
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplying low-molecular weight supramolecular gelators in an environmental setting – self-assembled gels as smart materials for pollutant removal
Babatunde O.
Okesola
and
David K.
Smith
*
Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK. E-mail: david.smith@york.ac.uk
First published on 31st May 2016
Abstract
This review explores supramolecular gels as materials for environmental remediation. These soft materials are formed by self-assembling low-molecular-weight building blocks, which can be programmed with molecular-scale information by simple organic synthesis. The resulting gels often have nanoscale ‘solid-like’ networks which are sample-spanning within a ‘liquid-like’ solvent phase. There is intimate contact between the solvent and the gel nanostructure, which has a very high effective surface area as a result of its dimensions. As such, these materials have the ability to bring a solid-like phase into contact with liquids in an environmental setting. Such materials can therefore remediate unwanted pollutants from the environment including: immobilisation of oil spills, removal of dyes, extraction of heavy metals or toxic anions, and the detection or removal of chemical weapons. Controlling the interactions between the gel nanofibres and pollutants can lead to selective uptake and extraction. Furthermore, if suitably designed, such materials can be recyclable and environmentally benign, while the responsive and tunable nature of the self-assembled network offers significant advantages over other materials solutions to problems caused by pollution in an environmental setting.
 Babatunde O. Okesola | Dr Babatunde O. Okesola graduated with a first class BSc in Chemistry and as faculty best graduating student from Federal University of Agriculture, Abeokuta (2010). He completed his PhD at the University of York (2015) and is now a postdoctoral researcher at Queen Mary University, London. He was recognised as a ‘Human of York’ (2013), awarded the University of York Kathleen Mary Stott (KMS) Prize for research (2014) and featured as one of the RSC's 175 Faces of Chemistry (2015). |
 David K. Smith | Prof. David K. Smith is Professor of Chemistry at University of York. His research focusses on applying fundamental understanding of supramolecular science, self-assembly and soft matter to the fields of nanomaterials and nanomedicine. He has received the RSC Corday Morgan Award for research excellence (2012) and a Higher Education Academy National Teaching Fellowship (2013) in recognition of his innovative education and outreach work. His Twitter handle is @professor_dave and he was recognised as one of the ‘20 Chemists worth Following’ by Chemistry and Engineering News (2014). He was featured as one of the RSC's Diverse 175 Faces of Chemistry (2014) and is a strong advocate of diversity in science. |
1. Introduction
Water is an abundant natural resource, yet the world is ‘thirsty’, with the scarcity of safe water being a serious challenge confronting humankind in the 21st century.1 Life without industrialization is unthinkable, but its rapid pace and the resulting impacts have seriously impaired global water quality. This has occurred through release of hazardous waste such as heavy metals, dyes, pharmaceuticals, petroleum products, pesticides and fertilizer into the environment.2 When these pollutants are discharged as untreated effluent into rivers, lakes, and oceans, they affect aquatic life and the food chain, and also predispose people to health-related problems including vomiting, cancer, neurological damage, liver failure and cognitive dysfunction. As such, both biological and physicochemical methods have been devised to combat water pollution, with adsorption of dissolved organic/inorganic pollutants onto solid materials being a key current methodology due to advantages such as simplicity, ease of operation and handling, regeneration, near-complete removal of pollutants and economic feasibility.3 The use of commercial activated carbon and other non-conventional adsorbents such as zeolites, chitosans, mineral clays, sawdust and waste biomatter to remove (e.g.) dyes and heavy metals from water is common practice. However, there are limitations such as lack of selectivity, generation of large amounts of toxic sludge, low pollutant uptake and costly regeneration processes. This means there is still significant interest in the development of innovative materials with applications in environmental remediation.This review explores self-assembled gel-phase nanomaterials.4 As described in more detail below, these are colloidal soft matter systems, which most usually consist of a sample-spanning nanoscale (or sometimes microscale) solid-like network within a liquid-like medium. They have the advantage of being highly solvated, yet have the rheological properties of solid-phase materials on analytical timescales. As such, they can potentially be manipulated as solids, while at the same time bringing their structuring into intimate contact with environmental liquid phases, maintaining high surface areas and rapid internal diffusion kinetics. Clearly nano-structured gels would be preferential to micro-structured gels in this regard. This review provides an overview of the applications of supramolecular gels in the purification of water contaminated with oil and solvent spills, dyes, heavy metals, toxic anions, and chemical warfare agents (Fig. 1).
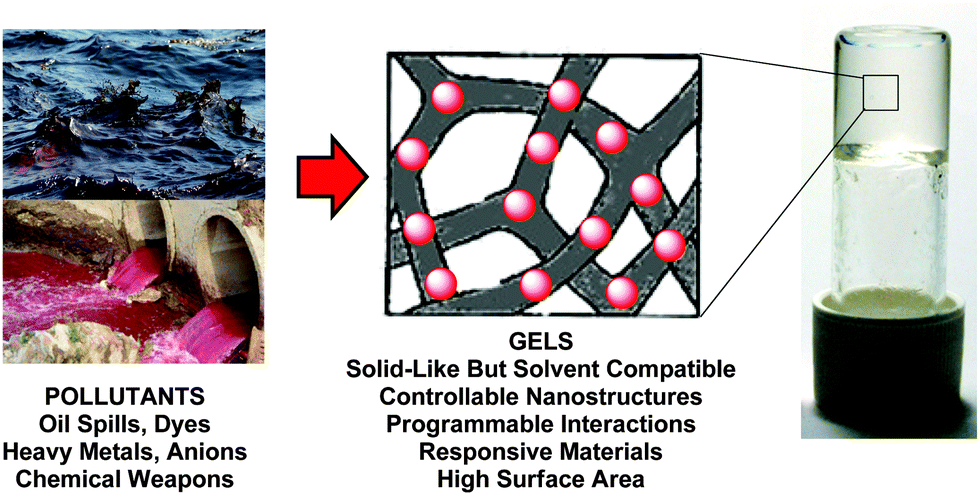 | ||
| Fig. 1 Self-assembled gel-phase soft materials for environmental remediation. | ||
Gels are easily recognised soft materials with a wide range of well-known applications – for example in cosmetics, pharmaceutical preparation, greases/lubricants, electrolytes, sealants and the food industry. The axiom that ‘if it looks like jell-o, then it must be a gel’5 is frequently used, and provides a useful layperson's definition. Many gels applied in everyday life are based on polymers which are entangled or crosslinked to constitute the solid-like network,6 however, an increasingly important class of gels are defined as low-molecular-weight supramolecular gels.4 Such gels are formed via non-covalent interactions (hydrogen bonds, van der Waals forces, π–π stacking, dipole–dipole, charge-transfer and coordination interactions, and solvophobic effects) which enable small molecules to reversibly self-assemble into supramolecular polymers that constitute the solid-like network (Fig. 2). These low-molecular-weight gelators (LMWGs), also sometimes referred to as molecular gelators, are based on organic compounds with molecular weights <2000 Da. They are known as organogelators (or oleogelators) if they self-assemble into gels in organic solvents (or oils), ionogels if they self-assemble in ionic liquids,7 and hydrogelators if they assemble into gels in water.
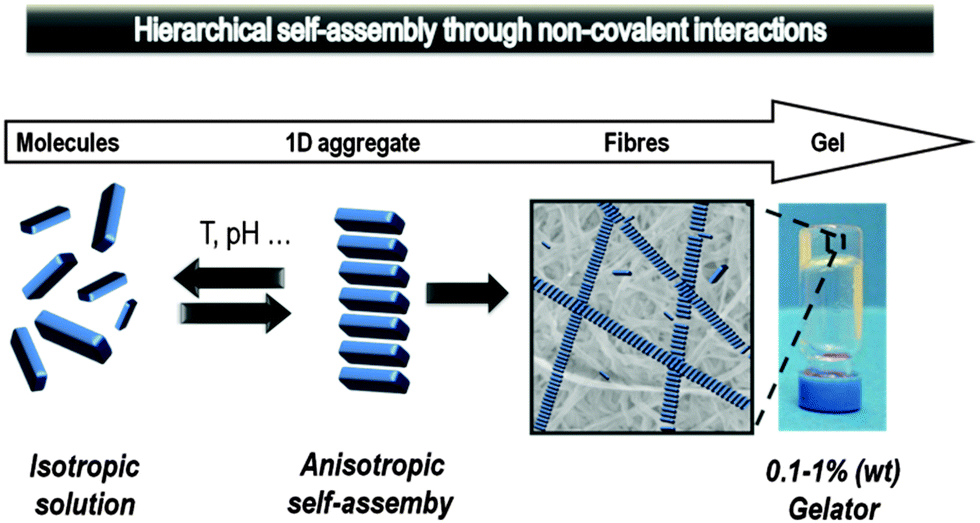 | ||
| Fig. 2 Bottom-up self-assembly of a supramolecular gel from low molecular weight building blocks. Figure reproduced from ref. 4d with permission. | ||
Although there has been intense recent research in this area, it is important to realise that the published origins of this type of gel can be found relatively far back in scientific history. For example, dibenzylidenesorbitol (DBS) was first reported as a gelator in 1891,8 and the gelation properties of 12-hydroxystearic acid (HSA) were discussed in 1912.9 Over the past century, industry has made some use of these classic LMWGs in a variety of applications, ranging from lubricating engine greases and additives for clarification of plastics, to the solid-like matrices used in glue sticks and personal care products.10 However, the use of LMWGs in environmental remediation has been less well explored by industry.
Over the 100 years since the initial reports of gelating systems, a wide range of molecular building blocks including peptides, ureas, sugars, steroids and bile acids, lipids, nucleobases, and simple alkanes have demonstrated their potential as LMWGs.5 Importantly, in supramolecular gels, the molecular-scale information incorporated into the LMWGs by organic synthesis is translated up into the nano-scale or micro-scale fibrillar network via hierarchical self-assembly. The resulting fibrils then bundle into fibres that tangle with one another to form an extended sample-spanning network capable of immobilizing a large volume of solvent (often >99%) under the influence of capillary forces (Fig. 2). A key feature of self-assembled gels is that they display the chemical functionality with which they are ‘programmed’. It should be noted that other morphologies occasionally underpin self-assembled gels, such as sample-spanning vesicular or plate-like morphologies.
Supramolecular gels are also highly responsive as a result of the non-covalent interactions which underpin them. Light, pH, heat–cool cycles, solvent changes, analyte/ligand addition, enzymes, oxidants/reductants and sonication have all been demonstrated to be capable of switching gelation on or off.11 In the simplest case, a gel is formed when a hot solution of a LMWG is cooled to a certain temperature, known as the sol–gel transition temperature (Tgel). Heating the gel again leads to its disassembly into a sol as a result of dissolution entropy overcoming the enthalpy associated with self-assembly.
In recent years, LMWGs in water or organic solvents have increasingly been investigated for high-tech applications in areas as diverse as nanomedicine, catalysis, drug delivery, light harvesting, tissue engineering, nanoelectronics, templating and sensing.12 The potential of self-assembled gels in these areas is attributed to their unique combination of responsiveness, programmability and biocompatibility.
2. Supramolecular gels: emerging nanosorbents for water purification
As part of global efforts tailored toward combating the rapid pace of environmental damage, supramolecular gels based on LMWGs have, as described in detail below, begun to attract attention as emerging materials for water purification. This article explores a range of potential environmental applications of supramolecular gels, and then discusses some of the general aspects of gel performance which can be modified and optimised. Importantly, the final part of this article provides a critical overview of the state-of-the-art, focussing on how researchers can ensure their research has maximum impact and helps move the field forwards.2.1 Supramolecular gels for oil spill remediation
Marine pollution mostly through accidental or intentional discharge of crude oil and petrochemicals is a serious environmental problem. Waterways have witnessed alarming oil spills, for example the 5 billion barrels of crude oil released in the Gulf of Mexico in 2010.13 This is problematic from both an economic perspective through wastage of valuable non-renewable oil, as well as the potential impacts on human health through consumption of sea foods obtained from oil-polluted seas, impacts on climate as a result of the accumulation of volatile hydrocarbons in the stratosphere, and devastating effects on the delicate balance of the marine ecosystem.14 Conventional methods for remediating oil spills include absorption, dispersion, bioremediation and solidification.15 Sorbents are solids that absorb the oil, dispersants emulsify the oil and solidifiers are polymeric materials that gel the oil. However, these approaches are often either inefficient, not economically viable for large scale application, or can themselves leave behind toxic residues which bio-accumulate through food webs. Low molecular weight organogels have potential application for congealing oil spills,16 in particular in cases where the gelator has the following key properties: (i) simple, low-cost synthesis, (ii) environmental compatibility, (iii) thermoreversibility facilitating oil recovery, and (iv) recyclability & reusability.Interestingly, the use of LMWGs with oil slicks has actually been known since the 1970s, however, these early industrial examples are rarely cited in the more recent, academically-driven literature. For example, in 1971, in situ formation of colloidal gel-forming ureas by reaction of amines and isocyanates was explored as a technology for oil-spill immobilisation, although a practical solution to the problem was not found at this point using this approach.17 Another early example of gelling oils with small organic molecules was demonstrated in the patent of Saito et al. from 1976.18 In the original patent, derivatives of N-acetyl amino acids were ad-mixed with non-polar organic solvents such as kerosene and stirred at elevated temperatures (120 °C). A stable gel was then formed within 2 min as the solution cooled to room temperature. As proof-of-principle for oil-spill remediation, a solution of N-lauroylglutamic acid-α,γ-di-n-octylamide (1 g) dissolved in benzene (5 mL) selectively gelled a heavy oil suspension (25 g) within 20 min in the presence of sea water. The benzene was also entrapped along with a small amount of water. The solidified oil was filtered off through wire gauze in order to separate it from the bulk sea water. It should be noted that in this early work, the requirement of 4 wt% gelator is not ideal, indeed more recent work has highlighted that LMWGs can be effective at concentrations well below 1 wt%.
In 1985, dibenzylidene-D-sorbitol (DBS) and its derivatives were patented as LMWGs to gel crude oil in the presence of seawater.19 To exemplify the method, a slick of kerosene on the surface of sea water was treated by spraying with a mixture of hydrophilic solvent such as N-methyl-2-pyrrolidone (NMP) and hydrophobic solvent such as liquid palm oil containing 5 wt% DBS and poly(oxyethylene) lauryl alcohol ether (1%) for 10 min. The oil phase was selectively gelled by the DBS, leading to recovery of 100% of the kerosene with only 1% water content. In another composition, 6 wt% of DBS derivative 1,3:2,4-di(p-methylbenzylidene)-D-sorbitol enabled 95% recovery of gasoline from sea water with only 0.3% water content. The immobilised oil was scooped off the surface of the seawater using a net and recovered by dilution or distillation. Once again, relatively high loadings of the LMWG were employed, furthermore dispersant solvents such as NMP are not biologically or environmentally desirable. Clearly, however, LMWGs have potential in oil spill decontamination, and could perhaps enhance oil transportation safety.
In more recent, elegant work, Bhattacharya and Krishnan–Ghosh used a simple amino acid amphiphile, N-lauroyl-L-alanine 1 (Fig. 3) to selectively gel aromatic and aliphatic hydrocarbons as well as commercial oils such as kerosene, petrol and paraffin in biphasic oil–water mixtures.20 A requisite amount (typically <1% wt/vol) of the gelator was added to the biphasic oil–water mixture either by dissolving the gelator in the solvent mixture by heating, or by adding it as a solution in ethanol. The oil-phase was selectively gelled leaving the aqueous phase unaffected, with the gels remaining stable for one week. Moreover, the presence of NaCl, CuSO4, KMnO4 and other potentially competitive impurities had no significant effect on gelation, indicative of effective self-assembly.
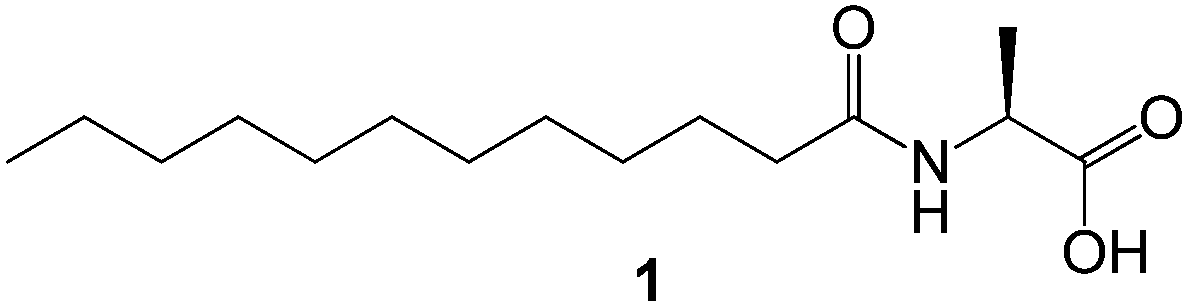 | ||
| Fig. 3 Chemical structure of N-lauroyl-L-alanine 1 used by Bhattacharya and Krishnan–Ghosh for gelation of commercial oils in biphasic mixtures.20 | ||
Dastidar and co-workers reported a series of two-component organic salts that demonstrated selective gelation of oils from oil–water mixtures.21 The presence of two-components introduces additional structural tunability into the system. One of the gelators selectively immobilised petrol, kerosene, cottonseed oil, sunflower oil and coconut oil in biphasic oil–water mixtures. A heat–cool cycle, with or without some methanol added, was employed to achieve gelation. Preferential dissolution of the gelators in the oil-phase mediated the phase-selective gelation. Once again, the presence of inorganic salts did not significantly affect gelation. Building on this concept, Dastidar and co-workers screened a combinatorial library of primary ammonium monocarboxylate salts derived from cinnamic acid derivatives 2 and n-alkyl primary amines 3 (Fig. 4).22 Some of the combinations exhibited selective gelation of petrol, kerosene and diesel from their mixtures with water. This clearly demonstrates the benefit of structural tunability in terms of mediating the solubility/gelation potential, and hence optimising performance.
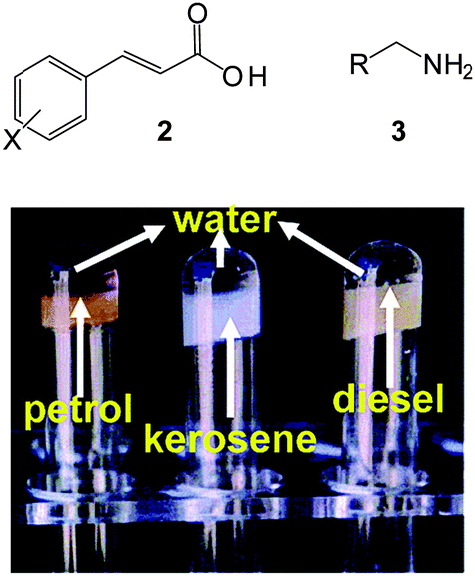 | ||
| Fig. 4 Chemical structures of cinnamic acid derivatives 2 and primary amines 3 and the photo of phase-selective gelation of commercial oils from their mixtures with water. Figure adapted from ref. 22 with permission. | ||
John and co-workers have been pioneers in highlighting the essential requirement of embracing the philosophy of green chemistry in the design and development of self-assembled gel-phase materials – of particular importance for systems intended for environmental remediation.23 The philosophy has 12 key principles, amongst them: the minimization of waste, benign synthetic procedures, and the use of renewable resources as feedstocks.24 Sugar-derived gelators may be constructed from renewable resources and in some cases are ideal candidates for eco-friendly oil-spill remediation – so-called ‘biorefinery-design’.23 As proof of concept, in 2010, John and co-workers reported sugar-based gelators that immobilized diesel, mineral oil, silicone oil, crude oil fractions (alkanes with n > 9 carbon atoms) and mixed hydrocarbon solvents (aromatic and aliphatic) from mixtures with water at room temperature.25 A solution of selected gelators 4 and 5 (Fig. 5) at high concentration in ethanol was added to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of oil and water. The gelator spontaneously migrated into the oil-phase and formed self-supporting gels at room temperature. The purity of the gelator, and the presence of impurities in the water or pH variation did not have any significant effect. It was hypothesized that hydrogen-bonding between LMWGs in the oil-phase underpinned self-assembly.
:1 mixture of oil and water. The gelator spontaneously migrated into the oil-phase and formed self-supporting gels at room temperature. The purity of the gelator, and the presence of impurities in the water or pH variation did not have any significant effect. It was hypothesized that hydrogen-bonding between LMWGs in the oil-phase underpinned self-assembly.
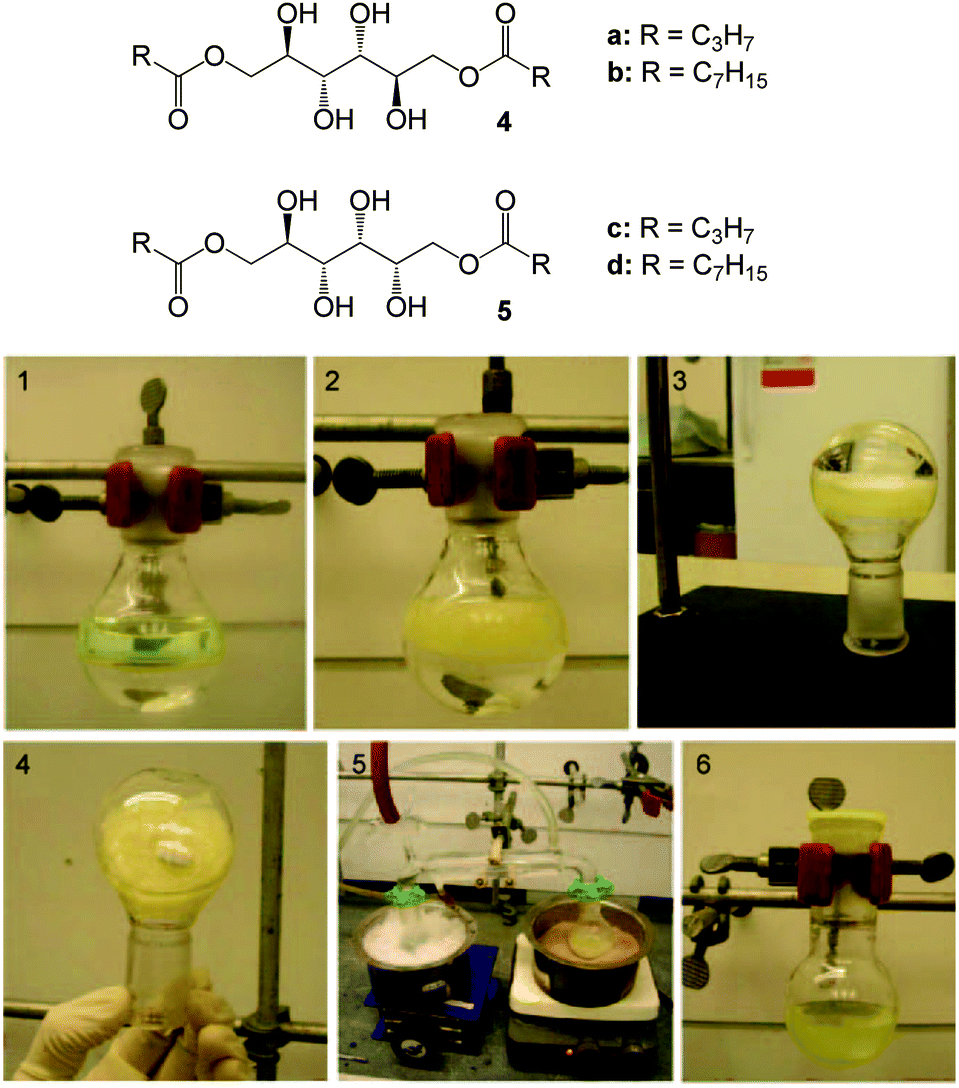 | ||
| Fig. 5 Chemical structures of sugar-based gelators 4 and 5 and images 1–6 showing the immobilisation of bulk diesel from its two-component mixtures with water, and recovery of the oil by distillation. Figure adapted from ref. 25 with permission. | ||
Phase-selective gelation systems, such as those described above, which involve a heat–cool cycle or require addition of co-solvent are not ideal for real-life applications. The heat–cool method is not applicable to oil recovery from open water sources, while the use of co-solvents could lead to further addition of relatively large volumes of undesirable (or even toxic) carrier solvents into the environment. With this in mind, Fang and co-workers reported a family of cholesterol-based gelators 6 (Fig. 6) that exhibited superior phase-selective gelation of xylene and/or kerosene from their mixtures with water at room temperature.26 The gelators demonstrated a remarkable gelation of the oil-phase simply by mixing – neither heating nor addition of a co-solvent was required. Gelator 6a demonstrated selective gelation of xylene at temperatures as low as 0 °C while 6b could only gel kerosene between 25–30 °C. The rheological properties of the gelator/xylene systems exhibited enhanced mechanical strength. Such systems clearly have promising characteristics although they are based on relatively elaborate molecular structures.
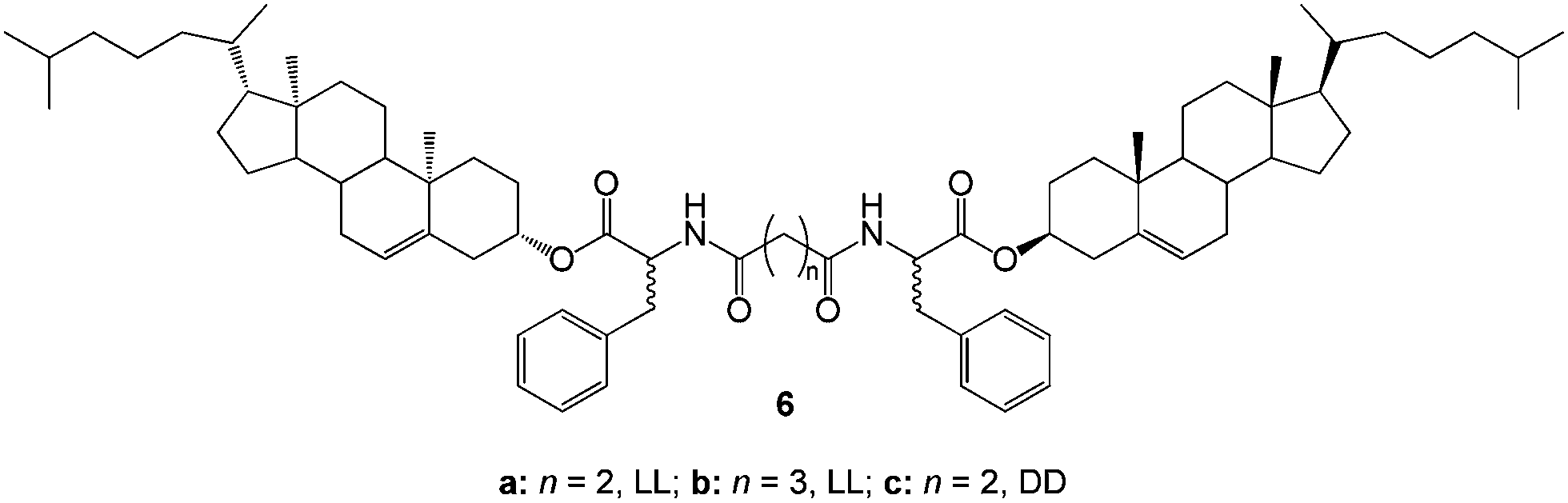 | ||
| Fig. 6 Molecular structures of the diacid amides of dicholesteryl L-phenylalaninate 6 employed by Fang and co-workers.26 | ||
In 2009, Fang and co-workers went on to report the ability of some related cholesteryl derivatives to selectively gel chloroform, tetrachloromethane, toluene or xylene from their mixtures with water at room temperature.27 Once again, neither heating nor co-solvents were required. The gelators exhibited significant resistance to salts. It should be noted that they did not demonstrate gelation of crude oil and it is important to be able to translate gelation events precisely into different solvents. However, the solvents studied here are also a threat to both the environment and human health.
Prathap and Sureshan reported a novel approach using a sugar-based organogelator.28 Gelator 7 (Fig. 7) was initially dissolved in a small volume of commercial petrol or diesel and aerially sprayed onto the oil–water biphasic mixture such that both carrier solvent and oil-slick were immobilised by the gel. The gelator immobilised the oil-layer at 2–4% wt/vol minimum gelation concentration for petrol but a much lower concentration of 0.75% wt/vol for diesel. The gels were strong and could be removed from the water with forceps and withstand stress, with the presence of NaCl and other ions not affecting gelation. Bearing in mind that supramolecular gels are generally weak, this magnitude of mechanical strength is relatively rare and highly desirable.
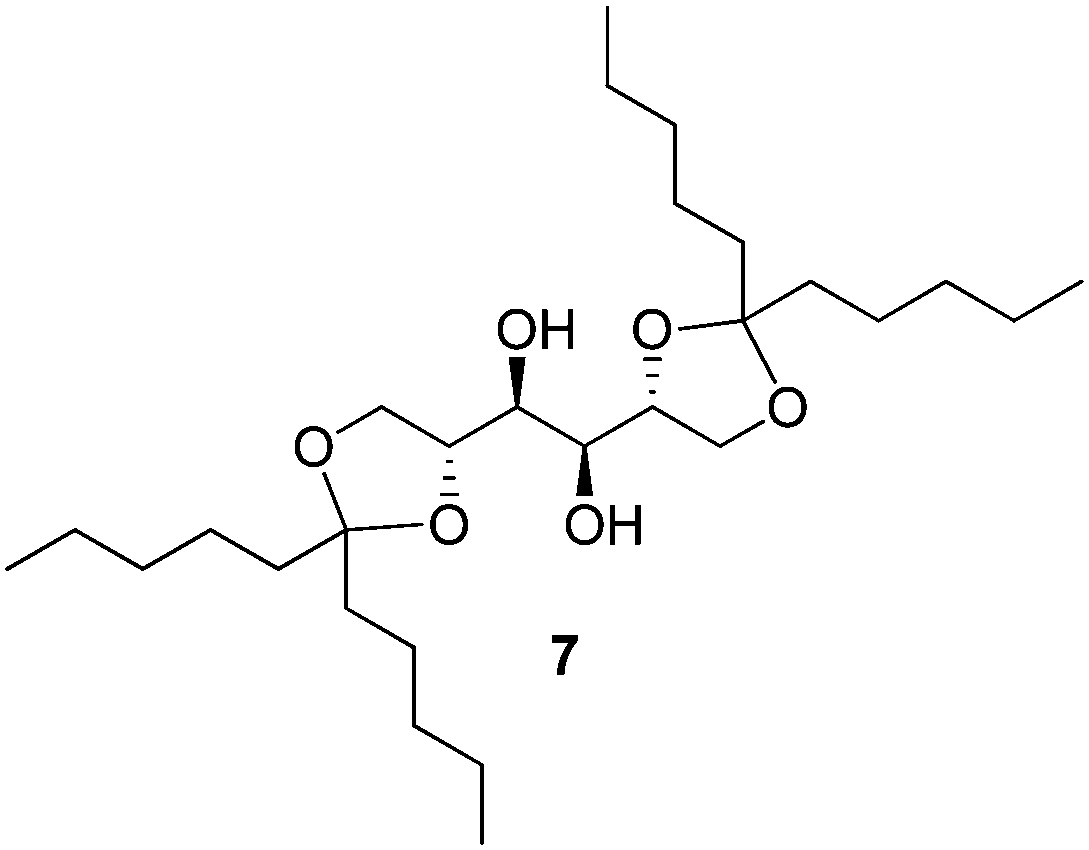 | ||
| Fig. 7 Gelator 7 employed by Prathap and Sureshan for the formation of mechanically robust gels in petrol and diesel. | ||
In 2014, Yan et al. noted that crude oils are complex and contain natural acids and other elements capable of impeding phase-selective gelation in crude oil–water biphasic mixtures.29 The authors employed a two-component gelation system comprising stoichiometric amounts of an n-alkylamine (n ≥ 8) and tetrazolyl derivatives 8–10 (Fig. 8) with hydrogen bonding interactions and the triazole N–H playing pivotal roles in self-assembly. The two-component gels were thermo-reversible, thixotropic and had some self-healing characteristics (Fig. 8). The two-component gelator could selectively immobilise crude oil which contained 1.68% sulfur and 3.12% acid content at room temperature, forming gels which were stable under gravity for more than two weeks.
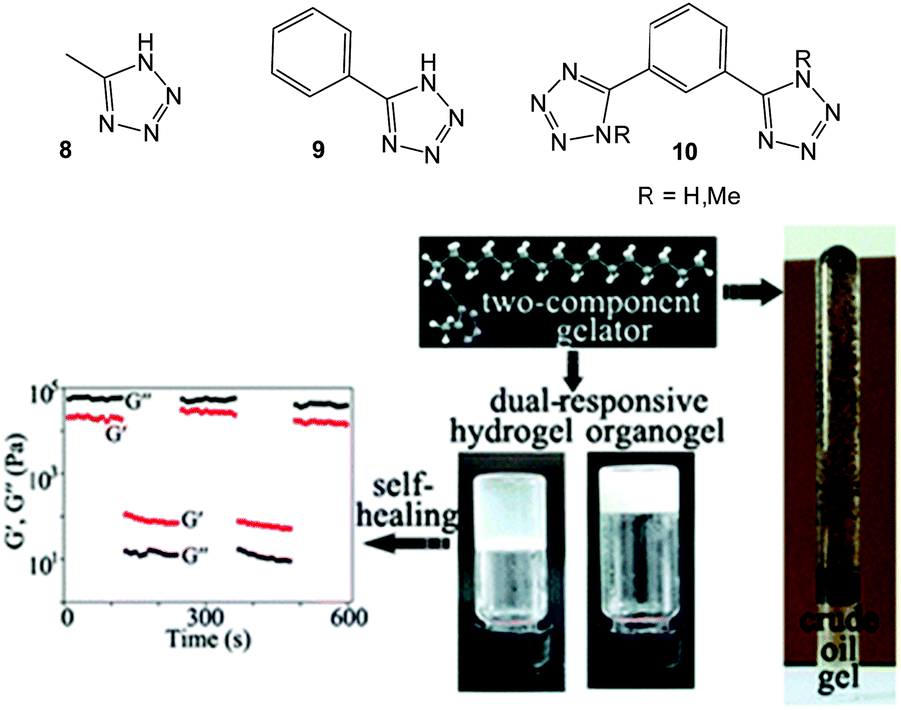 | ||
| Fig. 8 (top) Chemical structures of 8, 9, and 10; (bottom left) a loop test of hydrogel of L-octylamine via continuous step-stress measurement at 1 Hz; (bottom right) photos of dual responsive gels and inverted crude oil gel. Figure adapted from ref. 29 with permission. | ||
Fang and co-workers have recently demonstrated two simple gelators 11a–b (Fig. 9) based on N-acetylglucosamines which can be prepared in one synthetic step, and exhibit efficient and instant gelation of petrol, kerosene, diesel, silicone oil, pump oil and pure organic solvents and which they claim to be eco-friendly.30 Initially, moderately strong gels were formed in petrol and diesel using the traditional heat–cool method, with minimum gelation concentrations (MGCs) ranging from 0.2–3.5% wt/vol. To demonstrate practicality, a concentrated solution of the gelator in THF was added by syringe at the interface of 0.8:2 mixture of diesel/petrol and water at room temperature. Gelation occurred within 10 s, and full strength gels were formed within 45 s. The oils were recovered from the gels by simple distillation and the gelators reused more than three times without losing their ability. Gelation was still observed at 0–5 °C, demonstrating potential suitability for oil spill remediation in geographical regions with low temperatures. However, a simpler delivery mode, avoiding the use of co-solvent would be desirable.
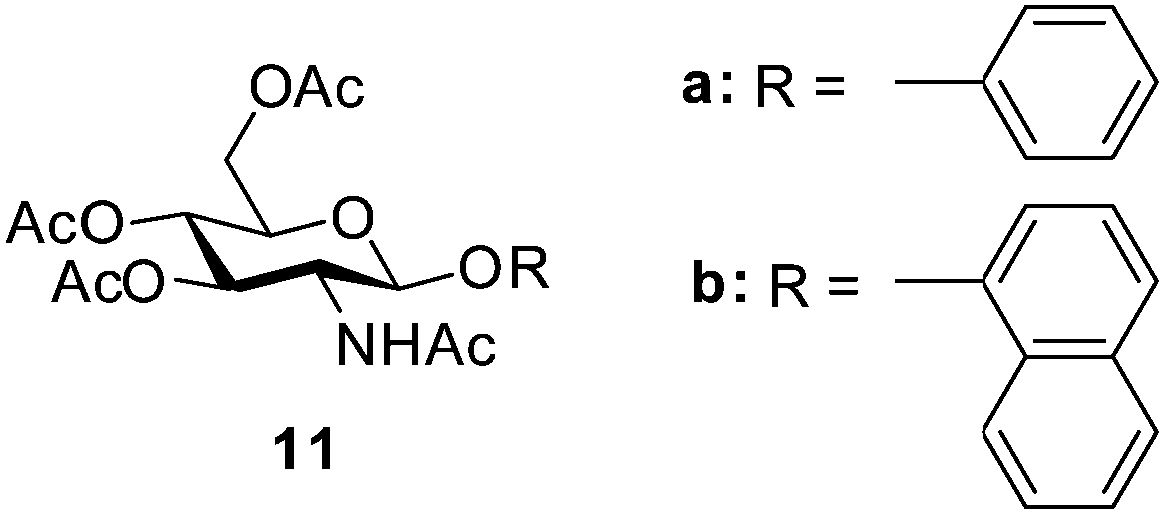 | ||
| Fig. 9 Structures of β-glycosides of N-acetyl glucosamine 11a and b.30 | ||
As explained earlier, 12-hydroxystearic acid (12-HSA) is one of the oldest LMWGs – it is bio-derived and has current applications in the lubrication and food industries.9,31 Its use as potential oil slick gelating agent, along with a wide range of related molecules, such as its primary amide derivative, was patented in 2012 by Viswanatha and Weiss.32 In addition to patenting these molecular structures for the desired application, the patent also includes methods for contacting the LMWG with the oil spillage, including encapsulation of the active agent dissolved in water miscible solvents such as alcohols or acetone, within water-soluble bags which release the agent on contact with the environment requiring remediation. The patent also discusses recovery of the remediated oil. It is noted that many of the gels formed can rapidly recover their rheological performance after exposure to shear forces. These researchers also explored the effect of high amplitude surface waves on the rheological properties of such gels – important for application in marine environments.33
In related academic research, Lee and Rogers demonstrated the use of 12-HSA xerogels to adsorb non-polar solvents and commercial oils from mixtures with water.34 Xerogels were prepared by evaporating acetonitrile from an organogel of 12-HSA, and a known amount was then suspended in the oil phase of the simulated oil spill. After 1 hour, the xerogel increased in weight by ca. 460 wt% in diesel and ca. 580 wt% in gasoline, against ca. 3.9 wt% and ca. 5.2 wt% in distilled water and sea water respectively. The adsorbed oil could be simply recovered and the xerogel re-used. Xerogels prepared by drying solvents other than acetonitrile from 12-HSA organogels were less effective – intriguingly demonstrating that drying from different solvents yields xerogels with different properties and performance, not just simple solids.
Jin and co-workers reported simple monoglyceride gelators for oil uptake, including diesel and kerosene, as well as olive oil and organic solvents.35 Minimum gelation concentrations were often <1% wt/vol and it was demonstrated that fuel oil could be collected by vacuum distillation. Such structures were also of interest because they are easy to synthesise and environmentally benign in their own right.
Qu and co-workers have modified aromatic rings with amino acids to yield gelators capable of phase-selective gelation of aromatic solvents, specifically dimethylbenzene, in the presence of water.36 However, it remains to be demonstrated that this system could work in more real-world conditions typical of spillages. Similar kinds of studies were performed by Feng and co-workers on alkyl-chain-modified phenylalanine derivatives.37
Das and co-workers developed a beta amino acid based gelator which was capable of phase-selective gelation of model oil spills from biphasic mixtures of oil and water.38 Diaz and co-workers applied tetrapeptide gelators modified with a side-chain azobenzene unit to achieve phase-selective gelation of oil–water mixtures at the same low concentration as the minimum gelation concentration in pure oil, although a heat–cool cycle was required for gelation.39 The process could be scaled up, and gasoline or diesel near-quantitatively recovered by distillation. In addition, when applied in an organic medium, these peptide gelators could remove water-soluble dyes such as crystal violet from aqueous solution – a topic discussed in more detail in the following section.
Yadav and co-workers reported enantiomeric gelators based on arabinose and noted that these simple self-assembling systems could gelate crude oil in biphasic mixtures mimetic of marine ocean spillages.40 Interestingly, this was a relatively rare example of crude oil immobilisation – more often diesel, petrol and kerosene are used in such studies (probably because of their easier availability). The authors also demonstrated that these gelators, in benzene, were capable of acting as microreactors for photochemical reactions. This suggests a potential future in which organic solvent waste may be remediated and then the resulting gels directly used as microreactors for chemical processes. This would add value to the chemical production chain by finding new applications for what would otherwise be waste, and potentially enabling a transition from waste to wealth.
It is clear that LMWGs have genuine potential for oil spill remediation. The ideal system, suitable to applied at scale in the real-world, must have:
• cheap, scalable synthesis,
• rapid gelation avoiding co-solvents or heat–cool cycles,
• recoverable oil,
• reusable gelators,
• environmentally-friendly bioderived gelators,
• stable gels down to low temperatures (ca. 0 °C),
• stable gels even in the presence of shear forces.
The examples discussed above meet some of these criteria, but there is still further optimisation to be done. Multi-component methodologies may help formulate systems which possess all of the desired attributes. As one of the most developed areas of applied environmental supramolecular gel chemistry, researchers with systems meeting all of the desired criteria should ideally demonstrate their technology in field tests, in order to progress this technology towards real-world application.
2.2 Supramolecular gels for sequestering dyes from water
Gels based on LMWGs can be easy to make and modify, their high nanoscale surface areas giving potential for direct interaction with solvated dyes, while their reversibility and responsiveness gives potential for recycling and reuse. They are highly solvent compatible, which allows effective contact between the polluted aqueous phase and the self-assembled nanofibres. Furthermore, they can be applied either in the solvated state (as gels) or in the dried state (as xerogels) – both of which leave the inherent gel-phase nanostructuring intact. Gels are also often amphiphilic in nature which can also assist them in forming interactions with dye molecules.42 The optimisation of some of these features is discussed in general terms later in the article.
In one of the earliest reports from 2007, Banerjee and co-workers reported the synthesis of phenylalanine based bola-amphiphilic hydrogelator 12 (Fig. 10) which forms sonication-induced hydrogels in the presence of divalent metals within the physiological pH range (6.5–7.2).43 The dried metallated xerogels demonstrated some uptake of crystal violet (a cationic dye), naphthol blue black (an anionic dye) and pyrene (a non-ionic dye). Ionic dyes were taken up more effectively than non-ionic and the maximum uptake was 84 mg g−1. Although the pH range in which gels formed was somewhat limited, nonetheless this study was a significant pioneering step into the field. Furthermore, pH variation enabled conversion of the gels into a precipitate or a sol (at low and high pH respectively), and was used to demonstrate controlled entrapment and release of a biological molecule, vitamin B12, suggesting further scope for controlled pollutant uptake and release.
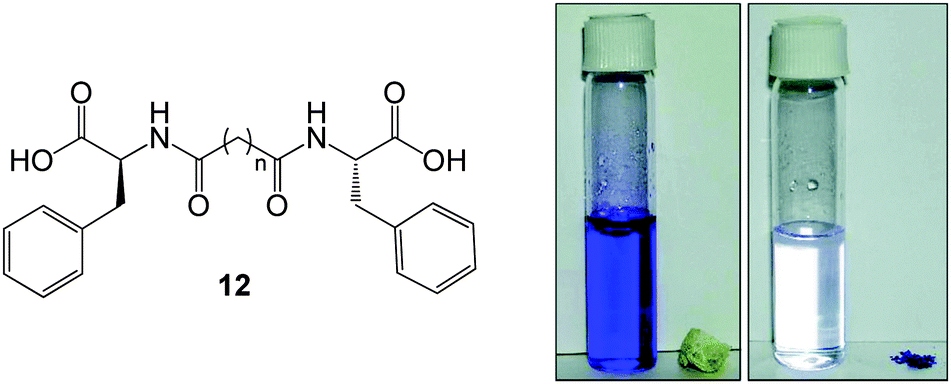 | ||
| Fig. 10 Chemical structure of phenylalanine-based bolaamphiphile 12 which forms hydrogels in the presence of divalent metals, and a photograph of copper(II)-modified xerogelator removing crystal violet from aqueous solution. Figure adapted from ref. 43 with permission. | ||
At about the same time, Kang and co-workers reported a silanated urea-functionalised terpyridine-based metallo-organogelator 13 (Fig. 11) which exhibited strong affinity and high uptake for the various model dyes from aqueous solution, namely basic blue 41, crystal violet and bromocresol green.44 The gelator formed stable organogels in acetonitrile with and without divalent metal ions such as Cu(II) and Zn(II) as a result of urea hydrogen bonding – a process followed by polycondensation of the triethoxysilane to yield additional gel stabilisation. The presence of metal ions bound to the terpyridine group did not have any effect on the morphology of the fibrillar network but did impact on the levels of dye adsorption, decreasing the uptake of cationic dyes, and increasing the uptake of anionic dyes. It was assumed in this case that the xerogel is neutral in the absence of metals but becomes cationic when doped with metal salts, hence electrostatic repulsion between the cationic dyes and the gel limits uptake, while the reverse is the case for the anionic dye. The hydrophobic interaction between the aromatic moieties of the gelator and that of the dyes was reasoned to be the principal driving force for dye adsorption.
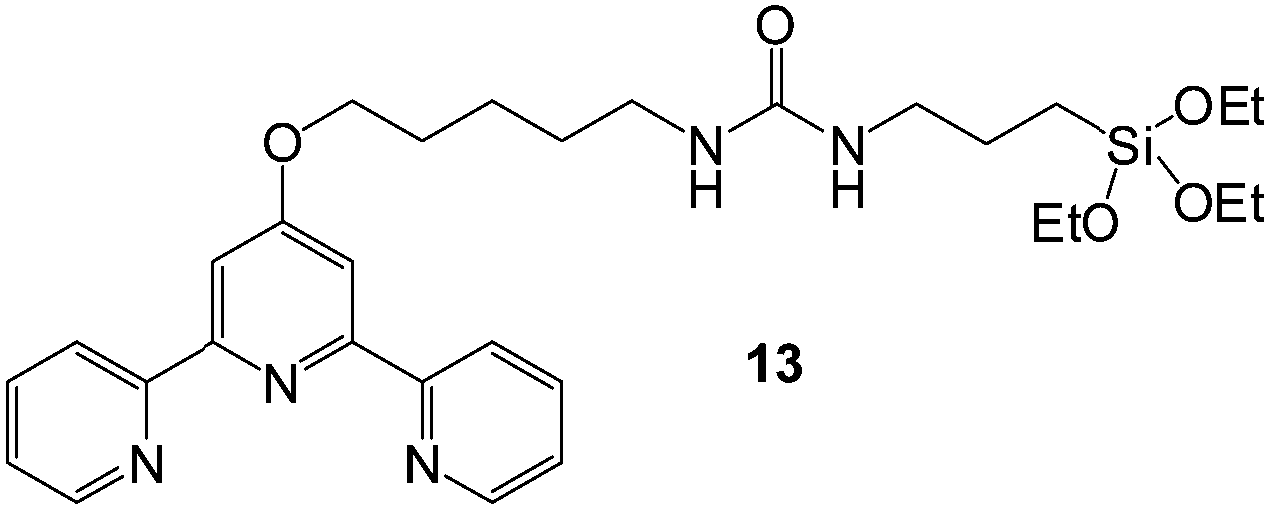 | ||
| Fig. 11 Chemical structure of terpyridine-based ligand 13 which forms organogels in the presence of metals, the xerogels of which are capable of dye adsorption.44 | ||
A family of dipeptide organogelators exemplified by 14–16 (Fig. 12) which selectively gelated aromatic organic solvents in the presence of water was reported by Das and co-workers.45 The authors demonstrated that structural variation controls fibre network morphology and dye uptake ability. To demonstrate the effect of molecular architecture on dye uptake, the xerogels of these dipeptide-based gelators were allowed to interact with aqueous solutions of crystal violet and rhodamine 6G. Although uptake was relatively slow and modest, it is noteworthy that this family of gelators did not require coordination with metal ions in order to be activated for this application. Furthermore, different gelators had different dye adsorption abilities – for example, the presence of a tryptophan moiety in gelators 14 and 15 was attributed to the better uptake of dyes than phenylalanine-based gelator 16.
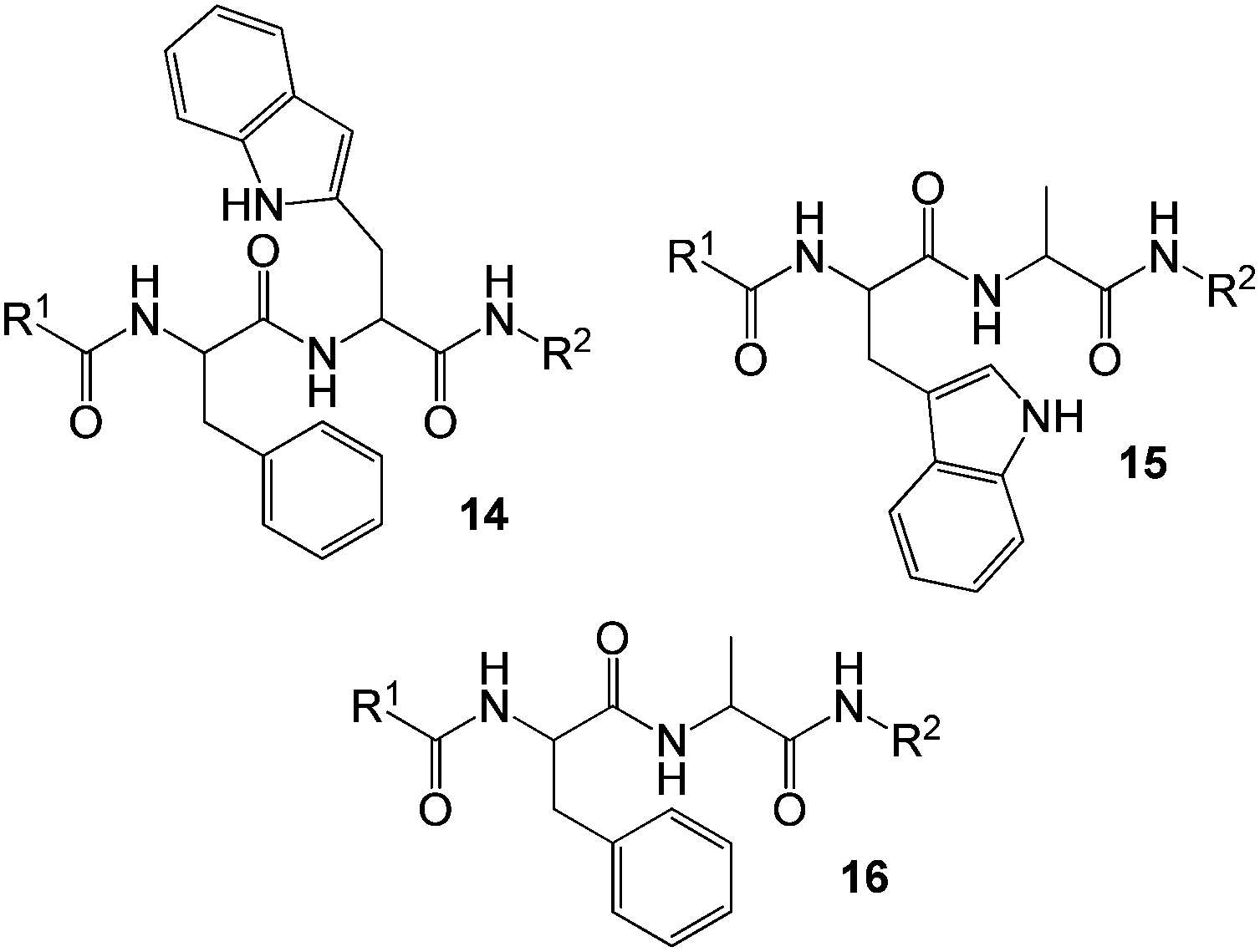 | ||
| Fig. 12 Chemical structures of dipeptide gelators 14–16 reported by Das and co-workers – those including tryptophan groups have the best dye adsorption characteristics.45 | ||
A library of tripeptide-based hydrogelators 17 (Fig. 13) was reported by Banerjee and co-workers, and in this case, the ability of their wet hydrogels to capture and remove rhodamine B, reactive blue 4 and direct red 80 from water was studied.46 Unlike the pH-responsive metallo-hydrogels reported earlier by these authors,43 the tripeptides formed thermoreversible hydrogels at basic pH values (11.5–13.5). Below pH 11.5 the gelators were insoluble. The hydrogels of gelator 17b demonstrated some removal of the dyes from water over extended periods. The spent gelators could be regenerated by dropwise addition of 1 M HCl, with the peptides precipitating out at pH 7.5, leaving the organic dyes in the aqueous medium – the gelator was then filtered-off, washed repeatedly with deionized water and dried. Although somewhat complex, this demonstrates some potential for recyclability and subsequent reuse.
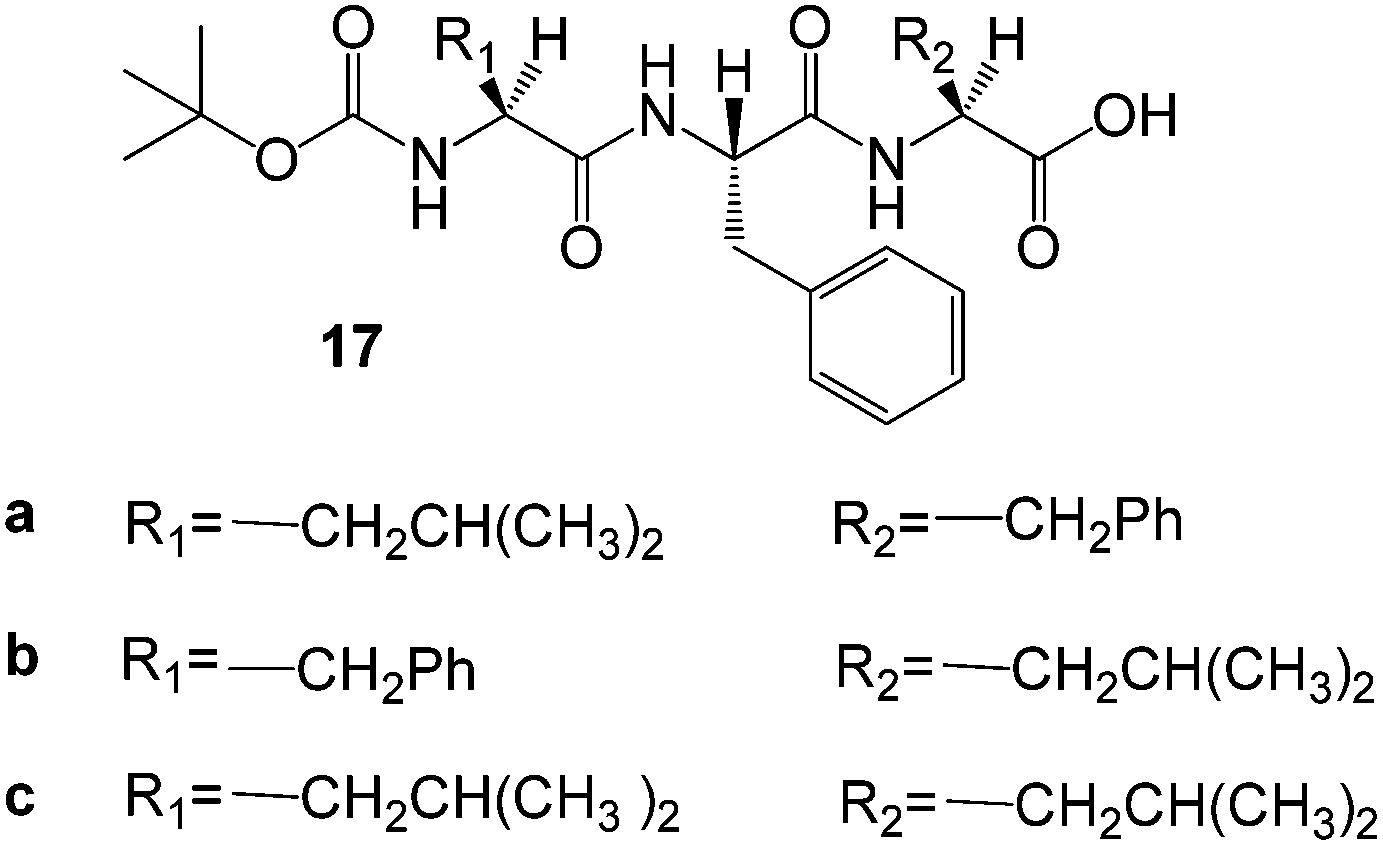 | ||
| Fig. 13 Structures of phenylalanine-based tripeptide gelators 17a–c.46 | ||
In 2010, Das and co-workers investigated the tunability and diverse potential of non-gelator precursor 18 to furnish, through simple synthetic modifications, a hydrogelator 19, an organogelator 20 and an ionogelator 21 (Fig. 14) – a remarkable family of gelators each of which is optimised for different solvent environments by the structural changes.47 It was observed that more than 90% of crystal violet and napththol blue black were adsorbed by the ionogel of 21 in 8 h and 20 h respectively. The organogels of 19 in toluene slowly adsorbed only 50% of these dyes in 6 h and 20 h respectively. It was suggested that the better performance of the ionogel may be a result of the ionic character of the ionic liquid – suggesting that the solvent phase within a gel can play an active role. However, it would be important to avoid solvent leaching from such gels if used in a true environmental setting.
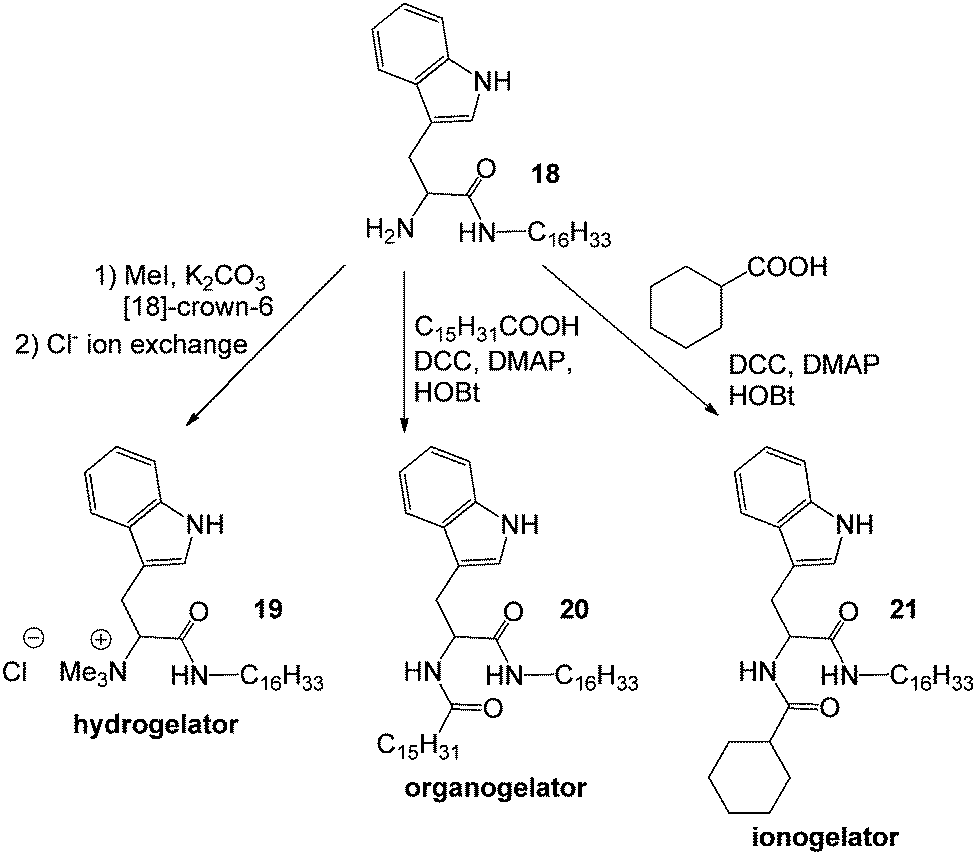 | ||
| Fig. 14 Tuning the amphiphilicity of a non-gelator 18 to a hydrogelator 19, an organogelator 20 and an ionogelator 21 by simple chemical modifications.47 | ||
A family of novel and structurally simple, pH-tunable hydrogelators (e.g., 22 and 23, Fig. 15) having a urea group and aromatic rings functionalised with nitro/nitrile and carboxylic acid functional groups was reported by Hayes, Escuder, Miravet and co-workers.48 Methylene blue was used as a model candidate for dye adsorption. Adsorption experiments were carried out in an unconventional way. A basic solution of gelators was added to an aqueous solutions of the dye, with slow acidification then being used to induce gelation in the presence of the dye. Possibly as a result of the experimental approach, the resulting hydrogels had effectively achieved extremely high adsorption efficiencies, with maximum dye uptakes of 1020 mg g−1 – much better than any of the other examples discussed so far. Intercalation was proposed to be the primary driving force for the adsorption of planar methylene blue into the hydrogel nanofibres, although acid–base interactions were considered to play an active supporting role in some cases. Interestingly, the inclusion of dye into the gel fibres had a significant impact on their length, which decreased from 10 μm to 600 nm when 20% methylene blue was added – suggesting dye intercalation somewhat limits the effectiveness of nanofibre self-assembly. To understand structure–property relationships and optimise dye uptake efficiency, analogues such as 24 were synthesised with bola-amphiphilic structures and an increased number of acid groups (Fig. 15).49 The resulting hydrogels were formed via a pH-switching protocol and had lower minimum gelation concentrations and enhanced mechanical strengths compared with the parent compounds. Interestingly, however, the hydrogels of 24 did not demonstrate dye adsorption capability at gelator concentrations >20 mM in contrast to 22 and 23, although at lower gelator concentrations, the hydrogels of 24 demonstrated more effective molar uptake of the dye due to the increase in the number of available sites for dye intercalation. It was reasoned that at elevated concentrations, the aggregation mode of 24 was significantly different and that the network pore sizes and dye permeability into the gel were adversely affected.
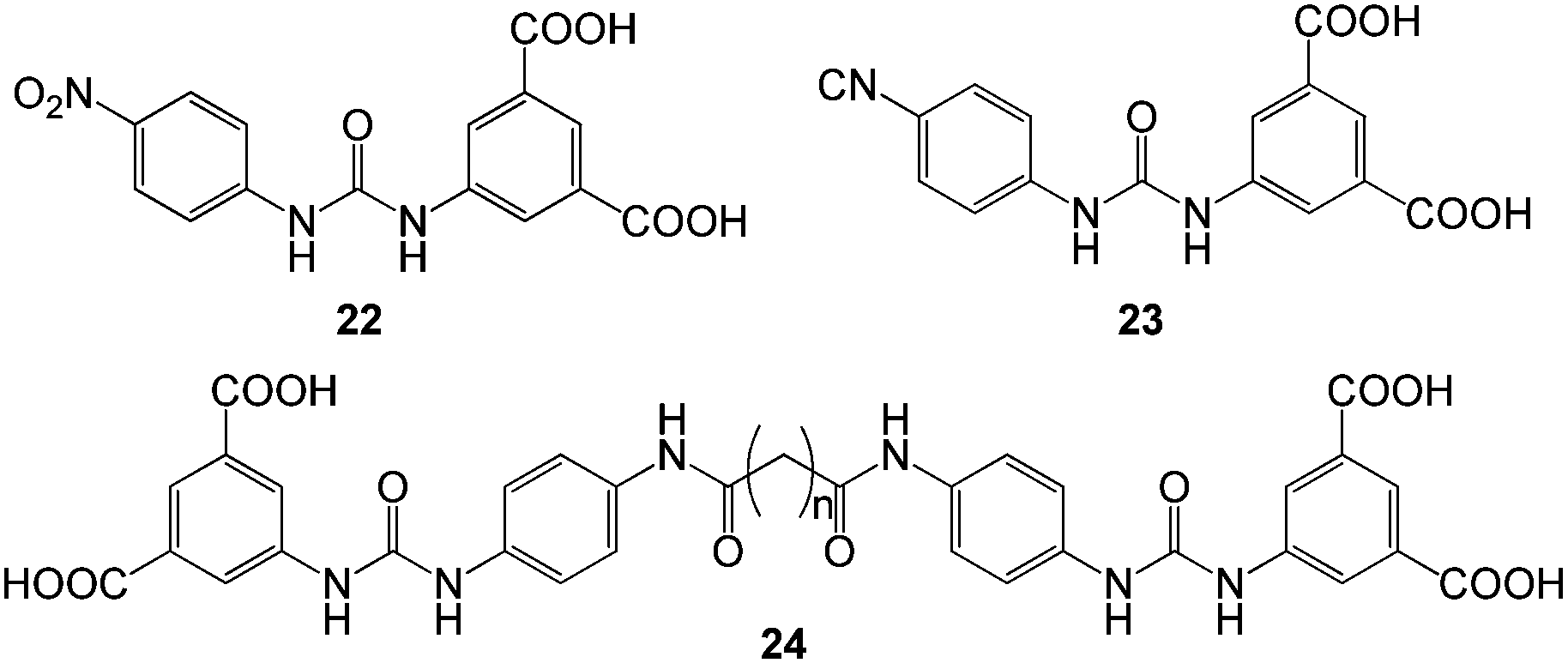 | ||
| Fig. 15 Chemical structure of urea based gelators 22–24 developed by Hayes, Miravet, Escuder and co-workers for dye adsorption.48,49 | ||
Ju and co-workers reported organogels in aromatic solvents based on triterpenoid–tripeptide conjugates.50 They studied the adsorption of dyes into these organogels, using positively-charged rhodamine 6G and acriflavine and negatively charged fluorescein and cresol red, observing adsorption of the cationic dyes. They also noted that cresol red could be deprotonated on interaction with the gel, suggestive of a significant increase in basicity of the gelator as a result of self-assembly – this type of pKa-modification effect on gel assembly is well-precedented from the work of Miravet and co-workers.51 It should be noted, however, that using organogels in aromatic solvents is not optimal for environmental applications.
In 2013, Sukul and Malik reported a thermoreversible two-component hydrogels comprising the purine nucleobase, adenine and tricarboxylic acids,52 with the interactions between adenine and each of the tricarboxylic acids producing hydrogels with different nanoscale morphologies. These hydrogels demonstrated some modest dye adsorption capability when they were contacted with aqueous solutions of methylene blue, crystal violet and Rhodamine 6G for 48 h, but the maximum uptake was only 6–8 mg g−1. Hydrophobic interactions were suggested to be responsible for adsorption.
Samai and Biradha also reported gels which relied on the presence of two-components – in this case ligands combining pyridine and benzimidazole units and metal salts (Cu(II) or Cd(II)).53 They noted that the anion played some role in controlling the gelation event in alcoholic solvents, and used their self-assembled materials for the extraction of methyl orange via uptake into the hydrophobic domains. However, dye uptake levels were, at best, ca. 10 mg g−1. In addition to dye adsorption, the xerogels were demonstrated to adsorb N2 at a high level of 58.95 cm3 g−1. Toxic or environmentally-relevant gases were not investigated, but the potential of xerogels in this different environmental application is clear.
Another effective two-component gelator was reported by Song and co-workers based on chiral amphiphilic lithocholic acid 25 and zwitterionic surfactant dodecyl (dimethyl) amine oxide 26 (Fig. 16).54 These components are individually incapable of achieving gelation, but the electrostatic complex formed on mixing the two components in the appropriate stoichiometric ratio assembles into a pH-responsive gel in water. The dried xerogel demonstrated very good adsorption of naphthol blue black (202 mg g−1 within 15 h). However, the nanofibres of the gel only adsorbed slight amounts of some other dyes such as methyl red, chrome azurol S and Rhodamine 6G. It was suggested that the adsorption of naphthol blue black is primarily driven by hydrophobic interactions between the nanofibres and the dye.
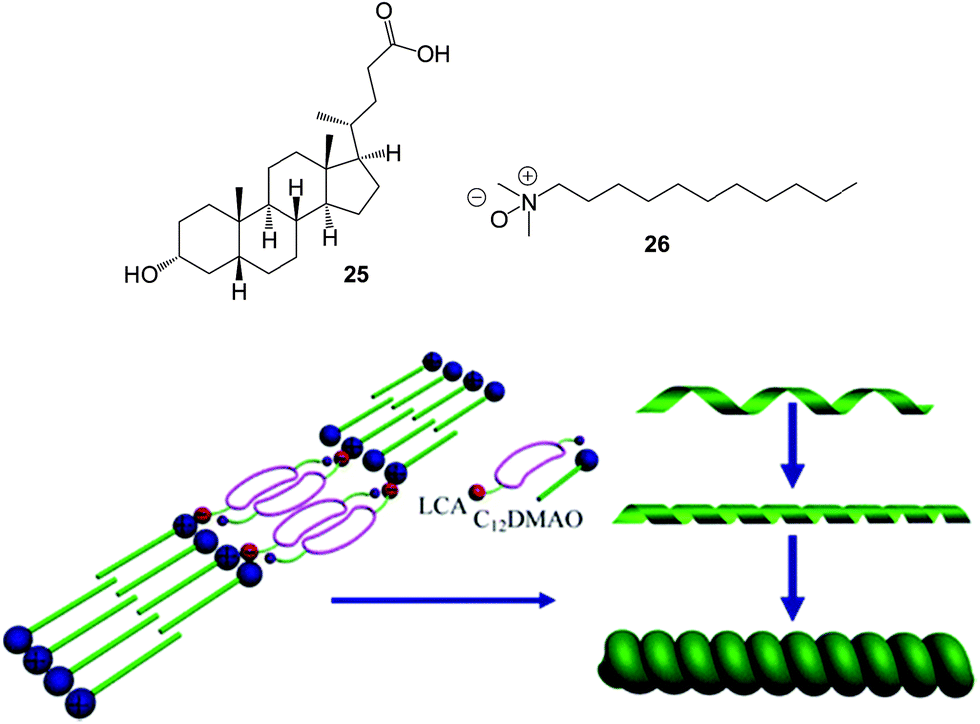 | ||
| Fig. 16 Chemical structures of amphiphilic lithocholic acid 25 and dodecyl (dimethyl) amine oxide 26 and schematic of the formation of the two-component hydrogelator and the subsequent formation of a chiral aggregate for dye extraction. Figure adapted from ref. 54 with permission. | ||
Song and co-workers have also demonstrated that pH-responsive hydrogels and metallo-hydrogels of lithocholate (LC) formed on mixing with different monovalent cations in water, exhibited extremely high adsorption capacities for cationic dyes; methylene blue (870–1100 mg g−1) and Rhodamine 6G (1102–1420 mg g−1) with very rapid uptake (20 min).55 These adsorption levels demonstrate the true potential of gel-type nanomaterials, which in this case are effectively achieving stoichiometric levels of dye uptake. Such adsorption levels are significantly better than what can be achieved by many conventional adsorbents.
Metallogels have also been reported by Dey and co-workers which, in xerogel form, were demonstrated to extract dyes such as methyl orange and rhodamine B from water.56
In 2013, a pH-responsive phenylalanine-modified C2-symmetric cyclohexane-based hydrogelator was reported by Feng and co-workers.57 The hydrogels were prepared by pH-switching with slow addition of acid leading to protonation of the carboxylic acids, and hence gelation. The solvated hydrogels were capable of very rapid adsorption of methylene blue within 2–3 min when mixed with aqueous solutions of the dye, making this one of the most kinetically efficient gels, as many systems take 20–40 h. Subsequent removal of the gel by filtration yielded clean water. Using the Langmuir isotherm model, the maximum amount of dye adsorbed was estimated as 47 mg g−1. As such, although uptake was very fast, only moderate loading levels were achieved. To demonstrate recyclability, the gelator was redissolved in basic water followed by acidification, and the dye recovered by repeated washing with chloroform. Using this simple approach, the hydrogel demonstrated higher recyclability (90%) than the conventional adsorbent – activated carbon (73.5%). However, repeated washing with halogenated solvents is not an environmentally benign protocol.
It is noteworthy that many of the systems employed as solvated hydrogels to remove dyes from water are either metal-containing gels, or are pH-dependent (normally because of the presence of a carboxylic acid unit which requires protonation for gelation). It is also noteworthy that some of the syntheses require multiple steps, and are not easily amenable to scale-up. With these problems in mind, we considered it important to develop pH-tolerant, easy-to-make hydrogelators. As a scaffold, we used the well-known and easy-to-make 1,3:2,4-dibenzylidenesorbitol (DBS) framework which already has extensive industrial applications,10b and modified it with acylhydrazide functional groups to give 27 (Fig. 17).58 Acylhydrazides have similar polarity to the carboxylic acids, which are often required to enable hydrogelation, but they do not have the same pH dependence. As such we reasoned this approach may provide gels which were more pH tolerant in a very simple two-step synthetic process. Indeed, DBS-CONHNH2 forms gels using a simple heat–cool cycle at environmentally-relevant pH values from ca. 2–12.
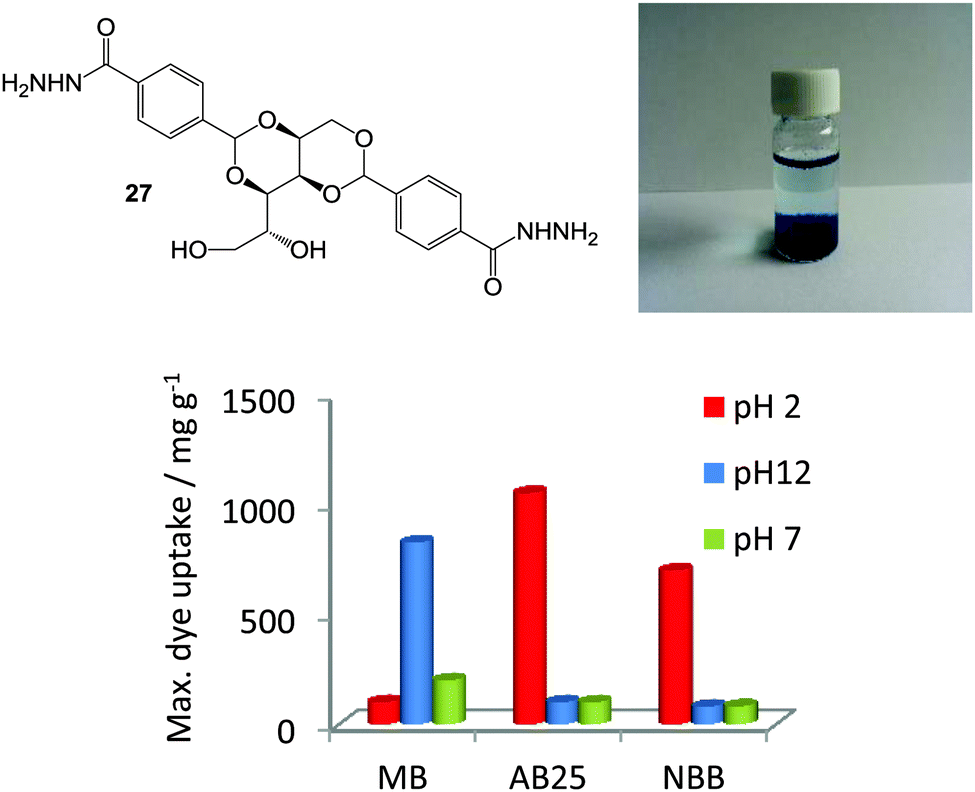 | ||
| Fig. 17 Chemical structure of DBS acylhydrazide (DBS-CONHNH2, 27), photograph of gel extracting methylene blue from basic water and the maximum dye adsorption onto 27 supramolecular gel under various conditions of pH. Figure adapted from ref. 58 with permission. | ||
Hydrogels of 27 exhibited very high levels of dye uptake (700–1050 mg g−1),58 with changes in the ambient pH controlling the amount of dye adsorbed in each case (Fig. 17). Interestingly, this hydrogel exhibited an ambidextrous ability to extract different types of dye under different pH conditions. Cationic methylene blue was efficiently adsorbed from basic pH conditions while anionic dyes, acid blue 25 and naphthol blue black were better adsorbed from solutions with acidic pH values. The hydrogel could also simultaneously adsorb a mixture of anionic dyes from acidic solution – reflective of real-world conditions where mixtures of dyes may be present in waste streams. Furthermore, the relative stability of the gel to pH enables its recyclability, as dyes could be removed from the gel simply by washing it with solutions of different pH values.
Recently, Zhang, Song and co-workers reported a simple, versatile organogelator based on an amide-functionalised monoacetal derivative of D-gluconic acid.59 They employed this system for a variety of different purposes, and amongst their studies, demonstrated that the xerogel could extract crystal violet from water at levels of ca. 28 mg g−1. They also noted that a tetrahydrofuran solution of the compound injected into the interface between pump oil and water could immobilise the pump oil. Once again, this demonstrates the potential of this approach, although some of the solvents used were not environmentally friendly.
In 2015, Yu and co-workers reported glucose-derived gelators which removed dyes such as crystal violet or rhodamine B from contaminated water,60 with the gelator either being used in the form of the xerogel, or as an organogel in benzylalcohol. It was noted that there was, in any case, some partitioning of dyes into benzylalcohol, but the gelator significantly enhanced dye extraction. It was suggested that dye extraction was facilitated by interactions with the hydrophobic chains responsible for self-assembly, although dye uptake remained relatively low at ca. 25 mg g−1. Traces of benzyl alcohol also leached into the purified water. When xerogels were used for dye extraction, some of these problems were avoided, and furthermore the xerogel could be recycled by washing with methanol. These gelators also removed toxic solvents such as aniline and nitrobenzene from contaminated water via formation of a gel in the organic phase on shaking.
A recent report from Karan and Bhattacharjee once again turned to metal-functionalised gels to achieve dye uptake. They employed Cd(II) and Zn(II) complexes of carboxymethyl-(3,5-di-tertbutyl-2-hydroxybenzyl)amino acetic acid.61 They demonstrated that selective uptake of cationic dyes such as methylene blue and crystal violet could be achieved from mixtures with anionic dyes such as methyl orange or fluorescein. The best uptake levels of 56 mg g−1 for crystal violet were relatively modest, but a key advantage of this system was that the gels had high mechanical strength, and exhibited self-healing properties.
Yu and co-workers recently reported supramolecular gels using surfactants based on imidazolium salts.64 These gels formed in binary DMSO/H2O solvents (a limitation) and achieved efficient and selective dye extraction from water. This gel had a plate-like morphology, less commonly seen in supramolecular gels,65 accompanied with excellent mechanical strength. The gel showed particular selectivity for anionic dyes such as eosin Y as a result of the cationic nature of the imidazolium group, and it was proposed that the gelator-dye complex aggregated and precipitated. The kinetics of dye uptake were very rapid, although the gel samples were relatively small. The gels showed excellent tolerance to high levels of salt and could be reused for at least 25 cycles if the gel surface was refreshed each time with deionized water. It should be noted that in this study, the gel loading was very high at 20 wt%, and the dye loadings of each addition were relatively low – factors that probably assist the recyclability observed, as does the fact that the dyes are only adsorbed onto the surface of the gel. Nonetheless, this represents an interesting, different mechanism of dye extraction.
In an early and elegant study, Yang and co-workers developed Fmoc-derived naphthalene-modified peptides (28) to extract methyl violet from aqueous solution (Fig. 18).67 They formed self-assembled gels in water containing 5–20% of DMSO and demonstrated that the gels were stable to relatively high pH values. In a key step they mixed these gels with an agarose polymer gel (PG) to help provide mechanical robustness and stability. This allowed these researchers to use these materials for dye extraction in both standing and stirring modes – for most supramolecular gels, stirring would not be possible without mechanical destruction of the self-assembled gel. The presence of agarose in the hybrid gel slightly slowed down the kinetics of dye adsorption (Fig. 18), but the mechanical benefits more than compensated for this. Fluorescence spectroscopy indicated direct interactions between the adsorbed dye and the self-assembled gel nanofibres, and control experiments showed agarose alone was incapable of dye adsorption. As such, this study demonstrated the way in which hybrid hydrogel materials can harness the inherent advantages of each individual hydrogel network – in this case dye-adsorbing LMWGs combined with a more robust PG.
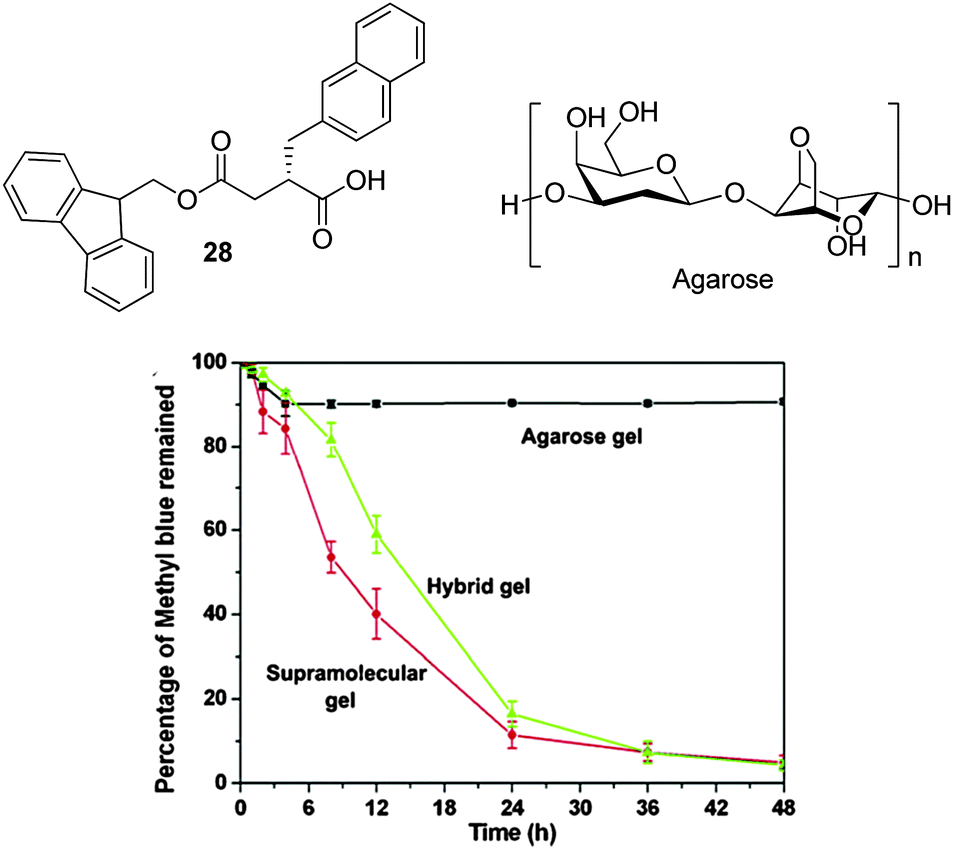 | ||
| Fig. 18 Low molecular weight gelator 28 and polymer gelator agarose, combined to yield a hybrid gel system for dye adsorption. Graph showing the kinetics of methylene blue adsorption by the individual gels and the hybrid system. Figure adapted from ref. 67 with permission. | ||
Yang and co-workers have stabilised DBS-derivative LMWGs in composite form by mixing with halloysite nanotubes loaded with Fe3O4 magnetic nanoparticles.68 These systems were mixed in alcoholic solvents to enable DBS assembly. Significant reinforcement of the rheological properties was observed, as well as the introduction of magnetic properties from the nanoparticles. Some ability to remove dyes, including congo red, methyl orange and malachite green from aqueous solutions was reported – although degrees of uptake were low (<10 mg g−1). Nonetheless, this study again demonstrated how hybrid materials can improve the physical characteristics of gels.
An alternative strategy to stabilise self-assembled gels is post-modification by covalent capture.66 In this approach, self-assembly is used as the initial step, allowing facile formation of the nanostructure, then in a subsequent step, the resulting nanostructures are ‘captured’ via covalent linking reactions on their surfaces. Zhang, Su and co-workers assembled supramolecular nanotubes from gelator 29 which have pyridyl groups on their surfaces and then crosslinked the dried nanotubes by reacting the nucleophilic pyridine groups with 1,4-bis(bromomethyl)benzene in acetonitrile (Fig. 19).69 Thermo-gravimetric analysis indicated that the modified nanotubes were stable up to about 310 °C in dried form. Scanning electron microscopy (SEM) demonstrated that the nanotubes remained intact during the post-modification step, but that there was some aggregation – possibly as a consequence of crosslinking between tubes. The uptake of various dyes was tested using these modified nanotubes, and rapid uptake with excellent loading levels up to 1290 mg g−1 was reported. In particular, anionic dyes were taken up very effectively, presumably as a consequence of direct interactions with the positive charges generated on the nanotube surfaces in the covalent capture step. The dyes could be released by soaking the nanotubes in saturated NaCl solution, and the regenerated materials could be reused for at least five cycles. It was argued that the crosslinked robust nature of these nanomaterials enhances their stability, robustness and potential for reuse.
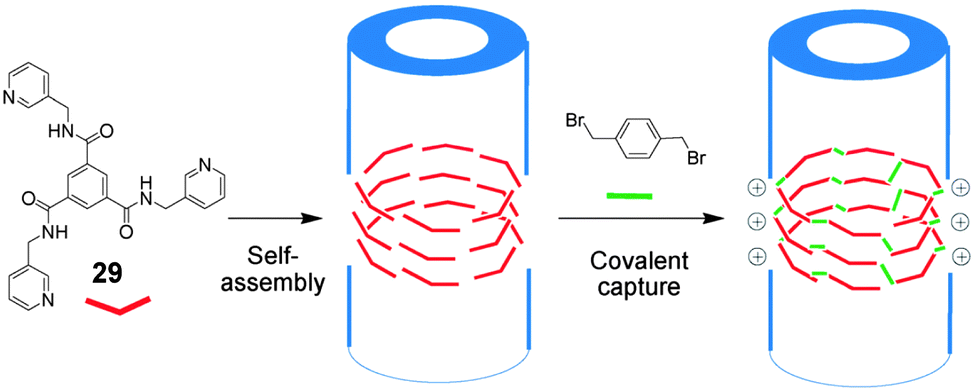 | ||
| Fig. 19 Self-assembling compound 29 and its covalent capture through surface crosslinking with 1,4-bis(bromomethyl)benzene. Figure adapted from ref. 69 with permission. | ||
2.3 Supramolecular gels for heavy metal uptake from water
In recent times, the use of metals to either trigger the gelation of LMWGs or tune the properties of the resulting gel has become a fast-growing area, with metallogels earning themselves a special name among the various classes of supramolecular gels.70 The high-tech applications of these materials have been reviewed by Tam and Yam,71 and are not discussed in further detail here, and there are many examples of gels acting as sensors for specific metal ions. Of relevance to this review, however, metallogelation has recently been extended to target the development of innovative water purification technologies with a view to removing specific metal ions from water sources. Key examples of this are explored in more detail here. Heavy metal ions are a particular problem in water supplies as a result of their ability to bind to biomolecules and modify/prevent their normal modes of action, leading to significant toxicity;72 as such, the focus here is on the binding and removal of these heavy metals. Many of these systems operate in a different manner to the pre-formed gels used for dye extraction in the previous section, with the gelation event only being triggered in the presence of metal ions, hence leading to effective separation of the heavy metal from the liquid like phase and into the nanoscale solid-like gel network.In 2012, Zhang and co-workers reported a family of hydrogelators 30a–e (Fig. 20) all of which were screened for gelation, both individually and in the presence of Pb2+ in water.73 Only 30b underwent efficient hydrogelation both in the presence and absence of Pb2+. It was argued that Pb2+ may cross-link the gelator via carboxylate group binding. Interestingly, selectivity was observed for Pb2+ amongst many other divalent metals, and in addition, the residual concentration of Pb2+ in the sample with 1:0.5 stoichiometry was as low as 76 μM. It was therefore suggested that this material might provide a basis for capturing and removing lead from contaminated water. The metallo-hydrogel demonstrated responsiveness to EDTA which could remove the metal ion via competitive binding, and potentially provide a simple mechanism for recycling and reuse. The gel also exhibited pH-switching and disintegrated to a sol under acidic conditions, with protonation of the carboxylic acid group breaking down the metal complex. Interestingly, the same researchers also reported that gels based on lead coordination could have additional environmental applications in terms of their ability to extract dyes, specifically methyl orange, from water.74
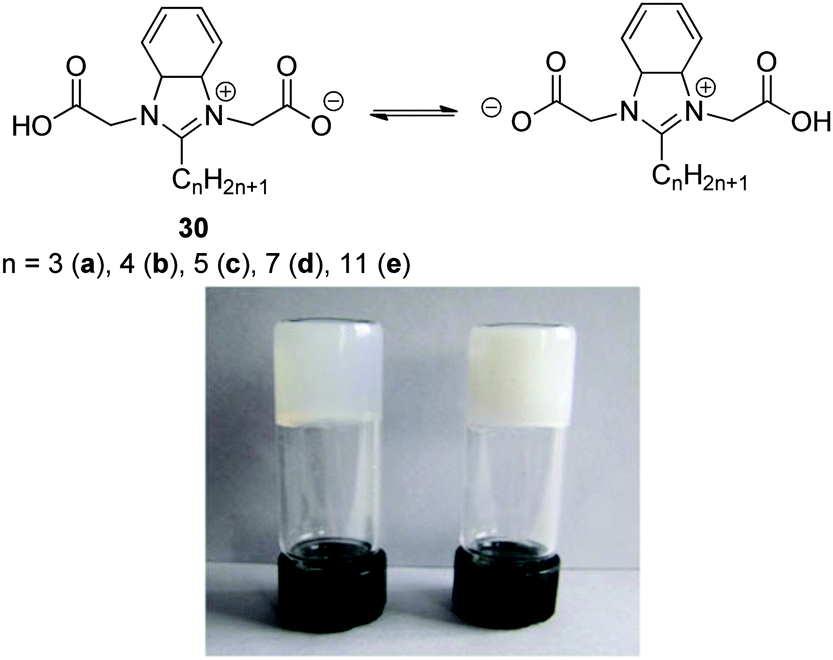 | ||
| Fig. 20 Chemical structures of ligand 30 and photograph of hydrogels of 30b in ratios with Pb2+ of 1:0.3 (left) and 1:1 (right). Figure adapted from ref. 73 with permission. | ||
At about the same time Schneider and co-workers designed a metal-binding hairpin peptide 31 (VKVKVKV-CGPKEC-VKVKVKV-NH2) which was capable of triggered hydrogel formation on complexation with monomethyl arsenous acid (MMA), ZnCl2, CdCl2, HgCl2 or Pb(NO3)2 (Fig. 21)75 The addition of stoichiometric amounts of metal ion to the peptide solution led to metal coordination, which yielded an amphiphilic β-hairpin that then self-assembled into rigid self-supporting hydrogels. Self-assembly is therefore predicated on the folding of the peptide triggered by the chelation of heavy metal ion to the two free cysteinyl thiols flanking the β-turn. Circular dichroism and mass spectrometry were used to confirm 1:1 metal–peptide stoichiometry, and TEM imaging showed that the hydrogels formed in the presence of MMA and Zn2+ are composed of elongated, and twisted fibrils with high order laminates. This is a very elegant approach to metal-triggered self-assembly, but given the laborious processes and synthetic costs of this peptide, it is important to reiterate that for sustainable water purification, low-cost materials are required.
 | ||
| Fig. 21 Image of 1 wt% peptide VKVKVKV-CGPKEC-VKVKVKV-NH2 (31) bulk samples in pH 7.4 buffer at 25 °C in the presence and absence of 1.0 equivalent of MMA, ZnCl2, CdCl2, HgCl2 and Pb(NO3)2. Figure reproduced from ref. 75 with permission. | ||
McNeil and co-workers have used mercury(II) to trigger the organogelation of a library of quinoxalinone-based molecules 32–35 (Fig. 22).76 Complexes of the ligands with Hg2+ form stable gels in organic solvents following a sonication and heat–cool cycle. Instantaneous gelation by simple mixing of aqueous solutions of Hg2+ with methanolic solutions of the ligands was also observed, while Co2+, Ni2+, Cd2+, Ba2+, Cu2+, Zn2+, and Ag+ did not induce gelation of 32. The selectivity of gelation between 32 and Hg2+ was attributed to the linear geometry of the resulting complex 36 (Fig. 22), which unlike other metal complexes, is able to support further self-assembly. To demonstrate potential for real-life application, water samples (river, tap and bottled) deliberately contaminated with Hg2+ were examined. Compound 32 was able to reduce 3800 ppm Hg2+ to 289 ppm by in situ gelation of contaminated water. As such this system can both detect Hg2+ contaminated water via spontaneous gelation and also mitigate against the pollution. Furthermore, compound 35 formed stable gels which contacted with each of the contaminated waters, even in the presence of chloride anions, which compete for binding to Hg(II) and limit the performance of compound 32.
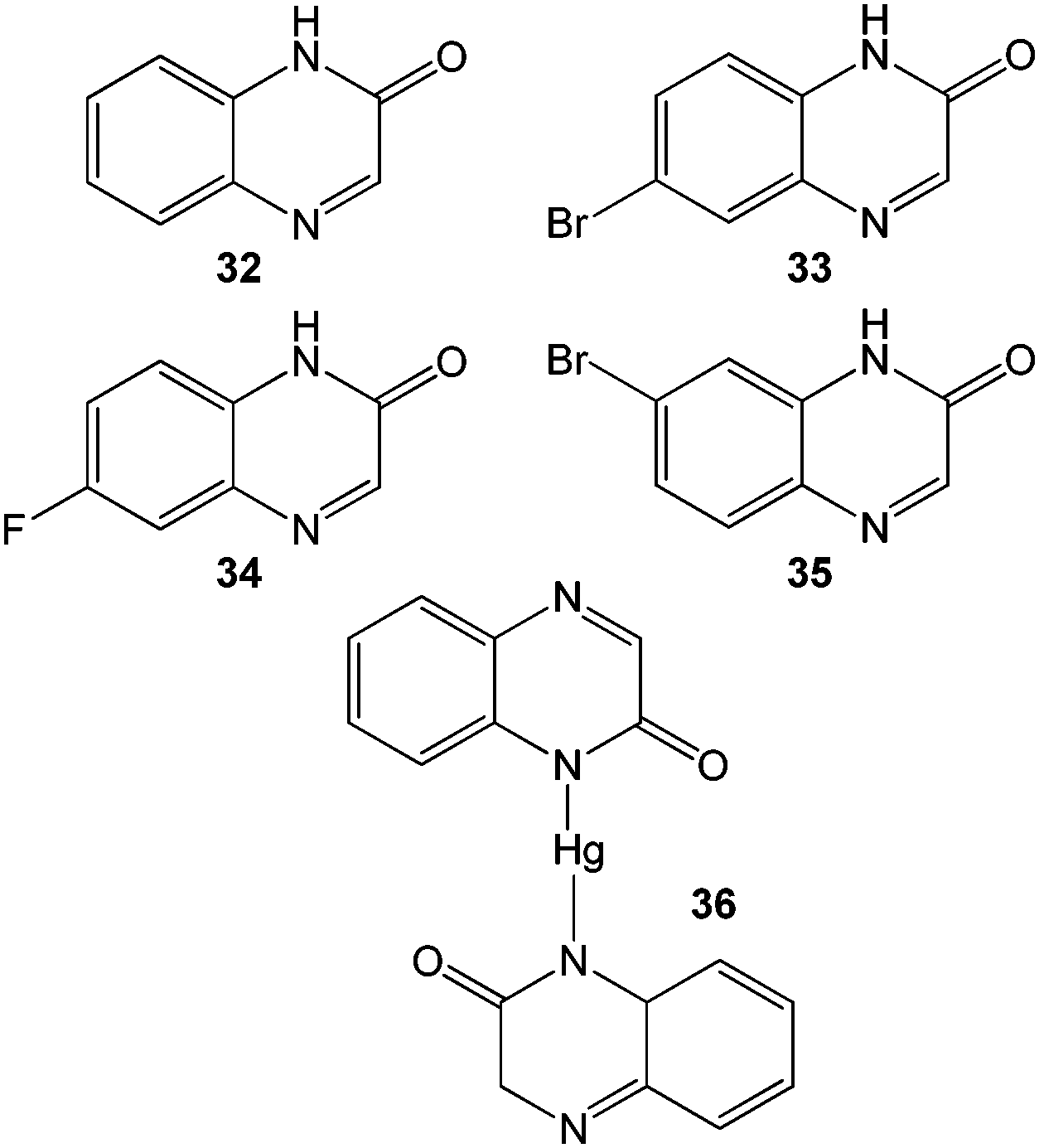 | ||
| Fig. 22 Chemical structures of quinoxalinone compounds 32–35 and hydrogelator complex 36 formed by complexation of compound 32 with Hg(II).76 | ||
In 2014, Mondal and co-workers demonstrated a dual-purpose gel-based approach for removing both toxic metals and dyes.77 In this case, when aqueous solutions of lead acetate and cadmium nitrate were separately added to a DMF solution of pyridine-pyrazole-based tris-amide gelator 37 (Fig. 23), gelation was observed on standing for few minutes. Adding more than 1.5 equivalents of metal ions to the gelator solution led to precipitation – demonstrating the importance of the stoichiometric complex in underpinning gel formation. However, it should be noted that DMF is not biocompatible. In another article, the authors also reported the ability of these gelators to respond to the presence of silver salts in water.78 Coordination of the metal ions with the pyridine-pyrazole was responsible for metallogel formation. Furthermore, the ligand transformed Ag+ ions into Ag nanoparticles. The xerogels incorporating Pb, Cd and Ag also demonstrated modest adsorption of dyes (ca. 14 mg g−1 of methyl orange) and nitrogen gas adsorption levels of 29 cm3 g−1. A similar report of a gel which formed in the presence of silver cations, and then adsorbed methyl orange dye has also been made by other researchers.79
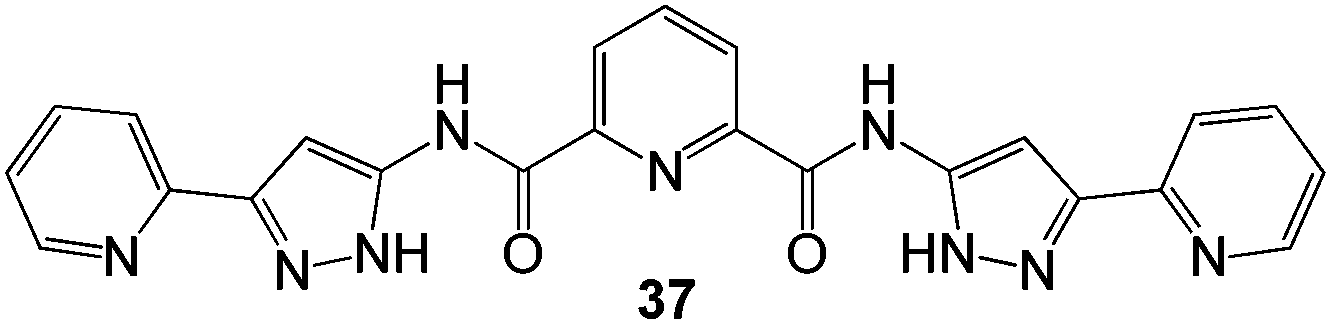 | ||
| Fig. 23 Chemical structure of pyridine-pyrazole based amide gelator 37.77 | ||
In a different approach, rather than using the metal to actively trigger a gelation event, we used a pre-formed gel for the removal of heavy metal ions from water.80 In particular, we reported that precious heavy metals such as gold, silver, palladium and platinum could be selectively recovered from aqueous metal ion mixtures typical of mine tailings or waste electronic and electrical equipment (WEEE). Furthermore, these metals ions were reduced to nanoparticulate form in situ and immobilized by the hydrogel nanofibres (Fig. 24). This tripartite function was achieved using hydrogels of DBS-CONHNH227. The ability of this gel to sequester precious metal ions from water was initially demonstrated by testing uptake of Au3+ and Ag+ from aqueous solutions, with analysis of the supernatant by ICP-MS and/or UV spectroscopy showing total uptake of the metals. The maximum uptake capacity of the gel was estimated to be as high as 2000 mg g−1 for Au and 900 mg g−1 for Ag – above these levels the gel collapsed. These loading levels are higher than those of many adsorbents reported in the literature and competitive with the very best. Interestingly, the gel changed colour to ruby red or yellow as Au3+ or Ag+ diffused in – colours characteristic of the nanoparticles (NPs) of these metals, suggesting that the hydrazide functional groups on the gel nanofibres reduce the metal ions and then act to cap metal nanoparticles (NPs) within the network – a rare example of a gel in which nanoparticle formation occurs spontaneously, without external reductant. It was further demonstrated that the gel could find application as a sensor for Au3+ and Ag+ ions in aqueous waste as it responded colorimetrically to ultra-trace (<5 nM) concentrations in water. As a consequence of their high reduction potentials, precious metals (Au, Ag, Pd and Pt) were selectively adsorbed from mixtures with earth-abundant metals (Zn, Cu, Fe and Ni) with lower reduction potentials (Fig. 24).
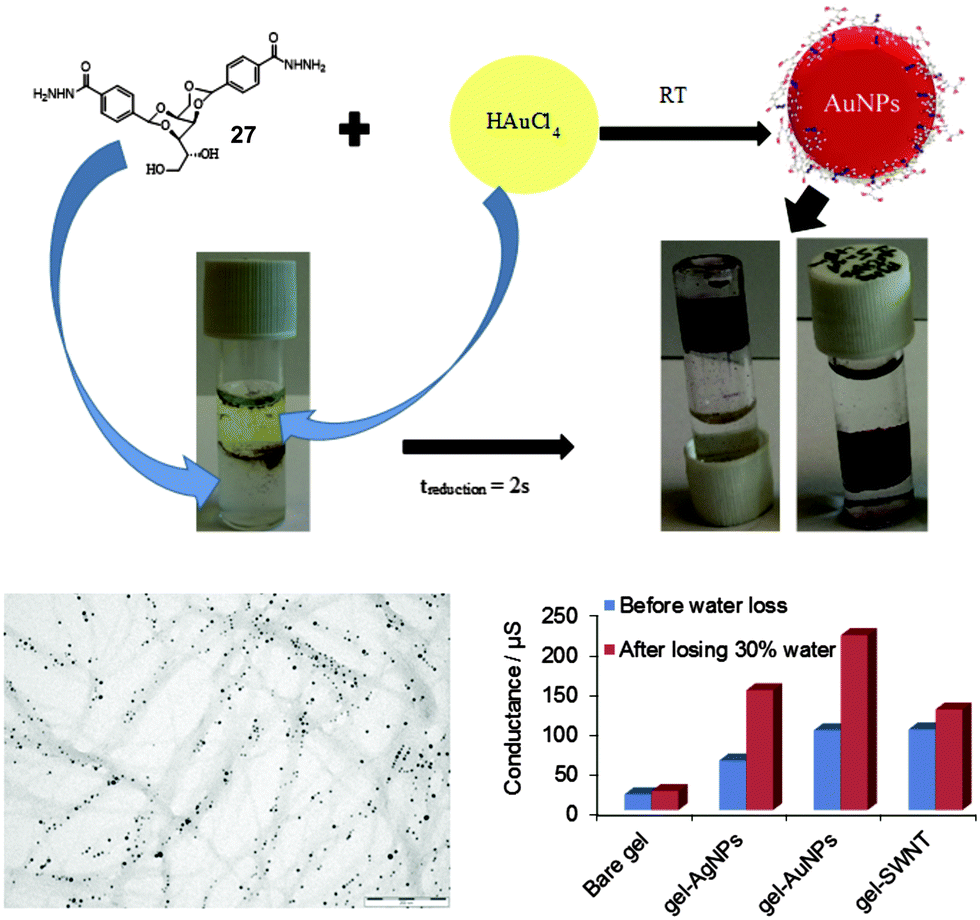 | ||
| Fig. 24 Scheme showing uptake of noble metal and in situ reduction to nanoparticles by hydrogel nanofibres of DBS-CONHNH2 (27), TEM image showing nanoparticles aligned along gel nanofibres and enhanced conductances of the nanoparticle-embedded gels compared with bare gel, and gels containing single walled nanotubes. Figure adapted from ref. 80 with permission. | ||
We reinforced the resulting nanometallogels by mixing with an agarose polymer gel to generate robust hybrid gels. We then demonstrated these materials had potential high-tech applications in their own right as a consequence of their inherent electrocatalytic and conductivity properties – ‘from waste to wealth’. Gel conductance was compared against a negative control (bare gel) and a positive control (single walled carbon nanotube-hydrogel hybrid). The hydrogels with embedded Au nanoparticles had higher conductances than both negative and positive controls as a result of organisation of metal nanoparticles on the fibrillar nano-scaffold. Furthermore, the AuNP–hydrogel hybrid was used to modify a carbon electrode and demonstrated to be capable of electrocatalysis, with a large reductive current being attributed to Au-catalysed O2 reduction. Therefore, the metal NPs embedded in a soft matter system can enable communication with electrodes. As such, these soft conductive nanomaterials could find application as bridges between the soft matter world of biology and the hard matter world of electronics, with the solid-like network and the liquid-like phase within the gel assisting the compatibility in each case.
In addition to extracting metal ions from aqueous waste there is also developing interest in the remediation of waste which contains metal nanoparticles. Although nanotechnology has enormous potential benefits, it is important to reflect that releasing nanoparticulate metals into the environment may endanger human health and the ecological sphere.81 For example, it has been reported that AgNPs have the tendency to undergo biochemical transformations in model systems relevant to human body.82 It would therefore be expedient for heavy metal nanoparticles to be immobilized or sequestered from the natural environment.
With this goal in mind, Barthélémy and co-workers demonstrated for the first the time the use of LMWGs to remove nanoparticle waste from aqueous solutions.83 Hydrogels of a fluorinated glycosyl-nucleoside amphiphile 38 (Fig. 25) were interacted separately with aqueous solutions containing quantum dots (QDs), gold nanoparticles (AuNPs) and titanium dioxide nanoparticles (TiO2-NPs). Fluorescence and UV-Vis spectroscopy were used to monitor the uptake of NPs. The optical signatures of the NPs were not observed in the supernatant liquids after 48 h in contact with the hydrogel. The mechanism by which this system works was not discussed in detail, although it was suggested that interactions of the NPs with the gel nanofibres may lead to a more stable state in which, as a result, the metal NPs become attached to the self-assembled fibres. Some support for this hypothesis was provided by TEM.
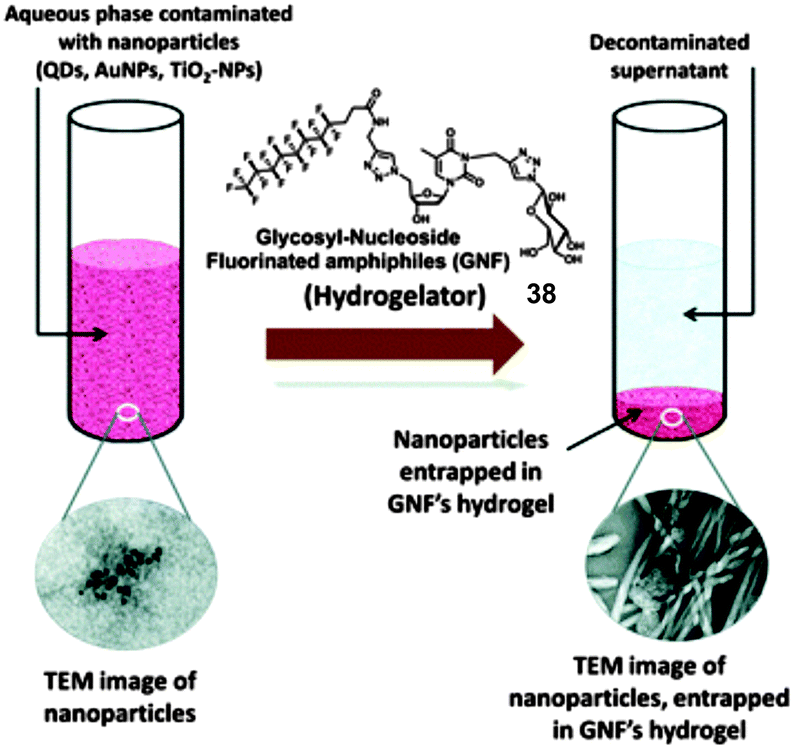 | ||
| Fig. 25 Schematic illustration of the removal of NPs from aqueous samples using fluorinated glycoside-nucleoside amphiphile hydrogelator 38 and TEM images of nanoparticles and GNF nanofibres with entrapped NPs. Figure adapted from ref. 83 with permission. | ||
2.4 Supramolecular gels for sensing and remediating anions in water
Anions are ubiquitous throughout biological systems and are present in natural water in appreciable levels. However, their direct impacts on the aquatic ecosystem and human health can be problematic. For example, pollutant anions such as phosphate and nitrate (which causes eutrophication of rivers), nitrite, (carcinogenesis, methaemoglobinaemia), fluoride (urolithiasis and fluorosis), perchlorate (explosive) and pertechnetate (radioactive) are of significant concern.84 Industrial, mining, refinery, and chemical storage sites pose risks of exposing anion pollutants to the environment. For example, chlorite, bromate and fluoride can be discharged from water treatment plants, perchlorate from military industries, pertechnetate from nuclear fuel reprocessing, cyanide from mining, and arsenate and selenite from irrigation. Elegant reviews on the supramolecular chemistry of anions have been published elsewhere85 and will not be expanded on here. Small molecules containing urea and amide functional groups can form gels as a consequence of their inherent self-complementary hydrogen-bond donor and acceptor ability.86 They also have excellent anion binding characteristics as a consequence of these hydrogen bond donor groups.87 As such, attempt to gelate such molecules in the presence of anions can lead to disruption of the gel network, as the anion competes for interaction with the hydrogen bond donors. This has effects on nano/microscale and also macroscopic properties of the gel. In innovative work, Steed and co-workers were the first to exploit this elegant mechanism to generate gels which demonstrate an on-off gelation response in the presence of anions.88 In general, these hydrogen bonded gels form in organic solvents, and subsequent studies have tested the response to various anions, also in organic solvents – as such this work is less relevant to the remediation of pollution from aqueous systems and is not discussed in further detail here. Further, for hydrogen bonding systems such as these, as a general rule, the most charge dense and basic anions, such as fluoride and phosphate are bound most strongly, and have the largest gel disruption effects. For this review, we are therefore particularly interested in gels which interact with anions under environmentally relevant conditions, and particularly interesting are systems which subvert these natural charge density based selectivity profiles, and can as a result exhibit responses to unusual anions.In early work from 2009, Jiang and co-workers reported a supramolecular hydrogel formed by Ag(I)–glutathione (GSH) coordination polymers (Fig. 26).89 The hydrogels demonstrated selective ‘naked-eye’ recognition of I−via its ability to trigger a gel–sol transition – but other more charge dense anions such as F−, Cl−, Br− and H2PO4− which commonly induce responses, could not initiate such a phase transition. As such, the gel exhibits an interesting, and unconventional selectivity preference. It was hypothesised that I− acts as a depolymerizing agent for Ag(I)–GSH, as a consequence of its relatively strong interaction with Ag+ and the subsequent formation of an insoluble AgI by-product. An assay was developed using this approach which allowed the quantitation of iodide in model waste water down to millimolar concentrations. Clearly this is an effective way of detecting iodide anions, but may be less useful for remediation, as the presence of iodide causes loss of the gel materials properties.
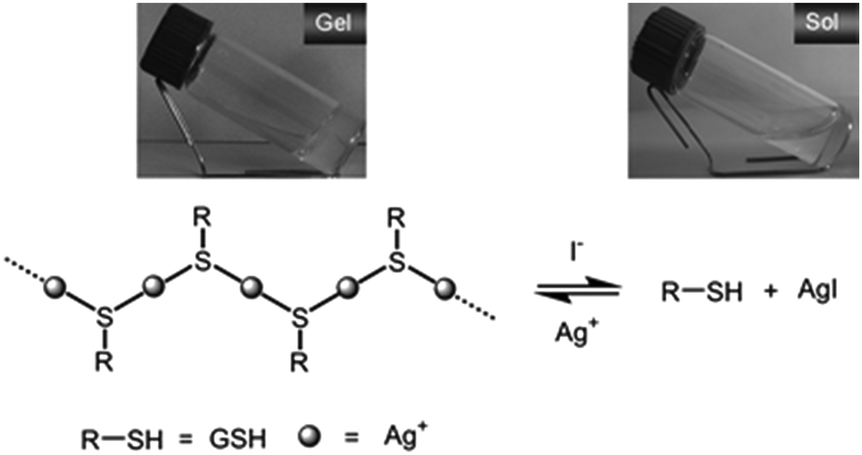 | ||
| Fig. 26 Illustration of reversible gel–sol state transition of supramolecular hydrogel of Ag(I)–GSH coordination polymers (where R–SH is glutathione, GSH) triggered by alternately adding I− into the hydrogel, or additional Ag(I) into the resulting sol. Figure reproduced from ref. 89 with permission. | ||
Yi and co-workers demonstrated colorimetric sensing of the environmentally-sensitive and toxic nitrite anion (NO2−) using a two-component gelator comprising naphthalimide undecanoic acid 39 and diaminoanthraquinone (DAQ, 40) in organic solvents (Fig. 27).90 The two-component gel selectively sensed NO2− when the anion was applied in aqueous solution, leading to collapse of the gel into a suspension. The colour of the gel also faded with significant quenching of fluorescent emission. Mass spectrometry confirmed that the observed changes were due to the formation of benzotriazole 41 resulting from reaction between diaminoanthraquinone 40 and the nitrite – as such this reaction-based sensing mechanism gives rise to the unusual anion selectivity.
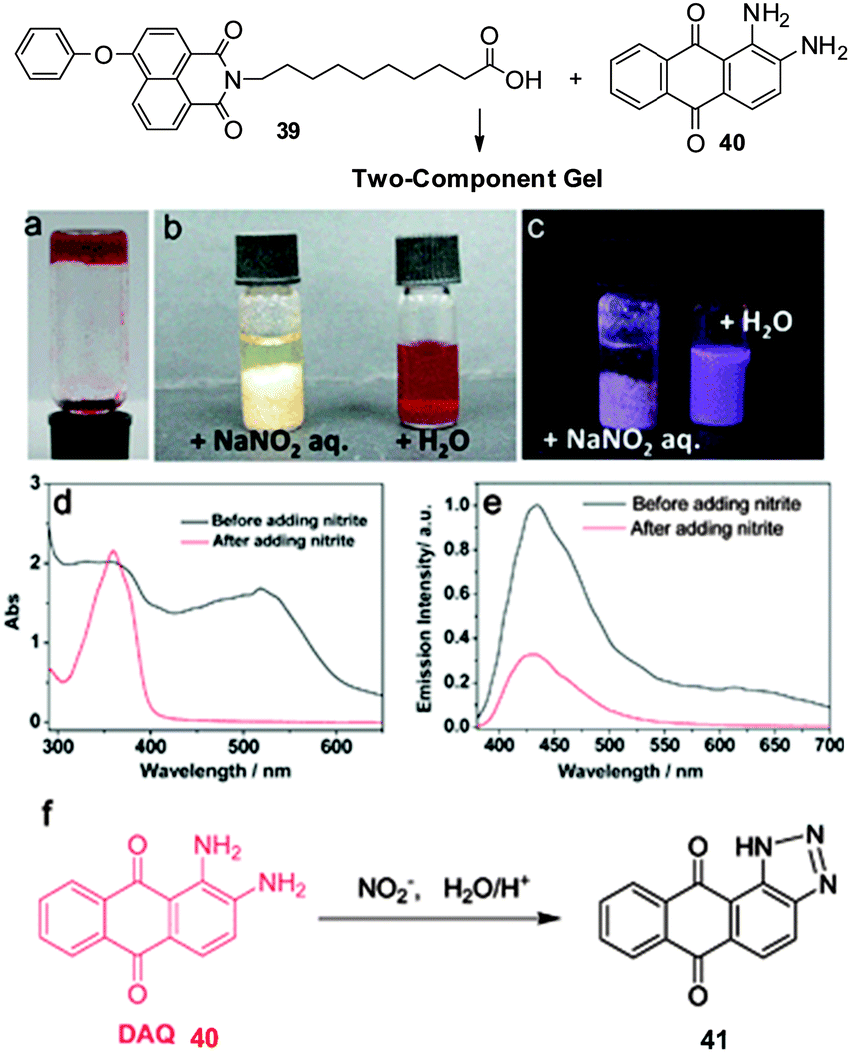 | ||
| Fig. 27 Chemical structure of two-component gel formed by naphthalimide undecanoic acid 39 and diaminoanthraquinone 40. Photo of gel 39 + 40 (25 mg mL−1 in acetonitrile) (a), and the gel with added NaNO2 solution (left) and pure water (right) (b) and with 365 nm light irradiation (c). (d) Absorption and (e) emission spectra of 39 + 40 before and after treatment with NO2−. (f) Reaction scheme between 40 and NO2−. Figure adapted from ref. 90. | ||
Gels capable of optically sensing the environmentally problematic cyanide anion have also been reported by Sun and co-workers.91 They used copper and zinc metallogels based on an L-glutamic acid Schiff base derivative which fluorescently respond to CN−. Specifically, the mixed-metal gel incorporating both copper and zinc responded to the presence of cyanide via a change in its optical properties as a result of competitive cyanide coordination to the copper yielding Cu(CN4)2−. Conversely, S2− bound the zinc selectively within this mixed-metal gel to form ZnS, and hence gave a different response. Other anions such as ClO4− and AcO− did not change the gel.
In terms of multiple anion sensing, Lin, Zhang and co-workers presented an elegant example of a sensor array created from metallogels which was able to detect a range of different and unusual anions (Fig. 28).92 These authors used a single gelator 42 which formed organogels, and modified it by coordination to a range of different metals (Cu2+, Fe3+, Hg2+, Cr3+ and Zn2+). The resulting metallogels exhibited different fluorescent responses to key anions: CN−, SCN−, S2− and I− in water, with detection limits ranging from 0.1 to 10 μM. Quantification and sensing of these anions is of environmental relevance. The mechanism of sensing involves competition between the organogelator and the anion for binding to the metal, with the resulting complexes having different fluorescence. Other anions such as F−, Cl−, Br−, AcO− N3−, H2PO4−, and ClO4− did not induce a response. These metallogels were formulated into an array, which was able to discriminate between target anions as a result of differential responses. The use of a gel facilitated the creation of the array in a potentially practical and easily handled format – avoiding the need to have multiple sensor solutions present.
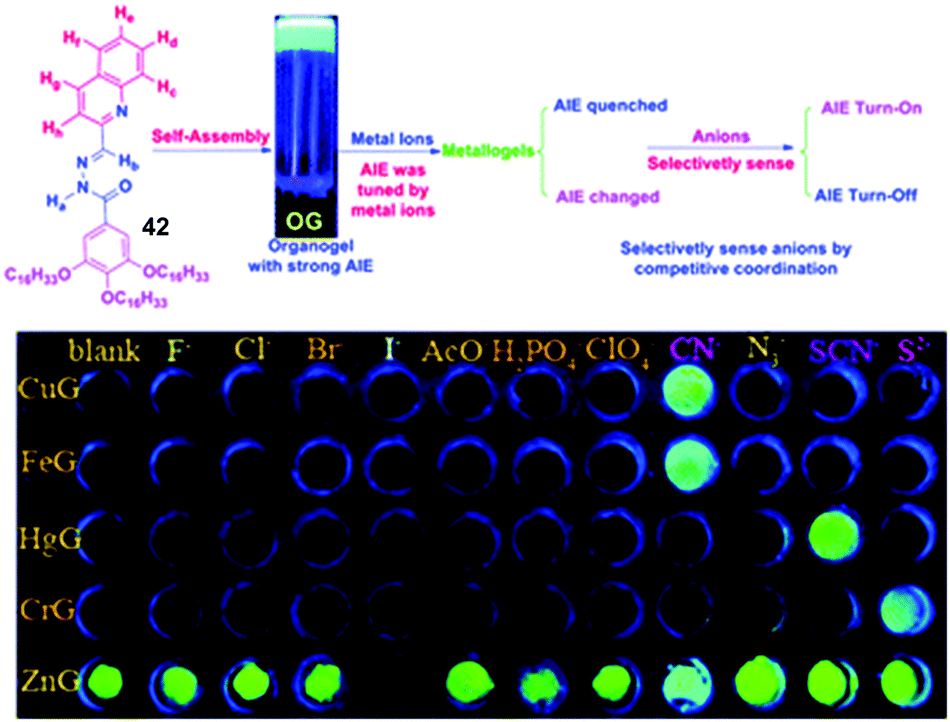 | ||
| Fig. 28 Gelator 42 self-assembles and has its assembly induced emission (AIE) tuned by the presence of metal ions. The emission of different metallogels is then tuned by the presence of certain anions, allowing the sensor to detect CN−, SCN− and S2−via an ‘array mode’ of sensing. Figure reproduced from ref. 92 with permission. | ||
However, the examples discussed so far are primarily sensors, which rely on the detection of environmentally relevant anions by a gel which changes its properties, or is even broken down, when exposed to anions – although ideal for sensing, this is not useful for active environmental remediation. Alternatively, the anion-induced transition of a potential LMWG from sol to gel state is possible and, importantly, this approach may have applications in environmental clean-up.
In early, elegant work from 2008, Mocerino, Ogden and co-workers demonstrated the hydrogelation of a proline-functionalised calix[4]arene 43 in the presence of specific anions – NO3− or Br− (Fig. 29).93 The gelation efficacy of 43 with these anions was attributed to the Hofmeister series – with kosmotropic anions (strongly hydrated, e.g. SO42−) promoting solution formation while more chaotropic anions (less hydrated, e.g. NO3−, Br−) stimulated self-assembly and the formation of stable hydrogels as the overall solubility of the complex was lower and it could more easily aggregate into solid-like fibres. However, if the anions were too chaotropic (e.g. I− or ClO4−), initial gelation was then followed by crystallisation. Inspired by this work, other researchers have also gone on to explore effects on gelation induced by the Hofmeister effect.94 None of these have focussed on using this concept in water purification technology, however, it may be plausible to extend the scope to water clean-up with the goal of designing gelators with the optimal solubilities to remove anions with specific polarities.
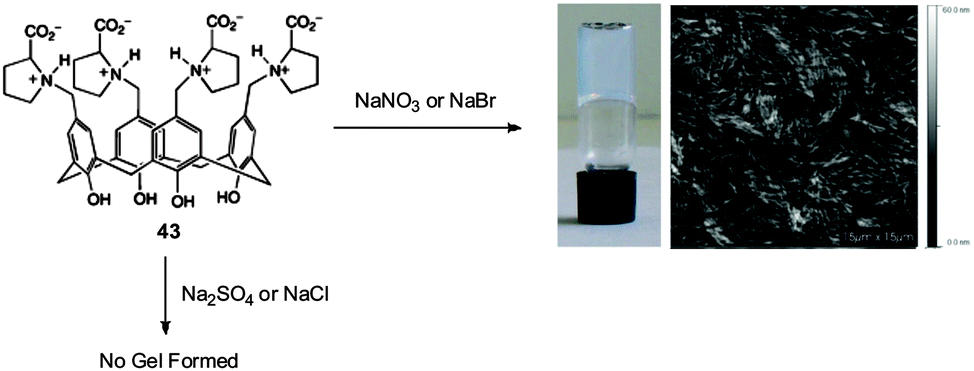 | ||
| Fig. 29 Proline-functionalised calix[4]arene 43 which forms nanofibrillar gels in the presence of nitrate or bromide anions – gel and AFM image shown correspond to compound 43 in the presence of La(NO3)3. Figure adapted from ref. 93 with permission. | ||
Zhang and co-workers demonstrated that protonated melamine 44 forms stable hydrogels only in the presence of oxoanions such as nitrate, phosphate, sulfate and ATP (Fig. 30).95 In this case, hydrogelation could also be switched off by increasing pH, leading to deprotonation of the gelator and loss of the positive charge required to interact with the anion in the competitive aqueous solvent. Gelation of this type, induced by anion complexation may be of use for removal of anions from aqueous waste – nitrate and phosphates are particular targets of interest in this regard given the problems of eutrophication in ground water.
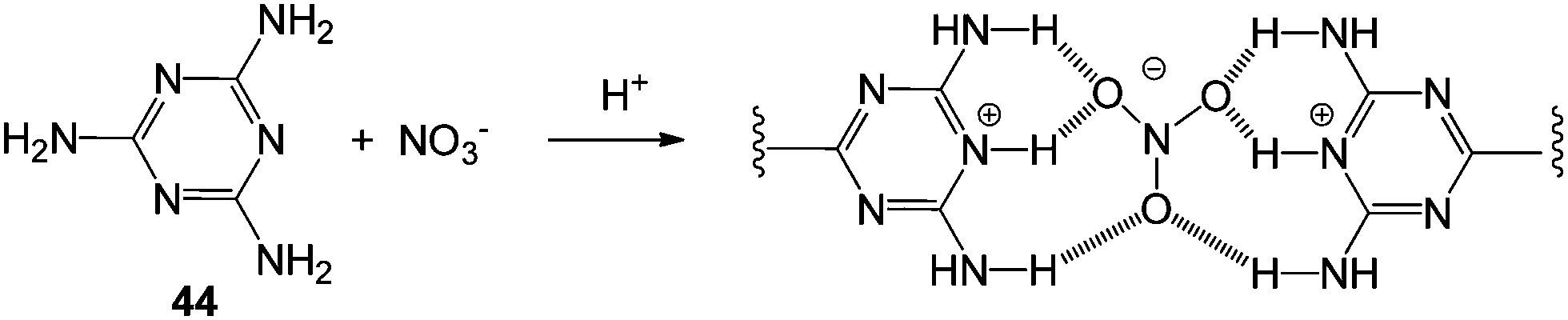 | ||
| Fig. 30 Melamine 44 binds the nitrate anion and forms gels when protonated – scheme shows the proposed hydrogen-bonding interactions of the complex which underpins the gel.95 | ||
Banerjee and co-workers reported a pyridine-functionalised amino amphiphile LMWG 45 which only formed gels in mixed aqueous media in the presence of hydrogen chloride gas (Fig. 31).96 Interestingly, the chloride ion was found to be very selective for gelation to be observed, with gelation attempts using HBr, HI, HNO3, H2SO4, HCOOH and CH3COOH all failing. It was proposed that the size of the chloride anion may play a key role in mediating the assembly of the amphiphilic system into higher order assemblies. It was suggested that this might provide an interesting route for removing hazardous HCl gas from the environment – and this was demonstrated using a binding and gel-trapping experiment. Of course, acid gases can more generally be removed, in bulk, by passage through basic media, but this remains an interesting and intriguing selective switch-on gelation mechanism which may have use in environmental sensing/detection applications.
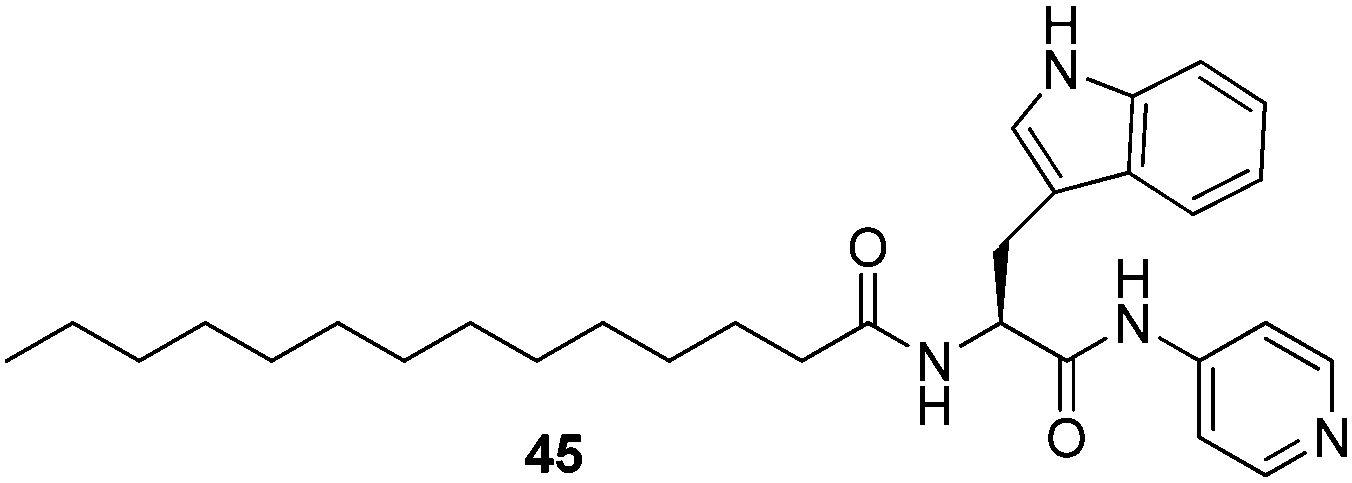 | ||
| Fig. 31 Pyridine-functionalised 45 which forms gels in mixed aqueous solvents in the presence of HCl.96 | ||
In a clever approach, McNeil and co-workers developed a gelator which was switched on in the presence of nitrite anions.97 Their approach involved using a nitrite-mediated reaction to form the gelator scaffold. They employed the Griess reaction of an aromatic amine 46 to generate a diazonium ion, and then intercepted this product with sodium 6-hydroxynaphthalene-2-sulfonate 47 to form azobenzene derivative 48 which was capable of forming a gel (Fig. 32). Vials containing 46 (suspended in 4 M H2SO4) were treated with nitrate-anion spiked water samples (to exemplify environmental waste) for 10 min, followed by adding 47 (in borax buffer). The resulting samples were heated to dissolve all solids and then allowed to cool to room temperature. In the nitrite-spiked samples, red azobenzene derivative 48 was formed, which led to gelation. This system therefore not only detects nitrite ions through a colour change (like the previous example in Fig. 27), but it also switches on gelation, which leads to potential remediation of the anion.
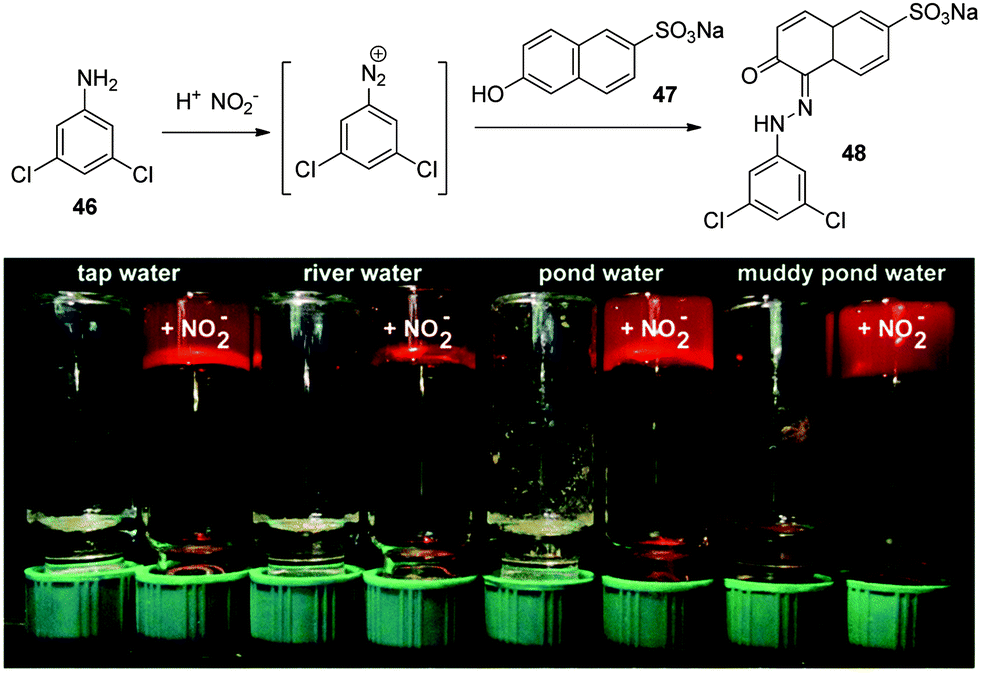 | ||
| Fig. 32 Top: Nitrite-induced formation of gelator scaffold 48 by conversion of precursor 46 into a diazonium ion and subsequent reaction with compound 47. Bottom: Photographs showing the ability to sense the presence of nitrite anions through a red response, and potentially remediate the anion by a gel formation mechanism. Figure adapted from ref. 97 with permission. | ||
Clearly, although there has been significant work on anion sensing, in general anion remediation still remains less well developed than some other environmental applications of gels, with only a limited number of examples as outlined above. Indeed, anion-triggered gelation is much rarer than that achieved by metals. This is reminiscent of the early years of supramolecular chemistry, where anion binding was a poor relation to the more widely studied field of cation binding – as such there is very significant scope for development here. Key advances in this field will focus on anions which would be difficult to bind and remove in other ways, and/or gels which, like those presented in this review, exhibit unusual selectivity preferences and subvert the normal expectations of binding strength based simply on charge density. Binding anions strongly and selectively in water remains a challenging target for supramolecular chemistry in general, and translating this knowledge into the field of switch-on gelation would therefore be of significant interest.
2.5 Supramolecular gels for sensing and remediating chemical warfare agents (CWAs)
A recent development using supramolecular gels for environmental sensing and remediation applies them to detect and/or decontaminate chemical warfare agents (CWAs), especially organophosphorus nerve agents, such as 49 (Tabun) 50 (Sarin) and 51, (Soman) (Fig. 33), which are the most toxic amongst all classes of CWAs and extremely unsafe to retain in the environment.98 Currently, high-temperature incineration is most commonly used for decontamination, however, supercritical water-induced oxidation, caustic bleaching, metal-catalysed hydrolysis and bioremediation are emerging methods to remediate CWAs. Limitations such as high energy costs, generation of secondary pollutants, and instability of enzymes under harsh conditions remain with these methods. Recently, Gale and co-workers have been interested in harnessing supramolecular interactions to bind CWAs, and this is an interesting approach to the problem of CWA detection and decontamination – often simulants of nerve agents, such as 52 (dimethoxymethylphosphonate, DMMP) and 53 (diethoxychlorophosphate, DCP) are used to probe the binding event in a lower-risk experimental approach (Fig. 33).99 As a phosphonate, DMMP 52 has limited reactivity, while the presence of a Cl leaving group on DCP 53 makes it more reactive than DMMP, whilst being more easily handled than genuine nerve agents such as 49–51. | ||
| Fig. 33 Chemical structures of organosphophorous chemical warfare agents Tabun 49, Sarin 50, Soman 51 and their simulants – dimethylmethylphosphonate 52 (DMMP), and diethylchlorophosphate 53 (DCP).99 | ||
An early example of a gel which could interact with, and sense CWAs, was reported by Lee and co-workers.100 Urea-functionalised 2-(2′-hydroxyphenyl)benzoxazole 54 (Fig. 34) formed stable organogels in carbon tetrachloride and cyclohexane and exhibited enhanced fluorescence emission. This was attributed to the tautomeric shift between the enol and keto tautomers of the 2-(2′-hydroxyphenyl)benzoxazole unit – the keto tautomer predominates in the gel because it strengthens the interactions responsible for self-assembly. Addition of liquid nerve agent simulant 53 (DCP) to the gel of 54 in CCl4, led a gel–sol transition (Fig. 34). It also induced to spectroscopic changes which were proposed to result from hydrogen bond interactions between the simulant and self-assembled 54. In the case of exposure to the vapour, the gel remained intact. As in the area of anion/gel chemistry, this is an example of sensing, of potential environmental relevance, but not remediation.
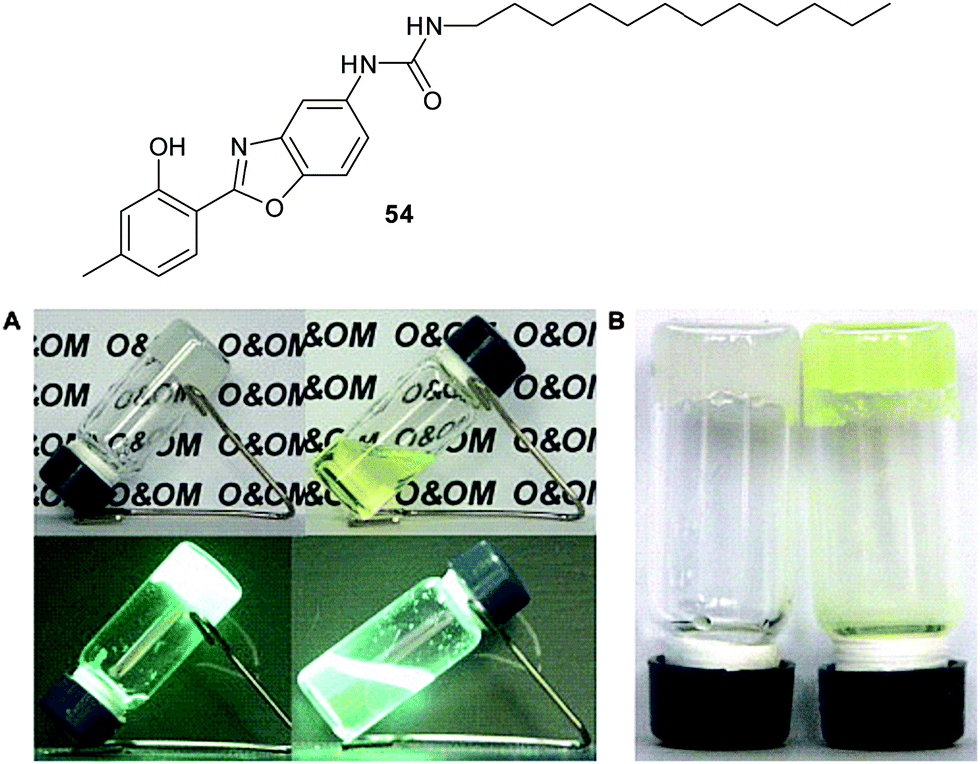 | ||
| Fig. 34 Chemical structure of gelator 51 and photographs of CCl4–gel of 51 (1% (w/v)) (A) before (left) and after (right) adding DCP liquid (100 equiv.) under ambient (top) and UV (bottom) light and (B) before (left) and after (right) exposure to DCP vapour. Figure adapted from ref. 100 with permission. | ||
In elegant research, Gale and co-workers attempted the first gel-phase remediation of CWAs by developing a family of cyclohexyldiamide gelators 55 capable of forming stable gels in organic solvents (Fig. 35).101 The compounds were able to gelate trimethylamine and nerve agent simulant DMMP (52) at concentrations as low as 3 mg ml−1. This suggests that nerve agents may be immobilised by this type of gelator. In contrast, more reactive nerve agent simulant DCP (53) could not be gelled by 55, but instead on addition of DCP to the surface of pyridine-based gels, a gel–sol phase transition occurred accompanied by a colour change from colourless to red (Fig. 35). It was noted that HCl and diethylhydrogenphosphate, the by-products of DCP hydrolysis did not disrupt these gels – as such it was concluded that the CWA-simulant was directly reacting with the gel fibres, not simply being hydrolysed – a mechanism for this process was proposed. The colour change was attributed to complex formation between the gelator, pyridine and DCP. This could therefore find potential applications in sensing CWAs and/or structurally-related pesticides.
 | ||
| Fig. 35 Chemical structure of cyclohexyl diamide 52 and photographs of gels containing compound 55d (20 mg) and DMSO (1 mL), pyridine (1 mL) and TEA (1 mL). (b) The addition of DCP (0.1 mL) to the surface of gel samples. (c) 1 h 48 min after DCP addition. (d) 2 h 58 min after DCP addition. (e) 5 h 5 min after DCP addition showing gel breakdown and intense red colour. Figure adapted from ref. 101 with permission. | ||
In 2015, these authors also reported a two-component gelator constituted from the decylammonium anthracene-9-carboxylate salt 56, which self-assembled in cyclohexane (Fig. 36).102 It was demonstrated that when aliquot solutions of CWA stimulus, soman (GD, 51) and its simulants (DMMP 52 and DCP 53), were added to the surface of the organogel, it was disrupted and underwent a gel–sol transition. With the use of 31P NMR and fluorescence spectroscopies, it was established that the gel–sol phase transition was driven by the polarity of the ambient solvent, intermolecular hydrogen bonding between the stimuli and the gelator, and rapid reaction process that can speed up the gel disruption, with different mechanisms operating in different cases. To demonstrate sensing potential, gels of 56 in cyclohexane were exposed to vapours of DCP and the time required for gel disruption was recorded. It took 680 s for the gel to respond to 0.01 ml of aliquot solution of DCP compared to just 280 s for 0.1 ml – indicative of dose-dependent sensing. Organogel disruption was further explored by incorporating a copper coil into the gel, which was then inverted over both the DCP sample and electrical positive/negative terminals. As the DCP vapour disrupted the gel, the copper coil fell-off and hence established a contact with the terminals, completing a circuit and switching on the connected LED. As such, this system demonstrated a simple low-tech manner in which a gel–sol response could create a practical detection system for sensing CWAs in air (Fig. 36).
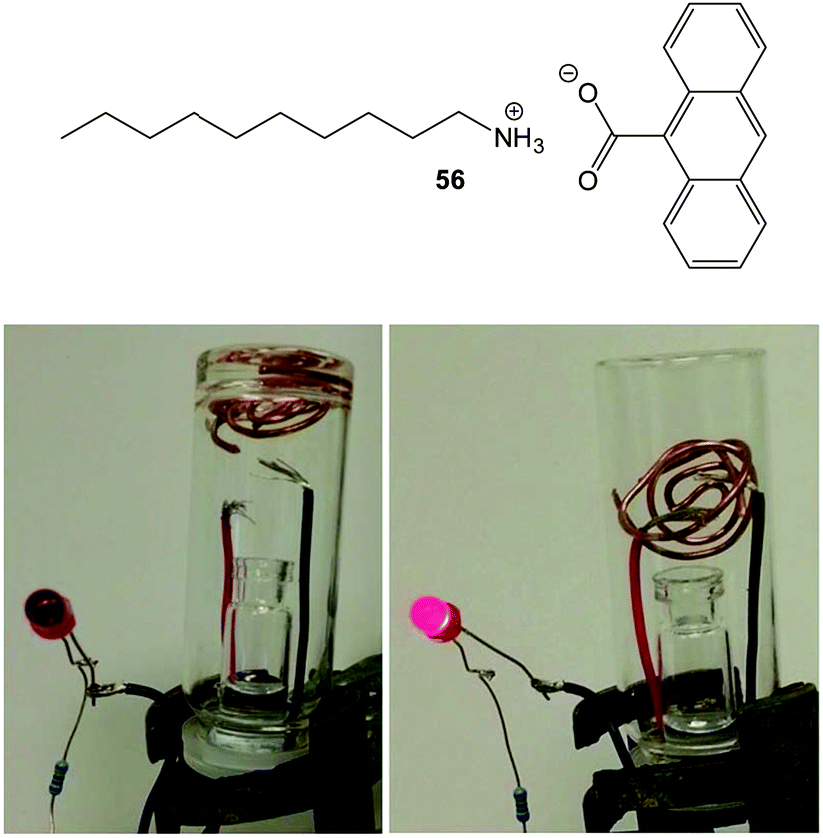 | ||
| Fig. 36 Structure of two-component gelator 56 and photographs: Left: Copper coil suspended in organogel above the sample of DCP and positive/negative contacts; Right: Organogel breaks down in response to DCP, resulting in the release of copper coil, completion of the electrical circuit and the LED switching on. Figure adapted from ref. 102 with permission. | ||
Gale and co-workers have also reported that DMMP is able to reinforce urea-based supramolecular gels, with the enantiopurity of the self-assembled gel having a significant impact on whether the gel is reinforced (in terms of yield strength and thermal stability) or damaged by the addition of the nerve agent simulant.103 The reinforcement of some of the gels on addition of small amounts of DMMP was proposed to be caused by the solvophobic effect, although in larger amounts, its hydrogen bonding potential also led to gel breakdown.
With remediation fully in mind, landmark research from Gale and co-workers recently demonstrated how DMSO organogels of their cyclohexyldiamides 55 (Fig. 35) mixed with 4-nitrobenzaldoxime 57 (reactive agent for decontamination of CWAs, Fig. 37) could be used to absorb, encapsulate and decontaminate CWA simulant DCP 50in situ.104 In this case, the hot organosol comprising gelator 55 (9.6–16.0 mg) and tetrabutylammonium 4-nitrobenzaloxime 57 was added to solutions (0.1–0.5 mL) of simulants (DMMP 52 and DCP 53), leading to the formation of gels that were stable for a long period of time (>24 h). Organogels incorporating DCP evolved a gas, suspected to be HCl, with a concurrent colour change from red to yellow; neither gas evolution nor a colour change were observed with less reactive DMMP. The same events were observed when solutions of the simulants were added to the surface of pre-formed organogels in sealed vials. If excess DCP was present, then the gels broke down into sols, releasing the excess oximate decontaminant – in this way high local concentrations of decontaminant solution can, in principle, be released in the presence of excess nerve agent, precisely at the time and location they are most required. This approach was further extended to sensing DCP vapour in air – an inverted vial containing an organogel placed over the simulant increased in colour intensity on exposure to DCP vapour as reactive decontaminant 57 reacted with the nerve agent simulant via a Beckman-type rearrangement to yield compound 58 (Fig. 37).
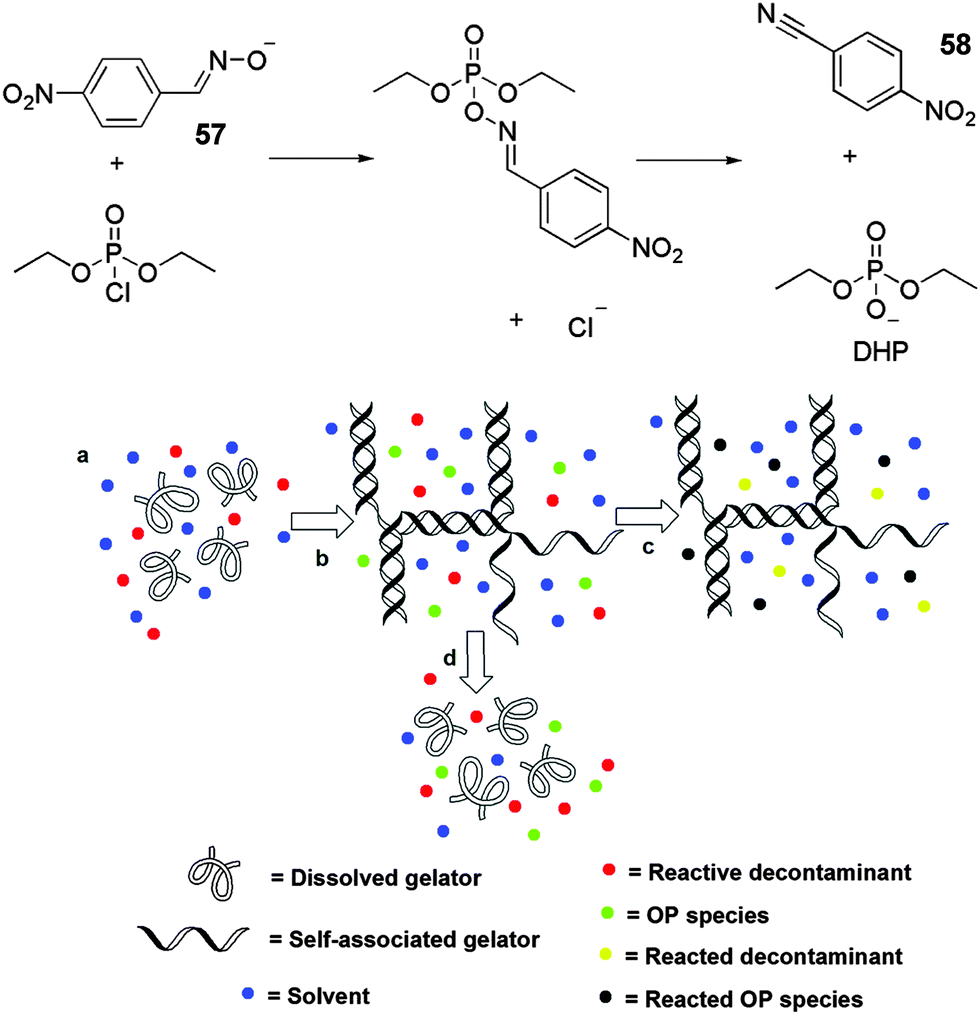 | ||
| Fig. 37 4-Nitroaldoxime 57 reactive nerve agent decontamination compound, the mechanism of its reaction with DCP to form 58, and a schematic of how a gel incorporating reactive decontaminant can respond to the presence of nerve agents and act to remediate against them. Figure adapted from ref. 104 with permission. | ||
It is worth noting that in addition to chemical weapons, sol–gel transitions can also be used to detect and potentially remediate other types of conventional weapons systems. For example McNeil and co-workers reported a system which switched-on gelation in the presence of the explosive triacetone triperoxide (TATP) as a result of oxidation of a thiol to a disulfide.105 However, the solvent in this work was methanol, and as such, it was reported primarily as a sensor. Gels which act as sensors for other important explosives such as trinitrotoluene (TNT) have also been reported.106 Clearly for the environmentally-relevant remediation of explosives, however, gelation in water would be desirable.
Supramolecular gels are intriguing media for these applications. It seems clear that they have genuine potential as rapid visual sensors for CWAs, which could be of real value in a field setting where contamination is suspected. In terms of remediation, it is unlikely that these systems would be useful for long term storage of CWAs, as gels can often be metastable over extended time periods. However, the simplicity of gel formation, and potential ease of handling, suggests that these materials may have applications for short-term remediation/collection of such waste before it can be disposed of more permanently. Furthermore, the incorporation of other remediation agents into gel-phase materials seems like a particularly promising strategy for achieving localised and rapid release of agents to mitigate against CWA waste.
3. Reflections on environmental applications of supramolecular gels and key design principles
As evidenced in the sections above, supramolecular gels are structurally diverse, as befits their range of potential applications. Nonetheless, there are some common design features which can be optimised, including: (i) solubility (ii) nanostructuring and nanoporosity (iii) molecular programming and molecular recognition, (iv) recyclability and reusability, and (v) environmental compatibility. Due to the importance of each of these parameters, their roles and relevance are described in more detail in the following subsections.3.1 Solubility
It has often been noted that gel-forming systems are finely balanced between being soluble and insoluble in the liquid-like medium of choice – and indeed an excellent understanding of solvent effects on gelation has emerged in recent years which reflects this view and suggests that gels form when solvent parameters are optimal for the gelator in question.107Many of the environmental applications described above, specifically the removal of dyes, metals and toxic anions ideally require hydrogel materials (or xerogels which have high compatibility with an aqueous phase). This will allow good penetration of the polluted aqueous phase into the gel nanostructure. Amphiphilicity – the incorporation of both hydrophilic and hydrophobic components into a single molecule is an excellent general way of achieving the finely balanced solubility required for gel formation in water. Indeed, many self-assembling hydrogels contain both hydrophobic and hydrophilic groups, and tuning the balance between them can allow the gelation potential to be optimised for the desired region of ‘solvent space’.107
In contrast, some of the other environmental applications described in this article require gelation in an organic phase – notably the immobilisation of oil spills or chemical weapons agents. Clearly, these organogelators will need to be optimised for self-assembly in a different region of solvent space, and will be somewhat less polar in character than hydrogels. This is particularly important for oil-spill remediation, because if the gelator has too high a degree of water solubility then it will partition between the oil-phase which should be gelated and the aqueous environmental phase which should not, and hence lose its effectiveness.
As a representative example of this general concept, as described earlier in this article, Das and co-workers tuned the amphiphilicity of a single non-gelator to obtain a hydrogelator, an organogelator, and an ionogelator.47 Similarly, as described above we have modified the industrially relevant DBS scaffold with carboxylic acid or acyl hydrazide groups in order to expand the scope of this gelator such that it can form true hydrogels in water.58,80,108 Most LMWGs are discovered serendipitously – a priori design remains challenging109 – as such, modification of key gelator fragments is a highly effective way of increasing the scope of their applications, and in the context of environmental applications, allowing them to operate effectively in the appropriate environmental medium.
Solubility can also directly impact on gelation kinetics, which is of key importance in these applications. For example in oil-spill immobilisation gelators are required which can form instantly in situ without the need for stimuli such as heating or sonication, and ideally without the presence of a co-solvent to assist their dissolution and subsequent self-assembly. Precise and careful optimisation of gelator solubility is therefore of key importance; if the energy barrier to getting the gelator into solution and subsequently self-assembling is too high, the gelator will not operate optimally. One effective way of achieving this is to use a two-component approach, in which once the individual components which have high solubility mix, they form a complex, which then has the optimal solubility for self-assembly and gelation. In this way, complex formation provides the required energy, and induces the solubility change, in order to transition the energy barrier associated with gelation. We have recently demonstrated how gelation kinetics of two-component gels can be tuned by simple structural modification.110 We believe that optimisation of gelation kinetics in (e.g.) crude oil using this general strategy could be a useful approach for developing improved oil-immobilisation agents for environmental use.
3.2 Nanostructuring and porosity
The hierarchical self-assembly of LMWGs is a sophisticated yet simple approach for fabricating supramolecular architectures. Importantly, the inherent nanostructuring increases the surface area of nanofibres; thus, the solid-like fibrillar network of a supramolecular gel is potentially a highly-accessible surface for very high pollutant uptake – as demonstrated by a number of the studies described above in which essentially stoichiometric uptake has been reported.As a consequence of their bottom-up assembly, self-assembled nanofibrillar networks with desired macroscopic properties can potentially be designed,111 with the morphology, and hence the mechanical properties of the fibrillar network, at least in principle, being engineered by controlling fibre and network formation.112
For a gel network, the storage or elastic modulus, G′ is dependent on the correlation length ξ (Fig. 38). As ξ, an important parameter determined by the average mesh size increases, the G′ of the gel decreases.113 It has also been noted that the presence of a polymer additive during the self-assembly of LMWGs can control the degree of branching by adsorbing on the growing tip of the fibre (and inducing a ‘crystallographic mismatch branching’ mechanism), and hence impact on the number of nodal points and the rheological performance.114 This therefore provides a simple mechanism by which the network morphology, and its mechanical performance, can be controlled.
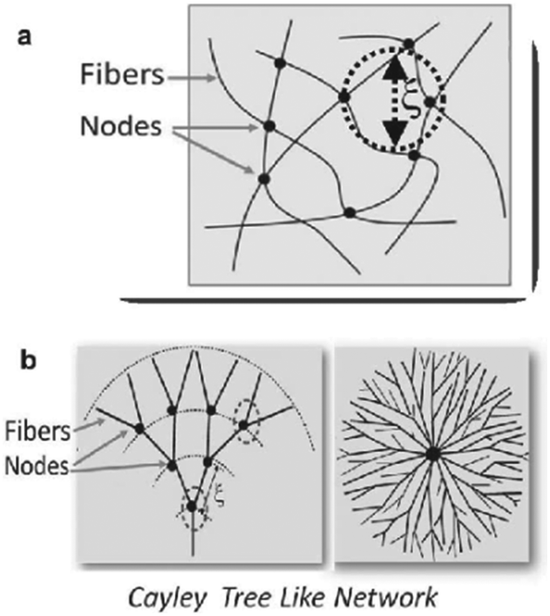 | ||
| Fig. 38 Illustration of fibre networks commonly observed in supramolecular materials. (a) Interconnecting fibre network consisting of fibres and joints (or nodes) with closed loops. (b) Cayley tree-like network (left) with open loops, and spherulites. Figure adapted from ref. 113 with permission. | ||
There are two different possible types of fibre network within gels: (i) single fibre networks and (ii) multi-domain spherulitic networks. Single fibre networks emerge when numerous fibre networks mutually interpenetrate and interlock to form a homogeneous overall network, which therefore exhibits good mechanical strength (Fig. 39a). On the other hand, a multi-domain fibre network is a collection of disjointed single fibre networks, which usually exhibits a high ξ value and hence has weak mechanical properties (Fig. 39b).
 | ||
| Fig. 39 Illustration of (a) single fibre (b) multi-domain fibre networks. Figure adapted from ref. 113 with permission. | ||
Due to the mechanical strength requirements for many environmental applications of supramolecular gels in water purification, and the need for the stability of the nanofibres in the presence of adsorbed pollutants, it is important that the networks of supramolecular gels be engineered to suit this purpose. Physical gel fabrication approaches such as supersaturation, supercooling, thermal processing, seeding and ultrasound have been widely employed to tune network and rheological properties of functional molecular gels.115 As noted earlier in the article, the strategy of co-formulating LMWGs with polymeric materials also offers an important strategy for enhancing the mechanical properties of self-assembled gels.66
Besides tuning the mechanical properties of nanofibres, the specific surface area of a supramolecular gel nanofibrillar network can also be tuned by simple chemical modifications which in turn impact on nanostructuring. For example, Feng and co-workers observed that the BET surfaces of the xerogels of C2-symmetric benzene-based gelator 59 (Fig. 40) decreased from 14.7 m2 g−1 to just 3.8 m2 g−1 when the carboxylic acid group on 59a was replaced with a terminal diglycol group to obtain gelator 59b.116 As a result of the relatively large surface area of the xerogel of 59a, the amount of cationic dye uptake of which it was capable was about 80 times that achieved by the xerogel of 59b. It is therefore evident that optimisation of nanostructuring is vital to develop the most effective gel-derived nanostructured materials for environmental remediation.
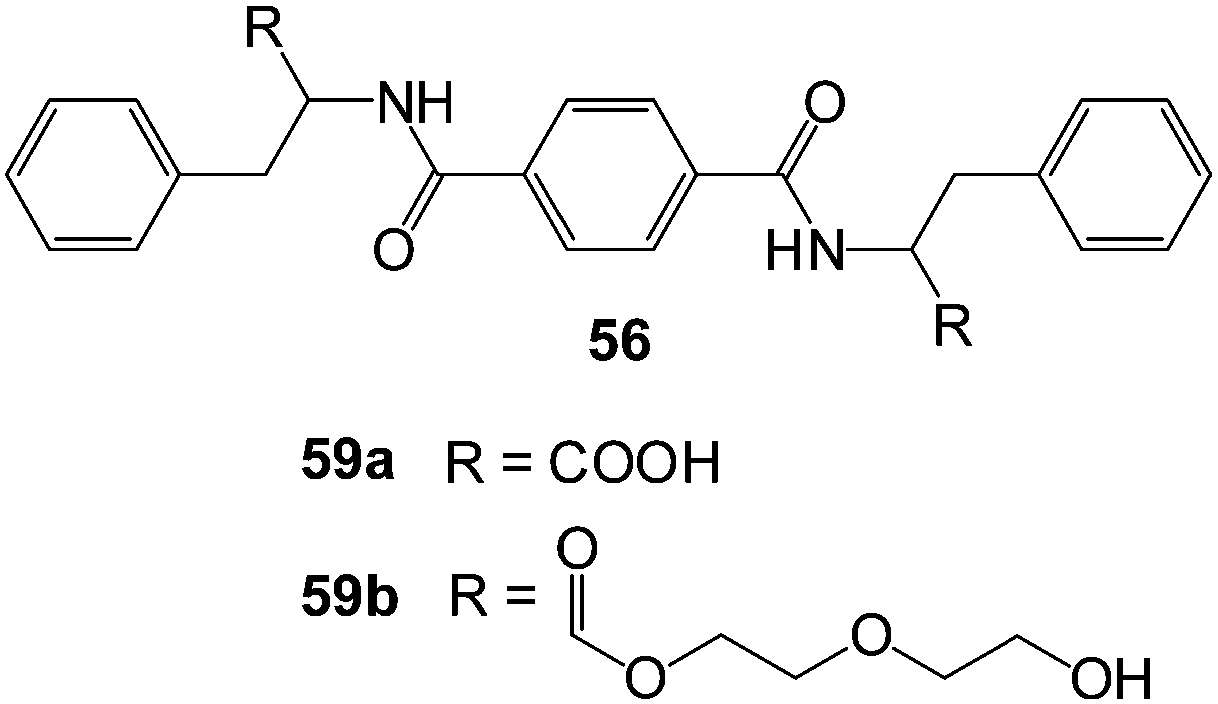 | ||
| Fig. 40 Chemical structures of C2-symmetric benzene-based gelators 59 – the nature of the R groups plays a key role in controlling network dimensions and porosity – and hence dye uptake.116 | ||
In most cases, environmental applications of gel matrices are diffusion-driven. High porosity and large internal surface areas with a large number of specific adsorption sites are fundamentally important characteristics of good adsorbents. When a LMWG is nanostructured by gelation in an appropriate solvent, a nanonet having a mesh with a range of diameters emerges. Extraneous materials can diffuse into, through and out of the network with the mesh size controlling the dynamics and mechanism of diffusion.117 The size of the mesh depends on gelator structure, gelator loading, solvent and the gelation process employed. Imaging techniques such as scanning electron microscopy (SEM), transmission electron microscopy and atomic force microscopy are commonly used tools for measuring the mesh size of molecular gels.118 However, these techniques have inherent limitations such as sample preparation causing damage or changes in the gel morphology. With the advent of environmental scanning electron microscopy (ESEM), it is possible to image a gel in its native (wet) state. Alternatively a gel can be converted it into an aerogel through drying under reduced pressure at low temperatures with potentially limited disruption of the nanoscale network.119
In the hunt for non-invasive techniques which are sensitive to gel network dynamics, the use of nuclear magnetic resonance (NMR) spectroscopy for the characterization of gels has witnessed enormous growth.120 Spin relaxation times, magic angle spinning (MAS), nuclear Overhauser enhancements (NOE) and pulse field gradient (PFG) are increasingly used to gain detailed insight into the molecular organization, specific interactions and internal mobility of constituents in a gel. Adams, Iggo and co-workers have recently demonstrated the use of PFG-NMR spectroscopy to analyse the mesh size in micellar solutions and in molecular hydrogels formed upon addition of Ca2+ to a solution of naphthalene diphenylalanine.121 Dextran guests of various masses and hydrodynamic radii (2Rh) were used to probe diffusion. Interestingly, for dextran with a nominal mass, Mr < 500 kDa and 2Rh < 40 nm, the diffusivity was only slightly restricted compared to its dilute aqueous solution, but a larger dextran (3300 kDa, 84 nm) was almost completely retarded by the gels – consistent with the mesh size of the gel (40–100 nm).
It is worth noting that for rheological strength a small mesh size is required, whereas for optimal diffusion and dynamics a larger mesh size would be preferred. As such, there is a trade-off to be had within supramolecular gels for optimisation of the network morphology in order to maximise performance in the desired environmental application.
3.3 Molecular programming and molecular recognition
Low molecular weight gels are archetypal supramolecular systems reflected by the precise way in which molecular-scale information is translated up to the nanoscale by self-assembly mediated through non-covalent interactions. When supramolecular gels are employed in environmental applications, an additional level of hierarchical supramolecular chemistry is often introduced, in that the self-assembled nanofibres must, in their own right, interact further with the target pollutant of interest. In many cases this further interaction is mediated through precise non-covalent interactions. Indeed, such interactions have been a key feature of many of the examples described earlier in this article. As an aside, it is worth noting that in general terms, the use a supramolecular approach to achieve environmental control is a topic of considerable, and increasing, interest.122There is therefore a vital need to programme the molecular structures of the self-assembled nanofibres such that they can best interact with the desired targets. This may involve the incorporation of groups on the periphery of the gelator which are capable of specific interactions, such as acid-amine interactions, hydrogen bond recognition, ligand–metal interactions etc. This is clearly a simple mechanism by which the application of LMWGs as supramolecular gels can be tuned towards a specific environmental target of interest.
3.4 Recyclability and reusability
It is vital that materials developed for environmental applications should either be recyclable or reusable – indeed, the whole life-cycle of materials is of key importance.24 In general terms, as self-assembled materials held together by relatively weak non-covalent interactions, gels can be relatively easily disassembled – although this is not always the simplest methodology for achieving materials reuse.A variety of approaches have been taken to achieve reuse and recyclability of these materials as described in detail earlier in the article. In demonstrations of solvent or oil-spill remediation technology, the oil is often recovered by distillation – clearly this requires the input of energy, but also has the benefit of recovering the gelator in xerogel form (assuming its thermal stability) so that it can be reused. When used for the extraction of dyes, a number of researchers have demonstrated that variation of pH can achieve effective recycling, either by precipitation of the gelator, or by washing the dye off the gel support. In metal extraction, again pH variation can be a useful approach to achieve recycling and reuse, alternatively, washing the gel with a chelating ligand such as EDTA can be an effective way of achieving this goal. Obviously, when gels are used for the remediation of chemical weapons agents, it is unlikely that recycling is the major concern – and in such cases safe disposal, in stable form, or enabling the reaction of the nerve agents in a controlled way, would be the number one priority.
An alternative to recycling and reuse for the original application is to reuse the resulting material, after pollution remediation, in a new ‘value-added’ application – in this way waste can be recycled to potentially generate wealth. Earlier in the article, we demonstrated this approach from our own research, using gels to extract precious metals from model electronics waste, forming nanoparticle-embedded gels which exhibited interesting electronic properties in their own right.80
3.5 Envirocompatibility
If a gelator is to be applied in the wider environment, it is essential that it should be environmentally benign, as introducing additional problematic agents into the environment in an attempt to mitigate pollution would be highly undesirable. As such, gelators should ideally be biocompatible and/or biodegrade. One key advantage of using the self-assembly approach to gelation is that the nanofibres themselves are reversible in nature, and will disassemble under certain conditions. Furthermore, many of the small molecules used to underpin gelation are either biocompatible themselves, or have well-understood pathways for degradation into molecular constituents which are not of significant environmental concern.Ideally, gelators used for environmental applications should also be derived from feedstocks which are environmentally derived and hence sustainable. This has been referred to as the ‘biorefinery concept’ by George and co-workers,23 highlighting examples in which gelators can be wholly derived from natural sources or readily synthesised using only other reagents which are also naturally derived.
It is interesting to reflect that LMWGs actually have a relatively long history, for example 12-HSA and DBS, as noted earlier in this article.8,9,10b Those gelators, like many chemicals from the 19th century were derived from natural sources – it was only the advent of the petroleum industry, and more specifically polymer chemistry in the early parts of the 20th century, which led materials scientists to turn away from this approach and embrace the powerful functionality offered by polymeric materials. It is therefore interesting to reflect on the way in which modern chemistry, by refocussing attention on self-assembled low-molecular-weight gels, rather than polymer gels, is actually returning towards older approaches, and turning its back to some extent on the resource-intensive approaches of the 20th century.
4. Conclusions and future prospects
In conclusion, it is clear that there is significant interest and activity in the development of soft matter, self-assembled gels with potential environmental applications. In particular, the key advantages of having simple tuneable materials, with high solvent compatibility and porosity, means these systems have potential to solve some pressing environmental problems. A number of examples of high-impact research have been presented in this article, although there is still plenty of scope for new paradigms in environmental remediation to emerge – and this should be the focus of research at the highest level.Significant progress has been made in understanding the fundamentals of interactions between pollutant species and self-assembled nanoscale networks, but there is still considerable scope for the discovery of gels which can interact with different types of target. There is also a need for additional focus on enhanced design and control of network morphology and overall materials performance.
It is also clear, however, that there are very many potential gelators, and many analytes of potential environmental concern. As such, there is a risk that a large number of incremental studies of the type, ‘Gelator X interacts with pollutant Y’, will result. Furthermore, in some areas of research, such as oil-spill remediation, it is clear that many of the fundamental issues have been addressed, and as such, there is actually the most pressing need for field trials of promising systems. We suggest here that in terms of approaching genuine environmental applications, researchers must focus on looking for progress by careful bench-marking of their work in the context of their proposed application against other literature reports, and in particular, by optimising the following key aspects of their gel-phase materials:
• selectivity of pollutant uptake
• maximum pollutant uptake capacity
• kinetics of pollutant uptake
• materials physical properties and performance
• gel reuse and recycling
• environmental compatibility of the gel lifecycle
The best new materials should outperform literature examples across all, or at least most, of these characteristics. We would then strongly encourage researchers to move onto device manufacture, combined with testing and/or field trials.
Environmental applications of supramolecular gels are a fascinating and increasingly active area of research, with some of the very best research being performed in developing economies, where the need for such materials is intense. This reflects the way in which the globalisation of science can lead to an increased focus on using the scientific method to solve problems of genuine global importance. It is clear that with smarter supramolecular chemistry, and the application of a greater environmental awareness in the design and development of novel self-assembled materials, this area of chemistry is well-placed to play a role in providing innovative solutions to 21st century environmental problems.
Acknowledgements
We thank The Wild Fund (University of York) for financial support of B. O. O.'s PhD research. We thank our partners and young sons, Dexter (DKS) and John (BOO), for their patience during the preparation of this manuscript.Notes and references
- (a) A. Mishra and J. H. Clark, in Green Materials for Sustainable Water Remediation and Treatment, ed. A. Mishra and J. H. Clark, RSC, Cambridge, 2013, pp. 1–10 Search PubMed; (b) WHO/UNICEF, Investing in water and sanitation: Increasing access, reducing inequalities, World Health Organization, Geneva, 2014.
- (a) R. K. Sharma, A. Adholeya, M. Das and A. Puri, in Green Materials for Sustainable Water Remediation and Treatment, ed. A. Mishra and J. H. Clark, RSC, Cambridge, 2013, pp. 11–29 Search PubMed; (b) J. W. Readman, in An Introduction to Pollution Science, ed. R. M. Harrison, RSC, Cambridge, 2006, pp. 77–121 Search PubMed.
- (a) S. Babel and T. A. Kurniawan, J. Hazard. Mater., 2003, 97, 219–243 CrossRef CAS PubMed; (b) V. K. Gupta and Suhas, J. Environ. Manage., 2009, 90, 2313–2342 CrossRef CAS PubMed.
- (a) Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, Netherlands, 2006 Search PubMed; (b) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed; (c) J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC; (d) Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, RSC, Cambridge, 2014 Search PubMed; (e) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS PubMed.
- Polymer Gels: Fundamentals and Applications, ed. H. B. Bohidar, P. Dubin and Y. Osada, American Chemical Society, Washington DC, 2002 Search PubMed.
- P. C. Marr and A. C. Marr, Green Chem., 2016, 18, 105–128 RSC.
- M. J. Meunier, Ann. Chim. Phys., 1891, 22, 412 Search PubMed.
- R. Zsigmondy and W. Batchmann, Z. Chem. Ind. Kolloide, 1912, 11, 145–157 CrossRef.
- (a) C. J. Donahue, J. Chem. Educ., 2006, 83, 862–869 CrossRef CAS; (b) B. O. Okesola, V. M. P. Vieira, D. J. Cornwell, N. K. Whitelaw and D. K. Smith, Soft Matter, 2015, 11, 4768–4787 RSC.
- (a) M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC; (b) S. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- (a) N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC; (b) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed; (c) S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC; (d) K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- Y. Gong, X. Zhao, Z. Cai, S. E. O’Reilly, X. Hao and D. Zhao, Mar. Pollut. Bull., 2014, 79, 16–33 CrossRef CAS PubMed.
- L. Guterman, Science, 2009, 323, 1558–1559 CrossRef CAS PubMed.
- (a) D. Dave and A. E. Ghaly, Am. J. Environ. Sci., 2011, 7, 423–440 CrossRef CAS; (b) D. D. Prenderghast and P. M. Gschwend, J. Cleaner Prod., 2014, 78, 233–242 CrossRef.
- K. Liu, P. He and Y. Fang, Sci. China: Chem., 2011, 54, 575–586 CrossRef CAS.
- USEPA-Water quality office, Gelling crude oils to reduce marine pollution from tanker oil spills, Water pollution control research series 15080DJN 1/71, U.S. government printing office, Washington D.C. 20402, 1971.
- T. Saito, Y. Matsuzawa, S. Ninagawa, M. Honna, M. Takesada and M. Takehara, US Pat., 3969087 A, 1976 Search PubMed.
- T. Kobayashi, Y. Kawashima, M. Yoshimura, M. Sugiura, T. Nobe and S. Fujimoto, US Pat., 4502975, 1985 Search PubMed.
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185–186 RSC.
- (a) D. R. Trivedi, A. Ballabh and P. Dastidar, Chem. Mater., 2003, 15, 3971–3973 CrossRef CAS; (b) D. R. Trivedi, A. Ballabh, P. Dastidar and B. Ganguly, Chem. – Eur. J., 2004, 10, 5311–5322 CrossRef CAS PubMed.
- A. Ballabh, D. R. Trivedi and P. Dastidar, Chem. Mater., 2006, 18, 3795–3800 CrossRef CAS.
- (a) G. John, B. V. Shankar, S. R. Jadhav and P. K. Vemula, Langmuir, 2010, 26, 17843–17851 CrossRef CAS PubMed; (b) H. L. Hwang, S. R. Jadhav, J. R. Silverman and G. John, J. Chem. Educ., 2014, 91, 1563–1568 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS PubMed.
- J. Peng, K. Liu, X. Liu, H. Xia, J. Liu and Y. Fang, New J. Chem., 2008, 32, 2218–2224 RSC.
- M. Xue, D. Gao, K. Liu, J. Peng and Y. Fang, Tetrahedron, 2009, 65, 3369–3377 CrossRef CAS.
- A. Prathap and K. M. Sureshan, Chem. Commun., 2012, 48, 5250–5252 RSC.
- L. Yan, G. Li, Z. Ye, F. Tian and S. Zhang, Chem. Commun., 2014, 50, 14839–14842 RSC.
- S. Mukherjee, C. Shang, X. Chen, X. Chang, K. Liu, C. Yu and Y. Fang, Chem. Commun., 2014, 50, 13940–13943 RSC.
- T. A. Stortz, A. K. Zetzl, S. Barbut, A. Cattaruzza and A. G. Marangoni, Lipids Cereal Technol., 2012, 24, 151–154 CrossRef CAS.
- M. A. M. Viswanatha and R. G. Weiss, US Pat., WO2012047251, 2012 Search PubMed.
- V. A. Mallia, D. L. Blair and R. H. Weiss, Ind. Eng. Chem. Res., 2016, 55, 954–960 CrossRef CAS.
- P. Lee and M. A. Rogers, Langmuir, 2013, 29, 5617–5621 CrossRef CAS PubMed.
- D. Wang, J. Niu, Z. Wang and J. Jin, Langmuir, 2015, 31, 1670–1674 CrossRef CAS PubMed.
- S.-L. Yu, X.-Q. Dou, D.-H. Qu and C.-L. Feng, J. Mol. Liq., 2014, 190, 94–98 CrossRef CAS.
- G. Feng, H. Chen, J. Cai, J. Wen and X. Liu, Soft Mater., 2014, 12, 403–410 CrossRef CAS.
- M. Konda, I. Maity, D. Rasale and A. K. Das, ChemPlusChem, 2014, 79, 1482–1488 CrossRef CAS.
- J. Bachi, S. Oehm, J. Mayr, C. Cativiela, J. J. Marrero-Tellado and D. D. Diaz, Int. J. Mol. Sci., 2015, 16, 11766–11784 CrossRef PubMed.
- Rajkamal, D. Chatterjee, A. Paul, S. Banerjee and S. Yadav, Chem. Commun., 2014, 50, 12131–12134 CAS.
- F. I. Hai, K. Yamamoto and K. Fukushi, Crit. Rev. Environ. Sci. Technol., 2007, 37, 315–377 CrossRef CAS.
- N. Zweep and J. H. van Esch, in Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, RSC, Cambridge, 2014, pp. 1–29 Search PubMed.
- S. Ray, A. K. Das and A. Banerjee, Chem. Mater., 2007, 19, 1633–1639 CrossRef CAS.
- E. J. Cho, I. Y. Jeong, S. J. Lee, W. S. Han, J. K. Kang and J. H. Jung, Tetrahedron Lett., 2008, 49, 1076–1079 CrossRef CAS.
- S. Debnath, A. Shome, S. Dutta and P. K. Das, Chem. – Eur. J., 2008, 14, 6870–6881 CrossRef CAS PubMed.
- B. Adhikari, G. Palui and A. Banerjee, Soft Matter, 2009, 5, 3452–3460 RSC.
- S. Dutta, D. Das, A. Dasgupta and P. K. Das, Chem. – Eur. J., 2010, 16, 1493–1505 CrossRef CAS PubMed.
- (a) F. Rodríguez-Llansola, B. Escuder, J. F. Miravet, D. Hermida-Merino, I. W. Hamley, C. J. Cardin and W. Hayes, Chem. Commun., 2010, 46, 7960–7962 RSC; (b) D. M. Wood, B. W. Greenland, A. L. Acton, F. Rodríguez-Llansola, C. A. Murray, C. J. Cardin, J. F. Miravet, B. Escuder, I. W. Hamley and W. Hayes, Chem. – Eur. J., 2012, 18, 2692–2699 CrossRef CAS PubMed.
- B. C. Baker, A. L. Acton, G. C. Stevens and W. Hayes, Tetrahedron, 2014, 70, 8303–8311 CrossRef CAS.
- J. Lu, Y. Gao, J. Wu and Y. Ju, RSC Adv., 2013, 3, 23548–23552 RSC.
- (a) F. Rodriguez-Llansola, B. Escuder and J. F. Miravet, Org. Biomol. Chem., 2009, 7, 3091–3094 RSC; (b) F. Rodriguez-Llansola, B. Escuder and J. F. Miravet, J. Am. Chem. Soc., 2009, 131, 11478–11484 CrossRef CAS PubMed.
- P. K. Sukul and S. Malik, RSC Adv., 2013, 3, 1902–1915 RSC.
- S. Samai and K. Biradha, Chem. Mater., 2012, 24, 1165–1173 CrossRef CAS.
- S. Song, L. Feng, A. Song and J. Hao, J. Phys. Chem. B, 2012, 116, 12850–12856 CrossRef CAS PubMed.
- H. Wang, W. Xu, S. Song, L. Feng, A. Song and J. Hao, J. Phys. Chem. B, 2014, 118, 4693–4701 CrossRef CAS PubMed.
- A. Dey, S. K. Mandal and K. Biradha, CrystEngComm, 2013, 15, 9769–9778 RSC.
- Y.-T. Tang, X.-Q. Dou, Z.-A. Ji, P. Li, S.-M. Zhu, J.-J. Gu, C.-L. Feng and D. Zhang, J. Mol. Liq., 2013, 177, 167–171 CrossRef CAS.
- B. O. Okesola and D. K. Smith, Chem. Commun., 2013, 49, 11164–11166 RSC.
- X. Guan, K. Fan, T. Gao, A. Ma, B. Zhang and J. Song, Chem. Commun., 2016, 52, 962–965 RSC.
- X. Zhang, J. Song, W. Ji, N. Xu, N. Gao, X. Zhang and H. Yu, J. Mater. Chem. A, 2015, 3, 18953–18962 CAS.
- C. K. Karan and M. Bhattacharjee, ACS Appl. Mater. Interfaces, 2016, 8, 5526–5535 CAS.
- S. Kiyonaka, K. Sugiyasu, S. Shinkai and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 10954–10955 CrossRef CAS PubMed.
- S. L. Zhou, S. Matsumoto, H. D. Tian, H. Yamane, A. Ojida, S. Kiyonaka and I. Hamachi, Chem. – Eur. J., 2005, 11, 1130–1136 CrossRef CAS PubMed.
- N. Cheng, Q. Hu, Y. Guo, Y. Wang and L. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 10258–10265 CAS.
- A. R. Hirst, D. K. Smith and J. P. Harrington, Chem. – Eur. J., 2005, 11, 6552–6559 CrossRef CAS PubMed.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 289–293 RSC.
- J. Yang, H. Wang, Z. Song, D. Kong, X. Chen and Z. Yang, Colloids Surf., B, 2010, 80, 155–160 CrossRef PubMed.
- X. Zeng, Z. Sun, H. Wang, Q. Wang and Y. Yang, Compos. Sci. Technol., 2016, 122, 149–154 CrossRef CAS.
- M. Lin, H. Liu, P. W. Miller, J. Zhang and C.-Y. Su, New J. Chem., 2014, 38, 3755–3761 RSC.
- (a) K. Murata, M. Aoki, T. Nishi, A. Ikeda and S. Shinkai, J. Chem. Soc., Chem. Commun., 1991, 1715–1718 RSC; (b) M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC.
- (a) J. O. Duruibe, M. O. C. Ogwuegbu and J. N. Egwurugwu, Int. J. Phys. Sci., 2007, 2, 112–118 Search PubMed; (b) M. A. Hashim, S. Mukhopadhyay, J. N. Sahu and B. Sengupta, J. Environ. Manage., 2011, 92, 2355–2388 CrossRef CAS PubMed.
- T. Wei, J. Dang, Q. Lin, H. Yao, Y. Liu, W. Zhang, J. Ming and Y. Zhang, Sci. China: Chem., 2012, 55, 2554–2561 CrossRef CAS.
- H. Yao, X.-M. Yao, Q. Lin, J.-J. Li, Y. Guo, T.-B. Wei and Y.-M. Zhang, Chin. Chem. Lett., 2013, 24, 703–706 CrossRef CAS.
- P. J. Knerr, M. C. Branco, R. Nagarkar, D. J. Pochan and J. P. Schneider, J. Mater. Chem., 2012, 22, 1352–1357 RSC.
- (a) K. N. King and A. J. McNeil, Chem. Commun., 2010, 46, 3511–3513 RSC; (b) K. K. Carter, H. B. Rycenga and A. J. McNeil, Langmuir, 2014, 30, 3522–3527 CrossRef CAS PubMed.
- S. Sengupta and R. Mondal, J. Mater. Chem., 2014, 2, 16373–16377 RSC.
- S. Sengupta, A. Goswami and R. Mondal, New J. Chem., 2014, 38, 2470–2479 RSC.
- Y.-M. Zhang, X.-M. You, Y. Guo, P. Zhang, B.-B. Shi, J. Liu, Q. Lin and T.-B. Wei, Supramol. Chem., 2014, 26, 39–47 CrossRef CAS.
- B. O. Okesola, S. K. Suravaram, A. Parkin and D. K. Smith, Angew. Chem., Int. Ed., 2016, 55, 183–187 CrossRef CAS PubMed.
- (a) A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed; (b) M. Farré, K. Gajda-Schrantz, L. Kantiani and D. Barceló, Anal. Bioanal. Chem., 2009, 393, 81–95 CrossRef PubMed; (c) A. Patwa, A. Thiéry, F. Lombard, M. K. S. Lilley, C. Boisset, J.-F. Bramard, J.-Y. Bottero and P. Barthélémy, Sci. Rep., 2015, 5, 11387 CrossRef PubMed.
- J. Liu, Z. Wang, F. D. Liu, A. B. Kane and R. H. Hurt, ACS Nano, 2012, 6, 9887–9899 CrossRef CAS PubMed.
- A. Patwa, J. Labille, J.-Y. Bottero, A. Thiery and P. Barthelemy, Chem. Commun., 2015, 51, 2547–2550 RSC.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- (a) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS; (b) M. Wenzel, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2012, 41, 480–520 RSC; (c) S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC; (d) P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205–241 RSC; (e) N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS PubMed; (f) P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC.
- F. Fages, F. Vögtle and M. Žinić, Top. Curr. Chem., 2005, 256, 77–131 CrossRef CAS PubMed.
- (a) A. F. Li, J. H. Wang, F. Wang and Y. B. Jiang, Chem. Soc. Rev., 2010, 39, 3729–3745 RSC; (b) V. Amendola, L. Fabbrizzi and L. Mosca, Chem. Soc. Rev., 2010, 39, 3889–3915 RSC.
- (a) C. E. Stanley, N. Clarke, K. M. Anderson, J. A. Elder, J. T. Lenthall and J. W. Steed, Chem. Commun., 2006, 3199–3201 RSC; (b) J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC; (c) G. O. Lloyd and J. W. Steed, Soft Matter, 2011, 7, 75–84 RSC.
- J.-S. Shen, D.-H. Li, Q.-G. Cai and Y.-B. Jiang, J. Mater. Chem., 2009, 19, 6219–6224 RSC.
- Q. Xia, Y. Mao, J. Wu, T. Shu and T. Yi, J. Mater. Chem. C, 2014, 2, 1854–1861 RSC.
- J. Sun, Y. Lin, L. Jin, T. Chen and B. Yin, Chem. Commun., 2016, 52, 768–771 RSC.
- Q. Lin, T.-T. Lu, X. Zhu, B. Sun, Q.-P. Yang, T.-B. Wei and Y.-M. Zhang, Chem. Commun., 2015, 51, 1635–1638 RSC.
- T. Becker, C. Y. Goh, F. Jones, M. J. Mclldowie, M. Mocerino and M. I. Ogden, Chem. Commun., 2008, 3900–3902 RSC.
- (a) S. Roy, N. Javid, P. W. J. M. Frederix, D. A. Lamprou, A. J. Urquhart, N. T. Hunt, P. J. Halling and R. V. Ulijn, Chem. – Eur. J., 2012, 18, 11723–11731 CrossRef CAS PubMed; (b) V. J. Nebot, J. J. Ojeda-Flores, J. Smets, S. Fernández-Prieto, B. Escuder and J. F. Miravet, Chem. – Eur. J., 2014, 20, 14465–14472 CrossRef CAS PubMed.
- J.-S. Shen, Q.-G. Cai, Y.-B. Jiang and H.-W. Zhang, Chem. Commun., 2010, 46, 6786–6788 RSC.
- S. Basak, N. Nandi and A. Banerjee, Chem. Commun., 2014, 50, 6917–6919 RSC.
- D. M. Zurcher, Y. J. Adhia, J. D. Romero and A. D. McNeil, Chem. Commun., 2014, 50, 7813–7816 RSC.
- (a) N. B. Munro, S. S. Talmage, G. D. Griffin, L. C. Waters, A. P. Watson, J. F. King and V. Hauschild, Environ. Health Perspect., 1999, 107, 933–974 CrossRef CAS PubMed; (b) S. W. Wiener and R. S. Hoffman, J. Intensive Care Med., 2004, 19, 22–37 CrossRef PubMed; (c) L. M. Eubanks, T. J. Dickerson and K. D. Janda, Chem. Soc. Rev., 2007, 36, 458–470 RSC.
- (a) M. R Sambrook, J. R Hiscock, A. Cook, A. C Green, I. Holden, J. C. Vincent and P. A. Gale, Chem. Commun., 2012, 48, 5605–5607 RSC; (b) A. Barba-Bon, A. M. Costero, M. Parra, S. Gil, R. Martinez-Manez, F. Sancenon, P. A. Gale and J. R. Hiscock, Chem. – Eur. J., 2013, 19, 1586–1590 CrossRef CAS PubMed; (c) J. R. Hiscock, F. Piana, M. R. Sambrook, N. J. Wells, A. J. Clark, J. N. Vincent, N. Busschaert, R. C. D. Brown and P. A. Gale, Chem. Commun., 2013, 49, 9119–9122 RSC; (d) J. Hiscock, M. R. Sambrook, P. B. Cranwell, P. Watts, J. C. Vincent, D. J. Xuereb, N. J. Wells, R. Raja and P. A. Gale, Chem. Commun., 2014, 50, 6217–6220 RSC.
- T. H. Kim, D. G. Kim, M. Lee and T. S. Lee, Tetrahedron, 2010, 66, 1667–1672 CrossRef CAS.
- J. R. Hiscock, I. L. Kirby, J. Herniman, G. J. Langley, A. J. Clark and P. A. Gale, RSC Adv., 2014, 4, 45517–45521 RSC.
- J. R. Hiscock, M. R. Sambrook, J. A. Ede, N. J. Wells and P. A. Gale, J. Mater. Chem. A, 2015, 3, 1230–1234 CAS.
- F. Piana, M. Facciotti, G. Pileio, J. R. Hiscock, W. Van Rossom, R. C. D. Brown and P. A. Gale, RSC Adv., 2015, 5, 12287–12292 RSC.
- J. R. Hiscock, M. R. Sambrook, N. J. Wells and P. A. Gale, Chem. Sci., 2015, 6, 5680–5684 RSC.
- J. Chen, W. Wu and A. J. McNeil, Chem. Commun., 2012, 48, 7310–7312 RSC.
- K. K. Kartha, S. S. Babu, S. Srinivasan and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 4834–4841 CrossRef CAS PubMed.
- Y. Lan, M. Corradini, R. G. Weiss, S. R. Raghavan and M. A. Rogers, Chem. Soc. Rev., 2015, 44, 6035–6058 RSC.
- (a) D. J. Cornwell, B. O. Okesola and D. K. Smith, Soft Matter, 2013, 9, 8730–8736 RSC; (b) D. J. Cornwell, B. O. Okesola and D. K. Smith, Angew. Chem., Int. Ed., 2014, 53, 12461–12465 CAS; (c) D. J. Cornwell, O. J. Daubney and D. K. Smith, J. Am. Chem. Soc., 2015, 137, 15486–15492 CrossRef CAS PubMed.
- J. H. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS PubMed.
- S. S. Rohner, J. Ruiz-Olles and D. K. Smith, RSC Adv., 2015, 5, 27190–27196 RSC.
- J.-Y. Xiong, X.-Y. Liu, J.-L. Li and M. W. Vallon, J. Phys. Chem. B, 2007, 111, 5558–5563 CrossRef CAS PubMed.
- R. Wang, X.-Y. Liu, J. Xiong and J. Li, J. Phys. Chem. B, 2006, 110, 7275–7280 CrossRef CAS PubMed.
- J.-L. Li and X.-Y. Liu, Adv. Funct. Mater., 2010, 20, 3196–3216 CrossRef CAS.
- X. Y. Liu and P. D. Sawant, Angew. Chem., Int. Ed., 2002, 41, 3641–3645 CrossRef CAS.
- (a) J.-L. Li, X.-Y. Liu, R.-Y. Wang and J.-Y. Xiong, J. Phys. Chem. B, 2005, 109, 24231–24235 CrossRef CAS PubMed; (b) J.-L. Li, R.-Y. Wang, X.-Y. Liu and H.-H. Pan, J. Phys. Chem. B, 2009, 113, 5011–5015 CrossRef CAS PubMed; (c) R.-Y. Wang, X.-Y. Liu and J.-L. Li, Cryst. Growth Des., 2009, 9, 3286–3291 CrossRef CAS; (d) M. A. Rogers, A. J. Wright and A. G. Marangoni, Soft Matter, 2008, 4, 1483–1490 RSC; (e) M. A. Rogers and A. G. Marangoni, Cryst. Growth Des., 2008, 8, 4596–4601 CrossRef CAS.
- X. Dou, P. Li, D. Zhang and C.-L. Feng, Soft Matter, 2012, 8, 3231–3238 RSC.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- (a) V. J. Nebot and D. K. Smith, in Functional Molecular Gels, ed. B. Escuder and J. F. Miravet, Royal Society of Chemistry, Cambridge, 2014, pp. 30–66 Search PubMed; (b) J. A. Foster, D. W. Johnson, M.-O. M. Pipenbrock and J. W. Steed, New J. Chem., 2014, 38, 927–932 RSC.
- C. A. Lagadec and D. K. Smith, Chem. Commun., 2012, 48, 7817–7819 RSC.
- (a) Y. E. Shapiro, Prog. Polym. Sci., 2011, 36, 1184–1253 CrossRef CAS; (b) B. Escuder, M. Llusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS PubMed; (c) A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS PubMed; (d) V. J. Nebot, J. Armengol, J. Smets, S. F. Prieto, B. Escuder and J. F. Miravet, Chem. – Eur. J., 2012, 18, 4063–4072 CrossRef CAS PubMed.
- M. Wallace, D. J. Adams and J. A. Iggo, Soft Matter, 2013, 9, 5483–5491 RSC.
- M. T. Albelda, J. C. Frias, E. Garcia-Espana and H.-J. Schneider, Chem. Soc. Rev., 2012, 41, 3859–3877 RSC.
| This journal is © The Royal Society of Chemistry 2016 |