
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA practical guide to working with H2S at the interface of chemistry and biology
Matthew D.
Hartle
and
Michael D.
Pluth
*
Department of Chemistry and Biochemistry, Materials Science Institute, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA. E-mail: pluth@uoregon.edu
First published on 11th May 2016
Abstract
Hydrogen sulfide (H2S) is the most recently accepted endogenously produced gasotransmitter and is now implicated in a variety of physiological functions. In this tutorial review, our goal is to provide researchers new to the field of H2S chemical biology with practical considerations, pitfalls, and best practices to enable smooth entry into investigations focused on biological H2S. We present practical handling and safety considerations for working with this reactive biomolecule, and cover basic roles of H2S biogenesis and action. Experimental methods for modulating H2S levels, including enzymatic knockout, RNA silencing, enzymatic inhibition, and use of small molecule H2S donors are highlighted. Complementing H2S modulation techniques, we also highlight current strategies for H2S detection and quantification.
 Matthew D. Hartle | Matthew D. Hartle earned his BS in chemistry with an area of special interest in space and planetary science and engineering from the Colorado School of Mines in 2012, where he conducted undergraduate research with Prof. Stephen Boyes. He is currently working towards his PhD under the direction of Prof. Michael Pluth in the Department of Chemistry & Biochemistry at the University of Oregon. His research interests focus on using bio(in)organic models to investigate the chemical mechanisms of H2S binding and reaction with heme and other metal- and non-metal-containing biologically relevant scaffolds. |
 Michael D. Pluth | Michael D. Pluth earned his PhD in 2008 as an NSF predoctoral fellow under joint direction of Profs. Robert Bergman and Kenneth Raymond at UC Berkeley. Mike then moved to MIT, joining the research group of Prof. Stephen Lippard as an NIH Postdoctoral Fellow, where he investigated methods for biological nitric oxide detection. In 2011, Mike joined the faculty in the Department of Chemistry & Biochemistry at the University of Oregon as an Assistant Professor. Research in the Pluth lab is focused on different aspects of molecular recognition, with particular emphasis in developing chemical tools for H2S quantification and donation, as well as using bio(in)organic models to investigate modes of H2S action. |
Key learning points• Basic properties of biological hydrogen sulfide (H2S)• The role of H2S in the landscape of reactive sulfur, oxygen, and nitrogen species (RSONS) • Practical safety considerations for handling H2S and H2S donors • Methods for modulating biological H2S: enzymatic knockout, RNA silencing, inhibition, stimulation, small molecule donors • Methods for detecting and quantifying biological H2S |
Introduction
Hydrogen sulfide (H2S) has emerged as an important biological signaling molecule that plays diverse roles in human health and physiology. Despite its long history as a toxic gas and environmental pollutant, H2S now joins nitric oxide (NO) and carbon monoxide (CO) as a gasotransmitter – a small, enzymatically generated, gaseous molecule with a tightly regulated metabolism that impacts physiological functions.1–3 The innate chemical properties of H2S, including its redox activity, acidity, and high nucleophilicity, allow for reaction with different cellular targets as part of its signaling capacity. By comparison, disentangling this chemistry is in many ways more complicated than that of NO and CO.4 Misregulation of endogenous H2S is implicated in diverse physiological processes, including blood pressure regulation, immune response, and long term potentiation, as well as various diseases in the neuronal, gastrointestinal, circulatory, and endocrine systems.2 Aligned with our rapidly expanding understanding of biological H2S, new and impactful investigative tools for elucidating its genesis, translocation, and action have emerged and continue to be an active and fruitful research area.By contrast to biological studies focused on NO and CO, H2S-focused investigations are often faced with unique experimental challenges – some of which are due to the recent emergence of H2S chemical biology and others from the innate physical properties of H2S. For example, although CO, NO, and H2S are all gasses, common gas detection techniques, such as gas chromatography (GC) and chemiluminescence detectors provide robust methods for detection and quantification for CO and NO, respectively, whereas similar techniques have not been widely developed for H2S. The different protonation states of H2S, and its interaction with the redox-active biological sulfur pool, make analysis by GC or other common analytical methods challenging. Similarly, reaction-based imaging techniques, as well as synthetic donor scaffolds, are often more complicated than similar systems for NO or CO. Despite these challenges, recent studies have suggested the potential interplay between the biological chemistry of H2S and NO5 as well as CO,3 thus highlighting the interconnectivity of these three gaseous molecules. These interactions, as well as those with H2S and reactive sulfur, oxygen, and nitrogen species (RSONS) highlight the complexity of the biological landscape in which H2S is involved, and the synergistic relationship of H2S to other important and reactive small molecules in biology.
In this tutorial review, we highlight key points regarding H2S chemical biology and provide a brief survey of accessible tools and methods for H2S research at the interface of chemistry and biology. Our goal is not to provide a comprehensive overview of all investigative tools for H2S research, but rather to highlight currently available classes of tools and to provide important considerations to help guide new researchers into the rapidly developing field of H2S chemical biology.
Properties and enzymatic production of H2S
H2S is a weak acid with a first pKa of 7.0, and a second pKa, corresponding to the deprotonation of hydrosulfide anion (HS−) of >14.1 Under physiological conditions, HS− constitutes about 80% of the speciation, neutral H2S about 20%, and dianionic S2− less than 1%.1 The HS− and H2S protonation states provide water-solubility and lipophilicity, respectively, and HS− is a potent nucleophile that can react with different electrophilic cellular targets including RSONS. Additionally, the different protonation states afford different metal-ligation and redox properties. The complex redox landscape of biological sulfur, with oxidation states ranging from −2 in H2S to +6 in SO42−, means that H2S/HS− can be readily oxidized to other biologically-relevant reactive sulfur species. Indeed, the redox-labile/sulfane-sulfur pool may provide an important method of sulfide storage and transport, but also may provide new signaling pathways distinct from those associated with H2S alone (vide infra).6Enzymatic H2S biosynthesis stems primarily from cystathionine γ-lyase (CSE), cystathionine-β-synthase (CBS), and 3-mercaptopyruvate sulfurtransferase (3-MST)/cysteine aminotransferase (CAT). In each of these pathways, the sulfur atom incorporated into H2S is derived from the sulfhydryl group of either homocysteine (Hcy) or L-cysteine (L-Cys). Details of these pathways have been recently reviewed in significant detail and are summarized briefly here (Fig. 1).1–3 CSE catalyzes the formation of H2S and homolanthionine from 2 equiv. of Hcy, but can also work in concert with CBS to convert Hcy and Cys to H2S and cystathionine. CBS can also catalyze the condensation of Hcy with serine (Ser) to generate cystathionine and H2O. Upon reaction with CSE, cystathionine is converted back to L-Cys with concomitant formation of homoserine. This generated Cys can participate in the above pathways, can be hydrolyzed to Ser and H2S by CBS or CSE, or can be condensed with a second equivalent of Cys by the same two enzymes to form lanthionine and H2S. Cysteine is also a viable substrate for CAT, which generates 3-mercaptopyruvate (3-MP), which is then converted to pyruvate, NH3, and H2S by 3-MST. The 3-MST pathway also accounts for H2S formed from D-Cys, which is first converted to 3-mercaptopyruvate by DAO.
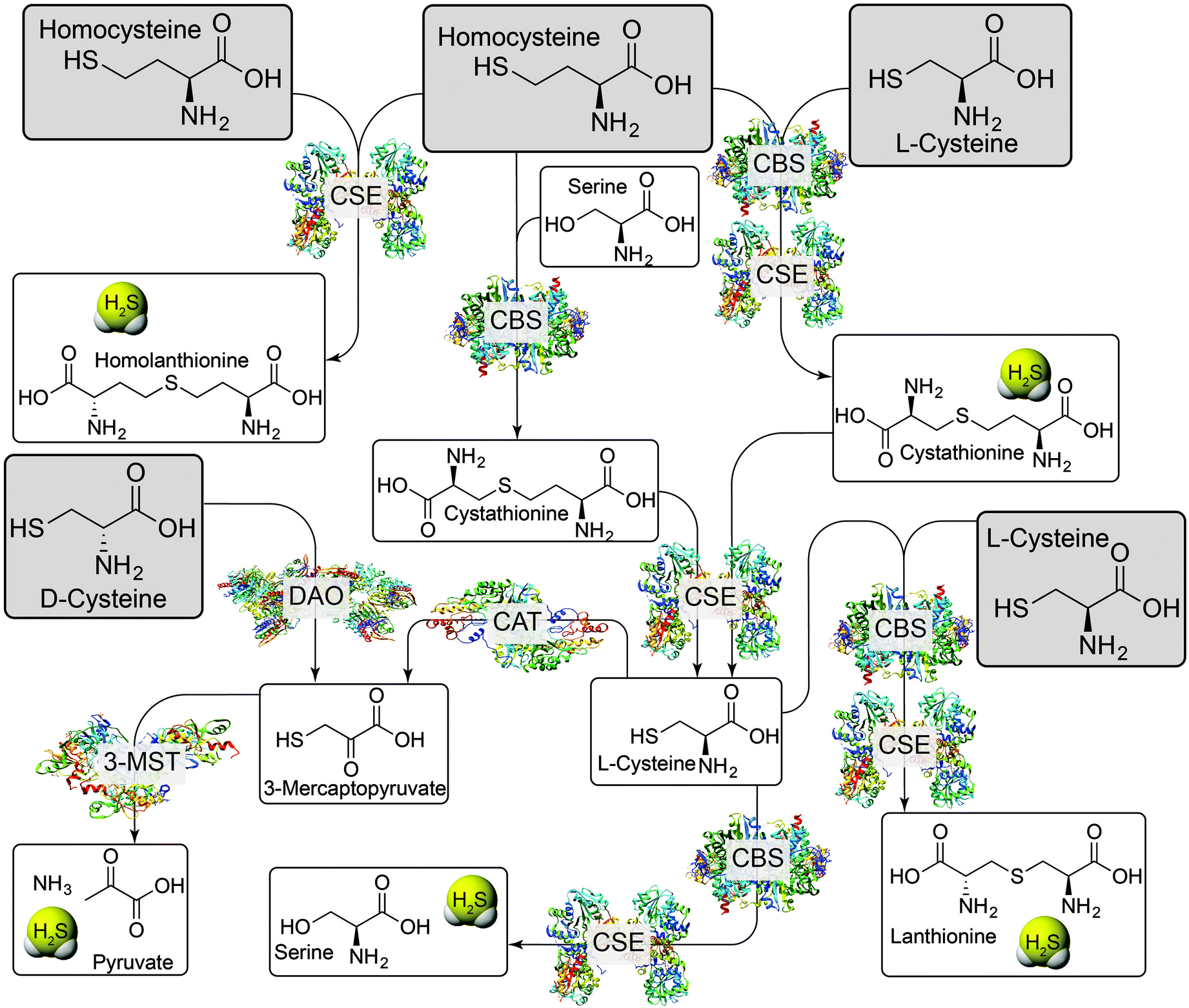 | ||
| Fig. 1 Enzymatic H2S production pathways. CSE: cystathionine γ-lyase; CBS: cystathionine β-synthase; CAT: cysteine aminotransferase; DAO: D-aminoacid oxidase; 3-MST: 3-mercaptopyruvate sulfurtransferase. Grey boxes indicate common substrates for H2S-producing enzymes. | ||
Practical handling and safety considerations
Hydrogen sulfide is a highly toxic gas, and adequate care must be given when handling H2S gas, sulfide salts, and synthetic H2S donors. Although the characteristic rotten egg odor is detectable as low as 0.01 ppm in air, odor alone should never be used as the primary method of safety monitoring. Rapid olfactory fatigue results upon exposure to concentrations greater than 100 ppm but also after continuous exposure to significantly lower levels. H2S concentrations as low as 20 ppm cause eye and lung irritation, 300–500 ppm levels result in serious eye damage, and 700 ppm or higher levels can result in unconsciousness, respiratory failure, and death.7 The combination of high toxicity with rapid olfactory fatigue necessitates handling in a well ventilated fume hood equipped with a commercially-available H2S alarm, which detects H2S gas and typically reports in the 5–10 ppm range, especially when using H2S gas directly or quantities of sulfide salts sufficient to provide a potentially toxic response. It is also practical, both from a safety and odor-mediation standpoint, to quench any H2S-containing solutions after use. Such quenching can be readily accomplished by preparation and use of a Zn2+ quenching solution, which rapidly reacts with free sulfide to generate insoluble ZnS. This quenching slurry can be prepared by adding 30 g Zn(OAc)2, 9 g sodium citrate, and 12 g NaOH to 1 L of H2O.7For experiments involving aqueous sulfide solutions, it is often more practical to use sulfide salts, such as NaSH or Na2S, rather than H2S gas directly. Despite their convenience, the commercial purity of NaSH or Na2S is often poor, and many samples contain significant quantities of elemental sulfur or polysulfides.4,8,9 In general, sulfide salts should be free-flowing white powders – any inclusion of yellow or other highly colored impurities should signal to the user that the sample does not have the appropriate purity and should be discarded. Because of the propensity of H2S/HS− to oxidize, especially in the presence of trace metal ions and oxygen, H2S solutions should be prepared in anaerobic buffer under a blanket of nitrogen or argon in clean, metal-free, glass- or plastic-ware. Such solutions should also be prepared in septum-sealed vials to prevent significant loss from H2S volatilization and also to enable easy transfer of sulfide solutions via gas-tight syringe. Because of this volatility and potential for oxidation, it is most practical to prepare sulfide-containing solutions immediately prior to use rather than to re-use solutions from previous experiments.
Interactions with the sulfane-sulfur pool
Complementing the signaling roles of biological H2S, significant evidence suggests that sulfane-sulfur compounds, such as polysulfides, persulfides, and other sources of reductant-labile sulfur, play important biochemical roles.6,8,9 The term “sulfane sulfur” refers to a sulfur atom with formally six valence electrons and no charge (S0), which is bound to one or more sulfur atoms.10 Upon reaction with cellular reductants or thiols, sulfane-sulfur compounds can release sulfide, thus providing a convenient source of H2S storage. Additionally, sulfane-sulfur species have distinct chemical reactivities that may contribute to additional modes of action. For example, persulfides (RSSH) are more nucleophilic than the corresponding thiols, and S-persulfidation of nucleophilic Cys residues in enzymes is known to modify enzymatic activity.1 As completely inorganic species, hydropolysulfides (HSSxH) are also important sulfane-sulfur species, as evidenced by a recent report demonstrating that 3-MST not only generates H2S, but also H2S3.8 The use of small molecule persulfides and persulfide-releasing motifs as H2S donors (vide infra) and as model systems is quickly providing more information on persulfide reactivity and its role in the intricate chemistry of RSONS.11 Although isolated persulfides are typically unstable in solution and have not yet found utility in biochemical investigations, caged persulfides, which release persulfides upon hydrolysis or reaction with nucleophiles, or persulfides generated in situ from reaction of GSSG with HS−, have been used to investigate different roles of persulfides in reactive sulfur species regulation and action.12One of the most basic methods for sulfane-sulfur detection and quantification is the cold cyanolysis assay, which utilizes the reaction of sulfane sulfur atoms with CN− at basic pH to form thiocyanate (SCN−). Addition of excess ferric iron results in the formation of [Fe(SCN)(H2O)5]2+, which can be readily detected and quantified by the characteristic absorbance at 460 nm (Scheme 1a).13 In practice, the cold cyanolysis method is usually used for purified proteins or samples in simple matrices and cannot differentiate between individual components, such as persulfides and polysulfides, in the sulfane-sulfur pool.
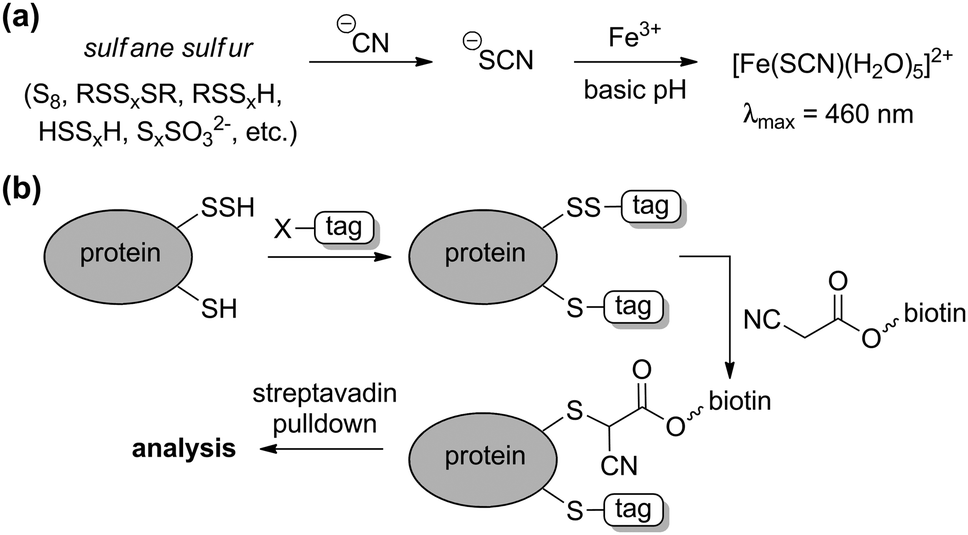 | ||
| Scheme 1 (a) Cold cyanolysis detection/quantification of sulfane sulfur. (b) General strategy for tag-switch labelling of protein persulfides. | ||
One significant limitation of the cold cyanolysis method is that persulfides are not differentiated from polysulfides or other sulfane-sulfur sources. As a step toward addressing this challenge, a tag-switch method to detect and assay persulfides was recently reported (Scheme 1b).14 In this method, all sulfhydryl groups, both from thiols (–SH) and persulfides (–SSH) are labelled with an electrophile, such as methylsulfonyl benzothiazole (MSBT), to generate the corresponding benzothiazole thioether and disulfide, respectively. Importantly, the resultant benzothiazole disulfide remains reactive toward specific carbon-based nucleophiles, allowing for conjugation with biotin tags. The biotinylated proteins can then be subjected to streptavidin pulldown and detection by standard Western blot or mass spectrometric techniques.14 Complementing these strategies, selective reaction-based fluorescent probes for polysulfides and sulfane sulfur have been reported and are the subject of a recent review.15 These, as well as other emerging tools, are poised to help differentiate between the genesis of H2S and other reactive polysulfides.
Methods for modulating H2S levels
The ability to modulate cellular H2S levels provides a cornerstone for investigating the actions of H2S in biology. Such control can be achieved by selective knockout and/or knockdown of H2S-producing enzymes, use of small-molecule competitive inhibitors or stimulators, or by administration of synthetic H2S donors. In model cell lines and organisms, supplementation with exogenous H2S can often provide protection (or rescue) from various (patho)physiological disease states associated with abnormal H2S biosynthesis. These insights suggest that synthetic H2S donors may not only provide important chemical tools for understanding biological H2S, but may also offer viable therapeutic potential for diseases associated with H2S misregulation.KO mouse models
Selective knockout (KO) of CSE, CBS, or 3-MST in different cell lines has been utilized in specific investigations, but the development of KO mouse models has provided a more broad platform on which to study the impacts of reduced enzymatic H2S generation.16 Homozygous CBS−/− KO in mice results in severe developmental growth problems, and few of the mice live past 4 weeks of age.17 The heterozygous CBS+/− KOs exhibit better viability and present hyperhomocysteinemia (high levels of homocysteine in the blood) but have not been used extensively in H2S-related investigations.17 By contrast, the CSE−/− mouse model18 has found significantly more utility in H2S-related investigations, including studies on the role of H2S in blood pressure regulation, angiogenesis, and neurodegeneration, as well as many other studies. A second CSE−/− mouse model exhibiting somewhat different phenotypes was also subsequently developed and used as an animal model for cystathionemia.19 Homozygous 3-MST−/− mice have been developed more recently and may also provide a useful animal model for H2S-related investigations.20 As a whole, the availability of KO mouse models provide a key tool in studying the impacts of H2S in contextually-rich biological environments and overcome some of the limitations of small molecule inhibitors or sulfide donors (vide infra).RNA knockdown methods
In addition to enzyme KO methods, gene knockdown by RNA interference (RNAi) methods has been used to silence CSE, CBS, and 3-MST. Importantly, these methods often provide a more simple approach to overcome the limitation of animal models or small-molecule inhibitors, and also offer a complementary approach for other enzyme modulation experiments. For example, small interfering RNA (siRNA) methods have been used to silence CSE and CBS.21 Unlike small-molecule inhibitors for 3-MST (vide infra), siRNA methods aimed at this enzyme have proven efficacious in reducing H2S production, leading to important insights into mitochondrial electron transport and cellular bioenergetics.22Small-molecule inhibitors
Small-molecule competitive inhibitors of H2S-producing enzymes are commonly used to decrease H2S synthesis in studies using isolated enzymes or cell culture models. On the basis of this widespread use, significant efforts have been made to discover or develop potent enzymatic inhibitors for CBS, CSE, and 3-MST. Although these studies have provided a useful suite of compounds for partial inhibition of H2S-producing enzymes, identifying inhibitors with specificity for one enzyme over another and low inhibitory constant (Ki) values remains a significant challenge.Commonly-used CSE inhibitors with IC50 < 1 mM are L-aminoethoxyvinylglycine (AVG, 1 μM), β-cyano-L-alanine (BCA, 14 μM), and propargylglycine (PAG, 40 μM) (Fig. 2).16,23 Each of these inhibitors also affect other proximal phosphate (PLP)-dependent enzymes at mM concentrations, making it likely that off-target inhibition is observed at practical concentrations. Inhibition of CBS has proven significantly more challenging, with fewer inhibitors with IC50 < 1 mM identified. Hydroxylamine (HA, 278 μM), aminooxyacetic acid (AOAA, 8.52 μM), and trifluoroalanine (66 μM) are among the most commonly used competitive inhibitors for CBS,23 but none of these show marked selectivity for CBS over CSE. Recent studies have used high-throughput screening methods, often in combination with H2S fluorescent probes (vide infra), to identify CBS inhibitors. Such screens have identified new lead compounds with good IC50 values in the low micromolar range, but many hits have not been compared directly with commonly-used inhibitors and their selectivity for CBS over CSE remains to be fully elucidated. Similar to the challenges with developing selective CBS inhibitors, selective inhibition of 3-MST has been unsuccessful to date.16 Given the overall difficulty in selectively inhibiting individual H2S producing enzymes, the identification and development of new inhibitors for H2S-producing enzymes is needed. Such inhibitors, if selective and functional at pharmacologically-reasonable concentrations, would provide important research tools for modulating cellular H2S levels.
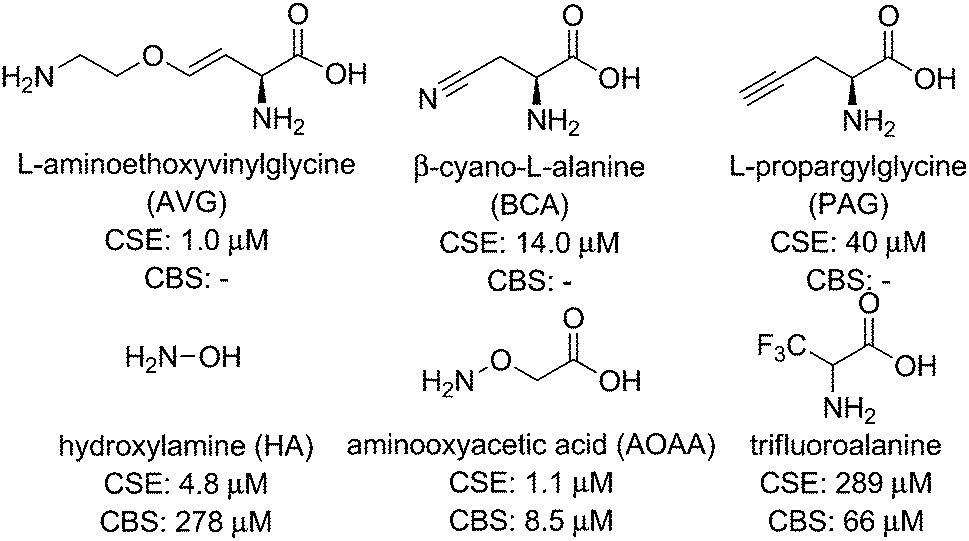 | ||
| Fig. 2 Selected common CSE and CBS inhibitors with associated IC50 values. | ||
Enzymatic stimulators
Complementing methods to reduce H2S synthesis using small-molecule inhibitors, H2S synthesis can also be increased by use of different enzymatic stimulators. The most common of such methods is use of S-adenosylmethionine (AdoMet), an allosteric activator of CBS, to increase enzymatic H2S production.24,25 In addition to CBS, CSE stimulation has also been demonstrated using cytokines and endotoxins, such as tumor necrosis factor α (TNF-α) or lipopolysaccharides (LPS), involved in immune response.26,27 By contrast to CBS and CSE stimulation, common stimulators of 3-MST are not readily available and remain an active area of research.Small-molecule H2S donors
Complementing methods to modulate endogenous H2S synthesis, the ability to increase endogenous H2S levels using exogenous sulfide sources provides an important research and pharmacological tool for studying the roles of biological H2S. Commonly-used sources of exogenous H2S include inorganic salts, such as NaSH and Na2S, as well as small-molecule synthetic donors. Although NaSH and Na2S provide convenient sources of sulfide, the large dose of H2S released upon addition to buffer does not match the slower, continuous, enzymatic production characteristic of endogenous H2S synthesis. Additionally, the large bolus of H2S often results in a toxicological response and is quickly oxidized/metabolized by the cellular environment. Motivated by these limitations, researchers have developed slow-release sulfide donors that better mimic the gradual H2S release of enzymatic synthesis. As our understanding of sulfide biology continues to evolve, one common observation is that slow-releasing H2S donors often elicit different cellular responses than inorganic sulfide salts.28 For example, sulfide salts and slow-releasing donors have been shown to be pro- and anti-inflammatory, respectively, in different models of inflammation, including sepsis.28 These differences highlight the importance of experimental design when using different sulfide sources, but also highlight the potential pharmacological importance of such slow-releasing donors. Here we provide a brief overview of the most commonly-used donor motifs along with their sulfide release mechanisms when known. We also refer interested readers to recent reviews on this topic.12,29Some of the most simple sulfide-donating motifs are organic polysulfides, which are often found in natural products. For example, diallyltrisulfide (DATS), isolated from garlic and other alliums, is a commonly used donor molecule, and other organic polysulfides such as varacin have also been implicated in H2S release.30 Because polysulfides are electrophilic, attack by GSH or other thiols results in formation of an intermediate persulfide, which after a second reaction with GSH releases H2S (Scheme 2a).31 Although DATS is the most commonly-used polysulfide donor, it is likely that other small molecule polysulfides are also potential platforms for H2S release.
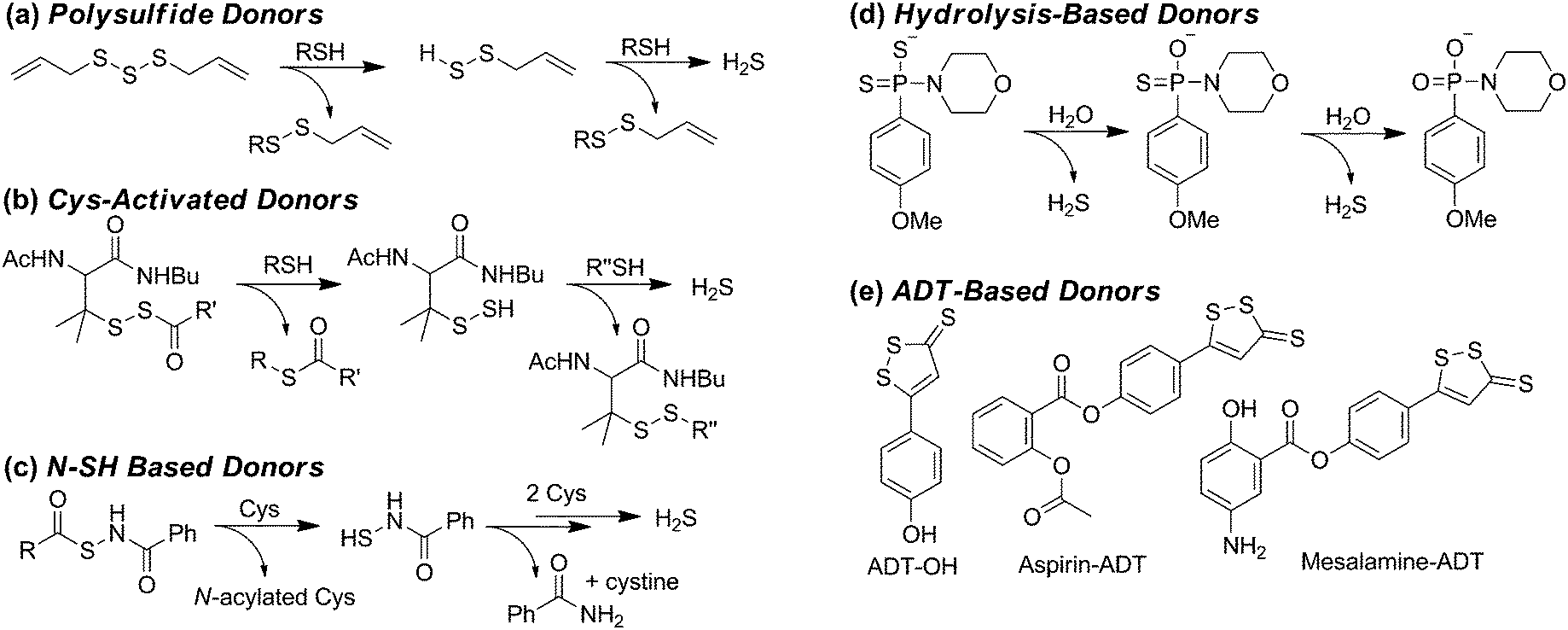 | ||
| Scheme 2 Common motifs for small-molecule H2S donors. (a) Organic polysulfides, (b) cysteine-activated H2S donors, (c) cysteine-activated H2S donors with N-SH motifs, (d) hydrolysis-based donors, and (e) anethole 1,2-dithiole-3-thione (ADT) derived donors. | ||
Inspired by disulfide exchange chemistry, chemists have also developed H2S donors that are activated by nucleophilic attack by endogenous thiols.32 For example, protection of the thiol in penicillamine derivatives with an acyldisulfide results in persulfide formation after nucleophilic attack by Cys (Scheme 2b).33 Once the persulfide is released, reaction with a second thiol generates a stable disulfide and extrudes H2S. Application of such donors to the treatment of myocardial ischemia/reperfusion (MI/R) injury in murine models results in reduced circulating levels of MI/R biomarkers suggesting these and similar donor motifs exhibit cardiac protection and may have potential therapeutic applications.33,34
Operating by similar Cys-activated H2S release mechanisms, donors containing N-mercapto (N-SH) motifs also generate a persulfide intermediate en route to H2S release (Scheme 2c).35 In these donors, thiol exchange between Cys and the donor generates an S-acylated Cys intermediate, which undergoes native chemical ligation to rearrange to N-acylated Cys and an N-mercaptobenzamide intermediate. This N-SH compound reacts with Cys to generate Cys persulfide, which reacts with a second equivalent of thiol to release H2S. The rate of H2S release from these scaffolds can be tuned by the addition of electron donating or withdrawing groups on the scaffold. Additionally, new H2S-donating materials activated by thiol activation are also emerging, indicating the potential viability of this strategy for incorporation into therapeutics such as wound dressing to encourage angiogenesis.36
In addition to donor activation by nucleophilic attack of thiols, other common donor constructs are activated by hydrolysis. For example, one of the most commonly used synthetic donors, GYY4137,37 is a phosphino-dithioate derived from Lawson's reagent. GYY4137 undergoes slow hydrolysis in water to release H2S (Scheme 2d), although the efficiency of sulfide release remains low.38 The rate of hydrolysis is pH dependent and slow at physiological pH, contrasting the rapid release of H2S by inorganic salts. For example, treatment of precontracted aortic rings with NaSH results in relaxation in 20 to 30 seconds, whereas GYY4137 treatment requires nearly 10 minutes to achieve similar relaxation.39
Another common class of H2S donors is based on 1,2-dithiole-3-thiones, with the most often used being 5-(4-hydroxyphenyl)-3H-1,2-dithiole-3-thione (ADT-OH) (Scheme 2e).40 Although the mechanism of H2S release from these scaffolds remains to be fully elucidated, one benefit of this donor motif is the ease with which it can be tethered to different molecules through ester or hydrolytically-stable amide linkages.41 For example, linkage of a triphenylphosphonium cation imparts mitochondrial targeting for AP39, whereas ligation to common non-steroidal anti-inflammatory drugs (NSAIDs) has resulted in significant reduction in GI damage scores.40 The use of ADT derivatives in NSAID applications highlight the potential therapeutic action of H2S donors;40 however, insights into the H2S release mechanism from such donors is poised to make significant impacts into our understanding of the therapeutic action of these donor motifs.
All of the above donor motifs provide sources of sulfide in aqueous solution, but researchers interested in H2S/HS− using biomimetic compounds in organic solvents also have access to organic-soluble sources of sulfide. In practice, H2S gas is sufficiently soluble in organic solutions for most investigations; however, NaSH or Na2S are generally insoluble in organic solvents. To aid in such investigations, we recently reported a simple method to prepare analytically-pure NBu4SH, which provides access to an organic-soluble form of HS−.42 Such compounds offer a convenient source of HS− in organic solution, enabling the separation of HS− from H2S in different bio(in)organic model studies, which is otherwise not possible in aqueous solution.
H2S detection and quantification
Coincident with the increased biological importance of H2S, new methods for H2S quantification and detection are rapidly emerging. Ranging from binary presence/absence tests based on the formation of PbS from PbII salts to complex molecular architectures aimed at real-time H2S detection, analytical methods of H2S determinations comprise an important area of H2S research. We highlight here general classes of analytical tools for H2S research and describe their general benefits and pitfalls. On the basis of the rapid expansion of new methods for H2S detection and quantification, as well as revisions and refinements of current methods, we refer the interested reader to a number of recent reviews on these topics.15,43Quantification methods
Commonly used sulfide quantification methods typically require sample homogeneity, which is straightforward for biological fluids such as blood or serum, but significantly more complicated for naturally heterogeneous samples such as those from cell or tissue culture experiments. Spectrophotometric methods such as the methylene blue (MB) assay have constituted one of the classical methods of H2S quantification. This method leverages the FeCl3-catalyzed electrophilic aromatic substitution of p-dimethylamino aniline with sulfide to form MB, which has a characteristic absorbance at 670 nm and allows for sulfide quantification (Scheme 3a). Despite its widespread use, recent studies have demonstrated that the detection limit of the MB assay for sulfide is only 2 μM rather than previously-reported lower values, making it insufficiently sensitive to differentiate between sulfide levels in normal versus CSE−/− mice.44 Furthermore, the MB method is performed under highly acidic conditions, which can result in sulfur extraction from other biological sources and diminish the accuracy of sulfide levels measured with this method. Researchers new to the field of sulfide quantification will notice the large number of past studies using the MB method but are cautioned when comparing H2S levels measured with this method, especially when more accurate contemporary methods are available. For example, the monobromobimane (mBB) method, which utilizes the reaction of two equivalents of mBB with H2S under basic conditions to generate fluorescent sulfide dibimane (SdB), enables quantification of H2S by fluorescence HPLC with a detection limit of 2 nM (Scheme 3b).44 Because mBB reacts with both H2S and thiols, separation of the fluorescent signals by HPLC is required for quantification. Despite this limitation, a significant benefit of this method is that workflows have been developed to analytically separate the free, reductant-labile/sulfane-sulfur, and acid-labile sulfide pools, enabling more detailed investigations across distributions in complex systems.44 Additionally, the mBB method has been used more recently to also detect and quantify different poly- and persulfides, providing analytical information about these important reactive sulfur species.45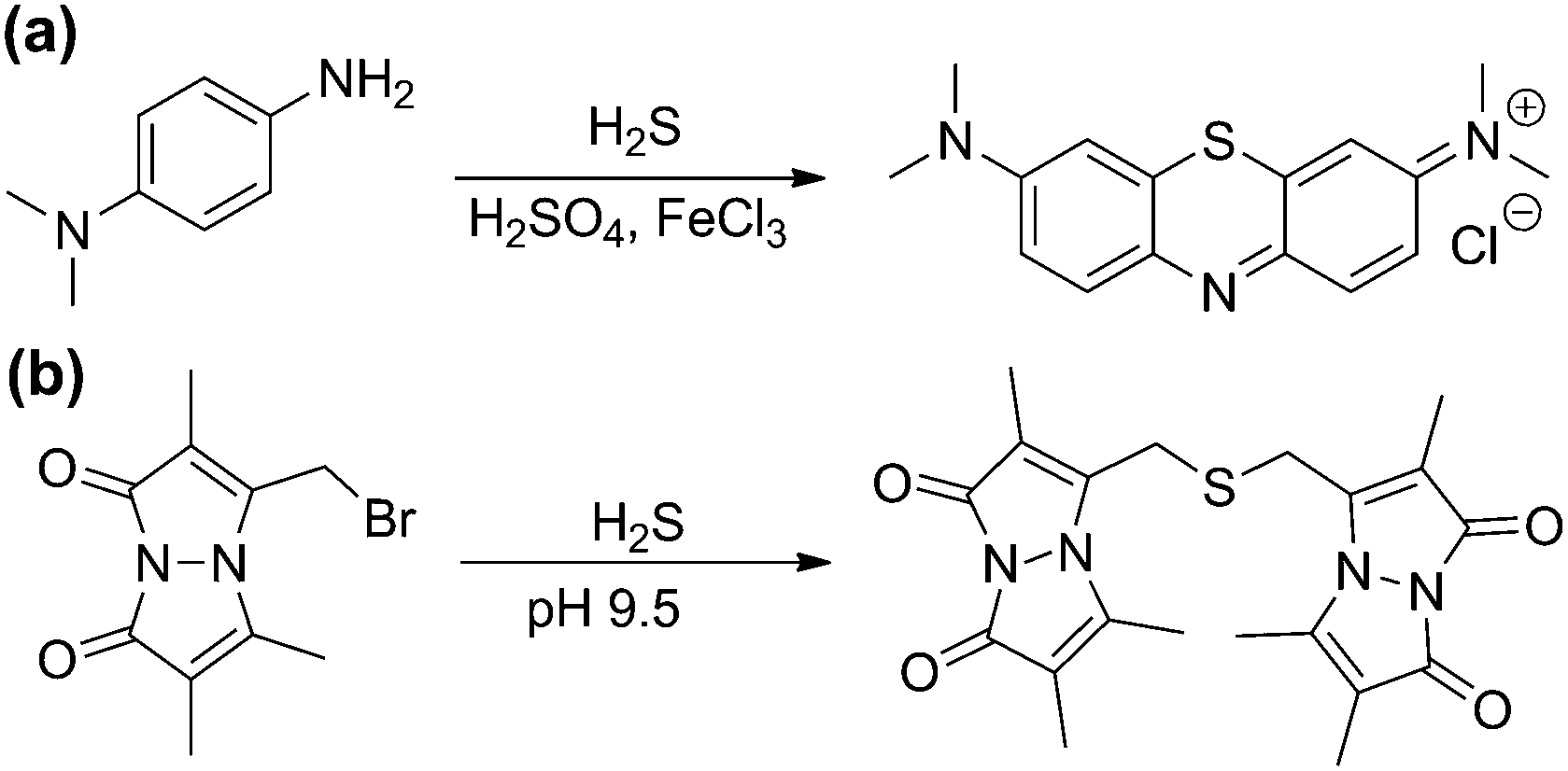 | ||
| Scheme 3 (a) Methylene blue (MB) and (b) monobromobimane (mBB) methods for H2S quantification. | ||
Complementing spectrophotometric H2S quantification methods are other analytical tools, such as sulfide selective electrodes. These tools can provide both the ability to quantify H2S, based on a calibration curve, and provide real-time H2S detection in solution. Readers interested in the differences in available sulfide electrodes are referred to a recent review focused on this topic.43 In general, sulfide electrodes employ an ion-selective membrane that allows H2S permeability for solution-based measurements. In the solution phase of the electrode, a strongly basic environment allows the dianionic sulfide ion to reduce Fe(CN)63− to Fe(CN)64−, which is subsequently re-oxidized at the platinum electrode to produce a current relative to H2S concentration. One advantage of sulfide electrodes is that they can be used directly in mammalian tissues;43 however, these electrodes cannot provide sub-cellular resolution of sulfide generation, storage, or transport. Even so, sulfide electrodes provide a convenient, time-resolved method for sulfide quantification or detection.43
Detection methods
The development, refinement, and application of reaction-based probes for H2S detection has expanded rapidly in the last few years. Such scaffolds typically couple various H2S-selective reactions with different chromophores to generate a colorimetric or fluorescence response. Because of this design strategy, such methods are irreversible and the resultant methods measure accumulated probe activation rather than real-time H2S dynamics. Despite these limitations, such constructs offer the potential to provide significantly higher spatiotemporal resolution than current H2S quantification methods and also provide access to live cell and tissue imaging experiments. The predominant strategy for developing reaction-based probes for H2S has been to utilize a fluorescence-quenching group on a fluorophore that can be modified or removed selectively by H2S. As is common with most small molecule fluorescent probes, different detection strategies exhibit somewhat different selectivity patterns and each strategy may be better suited for certain types of investigations. The interested reader is referred to recent reviews on different aspects of reaction-based H2S probe development.15The most common reaction-based detection strategy to date is H2S-mediated azide (R-N3) reduction (Scheme 4a).15 Based on the ease of appending azides onto fluorogenic scaffolds, over 70 papers in the last few years described H2S-detecting scaffolds using an azide to generate turn-on fluorescent probes (Scheme 4a).46–48 Reduction of other oxidized nitrogen functional groups, such as nitro groups, has also been utilized for H2S detection.48 For most fluorophores, the azide moiety quenches fluorescence by a push–pull energy transfer mechanism although other quenching mechanisms are also possible. Although the selectivity of azides for reduction by H2S rather than biological thiols is predominantly empirical, most azide-based probes provide good to excellent selectivity for H2S over other RSONS. A recent mechanistic investigation established that HS− is the active species involved in H2S-mediated azide reduction, and that sulfide is oxidized to sulfane sulfur, such as HSS−, during probe activation.49
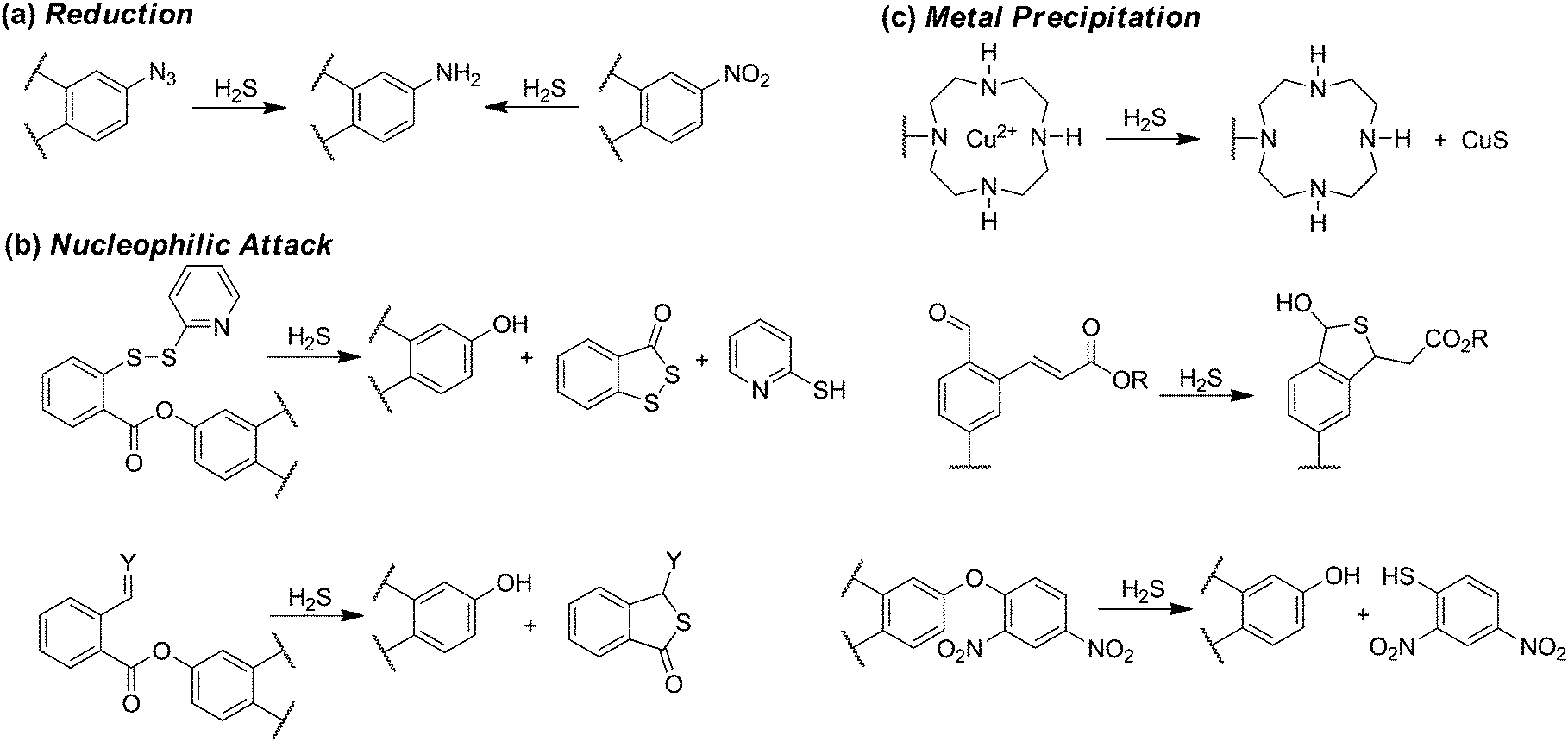 | ||
| Scheme 4 Common motifs in reaction-based probes for H2S, including reactions based on (a) reduction, (b) nucleophilic attack, and (c) metal precipitation. | ||
Further leveraging the high nucleophilicity of HS−, a variety of H2S sensing strategies have been developed in which sulfide attacks an electrophilic, fluorescence-quenching group appended on a fluorophore. Such scaffolds generally rely on the ability of H2S to participate in two sequential nucleophilic reactions to remove or modify the protecting group. One benefit of this strategy is that it imparts inherent selectivity because thiols, which can only participate in one nucleophilic attack, are unable to remove the protecting group.15 One consequence of this selectivity, however, is that in many cases thiols can react with the electrophilic scaffolds, thus effectively consuming the probe prior to reaction with H2S. This general strategy for H2S sensing has been used in activated disulfides, α-β-unsaturated ketones, and other activated electrophiles (Scheme 4b).50–52
In addition to exploiting the high nucleophilicity of HS− for sensing, the high metallophilic nature of sulfur has also been exploited.53 For example, ligation of a paramagnetic metal ion, such as Cu2+, to a fluorescent molecule results in fluorescence quenching. Subsequent reaction with H2S results in CuS formation and precipitation, thus releasing the fluorophore and producing a turn-on response. This strategy is inherently selective for sulfide over other biological thiols, but depending on the ligand characteristics may also be prone to metal reduction and release by cellular reductants or thiols, or exchange of the quenching paramagnetic metal ion with more prevalent diamagnetic ions like Zn2+.
Beyond H2S, polysulfides and persulfides are quickly becoming reactive sulfur species of interest. In addition to contributing to the biological chemistry associated with H2S, it is likely that these sulfane-sulfur species provide signaling pathways distinct to those associated with sulfide. Aligned with this importance, reaction-based probes for polysulfides and other sulfane-sulfur sources are now beginning to emerge.54,55 The development and refinement of these, as well as other, tools appear poised to provide complementary information on the different pools of biological sulfide and further inform on the role of H2S and sulfane-sulfur in the RSONS landscape.
Conclusions and outlook
Hydrogen sulfide is an increasingly important biological molecule joining NO and CO as a key signaling agent and adding a new level of complexity to the RSONS landscape. Importantly, many of the unanswered questions in the field lay at the interface of chemistry and biology, thus requiring collaborative investigations and innovation to develop, refine, and apply new investigative tools in this rapidly expanding area of research. Key challenges include developing selective inhibitors of H2S producing enzymes, generating new classes of sulfide donors that do not consume cellular thiols for activation or that can be programmed to release H2S in response to certain stimuli, and refining H2S imaging methods so that they do not irreversibly consume sulfide. As researchers progress toward these goals, the toolbox of available methods for studying biological H2S will continue to grow and become more accessible to researchers in adjacent fields (such as NO or CO biochemistry, redox biology, and pharmacology), thus broadening the potential impact of H2S-related investigations in these diverse scientific communities.Acknowledgements
We thank several funding agencies that have supported our work on H2S sensing (NIH, R01GM113030), bioinorganic chemistry (NSF, CHE-1454747), and related H2S investigations (Sloan Foundation).Notes and references
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- J. L. Wallace and R. Wang, Nat. Rev. Drug Discovery, 2015, 14, 329–345 CrossRef CAS PubMed.
- R. Wang, C. Szabo, F. Ichinose, A. Ahmed, M. Whiteman and A. Papapetropoulos, Trends Pharmacol. Sci., 2015, 36, 568–578 CrossRef CAS PubMed.
- P. Nagy, Z. Palinkas, A. Nagy, B. Budai, I. Toth and A. Vasas, Biochim. Biophys. Acta, 2014, 1840, 876–891 CrossRef CAS PubMed.
- M. M. Cortese-Krott, G. G. Kuhnle, A. Dyson, B. O. Fernandez, M. Grman, J. F. DuMond, M. P. Barrow, G. McLeod, H. Nakagawa, K. Ondrias, P. Nagy, S. B. King, J. E. Saavedra, L. K. Keefer, M. Singer, M. Kelm, A. R. Butler and M. Feelisch, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E4651–E4660 CrossRef CAS PubMed.
- T. V. Mishanina, M. Libiad and R. Banerjee, Nat. Chem. Biol., 2015, 11, 457–464 CrossRef CAS PubMed.
- M. N. Hughes, M. N. Centelles and K. P. Moore, Free Radical Biol. Med., 2009, 47, 1346–1353 CrossRef CAS PubMed.
- Y. Kimura, Y. Toyofuku, S. Koike, N. Shibuya, N. Nagahara, D. Lefer, Y. Ogasawara and H. Kimura, Sci. Rep., 2015, 5, 14774 CrossRef CAS PubMed.
- R. Greiner, Z. Palinkas, K. Basell, D. Becher, H. Antelmann, P. Nagy and T. P. Dick, Antioxid. Redox Signaling, 2013, 19, 1749–1765 CrossRef CAS PubMed.
- J. I. Toohey, Biochem. J., 1989, 264, 625–632 CrossRef CAS PubMed.
- T. S. Bailey and M. D. Pluth, Free Radical Biol. Med., 2015, 89, 662–667 CrossRef CAS PubMed.
- Y. Zhao, T. D. Biggs and M. Xian, Chem. Commun., 2014, 50, 11788–11805 RSC.
- J. L. Wood, Method Enzymol, 1987, vol. 143, pp. 25–29 Search PubMed.
- D. Zhang, I. Macinkovic, N. O. Devarie-Baez, J. Pan, C. M. Park, K. S. Carroll, M. R. Filipovic and M. Xian, Angew. Chem., Int. Ed., 2014, 53, 575–581 CrossRef CAS PubMed.
- V. S. Lin, W. Chen, M. Xian and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4596–4618 RSC.
- A. Papapetropoulos, M. Whiteman and G. Cirino, Br. J. Pharmacol., 2015, 172, 1633–1637 CrossRef CAS PubMed.
- M. Watanabe, J. Osada, Y. Aratani, K. Kluckman, R. Reddick, M. R. Malinow and N. Maeda, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 1585–1589 CrossRef CAS.
- G. Yang, L. Wu, B. Jiang, W. Yang, J. Qi, K. Cao, Q. Meng, A. K. Mustafa, W. Mu, S. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587–590 CrossRef CAS PubMed.
- I. Ishii, N. Akahoshi, H. Yamada, S. Nakano, T. Izumi and M. Suematsu, J. Biol. Chem., 2010, 285, 26358–26368 CrossRef CAS PubMed.
- N. Nagahara, M. Nagano, T. Ito, K. Shimamura, T. Akimoto and H. Suzuki, Sci. Rep., 2013, 3, 1986 Search PubMed.
- M. Nishida, T. Sawa, N. Kitajima, K. Ono, H. Inoue, H. Ihara, H. Motohashi, M. Yamamoto, M. Suematsu, H. Kurose, A. van der Vliet, B. A. Freeman, T. Shibata, K. Uchida, Y. Kumagai and T. Akaike, Nat. Chem. Biol., 2012, 8, 714–724 CrossRef CAS PubMed.
- K. Modis, C. Coletta, K. Erdelyi, A. Papapetropoulos and C. Szabo, FASEB J., 2013, 27, 601–611 CrossRef CAS PubMed.
- A. Asimakopoulou, P. Panopoulos, C. T. Chasapis, C. Coletta, Z. Zhou, G. Cirino, A. Giannis, C. Szabo, G. A. Spyroulias and A. Papapetropoulos, Br. J. Pharmacol., 2013, 169, 922–932 CrossRef CAS PubMed.
- J. D. Finkelstein, W. E. Kyle, J. J. Martin and A.-M. Pick, Biochem. Biophys. Res. Commun., 1975, 66, 81–87 CrossRef CAS PubMed.
- K. Modis, C. Coletta, A. Asimakopoulou, B. Szczesny, C. Chao, A. Papapetropoulos, M. R. Hellmich and C. Szabo, Nitric Oxide, 2014, 41, 146–156 CrossRef CAS PubMed.
- N. Sen, B. D. Paul, M. M. Gadalla, A. K. Mustafa, T. Sen, R. Xu, S. Kim and S. H. Snyder, Mol. Cell, 2012, 45, 13–24 CrossRef CAS PubMed.
- X. Y. Zhu, S. J. Liu, Y. J. Liu, S. Wang and X. Ni, Cell. Mol. Life Sci., 2010, 67, 1119–1132 CrossRef CAS PubMed.
- M. Whiteman and P. G. Winyard, Expert Rev. Clin. Pharmacol., 2011, 4, 13–32 CrossRef CAS PubMed.
- K. Kashfi and K. R. Olson, Biochem. Pharmacol., 2013, 85, 689–703 CrossRef CAS PubMed.
- M. Pluth, T. Bailey, M. Hammers, M. Hartle, H. Henthorn and A. Steiger, Synlett, 2015, 2633–2643 CrossRef CAS.
- G. A. Benavides, G. L. Squadrito, R. W. Mills, H. D. Patel, T. S. Isbell, R. P. Patel, V. M. Darley-Usmar, J. E. Doeller and D. W. Kraus, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17977–17982 CrossRef CAS PubMed.
- Y. Zhao, T. D. Biggs and M. Xian, Chem. Commun., 2014, 50, 11788–11805 RSC.
- Y. Zhao, S. Bhushan, C. Yang, H. Otsuka, J. D. Stein, A. Pacheco, B. Peng, N. O. Devarie-Baez, H. C. Aguilar, D. J. Lefer and M. Xian, ACS Chem. Biol., 2013, 8, 1283–1290 CrossRef CAS PubMed.
- Y. Zhao, C. Yang, C. Organ, Z. Li, S. Bhushan, H. Otsuka, A. Pacheco, J. Kang, H. C. Aguilar, D. J. Lefer and M. Xian, J. Med. Chem., 2015, 58, 7501–7511 CrossRef CAS PubMed.
- Y. Zhao, H. Wang and M. Xian, J. Am. Chem. Soc., 2011, 133, 15–17 CrossRef CAS PubMed.
- J. M. Carter, Y. Qian, J. C. Foster and J. B. Matson, Chem. Commun., 2015, 51, 13131–13134 RSC.
- L. Li, M. Whiteman, Y. Y. Guan, K. L. Neo, Y. Cheng, S. W. Lee, Y. Zhao, R. Baskar, C. H. Tan and P. K. Moore, Circulation, 2008, 117, 2351–2360 CrossRef CAS PubMed.
- B. E. Alexander, S. J. Coles, B. C. Fox, T. F. Khan, J. Maliszewski, A. Perry, M. B. Pitak, M. Whiteman and M. E. Wood, Med. Chem. Commun., 2015, 6, 1649–1655 RSC.
- G. Yang, X. Sun and R. Wang, FASEB J., 2004, 18, 1782–1784 CAS.
- J. Beltowski, Pharmacol. Rep., 2015, 67, 647–658 CrossRef CAS PubMed.
- M. Hammers, L. Singh, L. Montoya, A. Moghaddam and M. Pluth, Synlett, 2016 DOI:10.1055/s-0035-1560603.
- M. D. Hartle, D. J. Meininger, L. N. Zakharov, Z. J. Tonzetich and M. D. Pluth, Dalton Trans., 2015, 44, 19782–19785 RSC.
- T. Xu, N. Scafa, L. P. Xu, S. Zhou, K. Abdullah Al-Ghanem, S. Mahboob, B. Fugetsu and X. Zhang, Analyst, 2016, 141, 1185–1195 RSC.
- X. Shen, E. A. Peter, S. Bir, R. Wang and C. G. Kevil, Free Radical Biol. Med., 2012, 52, 2276–2283 CrossRef CAS PubMed.
- T. Ida, T. Sawa, H. Ihara, Y. Tsuchiya, Y. Watanabe, Y. Kumagai, M. Suematsu, H. Motohashi, S. Fujii, T. Matsunaga, M. Yamamoto, K. Ono, N. O. Devarie-Baez, M. Xian, J. M. Fukuto and T. Akaike, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7606–7611 CrossRef CAS PubMed.
- A. R. Lippert, E. J. New and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 10078–10080 CrossRef CAS PubMed.
- H. Peng, Y. Cheng, C. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672–9675 CrossRef CAS PubMed.
- L. A. Montoya and M. D. Pluth, Chem. Commun., 2012, 48, 4767–4769 RSC.
- H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2015, 137, 15330–15336 CrossRef CAS PubMed.
- Y. Qian, J. Karpus, O. Kabil, S. Y. Zhang, H. L. Zhu, R. Banerjee, J. Zhao and C. He, Nat. Commun., 2011, 2, 495 CrossRef PubMed.
- C. Liu, J. Pan, S. Li, Y. Zhao, L. Y. Wu, C. E. Berkman, A. R. Whorton and M. Xian, Angew. Chem., Int. Ed., 2011, 50, 10327–10329 CrossRef CAS PubMed.
- Y. Qian, L. Zhang, S. Ding, X. Deng, C. He, X. E. Zheng, H.-L. Zhu and J. Zhao, Chem. Sci., 2012, 3, 2920–2923 RSC.
- K. Sasakura, K. Hanaoka, N. Shibuya, Y. Mikami, Y. Kimura, T. Komatsu, T. Ueno, T. Terai, H. Kimura and T. Nagano, J. Am. Chem. Soc., 2011, 133, 18003–18005 CrossRef CAS PubMed.
- W. Chen, C. Liu, B. Peng, Y. Zhao, A. Pacheco and M. Xian, Chem. Sci., 2013, 4, 2892–2896 RSC.
- W. Chen, E. W. Rosser, T. Matsunaga, A. Pacheco, T. Akaike and M. Xian, Angew. Chem., Int. Ed., 2015, 54, 13961–13965 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |