schuhen and R. Duchateau, Organometallics, 2011, 30, 3346–3352 CrossRef CAS.
- K. Albahily, Y. Shaikh, E. Sebastiao, S. Gambarotta, I. Korobkov and S. I. Gorelsky, J. Am. Chem. Soc., 2011, 133, 6388–6395 CrossRef CAS PubMed.
- R. Boese, M. Düppmann, W. Kuchen and W. Peters, Z. Anorg. Allg. Chem., 1998, 624, 837–845 CrossRef CAS.
- R. Schubbe, K. Angermund, G. Fink and R. Goddard, Macromol. Chem. Phys., 1995, 196, 467–478 CrossRef CAS.
- W.-J. Guo and Z.-X. Wang, J. Org. Chem., 2013, 78, 1054–1061 CrossRef CAS PubMed.
- B. Prashanth and S. Singh, J. Chem. Sci., 2015, 127, 315–325 CrossRef CAS.
- R. L. Stapleton, J. Chai, N. J. Taylor and S. Collins, Organometallics, 2006, 25, 2514–2524 CrossRef CAS.
- I. V. Shishkov, F. Rominger and P. Hofmann, Organometallics, 2009, 28, 1049–1059 CrossRef CAS.
- B. F. Straub, F. Eisentrager and P. Hofmann, Chem. Commun., 1999, 2507–2508 RSC.
- P. Hofmann, I. V. Shishkov and F. Rominger, Inorg. Chem., 2008, 47, 11755–11762 CrossRef CAS PubMed.
- B. F. Straub, F. Rominger and P. Hofmann, Organometallics, 2000, 19, 4305–4309 CrossRef CAS.
- B. F. Straub, F. Rominger and P. Hofmann, Chem. Commun., 2000, 1611–1612 RSC.
- B. F. Straub, I. Gruber, F. Rominger and P. Hofmann, J. Organomet. Chem., 2003, 684, 124–143 CrossRef CAS.
- N. Nebra, C. Lescot, P. Dauban, S. Mallet-Ladeira, B. Martin-Vaca and D. Bourissou, Eur. J. Org. Chem., 2013, 984–990 CrossRef CAS.
- B. F. Straub and P. Hofmann, Angew. Chem., Int. Ed., 2001, 40, 1288–1290 CrossRef CAS.
- W. Keim, R. Appel, A. Storeck, C. Krüger and R. Goddard, Angew. Chem., Int. Ed., 1981, 20, 116–117 CrossRef.
- S. Collins, Coord. Chem. Rev., 2011, 255, 118–138 CrossRef CAS.
- P. J. Bailey, K. J. Grant and S. Parsons, Organometallics, 1998, 17, 551–555 CrossRef CAS.
- T. A. Peganova, A. V. Valyaeva, A. M. Kalsin, P. V. Petrovskii, A. O. Borissova, K. A. Lyssenko and N. A. Ustynyuk, Organometallics, 2009, 28, 3021–3028 CrossRef CAS.
- More correctly, Chart 1 shows the symmetry adapted linear combinations (SALC) of the atomic orbitals. The corresponding molecular orbitals retain the same symmetry elements, however their designation transforms either to A2 and B1 or A′′ and A′, respectively for the planar or lateral coordination of the metal atom. Here we used the point group symmetries for SALC to be independent of the symmetry of the resulting complex.
- H. Kondo, T. Sue, A. Kageyama, Y. Yamaguchi, Y. Sunada and H. Nagashima, J. Organomet. Chem., 2009, 694, 795–800 CrossRef CAS.
- T. Hayashida, Y. Yamaguchi, K. Kirchner and H. Nagashima, Chem. Lett., 2001, 954–955 CrossRef CAS.
- Y. Yamaguchi and H. Nagashima, Organometallics, 2000, 19, 725–727 CrossRef CAS.
- H. Nagashima, H. Kondo, T. Hayashida, Y. Yamaguchi, M. Gondo, S. Masuda, K. Miyazaki, K. Matsubara and K. Kirchner, Coord. Chem. Rev., 2003, 245, 177–190 CrossRef CAS.
- H. Kondo, Y. Yamaguchi and H. Nagashima, J. Am. Chem. Soc., 2001, 123, 500–501 CrossRef CAS.
- C. Bibal, M. Pink, Y. D. Smurnyy, J. Tomaszewski and K. G. Caulton, J. Am. Chem. Soc., 2004, 126, 2312–2313 CrossRef CAS PubMed.
- C. Bibal, Y. D. Smurnyy, M. Pink and K. G. Caulton, J. Am. Chem. Soc., 2005, 127, 8944–8945 CrossRef CAS PubMed.
- O. V. Gusev, T. A. Peganova, A. V. Gonchar, P. V. Petrovskii, K. A. Lyssenko and N. A. Ustynyuk, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 322–331 CrossRef CAS.
- O. J. Scherer and P. Klusmann, Z. Anorg. Allg. Chem., 1969, 370, 171 CrossRef CAS.
- M. Düppmann, W. Kuchen, W. Peters and W. Phosphorus, Sulfur Silicon Relat. Elem., 1997, 129, 53–58 CrossRef.
- A. D. Phillips, G. Laurenczy, R. Scopelliti and P. J. Dyson, Organometallics, 2007, 26, 1120–1122 CrossRef CAS.
- T. Singh, R. Kishan, M. Nethaji and N. Thirupathi, Inorg. Chem., 2012, 51, 157–169 CrossRef CAS PubMed.
- T. Hayashida and H. Nagashima, Organometallics, 2002, 21, 3884–3888 CrossRef CAS.
- P. J. Bailey, L. A. Mitchell and S. Parsons, J. Chem. Soc., Dalton Trans., 1996, 2839–2841 RSC.
- W. W. Seidel, W. Dachtler and T. Pape, Z. Anorg. Allg. Chem., 2012, 638, 116–121 CrossRef CAS.
- I. J. Munslow, A. R. Wade, R. J. Deeth and P. Scott, Chem. Commun., 2004, 2596–2597 RSC.
- C. F. Barboza da Silva, S. Schwarz, M. Galceran Mestres, S. Teijelo López and J. Strähle, Z. Anorg. Allg. Chem., 2004, 630, 1919–1923 CrossRef CAS.
- A. D. Phillips, O. Zava, R. Scopelitti, A. A. Nazarov and P. J. Dyson, Organometallics, 2010, 29, 417–427 CrossRef CAS.
- A. D. Phillips, K. Thommes, R. Scopelliti, C. Gandolfi, M. Albrecht, K. Severin, D. F. Schreiber and P. J. Dyson, Organometallics, 2011, 30, 6119–6132 CrossRef CAS.
- T. Glöge, D. Petrovic, C. Hrib, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2009, 4538–4546 CrossRef.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- T. Hayashida, H. Kondo, J. I. Terasawa, K. Kirchner, Y. Sunada and H. Nagashima, J. Organomet. Chem., 2007, 692, 382–394 CrossRef CAS.
- A. D. Bain, Prog. Nucl. Magn. Reson. Spectrosc., 2003, 43, 63–103 CrossRef CAS.
- Although we observe the formation of the cationic 16ē species in the solution of 3c in chloroform or dichloromethane proving them to be possible intermediates, the actual exchange mechanism in apolar solvents can be more complicated or even different from the one given in the simplified Scheme 2.
- K. Mashima, H. Kaneyoshi, S. Kaneko, A. Mikami, K. Tani and A. Nakamura, Organometallics, 1997, 16, 1016–1025 CrossRef CAS.
- A. W. Stumpf, E. Saive, A. Demonceau and A. F. Noels, J. Chem. Soc., Chem. Commun., 1995, 1127–1128 RSC.
- A. Demonceau, A. W. Stumpf, E. Saive and A. F. Noels, Macromolecules, 1997, 30, 3127–3136 CrossRef CAS.
- T. Hayashida and H. Nagashima, Organometallics, 2001, 20, 4996–4998 CrossRef CAS.
- The model reaction of 4b with 3 equiv. of TMSD in dichloromethane resulted in a complex mixture of products, one of each having the signal in 31P NMR at δ 78.3 perhaps corresponds to a carbene complex (a characteristic doublet in 1H NMR at δ 9.2 with JH–P = 16 Hz). To define the structures of all the species formed and their evolution with time in the ROMP catalytic cycle further mechanistic investigation is to be carried out.
- M. Bennett, T. N. Huang, T. Matheson, A. Smith, S. Ittel and W. Nickerson, Inorg. Synth., 1982, 21, 74–78 CrossRef CAS.
- N. A. Yakelis and R. G. Bergman, Organometallics, 2005, 24, 3579–3581 CrossRef CAS PubMed.
- Bruker, SAINT v7.23A, 2005 Search PubMed.
- G. M. Sheldrick, SADABS v2008/1, Bruker/Siemens Area Detector Absorption Correction Program, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Variable-temperature 1H NMR of 3b in toluene and the ΔG≠ calculations for the exchange of Eta and Etb in 3b in CD2Cl2. CCDC 1475876–1475879, 1494098 and 1494099. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03202h |
| This journal is © The Royal Society of Chemistry 2016 |