
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An operando emission spectroscopy study of Pt/Al2O3 and Pt/CeO2/Al2O3†
Valentina
Marchionni
ab,
Jakub
Szlachetko
ac,
Maarten
Nachtegaal
a,
Anastasios
Kambolis
a,
Oliver
Kröcher
 ad and
Davide
Ferri
*a
ad and
Davide
Ferri
*a
aPaul Scherrer Institut, CH-5232 Villigen PSI, Switzerland. E-mail: davide.ferri@psi.ch; Tel: +41 56 310 27 81
bETH Zurich, Institute for Chemical and Bioengineering, CH-8093 Zurich, Switzerland
cJan Kochanowski University, Institute of Physics, PL-25-406 Kielce, Poland
dÉcole polytechnique fédérale de Lausanne (EPFL), Institute of Chemical Science and Engineering, CH-1015 Lausanne, Switzerland
First published on 28th September 2016
Abstract
In situ time-resolved spectroscopic examination of catalysts based on well dispersed nanoparticles on metal oxides under transient conditions significantly facilitates the elucidation of reaction mechanisms. In this contribution, we demonstrate the level of structural information that can be obtained using high-energy resolution off-resonant spectroscopy (HEROS) to study 1.3 wt% Pt/Al2O3 and 1.3 wt% Pt/20 wt% CeO2/Al2O3 catalysts subjected to redox pulsing. First, HEROS is compared with XANES in a temperature programmed reduction experiment to demonstrate the increased sensitivity and time resolution of HEROS. Second, modulation excitation spectroscopy is exploited by redox pulsing to enhance the sensitivity of HEROS to structural changes by the application of phase sensitive detection (PSD) to the time-resolved HEROS data set. The HEROS measurements were complemented by resonant X-ray emission (RXES) and diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy measurements performed under identical conditions and in a single reactor cell in order to probe different aspects of the catalyst materials under the selected experimental conditions.
Introduction
Operando spectroscopy experiments have become crucial for the structural elucidation of functional materials at the very same time when they are working.1 This approach is common nowadays for catalysis science and is being extended to other fields where materials are subject to structural changes under operating conditions. Owing to the dynamic changes in the catalyst structure during reaction, fast data acquisition in the second to sub-second time resolution is often required when the catalyst material is analyzed under fast transient conditions in order to be able to capture reaction intermediates. Such experimental conditions may be required to simulate how the catalyst functions in the real application; on the other hand they also represent an experimental and analytical expedient to improve our perception of the relevant components of the catalyst that are involved in the process of interest.2,3 Among the large number of spectroscopic and diffraction methods available to observe a catalyst at work, X-ray absorption spectroscopy (XAS) has become an essential tool because of the intrinsic advantages of high energy X-rays, i.e. the element specificity and large penetration depth of X-rays, and of the nature of the information provided, i.e. the oxidation state and local coordination environment of the absorbing atom without long-range order requirements.4–6 A time resolution of a few milliseconds per spectrum has been demonstrated in both energy dispersive and quick X-ray absorption spectroscopy. However, limitations still exist that are mainly caused by the sensitivity of the measurement to materials that are intrinsically less inclined to reliable XAS interrogation in transmission mode because of their high dilution or because of the presence of highly absorbing elements. For these materials, e.g. noble metals supported on ceria, fluorescence detection could become indispensable, but its time resolution is greatly limited by the necessity to scan the X-ray energy and the sensitivity of state-of-the-art detectors. Scanning free emission/fluorescence methods probing the electronic structure of the material to follow transformations associated with the catalyst structure have received increasing interest.4,6 X-ray emission spectroscopy (XES) probes the occupied orbitals by recording the fluorescence radiation that is emitted when a core electron is removed by an incident photon and the inner shell vacancy is filled by an electron from a higher shell using a dispersive spectrometer based on perfect crystal Bragg optics. The process is termed resonant X-ray emission spectroscopy (RXES) when the incident X-ray energy is tuned across an absorption edge and the spectrum is much narrower than those acquired above edge excitation energies because the resonant scattering process is independent of the initial state broadening.7 The high-time resolution and quantitative information of the method were demonstrated in a recent RXES study to follow in situ the reduction of Ce4+ during CO oxidation on a ceria-supported platinum catalyst.8 Szlachetko et al.9 demonstrated the feasibility of high energy resolution off-resonant spectroscopy (HEROS) for catalysis studies to probe the unoccupied density of states with element specificity. HEROS collects high energy resolution XAS-like spectra in a single shot by recording an X-ray emission spectrum in the dispersive von Hamos geometry.10 Despite the fact that the energy of the incoming radiation is below the absorption threshold (off-resonant excitation), the electron is excited into an unoccupied state above the Fermi level by taking energy from the emitted photon, whose energy will thus be correspondingly reduced.9 The HEROS energy resolution is independent of the initial lifetime broadening, hence HEROS spectra exhibit more detailed information than conventional XAS spectra and are comparable to high energy resolution fluorescence detection (HERFD) XAS spectra.10 HERFD-XAS spectra exhibit higher resolution than normal transmission or the total fluorescence spectra and allow the identification of the bonding sites of reactants in supported metal catalysts.11–13 In addition, HEROS delivers self-absorption free data, because the incoming beam is fixed.14 Self-absorption occurs in conventional fluorescence mode XAS measurements, especially of concentrated samples, such as CeO2, when the induced fluorescence radiation is partially absorbed in the samples on its way out, and distorts the shape of the spectra.15 Therefore, besides the sub-second time-resolution, HEROS allows the quantification of structural changes.HEROS data acquisition is performed typically over repeated pulses in order to increase the signal-to-noise ratio. Beside the increased sensitivity to adsorbed intermediates due to data acquisition over repeated pulses,16 HEROS spectra are thus ideally suited to be treated with frequency response analytical tools such as phase sensitive detection (PSD).17 Then, the averaged time-resolved spectra are transformed into a set of phase-resolved spectra according to eqn (1):
 | (1) |
In a recent study,16 we used HEROS to follow in situ a reduced Pt/Al2O3 catalyst during alternate CO–O2 and H2–O2 pulses at 300 °C. This time-resolved high resolution experiment enabled the identification of an intermediate step during the oxidation and the recognition of different adsorbed species by the support of ab initio FEFF calculations. The oxidation step was found to be composed of the dissociative adsorption of oxygen followed by the partial oxidation of the Pt sub-surface. The intermediate state was attributed to chemisorbed oxygen in an atop position in the case of the CO–O2 experiment, whereas it was ascribed to chemisorbed oxygen on a Pt 3-fold site in the case of reduction by H2. The Pt–CO fingerprint in the HEROS spectrum was characterized by the broadening of the Pt L3-edge whiteline to the low energy side. The resulting shoulder at ca. 9.422 keV was missing in the spectrum obtained at the end of the reduction pulse using H2 decisively supporting the assignment.
Extending our previous work,16 in this study we demonstrate the utility of the combination of HEROS at the Pt L3-edge and PSD for time-resolved operando spectroscopy measurements of catalysts at realistic gas concentrations where structural changes may be small. Furthermore, we investigate the influence of CeO2 on the reducibility of a platinum catalyst in pulse experiments using hydrogen as the reducing agent. Identical resonant X-ray emission spectroscopy (RXES) measurements were carried out at the Ce L3-edge using the same spectroscopic cell in order to be able to examine the behavior of the oxygen buffer component of the catalyst in comparison with that of the precious metal analyzed by HEROS.
Experimental
The CeO2/Al2O3 support (20 wt% CeO2) was prepared by deposition–precipitation of Ce(NO3)3·6H2O on Al2O3 (Puralox, Sasol). Pt/Al2O3 and Pt/CeO2/Al2O3 were obtained by wet impregnation of the metal oxide supports with H2PtCl6 (Sigma-Aldrich). The final Pt loading obtained after calcination in static air at 500 °C for 2 h was 1.3 wt% (by ICP-OES). High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images were recorded using a FEI Tecnai F20ST microscope operated at 200 kV. The samples were prepared by evaporation of a 2-propanol suspension in air on a carbon-coated copper grid.Approximately 35 mg of powder catalyst (50–100 μm sieved fraction) were placed in the reactor cell between two quartz wool plugs. In order to compare the results of various spectroscopic techniques, a single reactor cell was used to avoid the need to discuss the effect of the reactor type. The cell was specifically designed for operando experiments using a combination of spectroscopic methods in a single experiment.27 The cell is designed to represent a plug flow reactor, has reduced dead volume in order to allow the fast exchange of gases and is therefore suitable for experiments under transient conditions. For the X-ray experiments, the cell was equipped with two 0.5 mm thick graphite windows with a diameter of 14.8 mm. For the equivalent diffuse reflectance infrared (DRIFT) spectroscopy experiments, a CaF2 window replaced one of the graphite windows. The gas manifold consisted of mass flow controllers (Bronkhorst) and two switching valves (Parker), which permitted the rapid variation of the gas feed to the cell for the purpose of the modulation experiments. Downstream the cell, a needle valve enabled a fine regulation of the pressure of the vent to avoid pressure sweeps during the switches. The outlet of the cell was connected to a mass spectrometer (Hiden) that was used to follow signals of m/z 2 (H2), 18 (H2O), 28 (CO), 32 (O2), and 44 (CO2). The total gas flow was maintained at 50 ml min−1. Under these flow conditions, we observed a delay of approximately 2 s between the valve switch and the onset of structural changes within the sample.
Prior to the modulation experiments, the calcined catalysts were subjected to temperature programmed reduction in 5 vol% H2/He from room temperature to 300 °C at 10 °C min−1 (H2-TPR). After reaching 300 °C, the gas feed was automatically and periodically switched at the same temperature while synchronously collecting time-resolved spectra. The modulation sequences consisted of alternate 1 vol% O2/He and 1 vol% H2/He pulses (O2–H2) or 1 vol% O2/He and 1 vol% CO/He pulses (O2–CO). The length of each pulse was 30 s, the total period (T) of a single sequence, consisting of one O2 and one H2 or CO pulse, was 60 s.
All synchrotron data were collected at the SuperXAS beamline of the Swiss Light Source (SLS, Switzerland).28 For high energy resolution off-resonant spectroscopy (HEROS) experiments, the incident energy was fixed at 11.55 keV, 14 eV below the Pt L3-edge (11.564 keV). The beam size was ca. 100 × 100 μm2. The HEROS spectra were recorded around the Lα1 X-ray emission (9.442 keV) line using a von-Hamos spectrometer10 employing a cylindrically bent Ge(800) crystal (diameter 25 cm) and a Pilatus detector. The acquisition time was 20 s per spectrum during the H2-TPR and the energy resolution was 0.5 eV. During the modulation experiments the acquisition time was decreased to 500 ms per spectrum and the energy resolution decreased to 1 eV; for a single modulation experiment 120 cycles were repeated, which equals to 2 h total acquisition time. The energy region between 9.435 keV and 9.440 keV, corresponding to the pre-edge range, was normalized to zero, while the intensity normalization to one was applied between 9.410 keV and 9.415 keV. To demonstrate the quality of information of HEROS, H2-TPR was repeated using the same set-up and reactor cell while recording quickXAS spectra.29 The spectra were collected at the Pt L3-edge (11.564 keV) in transmission mode using N2-filled ionization chambers in the middle of the catalyst bed. A Pt foil was mounted between the second and the third ionization chamber for absolute energy calibration. The Si(111) crystal of the quickXAS monochromator oscillated at 5 Hz frequency and the spectra were averaged over 1 min (only the spectra in the increasing energy direction were used). The X-ray absorption near edge structure (XANES) spectra were background corrected, normalized and fitted (linear combination fit, LCF) using the Demeter software package.30 The range for the LCF was between 20 eV below E0 and 30 eV above E0. The calcined catalyst, where Pt is fully oxidized, and the catalyst at the end of the H2-TPR, which is considered fully reduced, were taken as references for the LCF. The resonant X-ray emission spectroscopy (RXES) spectra at the Ce Lα1 emission energy (4.839 keV) were obtained at an incident energy of 5.726 (keV) using a von Hamos spectrometer employing a cylindrically bent Ge(400) crystal (diameter 25 cm) and a Pilatus detector.8 The distance between the detector and the sample was ca. 25 cm and was filled with a He bag to reduce absorption by air. The contribution of background counts and elastic scattering was subtracted from each spectrum by integrating the Pilatus images and the resulting spectra were normalized in the energy range 4.824–4.832 keV, where the RXES spectral shape was considered independent of the oxidation state. For normalization, the average intensity was determined in the selected energy range and each point of the spectra was divided by this value. As such, the procedure is independent of the energy step. The LCF was performed using the RXES spectra of the same sample under reference conditions to quantify the amount of Ce3+ and Ce4+. The fluorescence XANES and RXES spectra of the sample under static conditions in Ar at room temperature and in 5 vol% H2/Ar at 300 °C after the H2-TPR were taken as reference spectra (Fig. S1, ESI†). These RXES spectra were collected with an acquisition time of 300 s. A 5-element Si drift detector was used to simultaneously collect the fluorescence XANES data. The transmission Ce L3-edge XANES spectra of Ce(NO3)3 and CeO2 pellets served as references to quantify the amount of Ce3+ and Ce4+ in the fluorescence XANES spectra of the samples in the reference states using the LCF. All the XANES spectra were treated and analyzed similarly as mentioned above for the Pt XANES data.
The full sets of time-resolved data measured during a modulation experiment were processed by phase sensitive detection (PSD)17 using a Matlab script where the Simpson series was implemented to calculate the integral delivering the set of phase-resolved data.
Infrared spectra in diffuse reflectance mode (DRIFT) were collected by co-adding 4 interferograms at 2 cm−1 resolution and 80 kHz scanner velocity using the same reactor cell and a Vertex 80 spectrometer (Bruker) equipped with a liquid nitrogen cooled HgCdTe detector and a Praying Mantis mirror unit (Harrick). The background spectrum was acquired after reduction at 300 °C in 5 vol% H2/Ar before performing the CO–O2 modulation experiment consisting of 30 s pulses and 30 spectra per pulse. The area below the signal of CO adsorbed atop (2100–1900 cm−1) in the last DRIFT spectrum in the CO pulse was assumed to correspond to the maximum CO coverage (θ) at 300 °C.
Results and discussion
Temperature programmed reduction
The comparison between the results of H2-TPR obtained using HEROS and XANES is shown in Fig. 1. Fig. 1a and b present the HEROS spectra and Fig. 1c and d the Pt L3-edge XANES spectra collected in independent H2-TPR experiments on (a–c) Pt/Al2O3 and (b–d) Pt/CeO2/Al2O3. At room temperature, both catalysts can be considered fully oxidized. After calcination, there is no clear indication from high angle annular dark field scanning transmission electron microscopy (HAADF-STEM, not shown) of the formation of any type of Pt-containing entities such as nanoparticles in these catalysts, suggesting that Pt is finely dispersed on the Al2O3 and CeO2/Al2O3 supports and can be assumed to be in the form of Pt4+Ox. The clusters of round-shaped crystallites of ca. 5 nm diameter characterize the CeO2 phase (vide infra). Because the XANES whiteline of the Pt L3-edge corresponds to the 2p → 5d electron transition, the intense whiteline of the XANES spectra reflects the high oxidation state of Pt in the two samples (Fig. 1c and d). Since HEROS probes the empty density of d states, the high intensity of the HEROS whiteline reflects the high density of unoccupied 5d states (Fig. 1a and b), in agreement with the corresponding XANES data. However, it is evident that despite the lower signal-to-noise ratio of the spectrum, the normalized whiteline intensity of Pt/CeO2/Al2O3 is larger than that of Pt/Al2O3, while this difference is not obvious in the XANES spectra. This observation reveals possible structural differences between the two materials that cannot be evidenced by XANES. | ||
| Fig. 1 (a and b) HEROS and (c and d) XANES spectra collected during the H2-TPR on (a and c) Pt/Al2O3 and (b and d) Pt/CeO2/Al2O3 at (■) 25 °C and (○) 300 °C. (e) Fraction of oxidized Pt estimated from the linear combination fitting of the HEROS spectra and (f) of the XANES spectra of (●) Pt/Al2O3 and (○) Pt/CeO2/Al2O3 using the calcined and the reduced catalysts as references. | ||
During reduction under identical experimental conditions (Fig. 1a–d), the strong whitelines of both the HEROS and the Pt L3-edge XANES spectra gradually attenuate indicating reduction of the PtOx entities. Two features characterize the HEROS spectra. First, contrary to XANES, the HEROS spectra shift to higher energy upon reduction, which is in agreement with the lowering of the absorption edge position of XAS as expected from the energy conservation equation.9,16 This is more evident when considering the evolution of the edge energy of Pt/Al2O3; while the XANES edge shifts to lower energy (Fig. 2b), the HEROS spectra shift to higher energy (Fig. 2a). Secondly, HEROS appears to be more sensitive to the physico-chemical process than XANES as a result of its intrinsic higher spectral resolution. The HEROS whiteline of Fig. 1 first attenuates and then shifts to higher energy, while the XANES whiteline intensity decreases only from its initial value to the final one corresponding to the reduced state. Clearly, the shift cannot be resolved in XANES. This behavior is again better depicted in Fig. 2, where the temporal evolution of the energy position of the maxima of the whitelines of both HEROS and XANES spectra of Pt/Al2O3 is plotted. No energy shift is perceivable in the XANES spectra while the whiteline loses intensity in the whole temperature range of the H2-TPR (Fig. 2b). On the contrary, the maximum of the HEROS whiteline moves to higher energy above ca. 175 °C (Fig. 2a). This temperature corresponds to ca. 60% loss of oxidized Pt (Fig. 1e). If the edge energy is considered instead of the whiteline position (Fig. 2), the difference in the temporal dependence is not evident. An energy shift becomes measurable in both methods at ca. 150 °C, in coincidence with the beginning of reduction and 20–30 °C prior to the shift of the HEROS whiteline to higher energy described above. Therefore, equivalent information about Pt reduction is obtained from the HEROS and the XANES spectra. The edge energy shifts until ca. 9.427 keV and 11.564 keV in the HEROS and the XANES spectra, respectively. A tentative interpretation of this behavior is that the HEROS data indicate that the metallic phase becomes detectable through the shift of the whiteline only when a large fraction of PtOx has been consumed by hydrogen. Considering the fine dispersion of the initial oxidized PtOx species in the sample, this observation implies that HEROS may resolve the process of clustering of Pt atoms to form nanosized metallic particles better than XANES. Once a given cluster size is attained, or enough reduced Pt is present, HEROS can deliver an energy shift corresponding to what we interpret as the development of the metallic phase.
 | ||
| Fig. 2 Temporal dependence of (□) whiteline position and (●) edge energy of (a) HEROS and (b) XANES spectra of Pt/Al2O3 during the H2-TPR experiments of Fig. 1. | ||
After reduction at 300 °C, Pt nanoparticles of ca. 2.5 nm and 1.5 nm become visible in the HAADF-STEM images of Pt/Al2O3 and Pt/CeO2/Al2O3, respectively (Fig. 3). Because of the poor contrast between cerium and platinum, no metal particles can be easily visualized on CeO2. At this point, the XANES spectra of both catalysts (Fig. 1c and d) are characteristic of metallic Pt; thus, Pt appears to be mostly reduced. Similar to the spectra of the initial states, the HEROS spectra after reduction are different from the XANES spectra in the equivalent states. While it is obvious that the strong HEROS whiteline of the oxidized state at 9.423 keV is absent after reduction and is replaced by the whiteline of the reduced state at 9.426 keV, the spectrum of reduced Pt/CeO2/Al2O3 exhibits a stronger whiteline than that of Pt/Al2O3. It is tempting to assign this behavior to the presence of CeO2, and thus to the possibility that a fraction of Pt is still in oxidized form because of CeO2. Then, this fraction would also likely correspond to Pt in contact with CeO2, which unfortunately is not visible by electron microscopy. However, it should be noted that it is the whiteline of the reduced sample that demonstrates higher intensity, i.e. at the energy of the reduced metal, while nothing is visible at 9.423 keV. Based only on the HEROS spectra, it is difficult to state that a fraction of Pt may still be oxidized. Because the particle size31,32 and metal–support interactions33 can influence the intensity and shape of the whiteline of the spectrum of the metal, it is more likely that the difference observed in the HEROS spectra of the reduced states is due to the different particle size or the interaction with ceria. The effect of particle size is debated. Ichikuni et al.32 observed that the empty density of d states increases with the decreasing particle size, for Pt particles below 1.5 nm. On the contrary, Lei et al.31 observed a decrease in intensity near the edge and an increase in intensity beyond the edge with the decreasing particle size below about 5 nm for Pt/alumina. In order to be conclusive from a HEROS perspective, this topic should be matter of further and systematic investigations on a wide range of samples. It is also clear that this issue makes a quantitative comparison between the two samples not straightforward.
 | ||
| Fig. 3 HAAFD-STEM images of (a) Pt/Al2O3 and (b) Pt/CeO2/Al2O3 after reduction at 300 °C. | ||
Finally, Fig. 1e compares the evolution of the fraction of oxidized Pt during the HEROS H2-TPR experiment on the two catalysts. Reduction of Pt/Al2O3 sets in between 100 and 150 °C and is completed above 200 °C. The presence of CeO2 retards Pt reduction by ca. 20 °C demonstrating the existence of a direct interaction between Pt and the oxygen ions in CeO2.34 The corresponding XANES data of Fig. 1f are in qualitative agreement with the HEROS data. It should be highlighted that the H2-TPR profile measured by HEROS is more accurate than that obtained by XANES because of the faster acquisition time. This demonstrates that high quality HEROS spectra can be obtained with high time resolution that is not yet possible using conventional fluorescence detectors. This enables the study of highly absorbing materials as is the case of CeO2-containing catalysts and the bypass of issues associated with transmission XAS.
Transient measurements
The redox properties of the two catalysts and the influence of CeO2 on the redox behavior of Pt were probed using pulse experiments consisting of alternate pulses of 1 vol% O2 and 1 vol% H2 at 300 °C. HEROS spectra were collected at the Pt L3-edge of Pt/Al2O3 and Pt/CeO2/Al2O3. In order to correlate the reduction of Pt with the possible reduction of Ce4+ in the reduction pulse, independent RXES spectra were also collected at the Ce L3-edge of Pt/CeO2/Al2O3 to explicitly follow the oxidation state of the oxygen storage component.8 The averaged time-resolved HEROS spectra of both catalysts (Fig. 4a and b) exhibit a change in intensity around the whiteline indicating that, as expected, platinum is partially oxidized in the O2 pulse and reduced in the H2 pulse during the two modulation experiments. The signal-to-noise ratio of the spectra decreases significantly in the presence of CeO2, demonstrating the difficulty to obtain high quality time-resolved spectra in the presence of Ce. The signal-to-noise ratio of the time-resolved HEROS spectra would be of sufficient quality to extract the reduction–oxidation kinetics.16 Structural changes, hardly visible in the HEROS difference spectra (Fig. 4g and h), are enhanced by phase sensitive detection (PSD, Fig. 4d and e). It is evident that compared to the corresponding difference spectra obtained by subtracting the last spectrum of the reduction pulse from the last spectrum of the oxidation pulse, the data quality of the phase-resolved HEROS spectra is greatly improved primarily because noise is removed by the PSD algorithm.21 In the phase-resolved spectra, the main feature at 9.425 keV corresponds to the already visible changes around the whiteline in Fig. 4a and b due to reversible reduction and oxidation of Pt. However, additional features that cannot be recognized after simple inspection of the time-resolved data and the difference spectra because of the high noise level become visible. Two features are immediately visible in the phase-resolved spectra. The first is the signal around the energy region corresponding to the whiteline in the time-resolved data, while the second is a weaker but well-resolved signal of opposite sign at 9.428 keV that corresponds to the edge energy position that developed during H2-TPR. The presence and the opposite sign of the signals stand for the reversible oxidation–reduction of Pt. While the intensity of the phase-resolved spectra undergoes a major change, the form of the spectra changes as indicated by the presence of intersection points. The possibility to differentiate species from their kinetics based on the phase delay and to isolate their contribution in selected phase-resolved spectra is the real potential of PSD.23,25,35 When all phase-resolved spectra exhibit the same spectral features as the difference spectrum obtained from the spectra of two boundary states,21,22 as is the case for the phase-resolved RXES spectra of Pt/CeO2/Al2O3 (Fig. 4f), the spectra contain the contribution of only these two states. If a set of phase-resolved spectra deviates from the difference spectrum, more than two states exist suggesting the presence of additional or even intermediate species. This is the case for the phase-resolved HEROS spectra of Pt/Al2O3 (Fig. 4d) indicating that an intermediate state is present during the redox process stimulated by the O2–H2 pulses. In our previous study of Pt/Al2O3 during a H2–O2 modulation experiment,16 we assigned this intermediate state in combination with FEFF simulations to the initially chemisorbed O on a 3-fold Pt site (Pt3–O) that evolves to partially oxidized Pt at the end of the oxidation pulse. The identification of a feature at 9.426 keV at φPSD = 140° suggests that the nature of the intermediate species is the same in the present experiment.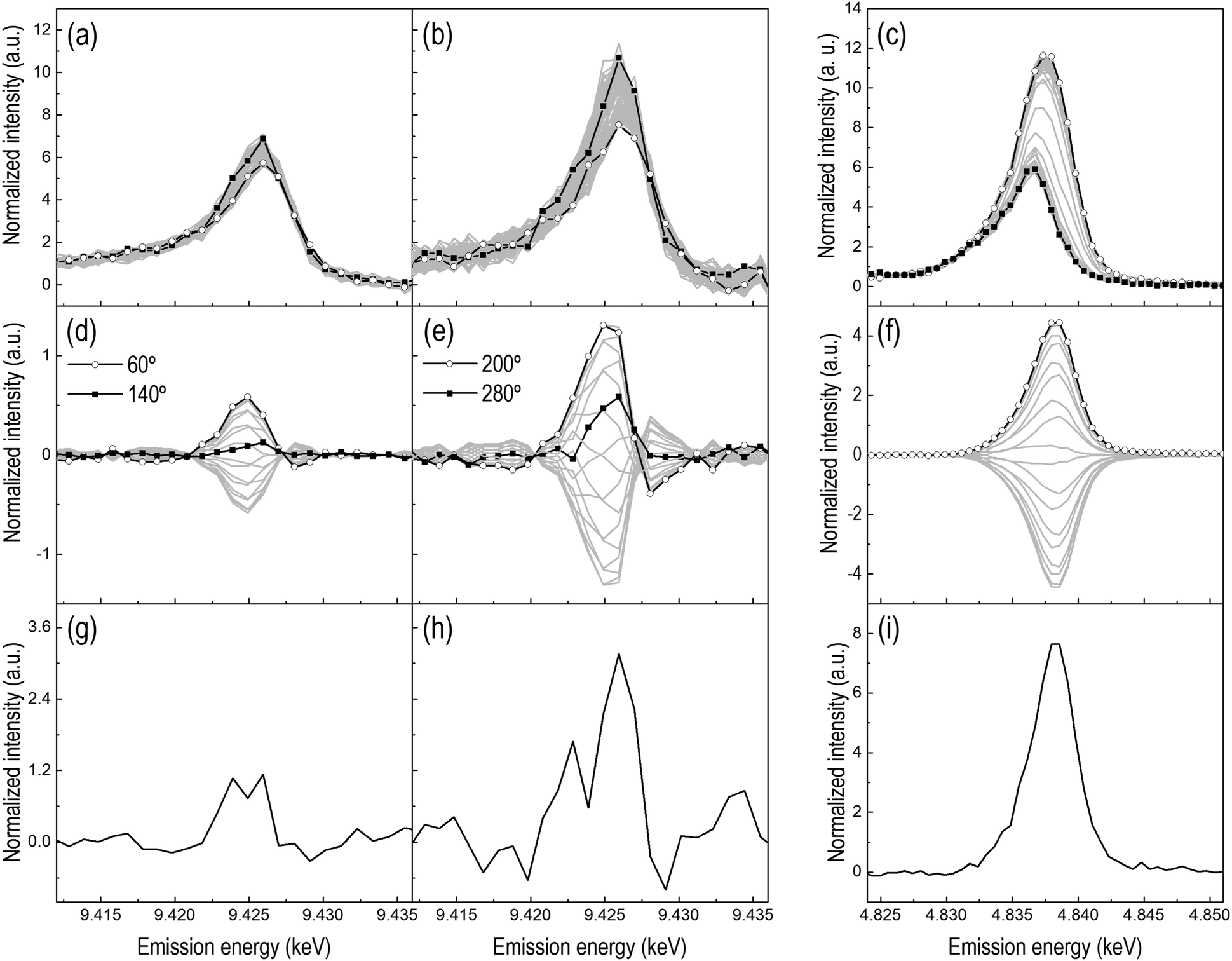 | ||
| Fig. 4 (a and b) Time-resolved and (d and e) phase-resolved HEROS spectra collected at the Pt L3-edge during the 1 vol% H2/He-1 vol% O2/He modulation experiment (period 30 + 30 s, 120 cycles; 500 ms per spectrum) at 300 °C on (a and d) Pt/Al2O3 and (b and e) Pt/CeO2/Al2O3. (c) Time-resolved and (f) phase-resolved RXES spectra collected at the Ce L3-edge during an identical experiment on Pt/CeO2/Al2O3 (period 30 + 30 s, 50 cycles; 1 s per spectrum). (a–c) Full symbols, last spectrum in O2; open symbols, last spectrum in H2. (g–i) Difference spectra obtained from time-resolved spectra. φPSD = 0–360° (20° step). | ||
The phase-resolved HEROS spectra collected on Pt/CeO2/Al2O3 (Fig. 4e) show higher intensity than the phase-resolved spectra of Pt/Al2O3 (Fig. 4d), suggesting that the structural changes in the presence of CeO2 are enhanced and that a larger fraction of Pt responds to the chemical stimulation. However, the whiteline intensity is probably biased by the influence of the particle size that we described above. This makes direct comparison between the spectra difficult. On the other hand, we note that the absolute intensity of the raw time-resolved HEROS spectra decreases in the presence of CeO2 because of the absorption by Ce (Fig. S2a and b, ESI†), but the corresponding phase-resolved spectra show slightly higher intensity than the phase-resolved spectra of Pt/Al2O3 (Fig. S2c and d, ESI†) hinting to the possibility that a larger fraction of Pt responds to the external stimulation in the presence of CeO2. Except for the intensity, the phase-resolved spectra of Pt/CeO2/Al2O3 exhibit a shape similar to that of Pt/Al2O3 spectra and a peak whose maximum intensity shifts gradually. The feature at 9.426 keV attributed to Pt3–O is visible at φPSD = 280°.
The averaged time-resolved RXES spectra of the Ce L3-edge recorded under identical experimental conditions are displayed in Fig. 4c. The spectrum with the whiteline at 4.836 keV corresponds to oxidized Ce4+. During the reduction pulse, the whiteline of the RXES spectra intensifies and shifts to 4.837 keV, which corresponds to the generation of Ce3+ species from reduction of Ce4+. The higher intensity of the whiteline of Ce3+ is caused by the fact that XES probes the occupied density of states. The corresponding phase-resolved spectra (Fig. 4f) are not much informative and contain information only on the fraction of Ce that responds to the external stimulation. All the phase-resolved RXES spectra possess the same shape but different intensity, no energy shift is observed and there are no intersections between the spectra. Therefore, as expected, the Ce L3-edge RXES spectra demonstrate the direct and reversible change between Ce4+ and Ce3+ without any perceptible intermediate state.
Fig. 5a reports the temporal evolution of the fraction of oxidized Pt obtained through the linear combination fit (LCF) of the time-resolved HEROS spectra collected during the H2–O2 modulation experiment on Pt/Al2O3 and Pt/CeO2/Al2O3. The time-resolution (500 ms per spectrum) is sufficient to follow the oxidation–reduction process in detail, which would not be straightforward in the fluorescence mode. Given the difficulty to judge the origin of the high intensity of the whiteline of Pt/CeO2/Al2O3, we show the data elaborated using the most oxidized and most reduced states of both samples at 300 °C as the initial and the final states, respectively. This does not allow us to make any quantitative comparison between the samples but provides a useful visualization of the rates of reduction and oxidation. Fig. 5b reports the fraction of Ce3+ obtained through LCF analysis of the RXES spectra. The intensity of the HEROS whiteline increases in the presence of O2 as Pt (partially) oxidizes, and decreases when Pt reduces in H2. For both catalysts the breakthrough of the H2 signal in the online mass spectrometric data (MS, Fig. 5d) in the reduction pulse coincides with the complete reduction of Pt. This occurs after 5 s on Pt/Al2O3 and 10 s on Pt/CeO2/Al2O3.
 | ||
| Fig. 5 Temporal profiles of (a) the fraction of oxidized Pt (HEROS) and (b) the fraction of Ce3+ (RXES) on (●) Pt/Al2O3 and (○) Pt/CeO2/Al2O3 during the 1 vol% H2/He-1 vol% O2/He modulation experiment at 300 °C (period 30 + 30 s, 120 cycles; 500 ms per spectrum). MS signals of (c) m/z 32 (O2) and (d) m/z 2 (H2). | ||
The temporal profile of the RXES data indicates that Ce4+ starts reducing after ca. 3 s in the reduction pulse, the signal reaches a steady state between 5 and 10 s after switching to H2 and no further reduction occurs. At this point ca. 27% Ce3+ has been produced and ceria cannot be reduced further. While in the subsequent O2 pulse the behavior of the two catalysts is similar and Ce3+ oxidation is delayed by ca. 5 s, the behavior in the reduction pulse reflects the chemical properties of the system, and thus the effect of the presence of CeO2. PtOx on Al2O3 and Ce4+ start reducing immediately after changing to H2, while the portion of Pt in the vicinity or in contact with CeO2 reduces towards the end of the sharp reduction of Ce4+, i.e. when the reduction of PtOx on Al2O3 in Pt/CeO2/Al2O3 and of Pt/Al2O3 is concluded. It is clear that Pt reduction extends slightly beyond the time needed to reduce Ce4+ completely at this temperature, suggesting that Ce4+ may reduce before PtOx in contact with CeO2. The slower reduction of Pt in the presence of CeO2 observed in both the H2-TPR and the transient H2–O2 modulation experiments reflects the intimate interaction between CeO2 and a portion of Pt,34 which modifies the ability of Pt to oxidize and reduce. Pt particles on Al2O3 will be less affected by CeO2 especially if located far from its agglomerates. The different size of the Pt particles deposited on Al2O3 in both catalysts after reduction at 300 °C confirms that CeO2 regulates the reduction of PtOx despite the low fraction of Pt in contact with it.
Fig. 6a shows the time-resolved HEROS spectra collected during a modulation experiment performed with 1 vol% of CO and O2 on Pt/Al2O3 at 300 °C. This experiment needs to be compared with the H2–O2 modulation experiment (Fig. 4a–d) to demonstrate the structural differences when using H2 or CO as a reducing agent in combination with PSD. Similar to the H2–O2 modulation experiment, the whiteline intensity increases in the O2 pulse and decreases in the reduction (CO) pulse. Except for the variation in the whiteline intensity, other differences between the spectra are not obvious compared to the spectra obtained at higher gas concentrations (Fig. S4, ESI†). Therefore, in the time-resolved data it is difficult to isolate the spectral signature of the three states identified in the previous study at higher gas concentrations, i.e. chemisorbed O and CO on metallic Pt in an atop position and partially oxidized Pt.16 PSD is suitable to extract the spectral components that vary according to and with the same frequency of the external stimulation (i.e. the frequency of the CO–O2 modulation), thus enhancing our perception of subtle structural changes. The phase-resolved spectra of Fig. 6b clearly contain information only on that portion of Pt that is stimulated by the CO–O2 modulation (note the significantly different y-axis scale). As discussed above, the inspection of the shape of the phase-resolved spectra suggests that the spectra clearly contain information on more than two boundary species.
 | ||
| Fig. 6 (a) Time-resolved and (b) phase-resolved HEROS spectra collected at the Pt L3-edge during the 1 vol% CO/He-1 vol% O2/He modulation experiment on Pt/Al2O3 at 300 °C (period 30 + 30 s, 120 cycles; 500 ms per spectrum). φPSD = 0–360° (10° step). Full symbols in (a), last spectrum in O2; open symbols, last spectrum in CO. | ||
The spectrum corresponding to φPSD = 320° shows two features. The whiteline signal at 9.426 keV is of opposite sign to the signal at 9.422 keV. The spectrum at φPSD = 150° is its mirror image. Therefore, these spectra contain the contribution of two species, i.e. the final oxidized state of Pt attained in the O2 pulse (9.426 keV) and the signal at 9.422 keV. This latter feature is rather evident in Fig. 6b, while in the phase-resolved spectra of the corresponding H2–O2 modulation experiments (Fig. 4d) this region is within the noise level. Hence, this signal must be assigned to the presence of CO because simultaneously the whiteline signal indicating oxidized Pt exhibits opposite sign, and is associated with CO adsorbed on metallic Pt. Together with the confirmation of the presence of adsorbed CO on Pt by DRIFT (see below and Fig. S5, ESI†), this assignment substantiates our previous findings.16
The phase-resolved spectrum at φPSD = 240° exhibits only a signal at 9.424 keV and no indication of the whiteline or of Pt–CO, thus suggesting the existence of a third species. Despite the noise, it is still possible to recognize in the temporal profiles of the signals at 9.424 keV and 9.426 keV emission energy of Fig. 7a that the signal at 9.426 keV increases immediately at the beginning of the oxidation pulse, whereas the signal at 9.424 keV grows more slowly, thus confirming the presence of an intermediate step. This intermediate forms clearly within the oxidation pulse and its emission energy corresponds to the signal of atop Pt-O that we observed previously.16Fig. 7b also displays the temporal profile of the HEROS signal at 9.421 keV that is attributed to Pt–CO. The magnitude of the changes is comparable with the level of noise and it is difficult to recognize a trend further in the CO pulse. However, it is clear that the intensity of the Pt–CO signal attains higher levels at the end of the CO pulse. The temporal profile of the Pt–CO surface coverage (θ) obtained in an equivalent DRIFT experiment (Fig. 7c and Fig. S5, ESI†) strongly supports the assignment of this HEROS feature to CO adsorbed on metallic Pt. Therefore, PSD enabled the identification of three different states of Pt in experiments with more realistic conditions of, for example, exhaust gas after treatment catalysts, and from time-resolved spectra of less straightforward interpretation. Finally, the online mass spectrometric data of Fig. 7d–f show the CO2 production in the two pulses and that the maximum of CO2 production was attained after ca. 7 s when the temporal profile of the intensity of the IR signal of Pt–CO reached a steady state and the outlet O2 concentration returned to zero. A sharp peak of CO2 appears immediately upon switching to O2 and simultaneous to the onset of the disappearance of the Pt–CO species and reaches a maximum after ca. 4 s. Pt/Al2O3 was more active at the O2 → CO switch than at the CO → O2 switch, changing from a partially oxidized Pt surface to a CO rich surface as it is also suggested by the delayed breakthrough of CO in the gas phase (ca. 10 s from the O2 → CO switch).36
 | ||
| Fig. 7 Temporal profiles of (a) the signals identified at (□) 9.424 and (●) 9.426 keV and (b) at 9.421 keV in the HEROS spectra collected at the Pt L3-edge during the 1 vol% CO-1 vol% O2 modulation experiment on Pt/Al2O3 at 300 °C. (c) Temporal variation of the surface coverage (θ) of CO linearly coordinated to metallic Pt (Pt–CO) obtained in an identical DRIFT experiment. (d)–(f) MS signals corresponding to m/z 44 (CO2), 32 (O2) and 28 (CO), respectively. | ||
Conclusions
Pt/Al2O3 was examined using time-resolved HEROS by exploiting the modulated excitation approach and phase sensitive detection (PSD). We have first made a comparison between HEROS and XANES for identical temperature programmed experiments to demonstrate the high time-resolution and sensitivity to small structural changes of HEROS compared to XANES. Identical modulation measurements were performed using resonant X-ray emission spectroscopy and diffuse reflectance infrared spectroscopy to interrogate the oxidation state of cerium and the CO surface coverage, respectively. Only the reversible reduction–oxidation of reduced Pt nanoparticles is an evident structural change in modulation experiments consisting of alternate pulses of H2 and O2 at 300 °C. Because of the difference in gas feed concentration, the sensitivity of HEROS to the Pt3–O intermediate species that was demonstrated previously at high gas feed concentration requires the application of PSD. Addition of CeO2 (Pt/CeO2/Al2O3 with 20 wt% CeO2) clearly increased the fraction of oxidized Pt in the H2–O2 modulation experiment and decreased the reduction rate. The increased sensitivity of HEROS induced by PSD was further confirmed in a CO–O2 modulation experiment on Pt/Al2O3, where it was possible to identify Pt–CO and the atop Pt–O intermediate already observed previously. The presented data demonstrate that the modulation approach significantly increases the sensitivity of HEROS, which facilitates the separation of spectral components and the identification of intermediate species. HEROS is a powerful in situ/operando spectroscopy method to probe the unoccupied density of states with high time-resolution.Acknowledgements
The authors are grateful to the Swiss National Science Foundation (SNF, project number 200021 138068/1), the Competence Centre for Materials Science and technology (CCMX) and the Paul Scherrer Institut for financial support. Beamtime allocation at the SuperXAS beamline is highly appreciated. Electron microscopy was accomplished at the Electron Microscopy Center at Argonne National Laboratory, a U.S. Department of Energy Office of Science Laboratory operated under Contract No. DE-AC02-06CH11357 by UChicago Argonne, LLC. The authors would like to thank Dr C. Proff for the electron microscopy images, Prof. M. Sikora for support with RXES data analysis, and Dr O. V. Safonova, Prof. J. Sa, Dr K. A. Michalow-Mauke and A. Marberger for help and support during measurements.References
- B. M. Weckhuysen, Chem. Commun., 2002, 97 RSC.
- R. J. Berger, F. Kapteijn, J. A. Moulijn, G. B. Marin, J. De Wilde, M. Olea, D. Chen, A. Holmen, L. Lietti, E. Tronconi and Y. Schuurmann, Appl. Catal., A, 2008, 342, 3 CrossRef CAS.
- A. M. Efstathiou and X. E. Verykios, Appl. Catal., A, 1997, 151, 109 CrossRef CAS.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736 CrossRef CAS PubMed.
- A. I. Frenkel, J. A. Rodriguez and J. G. Chen, ACS Catal., 2012, 2, 2269 CrossRef CAS.
- J. Singh, C. Lamberti and J. A. van Bokhoven, Chem. Soc. Rev., 2010, 39, 4754 RSC.
- J. Szlachetko, J. Sá, O. V. Safonova, G. Smolentsev, M. Szlachetko, J. A. van Bokhoven and M. Nachtegaal, J. Electron Spectrosc. Relat. Phenom., 2013, 188, 161–165 CrossRef CAS.
- R. Kopelent, J. A. van Bokhoven, J. Szlachetko, J. Edebeli, C. Paun, M. Nachtegaal and O. V. Safonova, Angew. Chem., Int. Ed., 2015, 54, 8728 CrossRef CAS PubMed.
- J. Szlachetko, M. Nachtegaal, J. Sá, J. C. Dousse, J. Hoszowska, E. Kleymenov, M. Janousch, O. V. Safonova, C. König and J. A. van Bokhoven, Chem. Commun., 2012, 48, 10898 RSC.
- J. Szlachetko, M. Nachtegaal, E. de Boni, M. Willimann, O. V. Safonova, J. Sa, G. Smolentsev, M. Szlachetko, J. A. van Bokhoven, J. C. Dousse, J. Hoszowska, Y. Kayser, P. Jagodzinski, A. Bergamaschi, B. Schmitt, C. David and A. Lücke, Rev. Sci. Instrum., 2012, 83, 103105 CrossRef CAS PubMed.
- O. Safonova, M. Tromp, J. A. van Bokhoven, F. M. F. de Groot, J. Evans and P. Glatzel, J. Phys. Chem. B, 2006, 110, 16162 CrossRef CAS PubMed.
- J. Singh and J. A. van Bokhoven, Catal. Today, 2010, 155, 199 CrossRef CAS.
- J. Singh, M. Tromp, O. Safonova, P. Glatzel and J. A. van Bokhoven, Catal. Today, 2009, 145, 300 CrossRef CAS.
- W. Blachucki, J. Szlachetko, J. Hoszowska, J. C. Dousse, Y. Kayser, M. Nachtegaal and J. Sá, Phys. Rev. Lett., 2014, 112, 173003 CrossRef CAS PubMed.
- S. Eisebitt, T. Böske, J. E. Rubensson and W. Eberhardt, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 14103–14109 CrossRef CAS.
- J. Szlachetko, D. Ferri, V. Marchionni, A. Kambolis, O. V. safonova, C. J. Milne, O. Kröcher, M. Nachtegaal and J. Sá, J. Am. Chem. Soc., 2013, 135, 19071 CrossRef CAS PubMed.
- D. Baurecht and U. P. Fringeli, Rev. Sci. Instrum., 2001, 72, 3782 CrossRef CAS.
- A. Haghofer, D. Ferri, K. Föttinger and G. Rupprechter, ACS Catal., 2012, 2, 2305 CrossRef CAS.
- V. Marchionni, M. A. Newton, A. Kambolis, S. K. Matam, A. Weidenkaff and D. Ferri, Catal. Today, 2014, 229, 80 CrossRef CAS.
- R. Kydd, D. Ferri, P. Hug, J. Scott, W. Y. Teoh and R. Amal, J. Catal., 2011, 277, 64 CrossRef CAS.
- D. Ferri, S. K. Matam, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, Phys. Chem. Chem. Phys., 2010, 12, 5634 RSC.
- C. F. J. König, J. A. van Bokhoven, T. J. Schildhauer and M. Nachtegaal, J. Phys. Chem. C, 2012, 116, 19857 Search PubMed.
- G. L. Chiarello and D. Ferri, Phys. Chem. Chem. Phys., 2015, 17, 10579 RSC.
- D. Ferri, M. A. Newton, M. Di Michiel, G. L. Chiarello, S. Yoon, Y. Lu and J. Andrieux, Angew. Chem., Int. Ed., 2014, 53, 8890 CrossRef CAS PubMed.
- D. Ferri, M. A. Newton, M. Di Michiel, S. Yoon, G. L. Chiarello, V. Marchionni, S. K. Matam, M. H. Aguirre, A. Weidenkaff, F. Wen and J. Gieshoff, Phys. Chem. Chem. Phys., 2013, 15, 8629 RSC.
- Y. Lu, S. Keav, V. Marchionni, G. L. Chiarello, A. Pappacena, M. Di Michiel, M. A. Newton, A. Weidenkaff and D. Ferri, Catal. Sci. Technol., 2014, 4, 2014 Search PubMed.
- G. L. Chiarello, M. Nachtegaal, V. Marchionni, L. Quaroni and D. Ferri, Rev. Sci. Instrum., 2014, 85, 074102 CrossRef PubMed.
- P. M. Abdala, O. V. Safonova, G. Wiker, W. van Beek, H. Emerich, J. A. van Bokhoven, J. Sá, J. Szlachetko and M. Nachtegaal, Chimia, 2012, 66, 699 CrossRef CAS PubMed.
- J. Stötzel, D. Lützenkirchen-Hecht, E. Fonda, N. De Oliveira, V. Briois and R. Frahm, Rev. Sci. Instrum., 2008, 79, 083107 CrossRef PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- Y. Lei, J. Jelic, L. C. Nitsche, R. Meyer and J. Miller, Top. Catal., 2011, 54, 334 CrossRef CAS.
- N. Ichikuni and Y. Iwasawa, Catal. Lett., 1993, 20, 87 CrossRef CAS.
- D. R. Short, A. N. Mansour, J. W. Cook Jr, D. E. Sayers and J. R. Katzer, J. Catal., 1983, 82, 299 CrossRef CAS.
- Y. Nagai, T. Hirabayashi, K. Dohmae, N. Takagi, T. Minami, H. Shinjoh and S. Matsumoto, J. Catal., 2006, 242, 103 CrossRef CAS.
- C. F. J. König, T. J. Schildhauer and M. Nachtegaal, J. Catal., 2013, 305, 92 CrossRef.
- D. Ferri, M. A. Newton and M. Nachtegaal, Top. Catal., 2011, 54, 1070 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp05992a |
| This journal is © the Owner Societies 2016 |