
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Man-made molecular machines: membrane bound
Matthew A.
Watson
and
Scott L.
Cockroft
*
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: scott.cockroft@ed.ac.uk
First published on 2nd March 2016
Abstract
Nature's molecular machines are a constant source of inspiration to the chemist. Many of these molecular machines function within lipid membranes, allowing them to exploit potential gradients between spatially close, but chemically distinct environments to fuel their work cycle. Indeed, the realisation of such principles in synthetic transmembrane systems remains a tantalising goal. This tutorial review opens by highlighting seminal examples of synthetic molecular machines. We illustrate the importance of surfaces for facilitating the extraction of work from molecular switches and motors. We chart the development of man-made transmembrane systems; from passive to machine-like stimuli-responsive channels, to fully autonomous transmembrane molecular machines. Finally, we highlight higher-order compartmentalised systems that exhibit emergent properties. We suggest that such higher-order architectures could serve as platforms for sophisticated devices that co-ordinate the activity of numerous transmembrane molecular machines.
 Matthew A. Watson | Matthew Watson obtained his Chemistry MSci degree at the University of Glasgow. In 2010 he joined the University of Edinburgh to undertake a PhD in bionanotechnology followed by a further period of postdoctoral research. During his time in the Cockroft group he has developed synthetic transmembrane molecular machines that operate far-from-equilibrium. He is interested in applying the principles of molecular machine operation to the construction of functional nanomaterials to address the challenges of our age. |
 Scott L. Cockroft | Scott Cockroft is a senior lecturer at the University of Edinburgh. He conducted PhD and postdoctoral work under the supervision of Prof. Christopher Hunter FRS (Sheffield, UK) and Prof. M. Reza Ghadiri (The Scripps Research Institute, California), respectively. The Cockroft group are researching molecular recognition in synthetic model systems, while seeking to harness these principles in the construction of molecular machines built from synthetic and biological components. In his spare time Scott enjoys sitting on stone walls contemplating life, while proudly displaying the Yorkshire rose on his chest. |
Key learning points(1) Illustrative examples of natural transmembrane molecular machines.(2) The conceptual basis of molecular machines, categorisation of machine behaviour. (3) The state of the art of synthetic molecular machines operating in solution and at interfaces. (4) The progress towards and the future of man-made transmembrane molecular machines. |
1. Introduction
Life depends upon the interplay of countless molecular machines.1 These natural molecular machines operate at length-scales where the dominant energetic factors are random thermal fluctuations. Indeed, proteins have been described as ‘kicking, screaming, stochastic molecules'.2 Membrane-spanning biological molecular machines such as adenosine triphosphate (ATP) synthase3 and the bacterial flagellar motor4 offer proven strategies for overcoming the constant buffeting of Brownian motion to perform work that facilitates the existence of life.5,6 Many biologically inspired mechanically interlocked and light-responsive molecules have been synthesised,7–9 some of which are capable of performing macroscopic work when arrayed on surfaces.10–14 While the importance of synthetic nanotechnology continues to be highlighted,5,15 significant advances have also been made using molecules of biological origin such as DNA.16 The characteristics of many man-made transmembrane systems17 are somewhat limited compared to the exquisite functionalities displayed by biological transmembrane machines.18 Nonetheless, the bleeding-edge is starting to deliver man-made nanomechanical transmembrane assemblies that integrate lessons from both evolved and designed systems to operate as true molecular machines under non-equilibrium conditions. Such advances offer great promise for the realisation of man-made transmembrane molecular machines that perform useful work.19Anchoring motor proteins within two-dimensional lipid membranes provides a means of countering the thermal forces that dominate behaviour at the molecular scale. One of the most studied class of biological transmembrane molecular machine are the ATP (synth)ase rotary motors (Fig. 1a).3,20 ATP (synth)ases have a rotaxane-like architecture21 in which a spindle (blue) is threaded through both an F0 motor in the membrane (green) and an F1 motor in the cytosol (red and grey). In addition, the motors are held in close association by a stator (orange and pink). This assembly operates in two reciprocal modes. In the ATP synthesis mode, the reaction between ADP (adenosine diphosphate) and Pi (phosphate) is catalysed at the active sites of the F1 motor. The catalytic cycle couples unidirectional rotation (driven by the flow of cations through the F0 motor down a transmembrane gradient) to a cycle of conformational changes in the F1 motor that involve sequential substrate binding, reaction, and ATP-dissociation.20 Conversely, in the reciprocal mode, a transmembrane cation gradient is generated by the hydrolysis of ATP.3
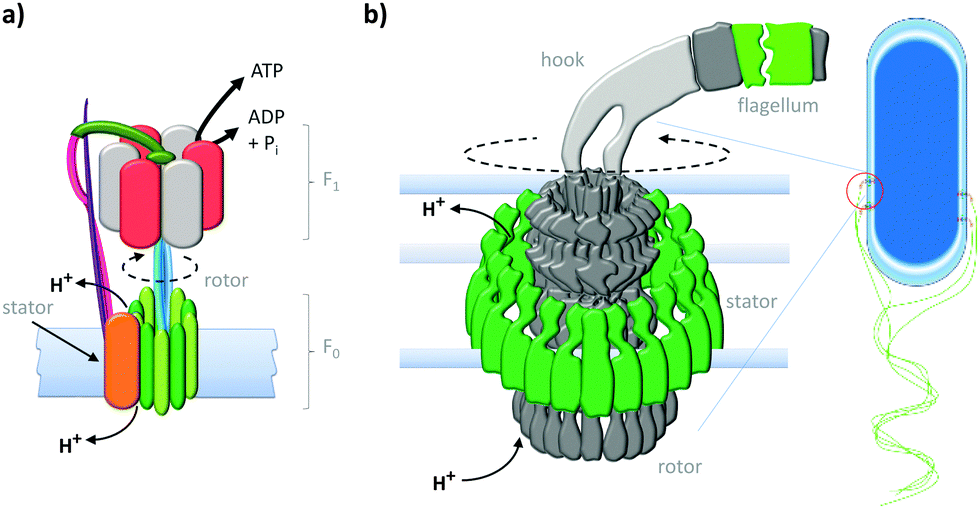 | ||
| Fig. 1 Natural transmembrane molecular machines. (a) Archetypal F1F0 rotary ATP (synth)ase.3,20 (b) Bacterial flagellar motor complex.22 | ||
The equally impressive bacterial flagella motor (Fig. 1b)22 also possesses a transmembrane rotaxane-like architecture and shares other mechanistic characteristics with ATP synthases. Flagellar motors utilise a ratchet mechanism to convert a transmembrane cation gradient into unidirectional motion that drives bacterial motility.22 The flow of cations through the stator (green) transiently changes the electrostatic interaction between the stator and the rotor to generate a torque that drives unidirectional rotation of the flagellum.22
2. Man-made molecular machines; in solution and at interfaces
Natural molecular machines exploit the principles of molecular self-assembly, and thereby provide lessons for the construction of synthetic molecular machines. The underlying operating principles of molecular machines have been thoroughly discussed.5,15 However, the categorisation of a candidate system as a machine can be contentious due to differing interpretations of what constitutes “work” (particularly when energy is partitioned subjectively between different components of a system). Nonetheless, we have suggested one means of categorisation in Fig. 2. Briefly, molecular shuttles flip between multiple states under thermodynamic control as a result of Brownian motion. The result is a time-averaged Boltzmann distribution between the states at equilibrium, with the position of the equilibrium being governed by the relative free energies of those states under differing conditions. A switch differs from a stochastic shuttle and can be defined as having machine-like properties due to the possession of at least two kinetically trapped states that can only be interconverted by the application of a specific stimulus. Work can be extracted from such a system when switching allows the return from a kinetically trapped state to a lower energy equilibrium state. In contrast, a true molecular machine can repetitively and asymmetrically cycle through different nanomechanical states by transducing potential energy into work.5,15 Notably, the changes between distinct states must not simply be the reverse of each other (as they are in a switch). Further distinctions can be drawn between autonomous and non-autonomous molecular machines. Non-autonomous machines rely on externally fluctuating conditions to ratchet asymmetrical cycles of work, while autonomous systems turn over chemical fuel, photons or other potential gradients to perform work continuously. In doing so, autonomous molecular machines generate a locally fluctuating potential, often manifested via ratcheted cycles of conformational change. The ratchet mechanisms that operate in both autonomous and non-autonomous machines can involve information23 or energy.24 Autonomous operation far from equilibrium is a defining characteristic of life exhibited by all biological molecular machines.19 Indeed, achieving comparable autonomous operation in synthetic systems remains a goal of utmost importance.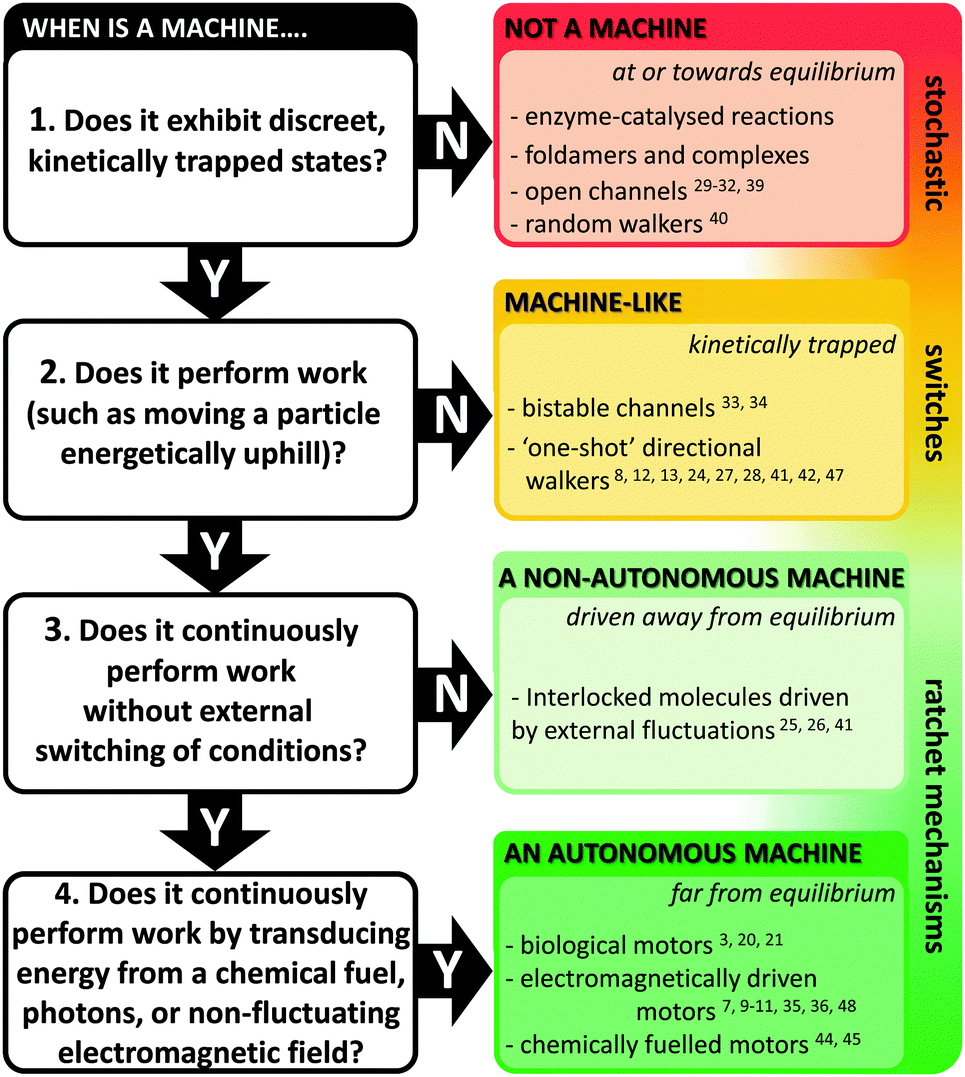 | ||
| Fig. 2 Flow chart for categorising machine and machine-like systems. Superscript numbers indicate examples cited in this review. | ||
A. Mechanically interlocked architectures
Synthetic mechanically interlocked structures such as rotaxanes (that share the same topology as ATP synthases and bacterial flagellar motors) have been designed that exhibit impressive and diverse machine-like behaviours. One recent example mimics the function of the ribosome (or perhaps more closely, biogenic non-ribosomal peptide synthesis).8 The system shown in Fig. 3 was based on a rotaxane in which a linear ‘track’ molecule (blue) was threaded through a macrocyclic host (green). The macrocycle could shuttle back and forth at random between the stoppered end of the track and the site of the first pendant amino acid group on the thread (orange) (Fig. 3i). The macrocycle was covalently linked to a peptide chain, the first residue of which was a cysteine protected by a trityl group (red, STrt). Following deprotection, the thiolate group of the cysteine residue underwent transacylation with the first amino-acid blocking group (orange), removing it from the track. A subsequent reaction between the terminal amine of the peptide chain on the macrocycle extended the peptide chain and regenerated the thiolate group (Fig. 3ii). This process was repeated until the final blocking amino acid was transferred to the growing peptide chain and the macrocycle dissociated from the track (Fig. 3iii and iv). Finally, the complete oligopeptide was cleaved from the macrocycle and the remaining protecting groups removed.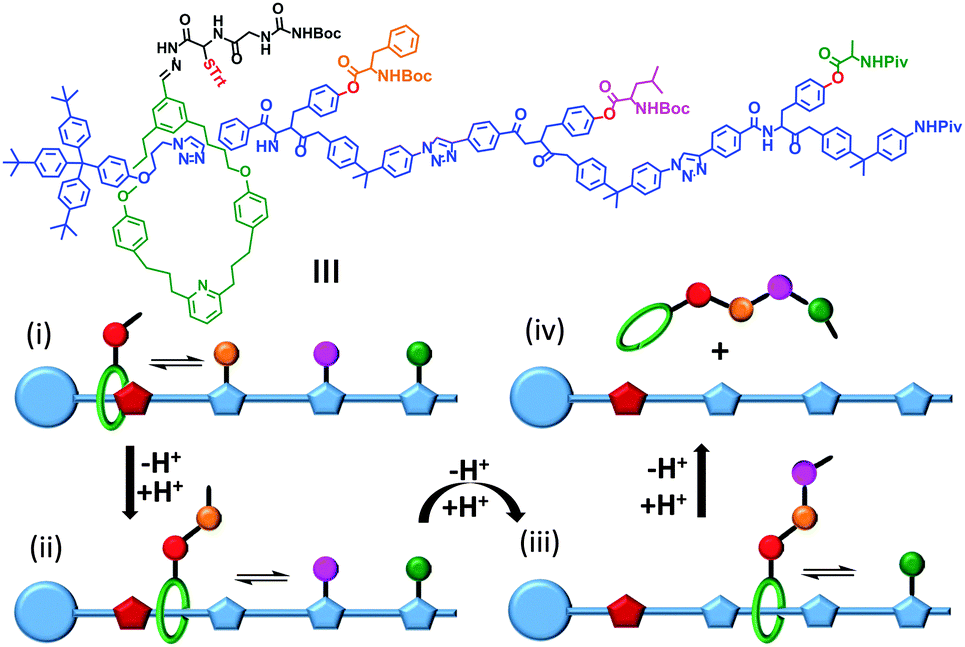 | ||
| Fig. 3 A rotaxane-based peptide synthesiser.8 Trt = CPh3, Boc = CO2t-Bu and Piv = OCOt-Bu. | ||
A topologically interlocked molecule has recently been employed as a rudimentary molecular pump (Fig. 4).25 The system was composed of a molecular axle that could accept up to two macrocycles (blue). When the positively charged viologen units on both the macrocycle and the axle were reduced to radical forms (purple) the repulsion between the macrocycle and the axle was decreased. The macrocycle was then able to thread onto the partially reduced viologen station on the axle. Following threading, the viologen units on the macrocycle and axle were re-oxidised using NOPF6. The electrostatic repulsion between the macrocycle and the positively charged end of the axle was sufficient to overcome the steric barrier presented by the isopropylphenyl group (green) such that the macrocycle became trapped on the stoppered alkyl chain. Repetition of the cycle of reduction, oxidation and reduction allowed a second macrocycle to be ‘pumped’ onto the alkyl chain in an overall energetically uphill process.
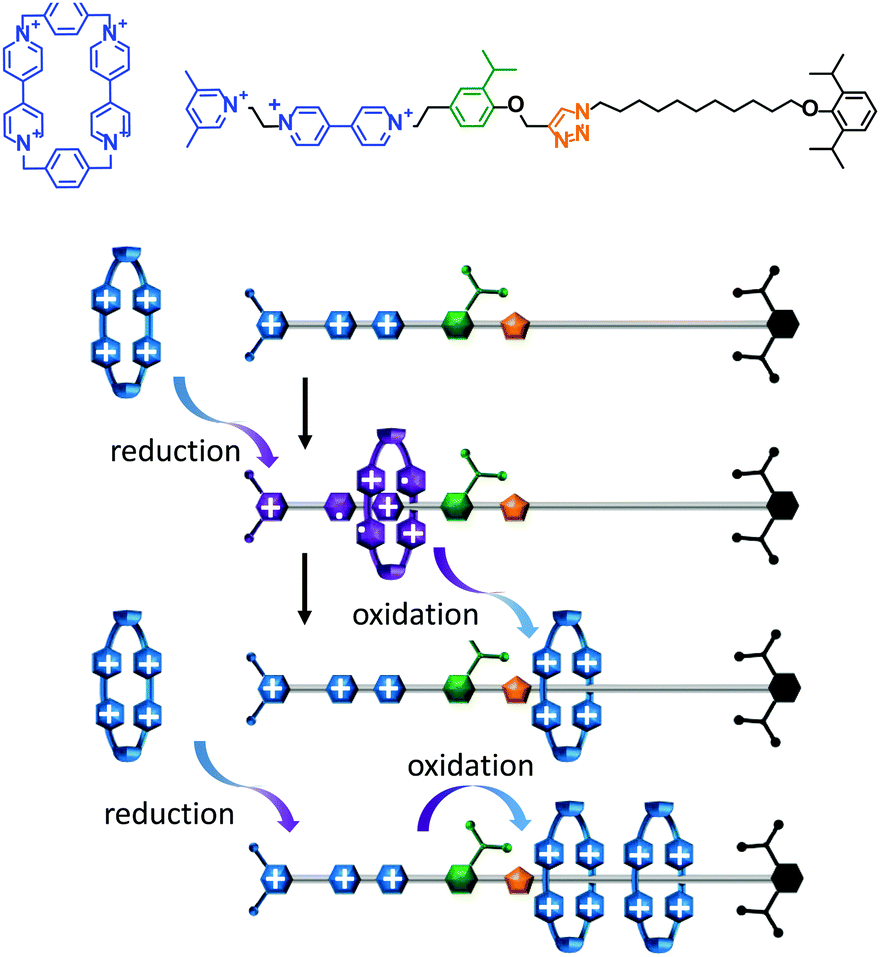 | ||
| Fig. 4 An artificial molecular pump driven by external switching of redox conditions.25 | ||
A key attribute of biological molecular machines is autonomous nanomechanical operation in repetitive cycles that dissipate energy supplied by a chemical or electromagnetic potential. One recent synthetic example that accomplished such dynamism is shown in Fig. 5.7 The system is composed of a 2,3-dinaphtho[24]crown-8 ether macrocycle (purple) and a molecular axle containing a photoisomerisable azobenzene moiety (E green, Z red), a cationic ammonium recognition site (blue) and a cyclopentyl terminus (grey). Under constant UV irradiation, the threading and dethreading proceeded with a directional bias (clockwise in Fig. 5). The macrocycle axle binding equilibria for both the E and the Z axle isomers lay towards the complex, but binding to the E-isomer was more favourable. Control experiments performed with double-stoppered compounds revealed that the barrier to macrocycle threading presented by the E-azobenzene moiety was lower than of the cyclopentyl group, while macrocycle unthreading over the Z-azobenzene moiety was less favoured compared to unthreading over the cyclopentyl group. Moreover, the E-complex was more sensitive to E/Z-photoisomerisation than the free E-axle, while the lifetime of the Z-complex was long enough that dethreading preferentially occurred over the cyclopentyl unit. The net result of the differing energetic barriers to threading/unthreading and the differing rates of isomer interconversion meant that threading and dethreading proceeded with a directional bias under constant photo-irradiation (clockwise in Fig. 5). Although the system operated autonomously in a cyclical manner with molecular-level directionality, this directionality was lost when summed over the random orientations of the freely tumbling assemblies in solution. However, if it were possible to directionally orientate such a system across a lipid membrane then this would constitute a man-made molecular pump.
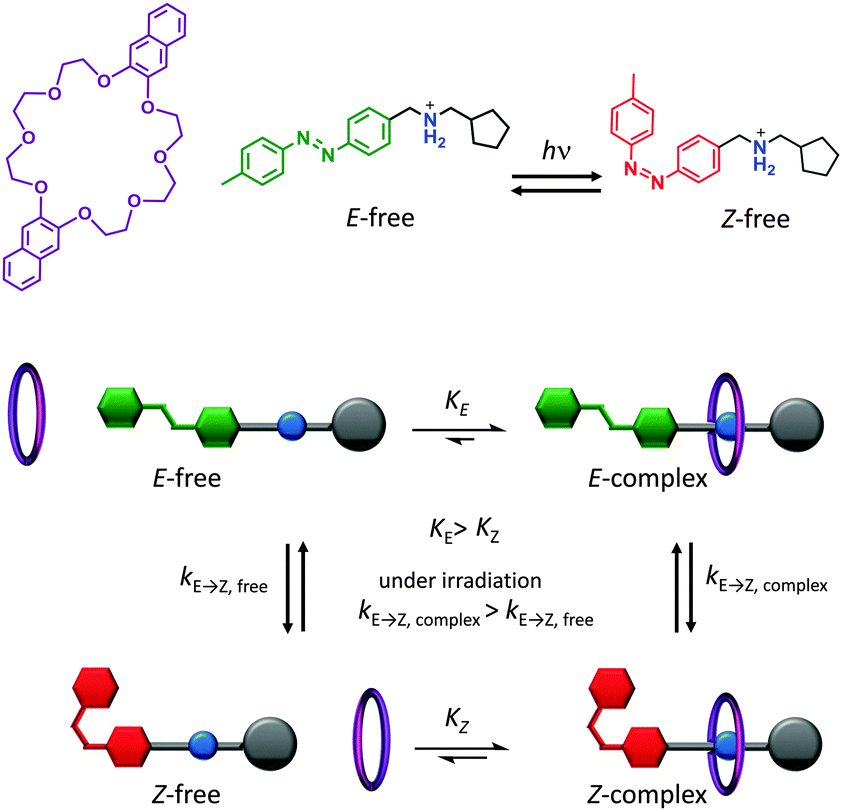 | ||
| Fig. 5 Light-driven autonomous cycle of unidirectional supramolecular assembly/disassembly.7 | ||
The challenge of extracting work from the controlled motion of mechanically interlocked species has been addressed by attaching them to surfaces. This reduces the degrees of freedom such that changes in the nanomechanical states of a large number of individual molecules may be manifested as a coherent change in the macroscopic properties of the surface. One demonstration of the extraction of macroscopic work from the collective nanomechanical movements of rotaxanes anchored to a surface is shown in Fig. 6.13 A bistable [3]rotaxane was prepared using two macrocycles (blue) that each featured a disulfide anchor (gold) threaded onto a stoppered thread containing two naphthalene stations (red) and two electroactive tetrathiofulvalene stations (green). The [3]rotaxanes formed a self-assembled monolayer on a gold cantilever via the disulfide anchors on the macrocycles. Oxidation of the tetrathiofulvalene stations to their cationic form (purple) using Fe(ClO4)3 resulted in electrostatic repulsion between the cationic tetrathiofulvalene stations and macrocycles. Lateral displacement of the cantilever beam of 35 nm was measured due to the force imparted by the collective contractions of the self-assembled monolayer.
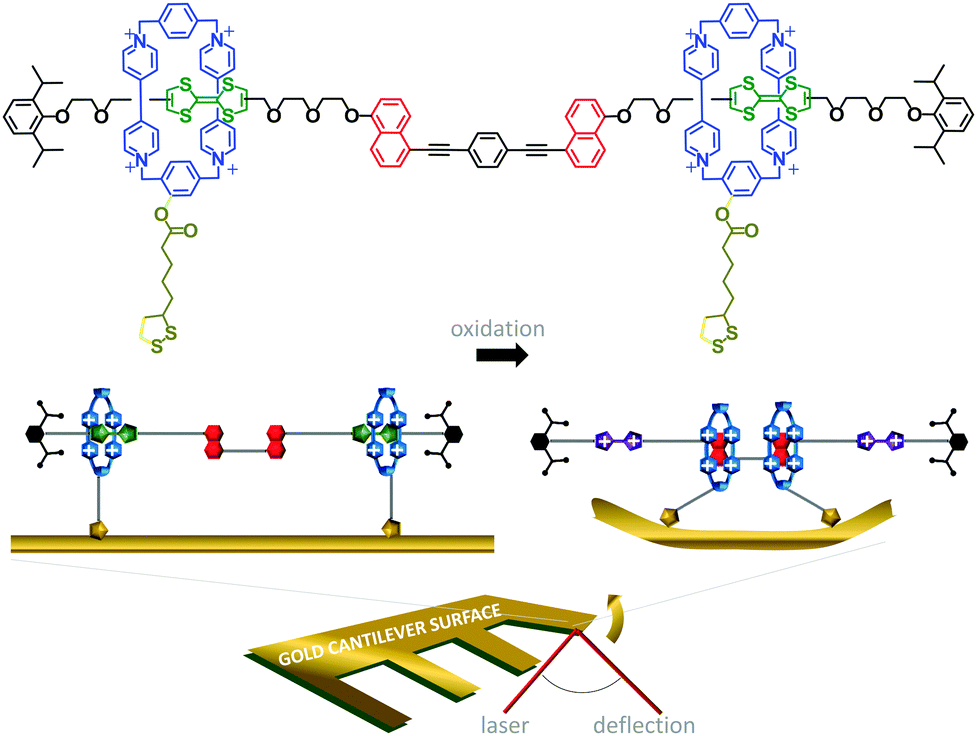 | ||
| Fig. 6 Cantilever actuation driven by redox switching of surface-bound rotaxane shuttles.13 | ||
B. Synthetic rotary motors
In contrast to switches, rotary motors operate by cycling through a number of distinct states in response to energetic stimulation or the availability of a suitable fuel. Such behaviour has been demonstrated in a fully synthetic catenated architecture (Fig. 7).26 The system was composed of two identical macrocycles (blue and purple) that were both threaded on a larger macrocycle containing four distinct binding sites (A to D). Each binding site exhibited different association constants for the threaded macrocycles due to their differing H-bonding abilities. Photoirradiation at 350 nm converted station A from the E to the Z isomer (1), while photoirradiation at 254 nm converted station B from the E to the Z isomer (2). Both stations A and B could be re-isomerised from Z to E (3) by heating to 100 °C for 24 hours, or by treatment with ethylenediamine at 50 °C for 48 hours or Br2 under ambient light at −78 °C for 10 minutes. Repeated, sequential application of stimuli (1), (2) then (3), resulted in the net unidirectional rotational movement of both small macrocycles with respect to the larger macrocycle. Although the system can be defined as performing local work, no work can be extracted as the rotors are freely tumbling in solution.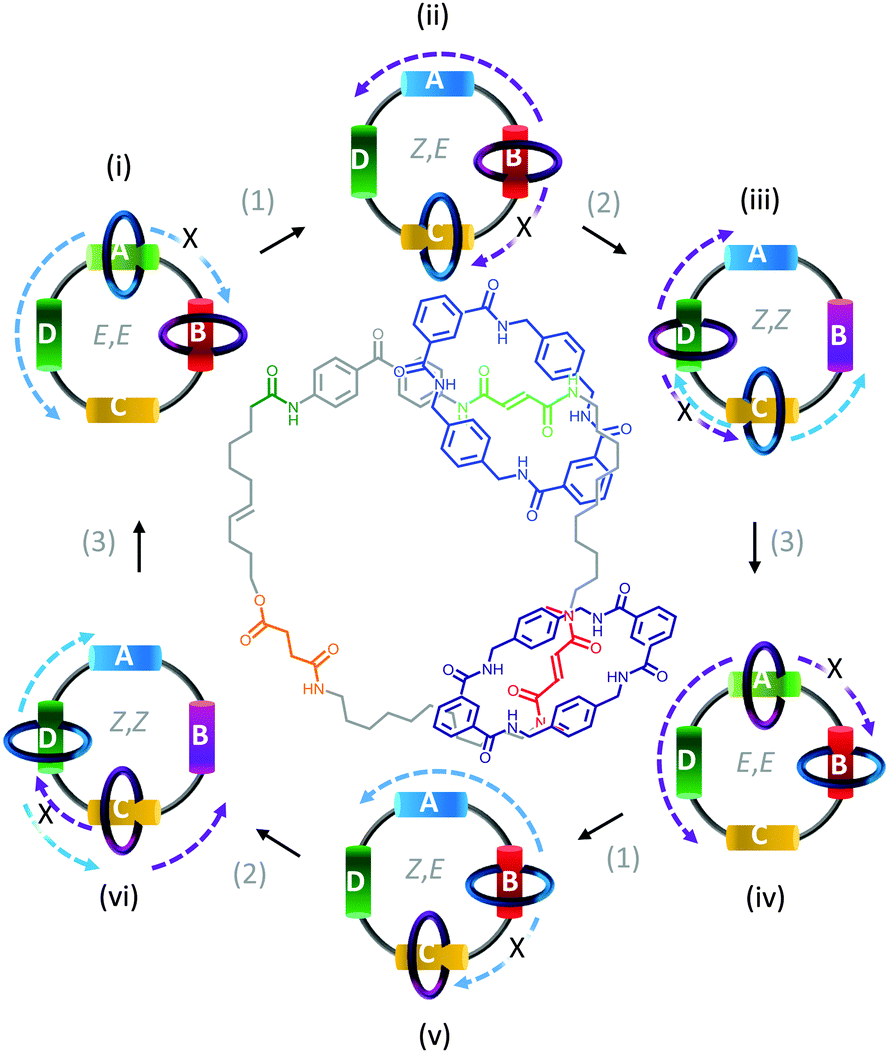 | ||
| Fig. 7 Unidirectional rotation of catenated rings driven by switching of external light and chemical conditions.26 | ||
Synthetic rotary motors have also been developed that can be driven by light (Fig. 8).9 These systems exploit repeated cycles of E/Z photoisomerisation of the central double bond and subsequent helicity inversion by thermal relaxation to convert between conformational states. Upon irradiation at 355 nm the central double bond photoisomerises from the most stable cis conformer (i) to a higher energy trans conformer (ii). Next, favourable helicity inversion facilitated by thermal fluctuations gives the stable trans conformer (iii). A second photo-excitation converts the compound to a higher energy cis conformer (iv) that undergoes thermal helicity inversion to return the system to the initial conformational state. Hence, under constant irradiation these motors rotate unidirectionally. Due to the diastereomeric relationship between each half of the molecule, retrograde motion does not occur because of the substantially higher barrier to re-isomerisation from the unstable state 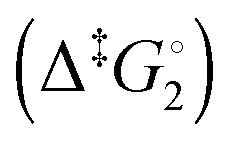 compared to the barrier height for helix inversion
compared to the barrier height for helix inversion 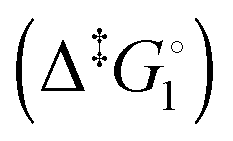 . Variation of the X, Y and R groups has yielded motors that rotate autonomously under constant irradiation at remarkable rates of up to 3 MHz!9 Although each motor molecule generates a local torque, work cannot be extracted from an ensemble of freely tumbling, randomly orientated molecules in solution. Once again, the key to the extraction of work from these molecular rotors was the involvement of a surface. In one study such motors were used as dopants in a chiral liquid-crystal film which caused the unidirectional rotation of microscale objects when photoirradiated.10
. Variation of the X, Y and R groups has yielded motors that rotate autonomously under constant irradiation at remarkable rates of up to 3 MHz!9 Although each motor molecule generates a local torque, work cannot be extracted from an ensemble of freely tumbling, randomly orientated molecules in solution. Once again, the key to the extraction of work from these molecular rotors was the involvement of a surface. In one study such motors were used as dopants in a chiral liquid-crystal film which caused the unidirectional rotation of microscale objects when photoirradiated.10
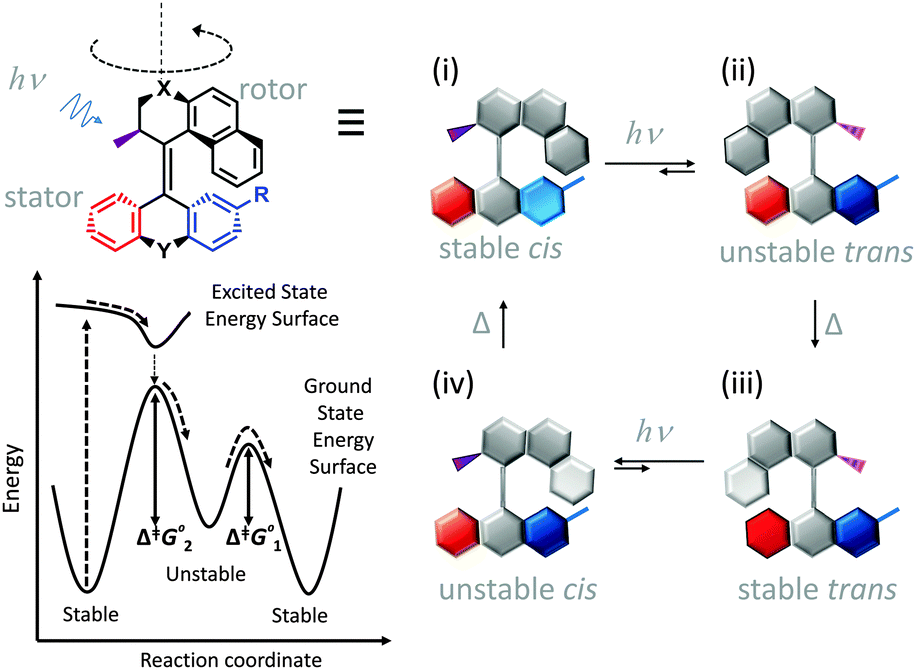 | ||
| Fig. 8 Light-driven unidirectional intramolecular rotation of a synthetic molecular motor.9 | ||
The unidirectional rotation of bisarylidene rotors has also been transduced into directional linear motion (Fig. 9).11 When a car-like molecule containing four bisarylidene rotors was stimulated with electrons from an STM tip, the concerted motion of the rotors caused it to move along the surface. The net movement of each molecule was determined by the relative stereochemistries of the four rotors. The R,R,S,S meso-isomer shown in Fig. 9 gave unidirectional translation, as did the S,S,R,R configuration. The R,R,R,R and S,S,S,S isomers moved randomly while R,S,R,S and S,R,S,R did not move due to the cancellation of directionally opposed motions.
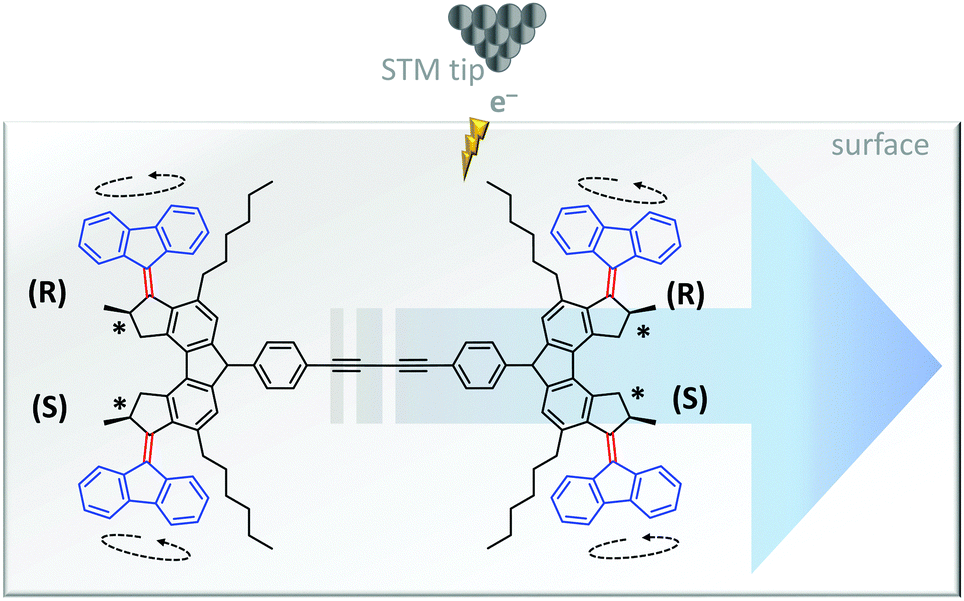 | ||
| Fig. 9 Unidirectional linear translocation of a molecule containing bisarylidene rotors.11 | ||
C. DNA-based molecular machines
Nature's molecular machines certainly exceed our own in terms of elegance, modularity and functionality. Bionanotechnological research has sought to exploit the principles of molecular recognition to repurpose biomolecules such as DNA and proteins as machine elements.16 Many of the achievements of wholly synthetic molecular machines, including programmable walkers27 and molecular synthesisers28 have also been accomplished by synthetic molecular machines based on DNA (sometimes years previously). Such DNA nanomachines exploit the programmable hydrogen-bonding interactions of canonical base pairing. One of the most impressive demonstrations of a DNA-based molecular machine is a peptide synthesiser based on a DNA walker (Fig. 10).28 The system operated in a stepwise fashion reminiscent of the naturally occurring ribosome. The DNA track (black) and the strands bearing the amino acid monomers (blue, red and green) were first assembled in solution, and the walker (purple) subsequently added. Next, the walker could hybridise to the first DNA foothold (blue) bringing the free amine on the walker and the succinyl ester of the first foothold into close enough proximity to initiate an acylation reaction. The reaction caused the walker to fully migrate onto the blue foothold to reduce strain in the system. In this configuration, a DNAzyme region of the walker (purple) was able to catalytically cleave an RNA linker sequence (black dot). Since the first foothold (blue) had now been shortened by cleavage, binding to the subsequent (red) foothold was favoured. Repetition of this sequence of events facilitated the autonomous synthesis of a tripeptide. Overall, the system compares favourably to the more recent fully synthetic peptide synthesiser introduced in Fig. 3; it is easily constructed and operates under isothermal conditions. However, in common with the synthetic peptide synthesiser (and unlike the biogenic ribosome) processivity is limited because the effective molarity of the reacting groups decreases with each amino-acid addition step. Furthermore, the fidelities of the man-made systems are limited by the increased flexibility of longer chains, which might allow monomers to be added in an unintended order.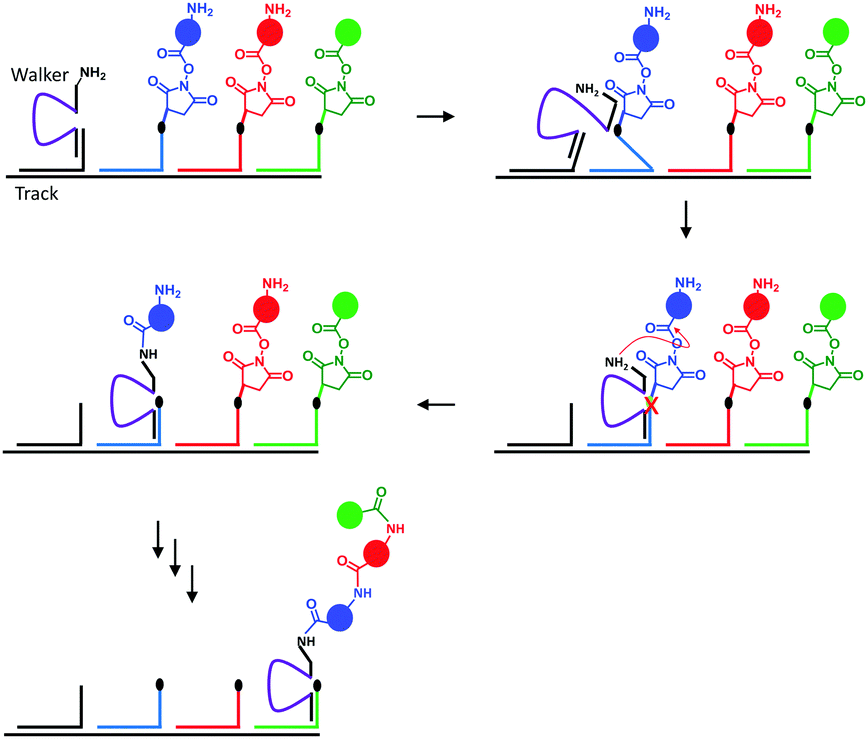 | ||
| Fig. 10 A peptide synthesised based on a DNA walker.28 | ||
3. Man-made transmembrane molecular machines
In biology, lipid bilayers provide a route to system complexity. Self-assembly facilitates the spatial organisation of discrete entities with specific orientations within a membrane. This includes selective, stimuli-responsive channels that operate at equilibrium, as well as active transmembrane pumps that operate far from equilibrium to transduce energy into useful work. Efforts to mimic the functionality of natural transmembrane systems have mirrored this progression in complexity; from passive (Section 3A) to selective channels (Section 3B), to light-driven (Section 3C) and nanomechanical transmembrane assemblies that can operate far from equilibrium (Section 3D).A. Artificial transmembrane channels
Lipid membranes provide an attractive platform for the construction of man-made molecular machines; bilayers are easily prepared and they are inherently biocompatible. Furthermore, they permit the spatial organisation of self-assembling biological and synthetic components in the realisation of man-made transmembrane devices. For example, both cyclic peptides (Fig. 11, left)29 and DNA (Fig. 11, right)30 have been used to create stable artificial nanopores across lipid bilayers. Both of these assemblies have dimensions close to those of natural protein channels and permit the passage of ions down a transmembrane potential or concentration gradient. The cyclic peptides (green) self-assemble into membrane-spanning tubes as they partition into the membrane. The assembly process was reasoned to occur by the stacking of aromatic side chains within the hydrophobic membrane interior, while the inside of the tubes present the polar amide backbone to the solvent. Similarly, the DNA origami nanopore was designed such that it was anchored into the bilayer via cholesterol-modified helix bundles (grey and red). The machine-like, voltage-sensitive gating (opening and closing) of the DNA-origami channels was also investigated. A channel variant bearing a strand that could readily adopt a channel-blocking conformation in response to high transmembrane potentials was observed to gate more than the original channel. Similarly, DNA origami tiles have been designed that modulate the flow of ions and biomolecules across channels at the ends of glass capillaries.31 Given the maturity of DNA nanotechnology,16,27 it can be envisioned that such approaches will soon develop beyond switchable channels to yield true transmembrane molecular machines capable of performing work (such as active transport).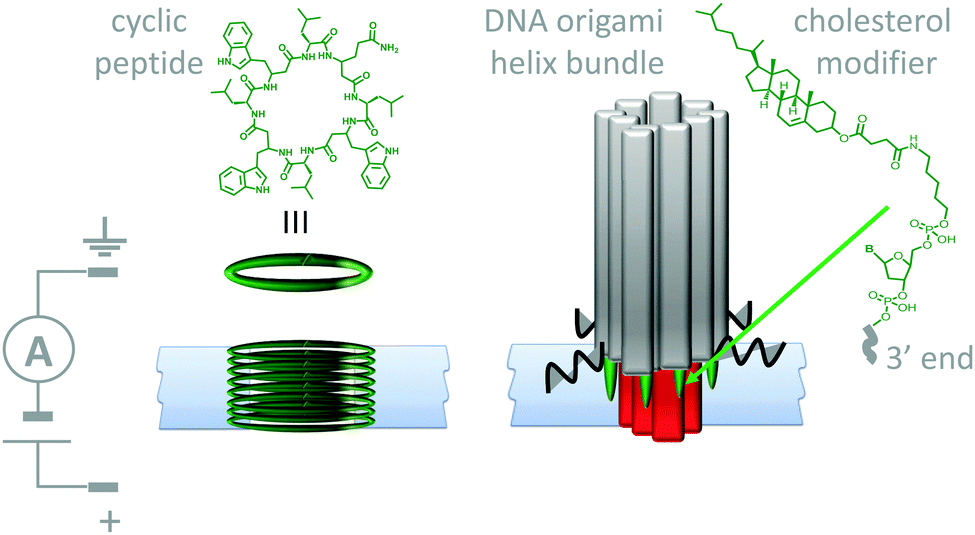 | ||
| Fig. 11 Artificial nanopores in lipid bilayer membranes made using cyclic peptides (left)29 and DNA origami (right).30 | ||
B. Ligand-responsive transmembrane channels
Biological nanopores have also inspired the development of systems in which micro- to nano-scale apertures in solid-state wafers separate fluid-filled compartments. These artificial apertures can be functionalised to give tuneable systems that respond to stimuli such as voltage, pH, temperature, light, and even specific ions and enantiomers.18 Furthermore, selective transport across a partition can be achieved by exploiting the interactions between an analyte and the molecules used to functionalise the aperture. Although this machine-like behaviour mimics cellular function, most approaches developed to date act as simple gates or switches that are incapable of performing work such as establishing a chemical gradient.The most sophisticated systems employing this top-down approach facilitate the simultaneous translocation and analysis of macromolecules. One such system mimics the nuclear pore complex (Fig. 12)32 and consisted of a nanopore formed in a polycarbonate wafer (grey) coated with gold and further modified with phenylalanine–glycine–nucleoporin proteins (blue). The membrane-like wafer acted as a selective filter that facilitated the diffusion of specific, free and cargo-bound transport factors (red) through the pore via favourable non-covalent interactions with the nucleoporins. In contrast, passive transmembrane diffusion of similarly sized molecules (green) that were not recognised by the nucleoporins was hindered. One obvious difference between many solid-state and biological channels is their size. This difference arises from the difficulty in reproducibly obtaining nanometre-scale features using top-down engineering techniques. Channel size is important because the interactions between an aperture and an analyte become more pronounced when they have similar dimensions, and hence the likelihood of departure from simple diffusive behaviour increases.
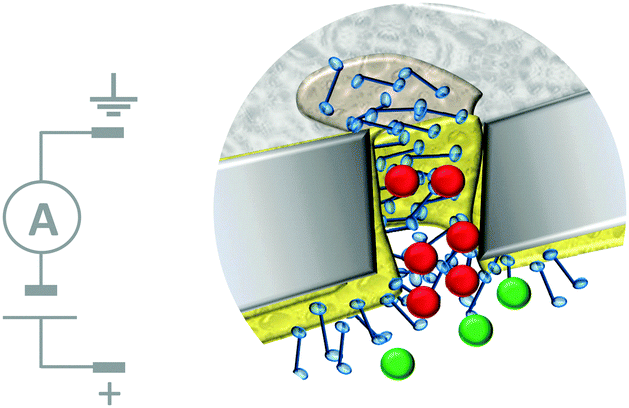 | ||
| Fig. 12 Artificial nanopore system that mimics the nuclear pore complex.32 | ||
Molecularly reproducible membrane-spanning devices can be obtained by repurposing biological pore proteins and modifying them as needed. For example, a protein chimera (Fig. 13) that combined a K+ channel protein (yellow) with a G-protein-coupled receptor (red) has been engineered.33 Analysis of ion currents flowing through the chimeric protein channels indicated that the ion channel opened upon binding of a known ligand (green) to the G-coupled-receptor. Thereby, artificial transmembrane signal transduction was achieved from a chemical signal without the molecule itself crossing the membrane.
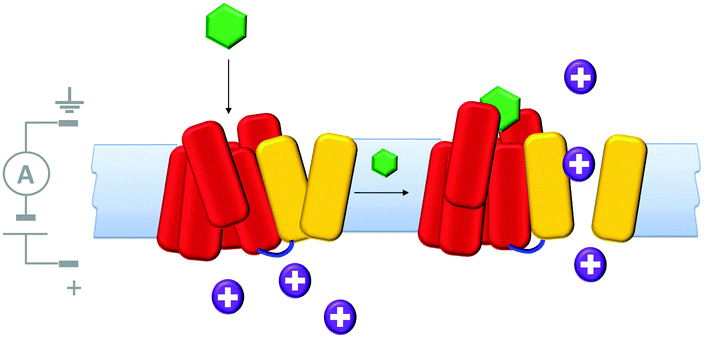 | ||
| Fig. 13 A man-made ligand-gated transmembrane ion channel formed from the chimera of a K+ ion channel (yellow) and G-protein-coupled receptor (red).33 | ||
C. Light-driven transmembrane devices
A number of devices that use light either as an external stimulus or as an energy source to drive transmembrane processes have been demonstrated. Synthetic photoactive functionalities that undergo conformational changes have been used in conjunction with biological channel proteins to facilitate optical control over membrane transport. One example of this approach involved engineering the mechanosensitive channel of large conduction (MscL) from E. coli (Fig. 14).34 The protein (yellow) was modified to have one cysteine residue per monomer allowing the incorporation of a photo-responsive cyanine group that could be reversibly switched between a neutral spiropyran form (red) and a charged zwitterionic merocyanine form (purple). When 366 nm UV light was shone on a membrane containing the chemically modified channels, a large-scale conformational change caused the channel to open, and permitting the flow of ions across the membrane under an applied transmembrane potential. The conformational change was fully reversed by ambient light at wavelengths >460 nm. Hence, light-actuated gating could be performed repeatedly by turning the UV light on and off.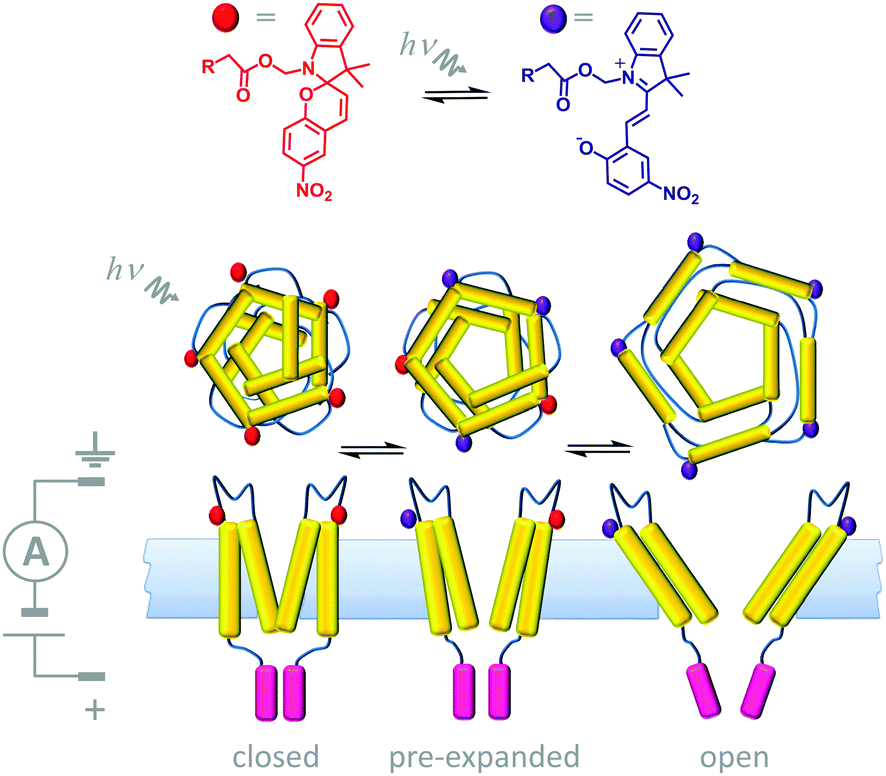 | ||
| Fig. 14 Semi-synthetic light-gated ion channel prepared from a chemically modified mechanosensitive channel protein.34 | ||
ATP synthase has also been used as a component in a variety of artificially assembled devices. The key to achieving non-equilibrium properties in the example shown in Fig. 15 was the addition of a diphenyl quinone (QS) and a photoactive carotene–porphyrin–naphthoquinone (C–P–Q) (Fig. 15).36 The synthetically functionalised membrane was shown to generate a transmembrane proton gradient in response to visible light, which could be exploited to drive the rotation of ATP synthase and the associated synthesis of ATP (as explained in Fig. 1a).
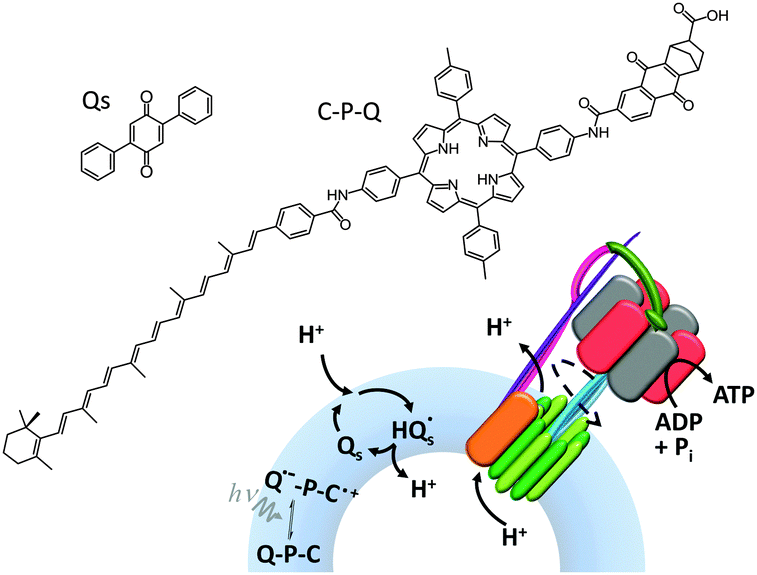 | ||
| Fig. 15 An artificial photosynthetic vesicle.36 | ||
A system that combines both ligand-mediated channel gating and the mimicry of transmembrane photosynthesis has been developed from wholly synthetic molecules (Fig. 16).35 The system comprised four p-octiphenyl chains (maroon), each bearing eight naphthalene-diimides (green). The complex self-assembled in lipid bilayer vesicles such that the π-surfaces of the naphthalene-diimides stacked to give a canted-barrel structure. In this geometry, irradiation of the complex at 638 nm caused electronic excitation that resulted in a transmembrane charge-separated state. The generated electrons and holes were used to drive a redox couple (reduction of quinones and oxidation of EDTA on opposite sides of the bilayer), maintaining the charge-separated state and generating a transmembrane proton gradient. Finally, the addition of an intercalating naphthalene derivative (yellow) switched the barrel into an open conformation, thereby permitting neutralisation of the accumulated proton gradient by the flow of hydroxide ions from outside the vesicle.
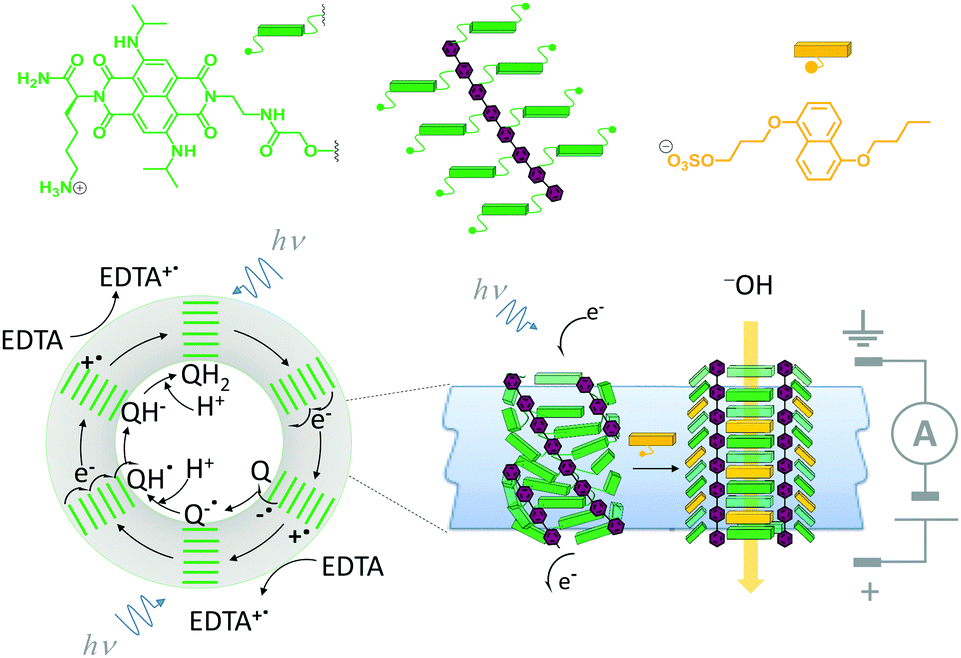 | ||
| Fig. 16 A self-assembled synthetic transmembrane electron pump that can also open in response to ligand binding.35 EDTA = ethylene diamine tetraacetate, Q = quinone. | ||
D. Nanomechanical motions in transmembrane devices
Although macroscopic mechanistic principles do not describe nanoscale processes,15 machine behaviour on the molecular level is most easily recognised where well-defined nanomechanical motions occur. Nanomechanical processes have been widely explored using biological nanopores. In particular, protein nanopores have found applications in single-molecule biophysics37 and in single-molecule DNA sequencing.38 In many cases nanopore-based detection methods are facilitated via nanomechanical processes such as the translocation of single-stranded DNA. For example, the connector protein associated with the ϕ29 DNA-packaging motor has been modified with a band of hydrophobic residues (yellow) such that it could form channels across lipid bilayers (Fig. 17).39 In this study, the DNA-packaging motor protein was not present, and the translocation of DNA through the channel was driven by the application of a transmembrane voltage.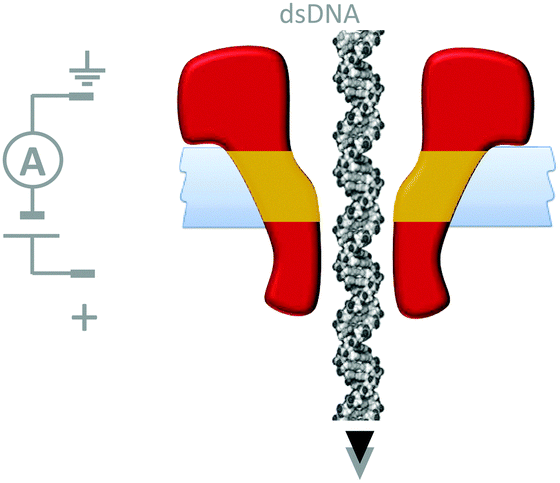 | ||
| Fig. 17 A membrane-adapted ϕ29 connector protein that permits the voltage-driven passage of double-stranded DNA across a membrane.39 | ||
Although open protein channels are not molecular machines, they can provide useful chassis for the construction of transmembrane nanomechanical devices. Mirroring earlier advances in the field of synthetic molecular walkers, the random walk of a small molecule inside a protein nanopore has been observed (Fig. 18).40 The approach employed a mutant α-haemolysin (α-HL) nanopore that contained five cysteine residues within the transmembrane stem. The molecular walker was formed in situ by the reaction of 4-sulfophenyl arsenous acid and 2-(2-methoxyethoxy)ethanethiol. Changes in the transmembrane ion current flowing through the pore revealed that the arsenous molecule walked stochastically by reversible formation of covalent bonds between the arsenic of the walker and the cysteine residues in the transmembrane stem. The motion of the walker was biased by the direction of the applied potential to move towards a thermodynamic sink.
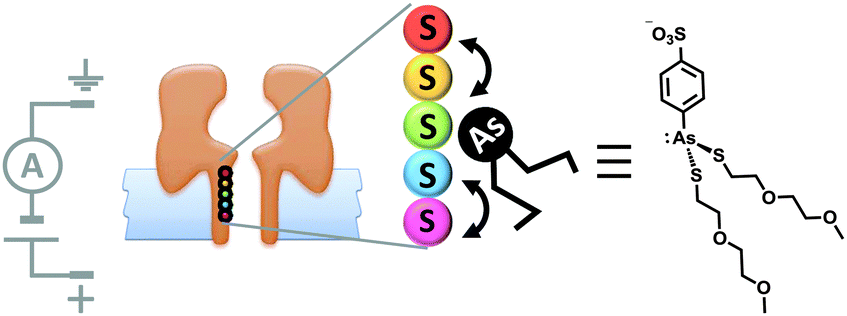 | ||
| Fig. 18 Stochastic walking of a small molecule within a transmembrane nanopore.40 | ||
Another example of machine-like behaviour was recently demonstrated using a DNA-modified cytolysin A nanopore (Cly A) that facilitated the selective transport of DNA across a membrane (Fig. 19).41 The mutant pore was covalently modified with single-stranded DNA ‘arms’ (black) that recognised a specific target DNA complex (yellow and purple) that was present on one side of the bilayer. Complexation to the arm increased both the negative charge density and the effective molarity in the vicinity of the pore compared to the free target. The combination of these two effects meant that the barrier to DNA translocation could be overcome at an applied voltage of +50 mV. Next, complexation of streptavidin (green circle) to the biotin group on the target sequence trapped the translocated DNA on that side of the membrane. A DNA strand (pink) was then able to displace the cargoes into solution via a strand displacement process. The free DNA arm was then able to diffuse back through the pore, allowing further cycles to occur. This system has several characteristics of a machine; it operates autonomously, cycling directionally through multiple nanomechanical states via an information ratchet mechanism,23 while also consuming a strand-displacing DNA fuel. Although DNA transport proceeded at a voltage that would normally be too low to overcome the energy barrier to translocation, DNA transport occurred in the same direction as the applied field (down the potential energy landscape). Thus, the translocation of DNA by this mechanism does not constitute work performed by the nanomechanical cycle (Qn. 2 in Fig. 2). Hence, the system can be considered as a catalytic translocase that lowers the activation barrier to a thermodynamically downhill process, rather than being an active transporter, pump or motor.
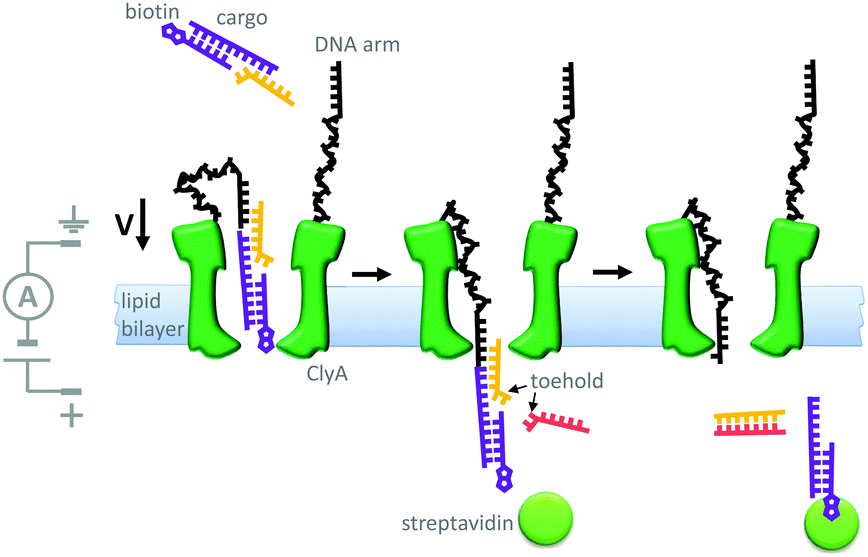 | ||
| Fig. 19 A transmembrane DNA transporter based on the cytolysin A pore protein.41 | ||
Indeed, motor proteins such as DNA polymerases have been employed in conjunction with biological pores to create transmembrane nanomechanical devices with machine-like properties. For example, the replication of DNA hairpins by ϕ29 DNA polymerase has been monitored at the single-molecule level using nanopores (Fig. 20).42 In this approach, a DNA-hairpin (black) was captured in an α-HL channel (blue) under an applied potential. The turn-over of dNTP fuel during extension of the self-primed hairpin by the polymerase (yellow) resulted in the captured DNA being ratcheted out of the pore against the applied transmembrane potential. The DNA hairpin contained an abasic region (red) that allowed the relative position of the strand in the pore to be determined from changes in the ion current flowing through a single channel.
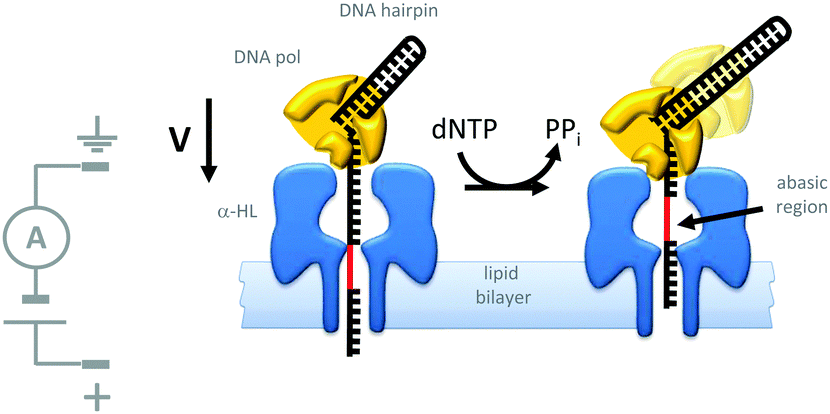 | ||
| Fig. 20 Polymerase-catalysed replication of DNA monitored at the single-molecule level using a nanopore.42 | ||
A similar approach has been used to study the ratcheting of a protein through an α-HL nanopore by the motor protein AAA+ unfoldase (ClpX in Fig. 21).43 The protein Smt3 (purple) was modified with a charged polypeptide tail (green) terminating in an ssrA recognition sequence (yellow). This modified protein was threaded through and captured within an α-HL nanopore by an applied transmembrane potential. The ssrA sequence was then recognised by the unfoldase (red) that was present on the opposite side of the lipid bilayer. Cycles of conformational change in the unfoldase driven by the turn-over of ATP allowed the captured protein to be unfolded and pulled through the pore (green to purple). The unfolding process was monitored via ratcheted changes in the ion current flowing through the pore under an applied potential.
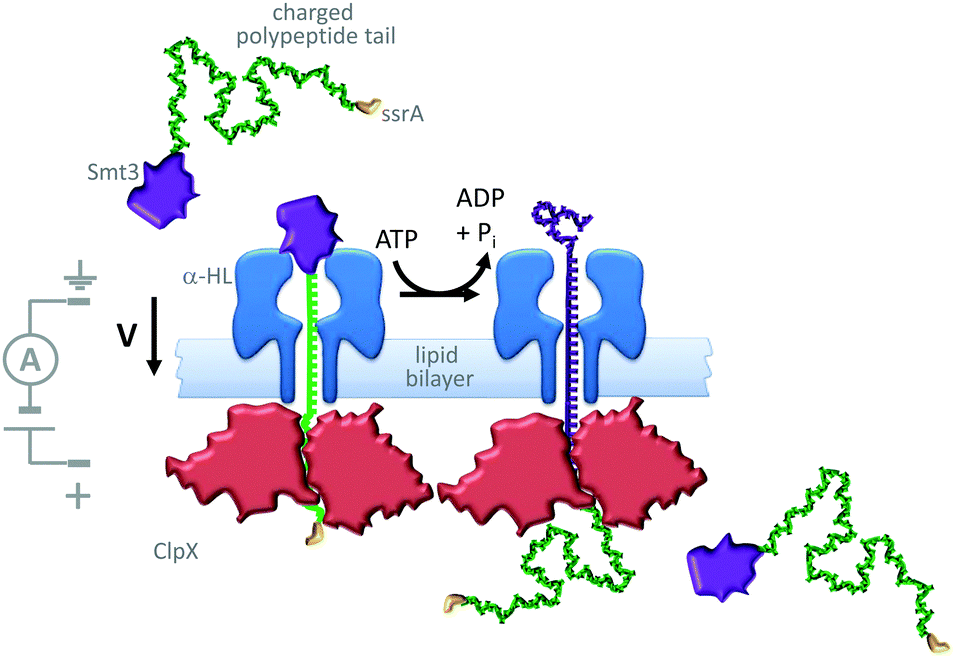 | ||
| Fig. 21 ATP-coupled transmembrane protein translocation through α-HL.43 | ||
Further efforts to create synthetic transmembrane molecular machines have employed protein chimeras. A recent, impressive example appended loops of the cochaperonin protein GroES (green) onto the α-HL nanopore (blue) (Fig. 22).44 Natively, GroES forms a complex with GroEL to yield a catalytically active chaperonin that turns over ATP to drive a series of conformational changes that mediate the folding of proteins (yellow) within the internal cavity of the GroES–GroEL complex. Since both the GroES–GroEL complex and the α-HL share a seven-fold symmetry, the α-HL–GroES chimera was able to template the assembly of an active GroEL heptamer (pink). The ATP-driven chaperonin function of this engineered transmembrane assembly was characterised using gel-shift experiments in the bulk, while dynamic activity was monitored at the single-molecule level using transmembrane ion currents.
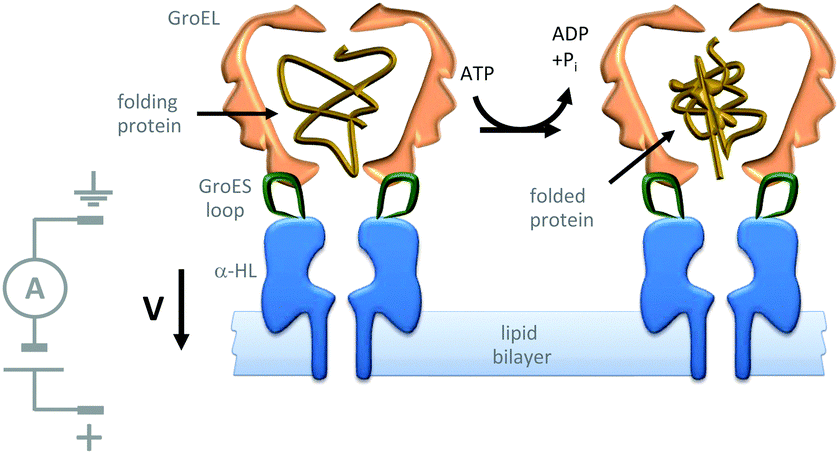 | ||
| Fig. 22 Single-molecule observation of the ATP-fuelled folding of a protein using an engineered transmembrane co-chaperonin.44 | ||
The assemblies described in Fig. 20–22 were used to monitor the machine behaviour of known motor proteins; a further milestone has been the realisation of systems in which machine behaviour is an emergent quality possessed by the entire assembly. This goal was recently attained in the form of a transmembrane nanoactuator that turned over DNA molecules to fuel cycles of autonomous reciprocating (back-and-forth) nanomechanical motion (Fig. 23).45 The approach built on the precedent set by a nanopore-based system capable of monitoring DNA polymerase activity with single-nucleotide resolution.46,47 The device was assembled by threading a DNA–PEG copolymer (black-red) capped with a streptavidin–biotin stopper (green circle) through an α-HL nanopore under an applied potential. The captured co-polymer was then trapped in place by hybridisation to a fully complementary DNA primer (purple-green) to form a rotaxane-like transmembrane architecture. In the presence of DNA polymerase, pyrovanadate, Mn2+ and DNA fuel molecules, ion-current recordings revealed asymmetric saw-tooth patterns. These patterns corresponded to an emergent cycle of back and forth movement in the threaded co-polymer at rates of up to one cycle per minute. Stepwise ‘downwards’ movement of the threaded co-polymer (left to right in Fig. 23) arose due to DNA polymerase-catalysed degradation of the primer fuel, while displacement of the degraded primer by the invasion of a full-length primer pulled the threaded co-polymer ‘upwards’ in a single step (against the applied potential). The asymmetric conformational cycle was co-ordinated via an information ratchet mechanism since strand displacement could only occur once the primer had been sufficiently degraded.
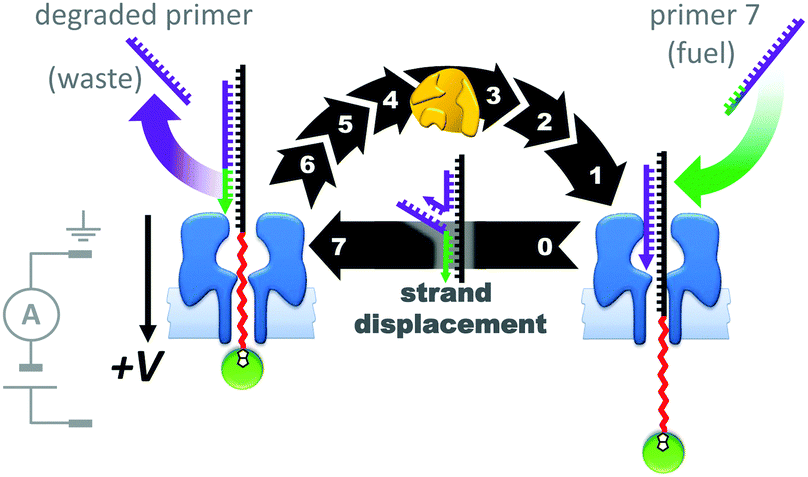 | ||
| Fig. 23 A transmembrane reciprocating nanoactuator that operates with a temporally asymmetric nanomechanical cycle.45 | ||
E. Multi-compartment transmembrane devices
Contrasting with the isolated devices described above, higher-order living organisms are hierarchically organised into multiple chemically distinct environments by lipid bilayers. This partitioning allows the generation of transmembrane potentials by molecular machines working in different compartments, giving rise to emergent collective properties. Artificial systems have been developed that mimic the multi-compartmental nature of living organisms. For example, aqueous droplets can be submerged in oil and arranged into networks in which bilayers form at the interfaces between the drops (Fig. 24). The addition of proteins that insert into the bilayers connecting these droplet allows ions and analytes to pass between the droplets in a designed manner. In addition, the approach allows these processes to be monitored by both electrical and optical methods. Applications have included a rudimentary light sensor that mimics optical, to chemical, to electrical signal transduction in animal sight (Fig. 24).48 The sensor consisted of three droplets containing bacteriorhodopsin that were each connected to a common electrode. These three droplets were interfaced with a central drop containing only buffer. A fifth drop containing α-HL (green) was also interfaced with the central drop and connected to a counter electrode. The bacteriorhodopsin and the α-HL were then able to insert into the bilayer interfaces with the central droplet. Photoirradiation caused the bacteriorhodopsin to pump protons across the bilayer, leading to a measurable electrical current arising from the flow of ions through the droplet circuit.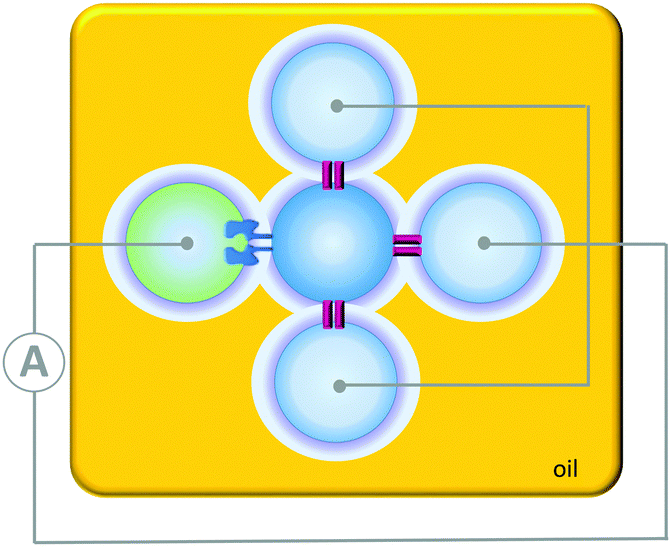 | ||
| Fig. 24 A simple circuit constructed from a droplet interface bilayer network connected by an α-haemolysin pore and bacteriorhodopsin.48 | ||
The droplet network approach has been further expanded by the generation of higher-order ‘multisomes’ that can operate in aqueous environments rather than in oil baths (Fig. 25).49 The assemblies were formed from multiple aqueous compartments that were organised, but not completely encapsulated within a larger droplet of oil. The aqueous droplets had distinct internal compositions (indicated by the different colours in Fig. 25) enabling distinct functionalities to be presented to the surrounding environment. In this manner, different droplets could respond to different stimuli in distinct ways. In addition, the aqueous droplets could be connected at their internal interfaces by the inclusion of transmembrane pores such as α-HL. Thus, these multisomes were able to engage with their environment by sensing, processing and responding.
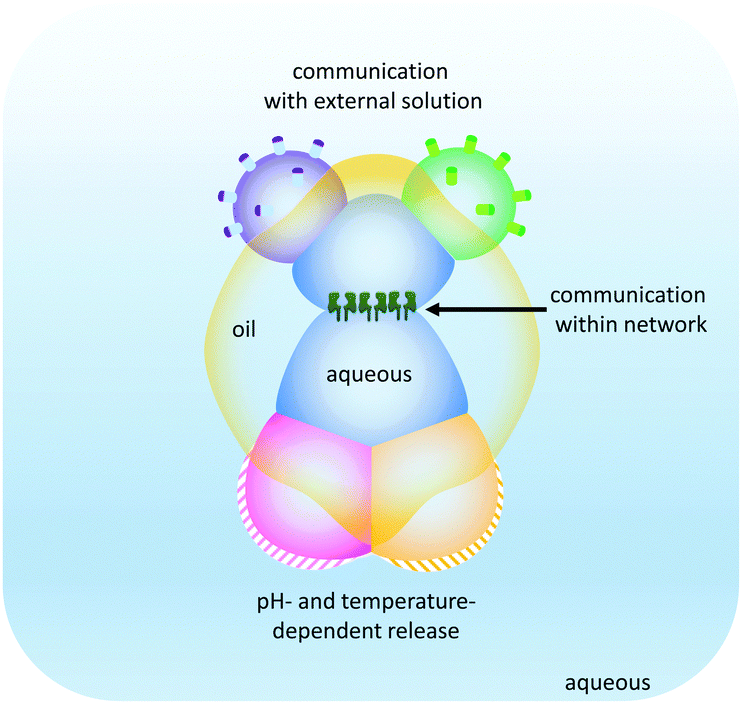 | ||
| Fig. 25 Droplet multisomes that operate in aqueous environments.49 | ||
The droplet bilayer approach has been combined with 3D-printing technology to produce heterogeneous droplet networks that mimic some of the properties of living tissues (Fig. 26).50 Sub-microlitre droplets were ‘printed’ in three dimensions within an oil droplet submerged in water (Fig. 26a). The resulting jelly-like material was held in place by a metal frame. Heterogeneous multi-compartment tissues could be printed that contained different concentrations (red and blue in Fig. 26a and b). The osmotic movement of water between droplets containing different salt concentrations in one- and two-dimensional tissues gave rise to assemblies with emergent three-dimensional morphologies (Fig. 26b). Furthermore, the inclusion of α-HL in specific droplets enabled the path of solute diffusion to be programmed in three dimensions (Fig. 26c). While these droplet-based systems do not constitute transmembrane molecular machines in their own right, they might serve as hosts for collectively operating transmembrane molecular machines in the future, paving the way to sophisticated multi-compartment devices with emergent properties.
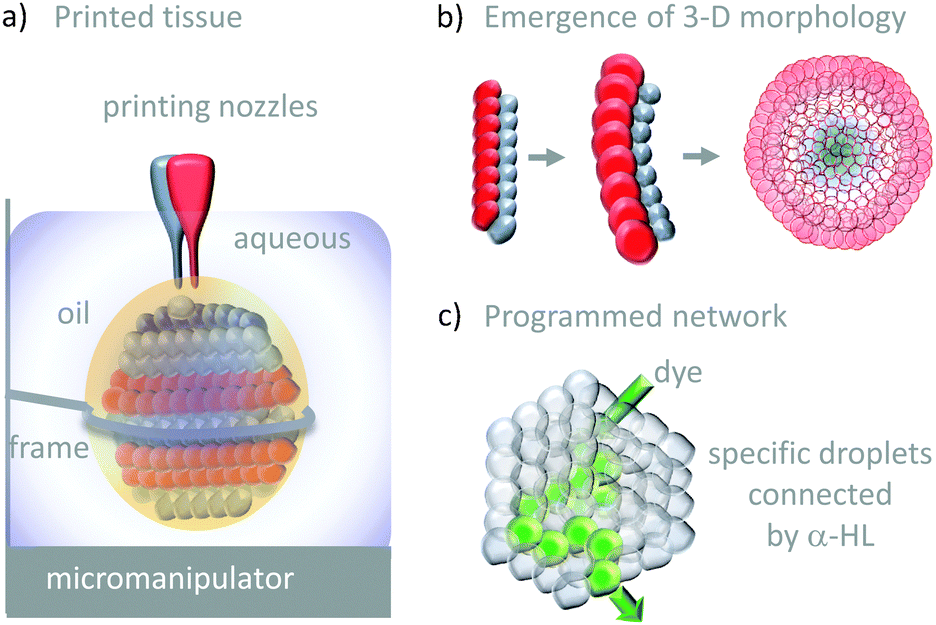 | ||
| Fig. 26 Tissue-like 3-D printed droplet networks.50 | ||
4. Conclusions and future directions
Many avenues are being pursued in the design of molecular machines. Biology has not only inspired the development of man-made molecular machines, but also provided insights into the mechanisms of energy transduction on the molecular level. Implementation of fundamental mechanisms such as ratcheting23,24 have led to the construction of minimal synthetic designs that are free of the molecular ‘baggage’ acquired over billions of years of biological evolution.5,15 Despite the simplicity and elegance of many synthetic systems,7,9,25,26 no macroscopic work can be extracted from freely tumbling, randomly oriented molecules.2 This fundamental Brownian limitation has been overcome by interfacing synthetic molecular machines with surfaces.10,11,13 In biology, similar entropic constraints are afforded to molecular machines that work along one-dimensional biopolymers or across two-dimensional membranes. Indeed, it is hard to imagine life without the nucleic acid polymerases,42,46,47 transmembrane photosynthesis,35 or ATP synthase.3 Substantial progress has been made since the first transmembrane channels were prepared,29 including the realisation of stimuli-responsive channels that mimic some biological functions.30–32 However, few artificial transmembrane systems are as robust or reproducible as their biological counterparts, or display the autonomous operation ubiquitous to the machinery of life. Thus, many researchers have repurposed biological molecules such as stable pore proteins33,34,41 and DNA30 to construct artificial transmembrane devices. Some of the most effective demonstrations of transmembrane machine behaviour have utilised motor proteins to attain assemblies that operate with autonomous nanomechanical cycles.44,45 While lessons continue to be learned from biology, principles from synthetic nanotechnology15 must be better integrated into the next generation of transmembrane architectures. Looking further ahead, multi-compartment devices49,50 might deliver hybrid synthetic/biological systems that coordinate the functions of numerous transmembrane machines to operate far from equilibrium,19 blurring the lines between life and the artificial.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EP/H021620-1) and ERC Starting Grant No. 336935, ‘Transmembrane molecular machines’ for financial support.Notes and references
- M. Schliwa and G. Woehlke, Nature, 2003, 422, 759–765 CrossRef CAS PubMed.
- R. D. Astumian, Phys. Chem. Chem. Phys., 2007, 9, 5067 RSC.
- S. Arai, S. Saijo, K. Suzuki, K. Mizutani, Y. Kakinuma, Y. Ishizuka-Katsura, N. Ohsawa, T. Terada, M. Shirouzu, S. Yokoyama, S. Iwata, I. Yamato and T. Murata, Nature, 2013, 493, 703–707 CrossRef CAS PubMed.
- X. Zhao, K. Zhang, T. Boquoi, B. Hu, M. A. Motaleb, K. A. Miller, M. E. James, N. W. Charon, M. D. Manson, S. J. Norris, C. Li and J. Liu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14390–14395 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2014, 10, 70–75 CrossRef PubMed.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef CAS PubMed.
- M. Klok, N. Boyle, M. T. Pryce, A. Meetsma, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2008, 130, 10484–10485 CrossRef CAS PubMed.
- R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef CAS PubMed.
- T. Kudernac, N. Ruangsupapichat, M. Parschau, B. Macia, N. Katsonis, S. R. Harutyunyan, K.-H. Ernst and B. L. Feringa, Nature, 2011, 479, 208–211 CrossRef CAS PubMed.
- J. Berna, D. A. Leigh, M. Lubomska, S. M. Mendoza, E. M. Perez, P. Rudolf, G. Teobaldi and F. Zerbetto, Nat. Mater., 2005, 4, 704–710 CrossRef CAS PubMed.
- T. J. Huang, B. Brough, C.-M. Ho, Y. Liu, A. H. Flood, P. A. Bonvallet, H.-R. Tseng, J. F. Stoddart, M. Baller and S. Magonov, Appl. Phys. Lett., 2004, 85, 5391–5393 CrossRef CAS.
- V. Balzani, A. Credi and M. Venturi, ChemPhysChem, 2008, 9, 202–220 CrossRef CAS PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- J. Bath and A. J. Turberfield, Nat. Nanotechnol., 2007, 2, 275–284 CrossRef CAS PubMed.
- S. Matile, A. Vargas Jentzsch, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC.
- X. Hou, W. Guo and L. Jiang, Chem. Soc. Rev., 2011, 40, 2385–2401 RSC.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed.
- J. Weber, Nat. Chem. Biol., 2010, 6, 794–795 CrossRef CAS PubMed.
- C. A. Schalley, K. Beizai and F. Vögtle, Acc. Chem. Res., 2001, 34, 465–476 CrossRef CAS PubMed.
- Y. Sowa and R. M. Berry, Q. Rev. Biophys., 2008, 41, 103–132 CrossRef CAS PubMed.
- V. Serreli, C.-F. Lee, E. R. Kay and D. A. Leigh, Nature, 2007, 445, 523–527 CrossRef CAS PubMed.
- A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. c. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124–2128 CrossRef CAS PubMed.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424, 174–179 CrossRef CAS PubMed.
- R. A. Muscat, J. Bath and A. J. Turberfield, Nano Lett., 2011, 11, 982–987 CrossRef CAS PubMed.
- Y. He and D. R. Liu, Nat. Nanotechnol., 2010, 5, 778–782 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS PubMed.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS PubMed.
- S. Hernández-Ainsa, K. Misiunas, V. V. Thacker, E. A. Hemmig and U. F. Keyser, Nano Lett., 2014, 14, 1270–1274 CrossRef PubMed.
- T. Jovanovic-Talisman, J. Tetenbaum-Novatt, A. S. McKenney, A. Zilman, R. Peters, M. P. Rout and B. T. Chait, Nature, 2009, 457, 1023–1027 CrossRef CAS PubMed.
- C. J. Moreau, J. P. Dupuis, J. Revilloud, K. Arumugam and M. Vivaudou, Nat. Nanotechnol., 2008, 3, 620–625 CrossRef CAS PubMed.
- A. Koçer, M. Walko, W. Meijberg and B. L. Feringa, Science, 2005, 309, 755–758 CrossRef PubMed.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS PubMed.
- G. Steinberg-Yfrach, J.-L. Rigaud, E. N. Durantini, A. L. Moore, D. Gust and T. A. Moore, Nature, 1998, 392, 479–482 CrossRef CAS PubMed.
- L. Ma and S. L. Cockroft, ChemBioChem, 2010, 11, 25–34 CrossRef CAS PubMed.
- D. Branton, D. W. Deamer, A. Marziali, H. Bayley, S. A. Benner, T. Butler, M. Di Ventra, S. Garaj, A. Hibbs, X. Huang, S. B. Jovanovich, P. S. Krstic, S. Lindsay, X. S. Ling, C. H. Mastrangelo, A. Meller, J. S. Oliver, Y. V. Pershin, J. M. Ramsey, R. Riehn, G. V. Soni, V. Tabard-Cossa, M. Wanunu, M. Wiggin and J. A. Schloss, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS PubMed.
- D. Wendell, P. Jing, J. Geng, V. Subramaniam, T. J. Lee, C. Montemagno and P. Guo, Nat. Nanotechnol., 2009, 4, 765–772 CrossRef CAS PubMed.
- G. S. Pulcu, E. Mikhailova, L.-S. Choi and H. Bayley, Nat. Nanotechnol., 2014, 10, 76–83 CrossRef PubMed.
- L. Franceschini, M. Soskine, A. Biesemans and G. Maglia, Nat. Commun., 2013, 4, 2415 Search PubMed.
- F. Olasagasti, K. R. Lieberman, S. Benner, G. M. Cherf, J. M. Dahl, D. W. Deamer and M. Akeson, Nat. Nanotechnol., 2010, 5, 798–806 CrossRef CAS PubMed.
- J. Nivala, D. B. Marks and M. Akeson, Nat. Biotechnol., 2013, 31, 247–250 CrossRef CAS PubMed.
- C.-W. Ho, V. van Meervelt, K.-C. Tsai, P.-J. de Temmerman, J. Mast and G. Maglia, Sci. Adv., 2015, 1, e1500905 Search PubMed.
- M. A. Watson and S. L. Cockroft, Angew. Chem., Int. Ed., 2016, 55, 1345–1349 CrossRef CAS PubMed.
- S. L. Cockroft, J. Chu, M. Amorin and M. R. Ghadiri, J. Am. Chem. Soc., 2008, 130, 818–820 CrossRef CAS PubMed.
- J. Chu, M. González-López, S. L. Cockroft, M. Amorin and M. R. Ghadiri, Angew. Chem., Int. Ed., 2010, 49, 10106–10109 CrossRef CAS PubMed.
- M. A. Holden, D. Needham and H. Bayley, J. Am. Chem. Soc., 2007, 129, 8650–8655 CrossRef CAS PubMed.
- G. Villar, A. J. Heron and H. Bayley, Nat. Nanotechnol., 2011, 6, 803–808 CrossRef CAS PubMed.
- G. Villar, A. D. Graham and H. Bayley, Science, 2013, 340, 48–52 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |