
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of the prebiotic molecule NH2CHO on astronomical amorphous solid water surfaces: accurate tunneling rate calculations†
Lei
Song
 and
Johannes
Kästner
*
and
Johannes
Kästner
*
Institute for Theoretical Chemistry, University of Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany. E-mail: kaestner@theochem.uni-stuttgart.de
First published on 6th October 2016
Abstract
Investigating how formamide forms in the interstellar medium is a hot topic in astrochemistry, which can contribute to our understanding of the origin of life on Earth. We have constructed a QM/MM model to simulate the hydrogenation of isocyanic acid on amorphous solid water surfaces to form formamide. The binding energy of HNCO on the ASW surface varies significantly between different binding sites, we found values between ∼0 and 100 kJ mol−1. The barrier for the hydrogenation reaction is almost independent of the binding energy, though. We calculated tunneling rate constants of H + HNCO → NH2CO at temperatures down to 103 K combining QM/MM with instanton theory. Tunneling dominates the reaction at such low temperatures. The tunneling reaction is hardly accelerated by the amorphous solid water surface compared to the gas phase for this system, even though the activation energy of the surface reaction is lower than the one of the gas-phase reaction. Both the height and width of the barrier affect the tunneling rate in practice. Strong kinetic isotope effects were observed by comparing to rate constants of D + HNCO → NHDCO. At 103 K we found a KIE of 231 on the surface and 146 in the gas phase. Furthermore, we investigated the gas-phase reaction NH2 + H2CO → NH2CHO + H and found it unlikely to occur at cryogenic temperatures. The data of our tunneling rate constants are expected to significantly influence astrochemical models.
1 Introduction
Formamide (NH2CHO), the simplest molecule containing a peptide bond, has attracted much attention in the field of astrochemistry owing to its potential role as a prebiotic precursor in the origin of life on Earth. It was first detected in a molecular cloud in 1971 by Rubin et al.1 Since then, formamide has been found on comets and in a variety of star-forming regions, such as in high mass young stellar objects (YSOs),2 outflow shock regions,3,4 and on the comet Hale–Bopp.5 Recently, López-Sepulcre et al.6 detected NH2CHO in five out of ten low- and intermediate-mass pre-stellar and protostellar objects as well as isocyanic acid (HNCO) in all ten sources under study. They found a tight and almost linear correlation between NH2CHO and HNCO abundance, which indicates the existence of a chemical relation between those two molecules.The formation sequence for complex organic molecules like NH2CHO can occur either in gas-phase or on the surface of dust grains in the interstellar medium.7–9 Consecutive hydrogenations of HNCO on the mantles of dust grains were proposed as a likely formation route to produce NH2CHO:
| H + HNCO → NH2CO | (1) |
| H + NH2CO → NH2CHO | (2) |
A gas-phase formation route of NH2CHO was investigated by Barone et al.12 using quantum chemical computations. They suggested the reaction
| NH2 + H2CO → NH2CHO + H | (3) |
The increased concentration of active species on the surface of dust grains lends weight to the surface formation route. The mantles of dust grains are predominantly composed of H2O in the amorphous phase combined with other molecules such as CO, CH4, NH3, and traces of other molecules like HNCO, and NH2CHO. Therefore, modeling the reactions on an ASW surface is probably close to the astronomical environment.13 The temperature is always low on the ASW surface, where quantum tunneling is expected to play an important role in chemical reactions. In addition, quantum tunneling is also very likely to happen in the hydrogenation reactions owing to the light reactant H atoms.14
In this work we study reaction (1) on an ASW surface using hybrid quantum mechanics/molecular mechanics (QM/MM) calculations. Combined with instanton theory, we provide tunneling rates of this reaction in the gas phase and on the ASW surface.
2 Methods
2.1 System preparation
The ASW surface was prepared by classical molecular dynamics (MD) simulations with NAMD.15 The initial sample is produced by VMD version 1.9.216 containing 9352 TIP3P water17 molecules. These were simulated in a slab of 85 Å × 85 Å and a thickness of approximately 36 Å. Periodic boundary conditions were applied along all three Cartesian axes with about 70 Å of vacuum between the slabs. This system was treated as a canonical ensemble, equilibrated at 300 K using a Langevin thermostat for 100 ps. After that, the thermostat was instantaneously quenched to 10 K and the system was left for 20 ps to produce a thermally equilibrated bulk amorphous water at low temperature. A hemisphere with a radius of 34 Å was cut out of the slab to be used in the following QM/MM calculations.A large sample of different binding sites on the surface was generated. The HNCO molecule was placed at 113 positions on a regular 2D-grid with a step size of 2 Å covering a circular area with a radius of 12 Å. In each of the 113 points, the molecule was placed 2 Å above the surface. Water molecules with at least one atom within 6 Å were treated by QM (typically about 23 molecules), water molecules within 12 Å were optimized (typically about 161). All other molecules of the hemispheric model were frozen.
2.2 QM/MM method
Both geometry optimization and tunneling rate calculations were performed using a state-of-art QM/MM approach.18,19 In this approach, the reactants H, HNCO and their closer water surroundings were treated with density functional theory (DFT) while more distant water molecules were described by the TIP3P force field.The hybrid QM/MM calculations18,19 were carried out with ChemShell,20,21 using an additive electrostatic embedding scheme, where the MM point charges polarize the QM electron density. We used B3LYP22/def2-SVPD23 to calculate the binding energies and binding site geometries. Different density functionals were tested and compared to coupled cluster reference values as outlined in Section 3.1. On the basis of this comparison, BHLYP-D324–26/def2-TZVP27 was used for barriers and rate calculations. The quantum chemical program package TURBOMOLE 6.628 was used for the QM part while DL_POLY29 built into ChemShell, was used for MM part. Force field parameters for H and HNCO (only the van der Waals parameters are used in QM/MM) were chosen in analogy to the CHARMM22 force field.30–32 The open-source optimizer DL_FIND33 was employed for geometry optimizations including the search for binding sites, the search for transition states with the dimer method34–36 and the determination of instanton paths using a modified Newton–Raphson approach.37,38
2.3 Instanton theory
Tunneling rates in this work were calculated using instanton theory39–44 in its semiclassical formulation.37,38,43,45–51 Instanton theory is based on statistical thermodynamics for the rate expression in which the partition function from a quantum mechanical ensemble is expressed via a Feynman path integral. Generally, this theory is only applicable below the crossover temperature Tc:52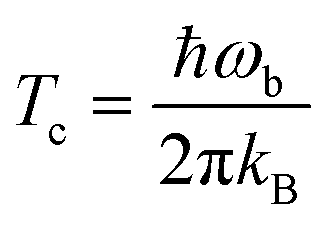 | (4) |
The Feynman paths were discretized to 40 images at T ≥ 135 K and 78 images at lower temperature. Convergence was checked rigorously, e.g. at 100 K doubling the number of images changed the rate constant by only 2%.
In order to make our calculated rate constants accessible to astromodellers, we fitted them to a rate expression proposed previously:75
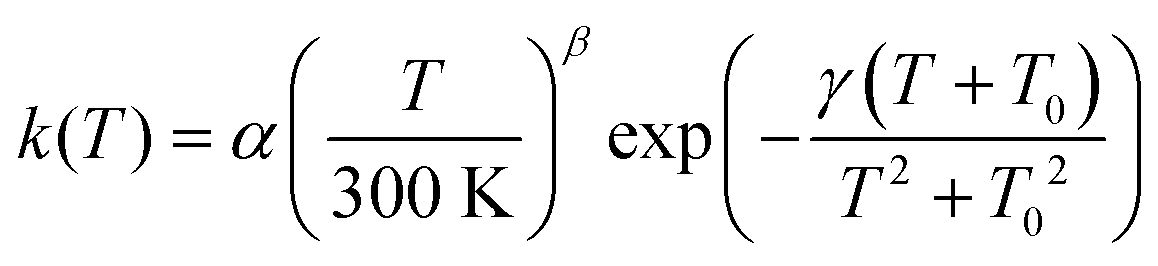 | (5) |
3 Results
3.1 Benchmark calculations
Benchmark calculations were performed to choose a proper DFT functional for the transition state search and tunnel rate calculations. We calculated the activation energy Ea for reaction (1) in the gas phase based on B3LYP-D322,26/def2-TZVPD23 optimized geometries using UCCSD(T)-F1276,77/cc-pVTZ-F1278 on a RHF reference in MOLPRO 2012.79 The resulting Ea of 32.7 kJ mol−1 was used as a reference and compared to the data from B3LYP, BHLYP, TPSS, TPSSH and PBE0 functionals with the def2-SVPD23 and def2-TZVP27 basis sets. All DFT calculations include D3 dispersion corrections.26 The results are compared in Fig. 1. The smallest deviation was found for the BHLYP-D324–26/def2-TZVP27 theory level which we selected as the proper quantum mechanical level for QM molecules.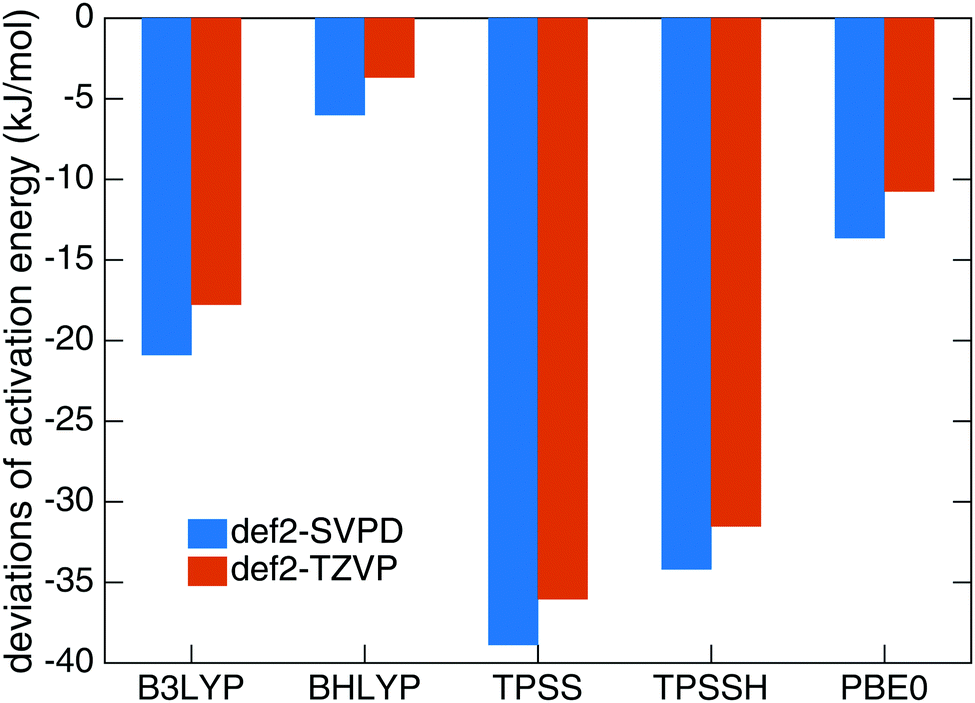 | ||
| Fig. 1 Deviations of activation energies of reaction (1) at different DFT levels with D3 dispersion correction from the results at UCCSD(T)-F12/cc-pVTZ-F12 level. | ||
3.2 HNCO binding sites and binding energies
Reaction (1) originates from HNCO bound to the ASW surface. We investigated different binding modes and their respective binding energies in our QM/MM setup using the B3LYP22/def2-SVPD23 level for the QM calculations. Geometry optimization was performed starting from 113 initial structures. Among those, 90 jobs finished successfully and provided four types of HNCO binding modes on the ASW surface as shown in Fig. 2. Panel (a) illustrates the major adsorption mode to which 48 out of the 90 cases belonged. In this case the H and O ends of the HNCO molecule act as H-bond donor and acceptor connecting to O and H atoms in the water ice, respectively. The N atom can also act as a H-bond acceptor while the H atom of the HNCO molecule still serves as a H-bond donor to connect to an O atom from the water. This case is depicted in panel (b) of Fig. 2 and accounts for 34 of 90 cases. The remaining 8 cases resulted in binding modes where either the N atom or H atom in the HNCO molecule connects to H or O of the surface, as shown in panels (c) and (d).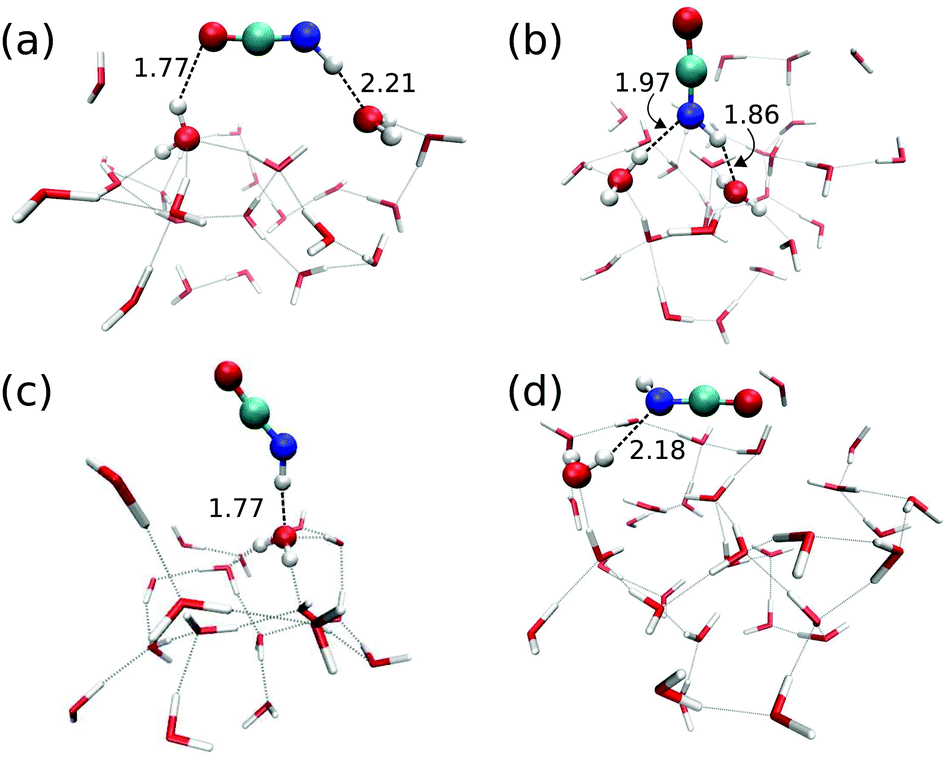 | ||
| Fig. 2 Four different HNCO binding modes on the amorphous solid water surface. Only QM molecules are shown, HNCO and all water molecules H-bonded to it are shown as ball-and-stick. Bond distances are given in Å. | ||
The binding energy of HNCO on the ASW surface was the energy required to disassemble the adsorbed HNCO from the surface into the gas phase. The minima of the ASW surface with and without HNCO in each of the 90 cases were calculated using the same QM, active and frozen water regions. Fig. 3 presents the distribution of binding energies from the 90 cases. It is obvious that the binding energy is very broadly distributed from 0 to about 100 kJ mol−1 with the largest fraction between 40 and 50 kJ mol−1. The tighter bound sites are expected to be occupied preferentially, which leads to a surface-coverage dependent binding energy. No clear correlation can be found between the binding modes distinguished in Fig. 2 and the binding energies. The rough surface of ASW leads to the significant spread of binding energies, which likely is of relevance for astrochemical modeling of adsorption and desorption processes. The binding energies are given in Fig. 3 without considering the vibrational zero point energy (ZPE). We calculated the ZPE for the four representative modes shown in Fig. 2. They reduce the binding energy by 8.0, 5.4, 2.3, and 7.7 kJ mol−1 for the modes a, b, c, and d, respectively. Thus, the influence of the ZPE on binding is small.
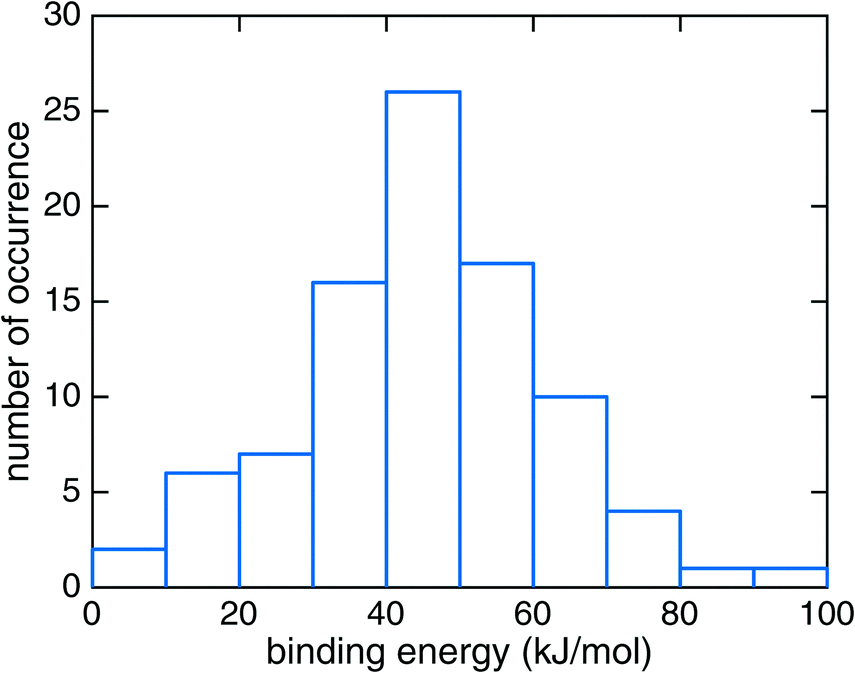 | ||
| Fig. 3 The distribution of HNCO binding energies on the amorphous solid water surface at the B3LYP22/def2-SVPD23 level of theory. | ||
3.3 Transition states
We investigated transition states for four different binding geometries with rather different binding energies. The resulting data are given in Table 1. The transition structures are labeled TS1 to TS4. Their binding energies differ between 27.9 and 80.3 kJ mol−1. The attack by a hydrogen atom at the N-site of HNCO requires the latter to be accessible. Thus, binding modes (a) and (c) of the ones depicted in Fig. 2 are most promising. TS1, TS3, and TS4 correspond to binding mode (a) while TS2 corresponds to binding mode (c). For the transition state search and the following tunneling rate calculations, we restricted the QM region to H + HNCO plus just three water molecules (5 for TS2, 4 for TS4), see Fig. 4. While the same set of atoms (12 Å) was optimized as in the investigations of the binding sites, the Hessian calculations were restricted to the QM region.| Gas TS | ASW | ||||
|---|---|---|---|---|---|
| TS1 | TS2 | TS3 | TS4 | ||
| HNCO binding energy | 48.1 | 27.9 | 80.3 | 52.1 | |
| N–H bond distance | 1.542 | 1.546 | 1.532 | 1.546 | 1.547 |
| ω b | 1339i | 1240i | 1271i | 1268i | 1262i |
| E a (ER mechanism) | 30.6 | 26.7 | 29.9 | 28.4 | 27.9 |
| E a incl. ZPE | 36.2 | 31.8 | 36.5 | 32.7 | 32.7 |
| T c | 307 | 284 | 291 | 290 | 289 |
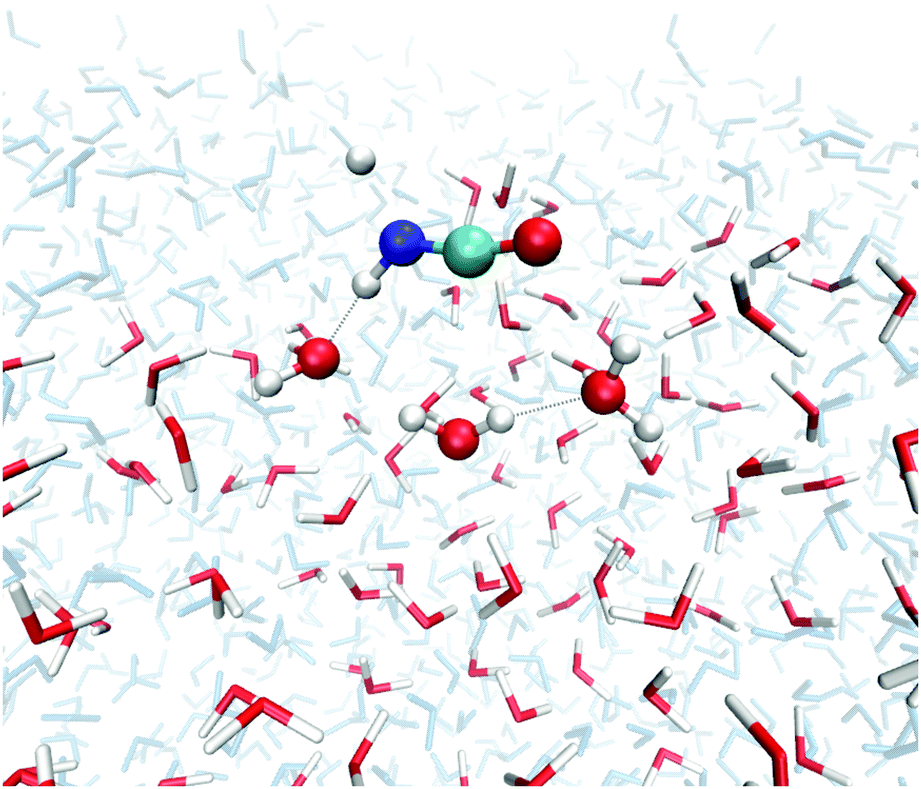 | ||
| Fig. 4 Optimized geometry of TS1 of reaction (1) on the ASW surface. In the TS search the QM region was restricted to the molecules shown as ball-and-stick models. All red/white water molecules were active, the blue/gray ones frozen. | ||
All data in Table 1 refer to a reactant state with HNCO adsorbed on the surface and H in the gas phase, i.e. to an Eley–Rideal-type (ER) surface reaction mechanism. Compared with the transition state in the gas phase, the ones on the ASW surface have slightly lower activation energies Ea. Without ZPE the four surface-bound activation energies are 3.9 to 0.7 kJ mol−1 lower than the gas-phase Ea, including ZPE they are between 4.4 kJ mol−1 lower and 0.3 kJ mol−1 higher. Note that despite the large spread in binding energies of the different adsorption sites, the associated activation energies are very similar. This indicates similar rate constants, which will be discussed in the following section. The N–H bond distances of the transition states on the surface are generally slightly longer than in the gas phase, see Table 1, indicating an earlier transition state on the surface.
The transition states TS1, TS3, and TS4 describe a movement of the hydrogen atom coming from the gas phase above the surface. By contrast in TS2, which originates from a structure like the one in Fig. 2(c), the hydrogen atom approaches the nitrogen site from closer to the surface, see also Fig. S1 of the ESI.† In this case, a well-defined pre-reactive minimum with H loosely bound to the surface was found. This corresponds to a possible reactant site for a Langmuir–Hinshelwood (LH) mechanism. The barrier with respect to the LH reactant state is 34.6 kJ mol−1 (37.9 kJ mol−1 with ZPE).
3.4 Tunneling rate constants
Starting from TS1 we calculated rate constants for reaction (1) following an ER mechanism on the ASW surface and compared them to the gas phase reaction treated at the same QM level of theory. The results are shown in Fig. 5. The red solid triangles correspond to the rate constants on the ASW surface, the blue solid circles to the ones of the corresponding gas-phase reaction. Instanton rate constant calculations are restricted to temperatures below Tc. At high temperature the surface-bound reaction is slightly faster than the gas-phase reaction; at low temperature the case is reversed and the gas-phase reaction becomes more efficient. Thus, there is no significant catalytic effect of the surface. However, the surface of course still has the effect of dissipating the excess energy of the reaction and increasing the local concentration of the reactants. Despite the lower barrier, the tunneling rate constant for the ASW-bound reaction is lower than the gas phase reaction at low temperature. This demonstrates again that besides the barrier height, the barrier width is important for the tunneling efficiency.71 The barrier shapes along the intrinsic reaction coordinates (IRC) are compared in Fig. 6, which clearly shows that the ASW-barrier is lower but broader than the gas-phase barrier which leads to the lower tunneling rate at low temperature.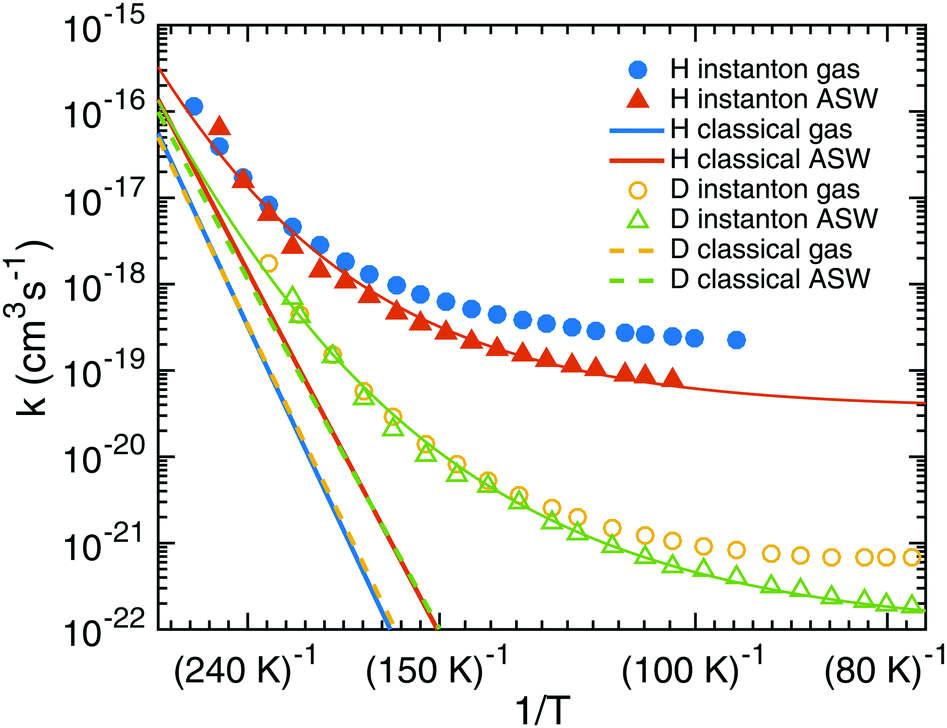 | ||
| Fig. 5 Instanton and classical rate constants for the reactions of H + HNCO → NH2CO and D + HNCO → NHDCO in gas and the ER process on the ASW surface. The thin lines represent fits using eqn (5). | ||
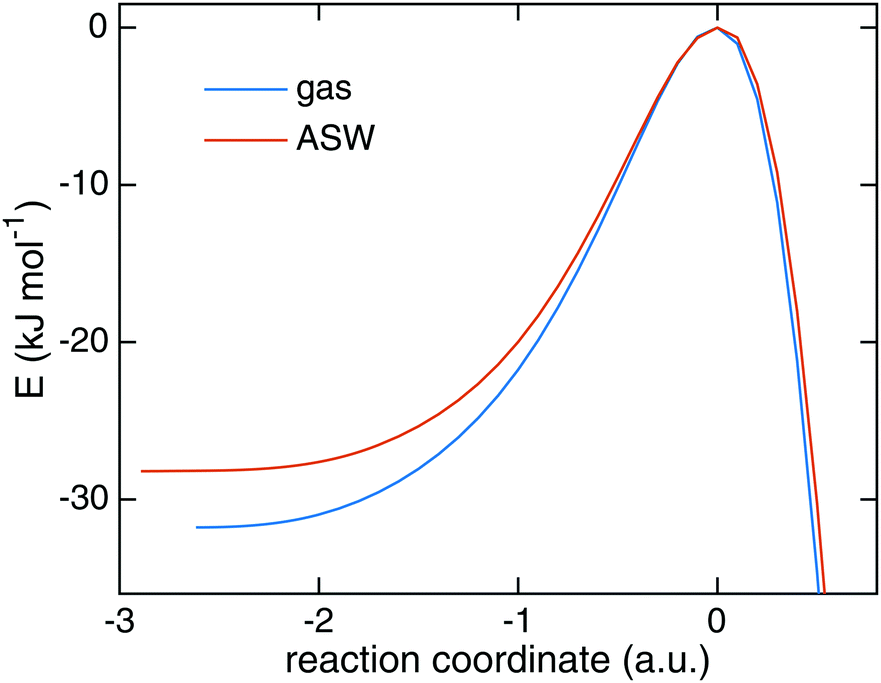 | ||
| Fig. 6 The minimum energy path of the reaction of H + HNCO → NH2CO in the gas phase and on the amorphous solid water surface. | ||
Our data allow the comparison between a structural model which contains the surface explicitly and a gas-phase model for the surface reaction. As discussed above, the barrier changes only very slightly due to the influence of the surface and quite independently of the binding site. The resulting rate constants are very similar. The surface, however, restricts the rotational motion of the reactant and the transition state. The change in the rotational partition function is included in the rate constants depicted in Fig. 5. One can model a surface by considering only the atoms HNCO + H explicitly but restricting the rotational motion, i.e. ignoring the change in the rotational partition function between HNCO and the transition state. This corresponds to the rotational restriction of both HNCO and the transition state on the surface. With such an approach, the rate constants obtained from a gas-phase model are even more similar to those obtained from the surface model, e.g., at 103 K we find a rate constant on the surface of 7.8 × 10−20 cm3 s−1, of 8.0 × 10−20 cm3 s−1 for the gas phase model with restricted rotation and of 2.4 × 10−19 cm3 s−1 for the gas phase model with full rotation. For the reaction under study a gas-phase model with restricted rotation results in sufficiently accurate surface rate constants.
Rate constants were fitted to eqn (5) to facilitate the use of our results in astrochemical models. The parameters are given in Table 2, the resulting curves are shown in Fig. 5 as thin red and green lines. They match the calculated rate constants reasonably well. We recommend using the fit in a temperature range close to the range that was used to produce it, i.e. 1000 K to ∼90 K for H + HNCO and 1000 K to ∼60 K for D + HNCO.
| Parameter | H | D |
|---|---|---|
| α (cm3 s−1) | 7.22 × 10−12 | 4.13 × 10−12 |
| β | 1 | 1 |
| γ (K) | 2856 | 2887 |
| T 0 (K) | 195.8 | 153.4 |
The red and blue straight lines in Fig. 5 correspond to the rate constants neglecting tunneling (but including quantized vibrations and, thus, the ZPE). Due to the smaller barrier, without tunneling the surface-bound reaction is always faster than the gas-phase reaction. Tunneling accelerates the reaction by many orders of magnitude at low temperature. Values for the rate constants with and without tunneling are given in Tables S2 and S3 of the ESI.† For example at 103 K, tunneling accelerates the gas-phase by a factor of 2 × 1010 and the surface reaction by a factor of 108. These values increase steeply with decreasing temperature.
The bimolecular rate constants reported above relate to an ER mechanism. At low temperature a LH mechanism is more likely. In that case we can assume HNCO to be stationary on the surface while the H atom diffuses with the hopping rate constant khop until it meets a HNCO site. Then it can either react or diffuse away again. The probability for reaction is kreact/(kreact + khop) where kreact is a unimolecular rate constant which we can calculate. It corresponds to the process of an encounter complex of H with HNCO reacting to NH2CO. Since H is bound very weakly on the surface, we were able to optimize such an encounter complex only for TS2. Its energy is 4.7 kJ mol−1 (1.4 kJ mol−1 with ZPE) below that of the separated reactants. The resulting rate constants are shown in Fig. 7. We fitted the parameters of eqn (5), which resulted in α = 3.56 × 1010 s−1, γ = 2503 K and T0 = 172.9 K. The parameter β was kept to 1 just like in the other fits.
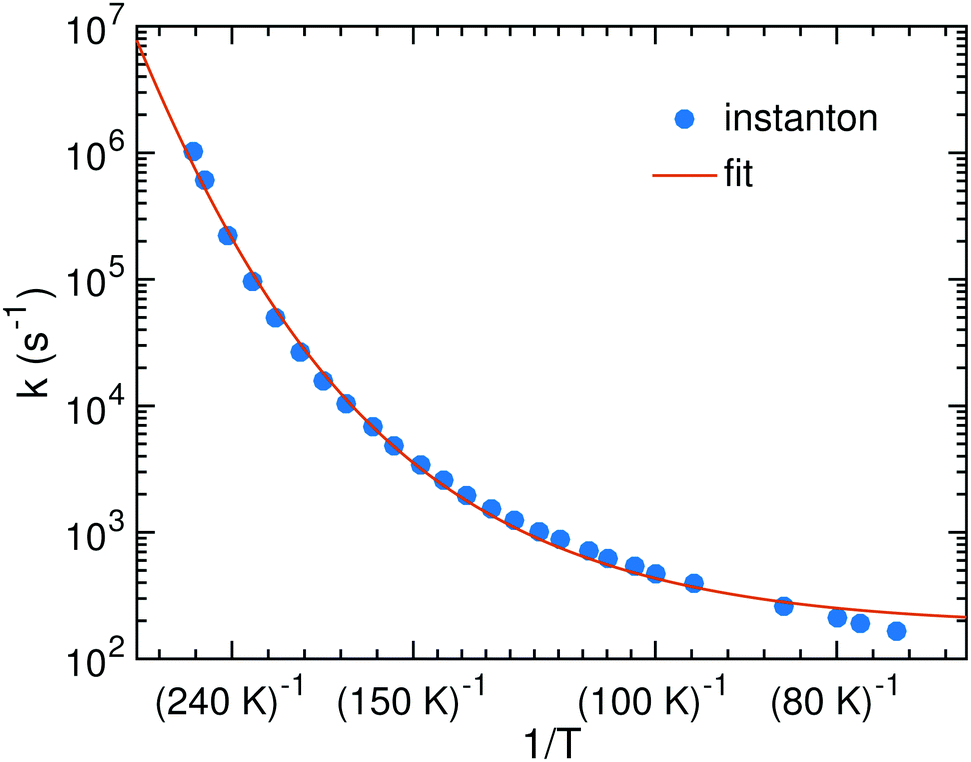 | ||
| Fig. 7 Instanton rate constants for the LH process of reaction (1) on the ASW surface. | ||
3.5 Kinetic isotope effects
In addition, we investigated the kinetic isotope effect (KIE) for reaction (1). For D + HNCO → NHDCO, the crossover temperature is reduced from 284 K to 218 K on the ASW surface and from 307 K to 235 K in the gas phase. In Fig. 5 instanton rate constants for the reactions with deuterium in the gas-phase are shown by yellow circles and on ASW by green triangles. Similar trends are visible as for the addition of protium to HNCO, but the rate constants are much smaller. As frequently observed for tunneling reactions, the KIE increases with decreasing temperature. At 103 K the KIE for the gas-phase reaction is 231, on the ASW surface it is 146. Even stronger KIEs can be expected at lower temperature. The KIEs without tunneling are much smaller as can be seen from Fig. 5, which indicates that the KIE is mostly caused by tunneling rather than by the difference in the ZPE.3.6 Alternative gas-phase reaction
To elucidate a possible role of reaction (3) for the formation of NH2CHO, we calculated the barrier for the initial reaction channel, the approach of NH2 to formaldehyde. We optimized the reactants and the transition state on the M06-2X80/def2-TZVP27 level using NWCHEM 6.681 and calculated single-point energies and vibrational frequencies on the UCCSD(T)-F1276,77/cc-pVTZ-F1278 level. The coordinates of the transition structure are given in the ESI.† In agreement with previous work,12 we found an almost submerged barrier on the potential energy surface, +2.7 kJ mol−1 compared to the separated reactants. Including the ZPE, however, resulted in a significant barrier of 17.8 kJ mol−1. The crossover temperature is 88.0 K. Thus, tunneling only plays a minor role above that temperature. The corresponding rate constant for reaction (3) at 100 K is k = 1.1 × 10−22 cm3 s−1 if tunneling is neglected and quite a similar value of k = 5.3 × 10−22 cm3 s−1 if tunneling is approximated via a symmetric Eckart barrier. Note that above the crossover temperature, instanton theory is not applicable. These rate constants can only serve as an upper limit to the full rate constant of reaction (3) since they only cover the entrance channel. The full reaction contains additional submerged barriers12 which might lower the rate even further. Nevertheless, even these upper bounds are significantly smaller than the rate constant of k = 2.4 × 10−19 cm3 s−1 for reaction (1) at the same temperature. Thus, we conclude that the gas-phase reaction (3) is expected not to play a significant role in the formation of NH2CHO.4 Conclusions
We investigated binding of HNCO to an ASW surface and subsequent hydrogenation. Different binding sites with a significant spread of binding energies were found. The activation barrier for the hydrogenation reaction turned out to be rather independent of the binding energy. We calculated the reaction rate constants for H + HNCO → NH2CO in the gas phase at temperatures of 289 K down to 95 K and on the ASW surface down to 103 K by combining the QM/MM method with instanton theory. Although the activation barrier for the surface reaction is 3.9 kJ mol−1 (4.4 kJ mol−1 including ZPE) lower than in the gas-phase, the ASW surface does not efficiently accelerate this reaction, but hinders it at temperatures below 240 K. It demonstrates that the width but not the height of the barrier dominantly affects the tunneling rate for this system. In addition, the deuterated reaction of D + HNCO → NHDCO has been investigated both in the gas-phase and on the ASW surface. According to the instanton calculations, the KIEs are 231 and 146 for the gas phase reaction and the surface reaction at 103 K, respectively and expected to be at least similarly strong at even lower temperature. The strong tunnel effect raises the rate constants to values which enable hydrogenation of HNCO on the surface of interstellar dust grains, making this a possible route for the formation of the pre-biotic molecule formamide. By contrast, the gas-phase route viareaction (3) seems inaccessible at low temperature.Acknowledgements
This work was financially supported by the European Research Council (ERC) under the European Unions Horizon 2020 research and innovation programme (grant agreement no. 646717, TUNNELCHEM). The authors also acknowledge support for CPU time by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no. INST 40/467-1 FUGG.References
- R. H. Rubin, G. W. Swenson, Jr., R. C. Benson, H. L. Tigelaar and W. H. Flygare, Astrophys. Lett., 1971, 169, L39 CrossRef CAS.
- S. E. Bisschop, J. K. Jørgensen, E. F. van Dishoeck and E. B. M. de Wachter, Astron. Astrophys., 2007, 465, 913–929 CrossRef CAS.
- T. Yamaguchi, S. Takano, Y. Watanabe, N. Sakai, T. Sakai, S.-Y. Liu, Y.-N. Su, N. Hirano, S. Takakuwa, Y. Aikawa, H. Nomura and S. Yamamoto, Publ. Astron. Soc. Jpn., 2012, 64, 105 CrossRef.
- E. Mendoza, B. Lefloch, A. López-Sepulcre, C. Ceccarelli, C. Codella, H. M. Boechat-Roberty and R. Bachiller, Mon. Not. R. Astron. Soc., 2014, 445, 151–161 CrossRef.
- D. Bockelée-Morvan, D. C. Lis, J. E. Wink, D. Despois, J. Crovisier, R. Bachiller, D. J. Benford, N. Biver, P. Colom, J. K. Davies, E. Gérard, B. Germain, M. Houde, D. Mehringer, R. Moreno, G. Paubert, T. G. Phillips and H. Rauer, Astron. Astrophys., 2000, 353, 1101–1114 Search PubMed.
- A. López-Sepulcre, A. A. Jaber, E. Mendoza, B. Lefloch, C. Ceccarelli, C. Vastel, R. Bachiller, J. Cernicharo, C. Codella, C. Kahane, M. Kama and M. Tafalla, Mon. Not. R. Astron. Soc., 2015, 449, 2438–2458 CrossRef.
- S. Raunier, T. Chiavassa, F. Duvernay, F. Borget, J. P. Aycard, E. Dartois and L. d'Hendecourt, Astron. Astrophys., 2004, 416, 165–169 CrossRef CAS.
- R. T. Garrod, S. L. Widicus Weaver and E. Herbst, Astrophys. J., 2008, 682, 283–302 CrossRef CAS.
- P. Redondo, C. Barrientos and A. Largo, Astrophys. J., 2014, 793, 32–39 CrossRef.
- M. T. Nguyen, D. Sengupta, L. Vereecken, J. Peeters and L. G. Vanquickenborne, J. Phys. Chem., 1996, 100, 1615–1621 CrossRef CAS.
- J. A. Noble, P. Theule, E. Congiu, F. Dulieu, M. Bonnin, A. Bassas, F. Duvernay, G. Danger and T. Chiavassa, Astron. Astrophys., 2015, 576, A91 CrossRef.
- V. Barone, C. Latouche, D. Skouteris, F. Vazart, N. Balucani, C. Ceccarelli and B. Lefloch, Astron. Astrophys., 2015, 453, L31–L35 Search PubMed.
- T. Hama and N. Watanabe, Chem. Rev., 2013, 113, 8783–8839 CrossRef CAS PubMed.
- J. Meisner and J. Kästner, Angew. Chem., Int. Ed., 2016, 55, 5400–5413 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- A. Warshel and M. Karplus, J. Am. Chem. Soc., 1972, 94, 5612–5625 CrossRef CAS.
- A. Warshel and M. Levitt, J. Mol. Biol., 1976, 103, 227–249 CrossRef CAS PubMed.
- P. Sherwood, A. H. de Vries, M. F. Guest, G. Schreckenbach, C. R. A. Catlow, S. A. French, A. A. Sokol, S. T. Bromley, W. Thiel, A. J. Turner, S. Billeter, F. Terstegen, S. Thiel, J. Kendrick, S. C. Rogers, J. Casci, M. Watson, F. King, E. Karlsen, M. Sjøvoll, A. Fahmi, A. Schäfer and C. Lennartz, J. Mol. Strut.: THEOCHEM, 2003, 632, 1 CrossRef CAS.
- S. Metz, J. Kästner, A. A. Sokol, T. W. Keal and P. Sherwood, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 101 CrossRef CAS.
- P. Stephens, F. Devlin, C. Chabalowski and M. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- TURBOMOLE V, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com Search PubMed.
- W. Smith, C. Yong and P. Rodger, Mol. Simul., 2002, 28, 385–471 CrossRef CAS.
- A. D. MacKerell Jr., D. Bashford, R. L. Bellott, R. L. Dunbrack Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586 CrossRef PubMed.
- A. MacKerell Jr. and N. K. Banavali, J. Comput. Chem., 2000, 21, 105 CrossRef.
- A. D. MacKerell Jr., M. Feig and C. Brooks III, J. Comput. Chem., 2004, 25, 1400 CrossRef PubMed.
- J. Kästner, J. M. Carr, T. W. Keal, W. Thiel, A. Wander and P. Sherwood, J. Phys. Chem. A, 2009, 113, 11856 CrossRef PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010 CrossRef CAS.
- A. Heyden, A. T. Bell and F. J. Keil, J. Chem. Phys., 2005, 123, 224101 CrossRef PubMed.
- J. Kästner and P. Sherwood, J. Chem. Phys., 2008, 128, 014106 CrossRef PubMed.
- J. B. Rommel, T. P. M. Goumans and J. Kästner, J. Chem. Theory Comput., 2011, 7, 690 CrossRef CAS PubMed.
- J. B. Rommel and J. Kästner, J. Chem. Phys., 2011, 134, 184107 CrossRef PubMed.
- J. S. Langer, Ann. Phys., 1967, 41, 108 CAS.
- W. H. Miller, J. Chem. Phys., 1975, 62, 1899 CrossRef CAS.
- S. Coleman, Phys. Rev. D: Part. Fields, 1977, 15, 2929 CrossRef.
- C. G. Callan Jr. and S. Coleman, Phys. Rev. D: Part. Fields, 1977, 16, 1762 CrossRef.
- S. C. Althorpe, J. Chem. Phys., 2011, 134, 114104 CrossRef PubMed.
- J. O. Richardson, J. Chem. Phys., 2016, 144, 114106 CrossRef PubMed.
- I. Affleck, Phys. Rev. Lett., 1981, 46, 388–391 CrossRef.
- S. Coleman, Nucl. Phys. B, 1988, 298, 178 CrossRef.
- P. Hänggi, P. Talkner and M. Borkovec, Rev. Mod. Phys., 1990, 62, 251 CrossRef.
- V. A. Benderskii, D. E. Makarov and C. A. Wight, Adv. Chem. Phys., 1994, 88, 55 CrossRef.
- M. Messina, G. K. Schenter and B. C. Garrett, J. Chem. Phys., 1995, 103, 3430 CrossRef CAS.
- J. O. Richardson and S. C. Althorpe, J. Chem. Phys., 2009, 131, 214106 CrossRef PubMed.
- Y. Zhang, J. B. Rommel, M. T. Cvitaš and S. C. Althorpe, Phys. Chem. Chem. Phys., 2014, 16, 24292–24300 RSC.
- M. J. Gillan, J. Phys. C: Solid State Phys., 1987, 20, 3621 CrossRef.
- S. Chapman, B. C. Garrett and W. H. Miller, J. Chem. Phys., 1975, 63, 2710 CrossRef CAS.
- G. Mills and H. Jónsson, Phys. Rev. Lett., 1994, 72, 1124 CrossRef CAS PubMed.
- G. Mills, H. Jónsson and G. K. Schenter, Surf. Sci., 1995, 324, 305–337 CrossRef CAS.
- G. Mills, G. K. Schenter, D. E. Makarov and H. Jónsson, Chem. Phys. Lett., 1997, 278, 91 CrossRef CAS.
- W. Siebrand, Z. Smedarchina, M. Z. Zgierski and A. Fernández-Ramos, Int. Rev. Phys. Chem., 1999, 18, 5 CrossRef CAS.
- Z. Smedarchina, W. Siebrand, A. Fernández-Ramos and Q. Cui, J. Am. Chem. Soc., 2003, 125, 243–251 CrossRef CAS PubMed.
- T. Qian, W. Ren, J. Shi, W. E and P. Sheng, Phys. A, 2007, 379, 491 CrossRef.
- S. Andersson, G. Nyman, A. Arnaldsson, U. Manthe and H. Jónsson, J. Phys. Chem. A, 2009, 113, 4468 CrossRef CAS PubMed.
- T. P. M. Goumans and S. Andersson, Mon. Not. R. Astron. Soc., 2010, 406, 2213–2217 CrossRef CAS.
- T. P. M. Goumans, Mon. Not. R. Astron. Soc., 2011, 415, 3129–3134 CrossRef CAS.
- T. P. M. Goumans, Mon. Not. R. Astron. Soc., 2011, 413, 2615–2620 CrossRef CAS.
- T. P. M. Goumans and J. Kästner, Angew. Chem., Int. Ed., 2010, 49, 7350–7352 CrossRef CAS PubMed.
- H. Jónsson, Proc. Natl. Acad. Sci. U. S. A., 2010, 108, 944–949 CrossRef PubMed.
- J. Meisner, J. B. Rommel and J. Kästner, J. Comput. Chem., 2011, 32, 3456 CrossRef CAS PubMed.
- T. P. M. Goumans and J. Kästner, J. Phys. Chem. A, 2011, 115, 10767 CrossRef CAS PubMed.
- D. M. Einarsdóttir, A. Arnaldsson, F. Óskarsson and H. Jónsson, Lect. Notes Comput. Sci. Eng., 2012, 7134, 45 Search PubMed.
- J. B. Rommel, Y. Liu, H.-J. Werner and J. Kästner, J. Phys. Chem. B, 2012, 116, 13682 CrossRef CAS PubMed.
- M. Kryvohuz and R. A. Marcus, J. Chem. Phys., 2012, 137, 134107 CrossRef CAS PubMed.
- J. Kästner, Chem. – Eur. J., 2013, 19, 8207–8212 CrossRef PubMed.
- S. Álvarez-Barcia, J. R. Flores and J. Kästner, J. Phys. Chem. A, 2014, 118, 78 CrossRef PubMed.
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 158 CrossRef.
- M. Kryvohuz, J. Phys. Chem. A, 2014, 118, 535–544 CrossRef CAS PubMed.
- J. Zheng and D. G. Truhlar, Phys. Chem. Chem. Phys., 2010, 12, 7782–7793 RSC.
- T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- K. A. Peterson, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2008, 128, 084102 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012, a package of ab initio programs, 2012, see http://https://www.molpro.net/ Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- M. Valiev, E. J. Bylaska, N. Govind, K. Kowalski, T. P. Straatsma, H. J. J. van Dam, D. Wang, J. Nieplocha, E. Apra, T. L. Windus and W. A. de Jong, Comput. Phys. Commun., 2010, 181, 1477–1489 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Geometric details, lists of calculated rate constants. See DOI: 10.1039/c6cp05727f |
| This journal is © the Owner Societies 2016 |