
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A 1 + 1′ resonance-enhanced multiphoton ionization scheme for rotationally state-selective detection of formaldehyde via the à 1A2 ← ![[X with combining tilde]](https://www.rsc.org/images/entities/h2_char_0058_0303.gif) 1A1 transition†
1A1 transition†
G. Barratt
Park
*ab,
Bastian C.
Krüger
a,
Sven
Meyer
a,
Alec M.
Wodtke
ab and
Tim
Schäfer
a
aInstitute for Physical Chemistry, University of Göttingen, Tammannstraße 6, 37077 Göttingen, Germany. E-mail: barratt.park@mpibpc.mpg.de
bMax Planck Institute for Biophysical Chemistry, Göttingen, Am Faßberg 11, 37077 Göttingen, Germany
First published on 20th July 2016
Abstract
The formaldehyde molecule is an important model system for understanding dynamical processes in small polyatomic molecules. However, prior to this work, there have been no reports of a resonance-enhanced multiphoton ionization (REMPI) detection scheme for formaldehyde suitable for rovibrationally state-selective detection in molecular beam scattering experiments. Previously reported tunable REMPI schemes are either non-rotationally resolved, involve multiple resonant steps, or involve many-photon ionization steps. In the current work, we present a new 1 + 1′ REMPI scheme for formaldehyde. The first photon is tunable and provides rotational resolution via the vibronically allowed à 1A2 ← ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 1A1 transition. Molecules are then directly ionized from the à state by one photon of 157 nm. The results indicate that the ionization cross section from the 41 vibrational level of the à state is independent of the rotational level used as intermediate, to within experimental uncertainty. The 1 + 1′ REMPI intensities are therefore directly proportional to the à ← absorption intensities and can be used for quantitative measurement of -state population distributions.
1A1 transition. Molecules are then directly ionized from the à state by one photon of 157 nm. The results indicate that the ionization cross section from the 41 vibrational level of the à state is independent of the rotational level used as intermediate, to within experimental uncertainty. The 1 + 1′ REMPI intensities are therefore directly proportional to the à ← absorption intensities and can be used for quantitative measurement of -state population distributions.
I. Introduction
Resonance-enhanced multiphoton ionization (REMPI) spectroscopy is a valuable tool for state-selective detection of atoms and molecules. The high sensitivity of the technique permits its use in a wide variety of dynamics experiments, including ultrahigh vacuum (UHV) molecular beam scattering studies, in which the scattered molecules may be widely distributed in space, time, and quantum state. Newer classes of experiments, such as state-selective ion imaging,1,2 foster the increasing relevance of REMPI-based methods. However, the application of REMPI spectroscopy to new molecular systems of interest requires the availability of a well-characterized multiphoton ionization scheme.In our own work, we are motivated to develop detection schemes for state-specific molecular beam surface scattering experiments. In recent decades, such experiments—primarily involving diatomic molecules—have provided rich information about the mechanisms of energy exchange between molecules and surfaces.3 In direct scattering experiments, the scattered molecules are typically highly rotationally excited and may exhibit rotational rainbows that convey key information about molecule–surface interactions.4 However, such effects have never been experimentally investigated in an asymmetric top molecule, where there is the possibility of distinct degrees of excitation in the Ja, Jb, and Jc components of angular momentum. Furthermore, in polyatomic molecule surface scattering experiments, it has yet to be demonstrated whether distinct vibrationally inelastic energy exchange mechanisms5,6 can occur mode selectively.
Formaldehyde is one of the most thoroughly spectroscopically characterized polyatomic molecules, and thus serves as an ideal candidate for such dynamical studies.7 The extensively characterized, sharply resolved structure of the à ← transition provides the opportunity for rovibrationally state-specific excitation and detection. In the gas phase, formaldehyde has already served as an important model system for the understanding of quantum-state specific photodissociation dynamics,8 and was the prototype system for the elucidation of the roaming mechanism.9 The dynamics of formaldehyde at surfaces are also fundamentally important because formaldehyde is an intermediate in the production of syngas from methane in steam reforming. However, the surface dynamics are more complex and less well understood.
We are aware of five previous studies that describe tunable REMPI spectroscopy of formaldehyde.10–14 The REMPI schemes used in these studies are shown schematically in Fig. 1, where they are compared with the scheme used in the current work. Most of these studies have made use of resonances with the (n,3p) Rydberg states, which lie 7.9–8.4 eV above the ground state and were first observed in VUV absorption15–17 and inelastic electron scattering experiments.18 (In this state label, “n” denotes the non-bonding 2b2 orbital from which an electron is removed and “3p” denotes the Rydberg-like orbital into which the electron is excited. Where applicable, x denotes the out-of-plane axis and z denotes the C2 symmetry axis.) Bomse and Dougal10 report 3 + 2 REMPI spectra of the (n,3pz) 1B2 and (n,3py) 1A1 states. Sun et al.11 report doubly-resonant 1 + 2′ + 1(′) REMPI, in which the first color is resonant with a transition to the à 1A2 or ã 3A2 state, and the second color is two-photon resonant with the (n,3px) 1A2 ← à or (n,3px) 3A2 ← ã transition. Liu and coauthors12 report a simplified 2 + 1 REMPI scheme, which enables the characterization of all three singlet (n,3p) states. Meisinger et al.13 expand on this work, using a 1 + 1′ + 1(′) scheme to probe the rotational structure of the (n,3px) 1A2 state, via double-resonance with rotationally resolved transitions to the intermediate à state. Finally, Li et al.14 report a 1 + 3 REMPI scheme to study the magnetic dipole allowed 420 band of the à ← transition. (We use the notation 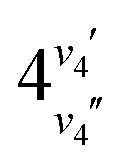 to denote the number of quanta of out-of-plane wagging vibration ν4 in the upper (v4′) and lower (v4′′) electronic state, respectively.)
to denote the number of quanta of out-of-plane wagging vibration ν4 in the upper (v4′) and lower (v4′′) electronic state, respectively.)
 | ||
| Fig. 1 The lowest-lying observed singlet electronic states of formaldehyde are shown. Previously reported tunable REMPI schemes are shown schematically with colored arrows. The current scheme is shown on the right. The dotted line represents the ionization potential at 10.87 eV. | ||
The previously reported tunable REMPI schemes have all been used for spectroscopic characterization of electronically excited states of the formaldehyde molecule (motivated in one case12 by vibrationally state-specific generation of the H2CO+ cation). In the current work, however, the goal is to develop a scheme for sensitive, rovibrationally state-selective detection of -state formaldehyde, compatible with UHV molecular beam scattering experiments. For this application, the REMPI schemes discussed above are unsatisfactory for several reasons. First, most of these schemes employ resonances with high-lying, rapidly predissociating (n,3p) Rydberg states. The resolution provided by these schemes is severely limited by lifetime broadening of the intermediate state. The longest-lived of the (n,3p) intermediate levels is the out-of-plane (n,3px) 1A2 state. The rovibrational levels of this state exhibit lifetimes on the order of 0.5–4 ps, which give rise to Lorentzian lineshapes with FWHM of 1.2–11 cm−1.13 Because the B rotational constant is ∼1.3 cm−1, REMPI schemes that employ such an intermediate state can provide only an approximate measurement of the rotational contour, even at low rotational temperature. Two-color double-resonance schemes, involving sequential (n,3p) ← à ← excitation,11,13 can provide rotational selectivity via the initial à ← step, but such schemes require two different laser frequencies to be scanned independently, which limits the rate of data acquisition and complicates the interpretation of relative intensity information. The 1 + 3 REMPI scheme employed by Li et al.14 avoids the use of (n,3p) Rydberg states, but requires the use of high laser intensity to drive the three-photon ionization step, and would give rise to severe power broadening when applied to vibronically dipole-allowed à ← transitions. Finally, all of the previously reported formaldehyde REMPI schemes require ≥3 photons, which limits the sensitivity. The applicability to molecular beam scattering experiments, where detection of small number densities is critical, is therefore limited.
In the current work, we describe a 1 + 1′ scheme for quantitative measurement of rotational distributions in formaldehyde. The first photon energy is tunable and can be scanned over the à ← 410 transition at around 353 nm (3.50 eV), providing state selectivity. The à state intermediate is subsequently ionized by one photon of 157 nm vacuum ultraviolet (VUV) laser radiation. A single photon of 157 nm (7.90 eV) is insufficient to ionize molecules from the ground state, but the combined two-photon energy 3.50 + 7.90 = 11.4 eV exceeds the 10.87 eV ionization potential.15 Other vibrational levels of the à state may also be used as intermediates. We choose the 410 transition for the current study because it has a strong vibronic intensity, and because the low-lying 41 upper level has a relatively long predissociative lifetime and lies in a region of low vibrational level density. We are aware of one previous study by Andreyev et al.19 in which formaldehyde was ionized directly from the à state. These authors excite the à ← transition using a non-tunable N2 laser at 337.1 nm and measure the lifetime of the excited state by ionizing with one photon of 160 nm radiation from an H2 laser. We extend this work to the measurement of state distributions by using a tunable dye laser.
In order to make quantitative use of our REMPI intensities, it is important to understand the dependence of the signal on both excitation steps. For some systems, such as ionization from the A 2Σ+ state of NO or the B 1Σ+ state of CO, the ionization cross section is found to be essentially independent of the rotational state used as intermediate.20,21 However, in other cases, moderate to strong dependence of the cross section on rotational state has been observed, such as in ionization from the E,F 1Σg+ state of H2 and the A 2Σ+ state of OH, respectively.22,23 In the current work, we observe that over a wide range of rotational energy, 157 nm ionization from the 41 level of Ã-state formaldehyde exhibits no strong dependence on the rotational quantum numbers. Apart from isolated instances in which specific rotational levels of the à 41 intermediate exhibit anomalously short predissociation lifetimes, the intensities in the 1 + 1′ REMPI spectrum are—to within the uncertainty of our experiment—directly proportional to the à ← absorption cross sections.
II. Experimental
The molecular beam apparatus used in this work, which was designed for surface scattering experiments, has been described previously.24 However, the orientation electrodes described in ref. 24 were removed for the current work. A diagram of the experimental configuration is given in Fig. 2. A pulsed beam of formaldehyde was generated by cracking solid paraformaldehyde (97% purity) in a sample holder inside the nozzle of a home-built pulsed solenoid valve, described in ref. 25. The tip of the nozzle was maintained at a temperature of 125 °C (to prevent re-polymerization) and the sample holder was heated to a cracking temperature of ∼70 °C. The cracked formaldehyde was passed through a sample of MgSO4 (99% purity) to remove water, and through a sintered stainless steel filter (20 micron pore size). The nozzle is backed by typically 8–12 bar of He or H2, resulting in a rotational temperature of Trot = 2–3 K or 6–9 K, respectively. Interconversion between the ortho (odd Ka′′) and para (even Ka′′) nuclear spin species26 does not occur on the timescale of our molecular beam expansion. Therefore, collisional cooling from Ka′′ = 1 → 0 is not allowed and rotational levels with J′′ ≤ 4 and Ka′′ = 0 or 1 are populated in the incident beam. The molecular expansion is sent through a 2 mm electroformed skimmer (Ni Model 2, Beam Dynamics, Inc.) located 6 cm from the nozzle and through a second 2 mm aperture, located 20 cm downstream before entering the UHV chamber, where the REMPI experiments are performed. For comparison with our REMPI spectra, we have collected laser induced fluorescence (LIF) spectra of the unskimmed molecular expansion in the source chamber of the apparatus, following the method described in ref. 25. | ||
| Fig. 2 The experimental configuration of the molecular beam surface scattering chamber is pictured. The chamber is equipped with a custom heatable nozzle for generation of formaldehyde molecular beams, and with a 1 + 1′ REMPI detection setup. The right-hand panel shows an expanded view of the detector. Following laser ionization, molecular cations are collected and focused by a pair of ring electrodes at −200 and −900 V and are subsequently guided onto the MCP detector by a repeller plate at +70 V. | ||
The resonant à ← photon for our 1 + 1′ REMPI scheme was obtained from a pulsed dye laser (Sirah Cobra Stretch SL, operating with LDS 698 dye), pumped at 10 Hz by a Nd:YAG laser (Continuum Powerlite 8010). The dye laser output was frequency doubled in a β-barium borate crystal (44° cut angle) to obtain tunable UV radiation with 10 ns pulse duration and typically 3–7 mJ pulse energy over the 340–360 nm range. The beam was collimated in a telescope to an area of ∼3 mm2. A small portion of the dye laser fundamental was coupled into a high-precision wavemeter (HighFinesse, WS7) for frequency calibration. The calibration uncertainty is ∼0.02 cm−1.25 The 157 nm VUV photon for the ionization step was obtained from a F2 laser (GAM EX350) with 20–26 ns pulse duration and 6–11 mJ pulse energy. Mild focusing of the 19 mm × 7 mm VUV beam to an area of ∼5.5 mm2 leads to significant improvement of the 1 + 1′ REMPI signal.
The output of the F2 laser passes through an evacuated flight tube, which is maintained at a pressure of ∼10−2 Torr so that absorption by O2 is negligible. The VUV radiation enters the molecular beam chamber through a CaF2 window. The dye laser enters the chamber from the other side and the lasers are counter-propagated, intersecting a molecular beam at a 90° angle to the molecular beam propagation axis. To achieve the most accurate relative intensities, we set the delay between the center of the two laser pulses to ∼5 ns. This delay is short enough to avoid losing signal from levels that have short lifetimes due to predissociation, but sufficiently long to avoid signal instability due to trigger jitter in the F2 laser. Ions are collected by a set of ion optics and detected on a 2-stage microchannel plate assembly (Hamamatsu F1552) in chevron configuration. In order to avoid saturating the detector with scattered laser radiation, the MCP voltage is pulsed from a rest voltage of 1000 V to a detection voltage of 1600–1800 V, approximately 100 ns after the arrival of the laser pulses, using a HV pulse generator (DEI PVX-4140).
We measured the power dependence of the REMPI signal on both lasers. We automated the data acquisition with a Labview program that simultaneously records the shot-by-shot signal on the MCP together with the power of both lasers. The pulse energy of each laser was measured after the laser beam passes through the chamber. The dye laser power was controlled using a variable neutral density filter (Thorlabs NDC-50C-2M), and the F2 laser power was varied by turning off the pump that evacuates the flight tube and slowly flowing in air. The VUV power meter is situated at the end of a short flight tube on the opposite side of the chamber from the F2 laser, and the pressure in this flight tube is in equilibrium with the pressure in the ingoing flight tube. In order to account for additional power attenuation between the chamber and the power meter, we correct the measured power by assuming Beer's law absorption with the relationship,
 | (1) |
III. Results and discussion
A. Simulated absorption spectra
For comparison with our measured spectra, we calculate the frequency and absorption intensity of each line using a Watson asymmetric top Hamiltonian, with the parameters of ref. 7 for the à 41 state and the parameters of ref. 27 for the ground state. Due to the presence of equivalent hydrogen nuclei in H2CO, the nuclear spin wavefunctions in the ground vibrational state obey ortho (odd Ka′′ levels) and para (even Ka′′ levels) nuclear spin statistics. Because interconversion of the ortho/para nuclear spin species26 does not occur on the timescale of our supersonic expansion, we fix the ortho![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :para population ratio to the 3:1 high temperature ratio, and we calculate the partition functions for the ortho and para manifolds independently. We do not observe a significant temperature difference between the ortho and para manifolds, and we have set Tortho = Tpara in all of the simulations shown in the current work. Comparison of the R- and P-branch intensities to the simulation (Fig. 5) confirms that the spectra are collected under conditions in which we do not observe significant saturation effects on the relative line intensities.
:para population ratio to the 3:1 high temperature ratio, and we calculate the partition functions for the ortho and para manifolds independently. We do not observe a significant temperature difference between the ortho and para manifolds, and we have set Tortho = Tpara in all of the simulations shown in the current work. Comparison of the R- and P-branch intensities to the simulation (Fig. 5) confirms that the spectra are collected under conditions in which we do not observe significant saturation effects on the relative line intensities.
B. Power dependence of transition intensity
The dependence of the REMPI signal on the laser pulse energy for both the resonant (à ←) step and the ionization step are shown in Fig. 3. With the ∼7 mJ of UV power available to us from our frequency-doubled dye laser, the strong rotational transitions of the à ← 410 band can be easily saturated (see Fig. 3(A)). However, the spectra reported in this work were recorded in the unsaturated regime where intensity is proportional to the absorption cross section. We fit the saturation curve in Fig. 3(A) to an equation of the form | (2) |
 | ||
| Fig. 3 The integrated 1 + 1′ REMPI ion signal of the à ← (410) transition is plotted as a function of the pulse energy of both lasers. The rotational transition that was used is labeled at the bottom of each panel. In the left panel (A), the F2 laser pulse energy is held constant at 7.1 mJ, and in the right panel (B), the frequency-doubled dye laser pulse energy was held constant at 5.3 mJ. For each shot of the experiment, the pulse energy of each laser was measured behind the chamber, but is corrected for the measured attenuation that occurs between the chamber and the detector. The pulse fluence is estimated by dividing the corrected pulse energy by the cross-sectional beam area. The resonant UV laser beam is collimated to an area of ∼3 mm2 and the 157 nm F2 laser beam is focused to an area of ∼5.5 mm2. Each data point shown in red represents the average signal obtained from seven repetitions of the experiment. The black curve shown in panel (A) is a fit of the data to eqn (2), with F0 = 450 ± 13 J m−2, and the black line shown in panel (B) is obtained from a linear best fit. | ||
We can compare the results of our saturation curve measurement to a prediction based on the measured cross section. Co et al.28 report an integrated cross section of 1.23 × 10−11 m2 molecule−1 s−1 for the à ← 410 transition at 300 K. This corresponds to an Einstein B coefficient of 1.05 × 1015 m3 J−1 s−2 for the 111 ← 000 transition, which implies saturation at F0 ≈ 69 J m−2. Because experimental characterization of the beam profile and focusing conditions is difficult, there is significant uncertainty in such a comparison. Therefore, the fact that our best-fit F0 parameter from the saturation curve measurement is within an order of magnitude of the value obtained from the absorption cross section indicates reasonable agreement.
We observe a linear dependence of the REMPI signal on F2 laser power, as shown in Fig. 3(B), which indicates that the ionization step is not saturated in our experiment. The 157 nm cross section for photoionization from the à state of formaldehyde is unknown. However, if we assume a typical value of ∼10−19 cm2,29 this would suggest saturation at a laser fluence of ∼1.3 × 105 J m−2, which is two orders of magnitude higher than the maximum fluence used in our experiment.
C. Effect of intermediate state lifetime on the relative intensities
The à state of formaldehyde exhibits an unusually strong, non-systematic rotational dependence of the predissociation rate. This effect has been interpreted in terms of rotationally state-specific internal conversion to rapidly dissociating-state levels.30 The fluorescence lifetimes of single rovibrational levels of the à 41 state have been reported by Shibuya and Lee,31 and by Henke et al.32 Shibuya and Lee31 report lifetimes as short as 40 ns for the Ka′ = 10 manifold. Henke et al.32 report lifetimes ranging from 4.7 μs down to 100 ns, but they observe a handful of rotational levels in their cold molecular jet experiment with lifetimes that were too short to measure (<100 ns). In order to evaluate the extent to which lifetime will affect the relative intensities in our REMPI experiment, it is important to know precisely how short these lifetimes are.
We measured the lifetimes of the five shortest-lived intermediate rotational levels that are accessible to us in our rotationally cold molecular beam by recording the 1 + 1′ REMPI signal as a function of delay between the laser pulses. Representative data for the shortest-lived states are shown in Fig. 4 and the results are tabulated in Table 1. Our measured lifetimes agree (to within a factor of two) with those reported by Henke et al.,32 except in the case of the  level, which we find to be the shortest-lived rotational level in the cold molecular beam spectrum. We believe that the 818 ns lifetime reported by Henke et al. is actually the lifetime of the 211 state, observed via the r,pQ0,2(2) line, which is only partially resolved from r,pQ0,1(1).
level, which we find to be the shortest-lived rotational level in the cold molecular beam spectrum. We believe that the 818 ns lifetime reported by Henke et al. is actually the lifetime of the 211 state, observed via the r,pQ0,2(2) line, which is only partially resolved from r,pQ0,1(1).
 | ||
| Fig. 4 The population decay of the two shortest-lived rotational levels (JKaKc = 110 and 312) of the à (41) state that are accessible in our rotationally cold molecular beam. The REMPI signal intensity is plotted as a function of delay between the laser pulses (red markers), and a single exponential fit to the data is shown (black curve). The resonant transition used for the measurement is labeled in each panel. The non-zero baseline is caused by incomplete resolution from neighboring lines with much longer lifetimes (∼1 μs). | ||
 . Lifetimes from ref. 32 are listed for comparison
. Lifetimes from ref. 32 are listed for comparison

D. 1 + 1′ REMPI spectra of the à ← ![[X with combining tilde]](https://www.rsc.org/images/entities/h3_char_0058_0303.gif) (410) band in a cold molecular beam
(410) band in a cold molecular beam
In Fig. 5, we compare the rotationally cold (Trot = 8 K) à ← (410) spectrum of formaldehyde observed via LIF in our source chamber and via 1 + 1′ REMPI in our UHV surface chamber. The simulated absorption spectrum (Section IIIA) is shown for comparison. The overall pattern of relative intensities observed in the two spectra is in good agreement, which is a indication that the REMPI signals we obtain are proportional to the à ← absorption cross sections, and that we do not observe rotationally-dependent enhancement of the REMPI signal over this region of the spectrum. However, transitions terminating on the short-lived rotational levels listed in Table 1 (marked with asterisks in Fig. 5) are not observed in the LIF spectrum, in which the fluorescence signal was integrated over a window that started 45 ns after the laser excitation (to avoid integrating over laser scatter on the detector). The short-lived levels are all present with approximately the correct relative intensity in our 1 + 1′ REMPI spectrum, in which the laser pulses were partially overlapped, with the center of the F2 laser pulse arriving a few ns after the center of the resonant dye laser pulse. However, we stress the importance of ensuring a short delay time between the laser pulses. Only the transition at 28320 cm−1, terminating on the shortest-lived 110 level, exhibits significant attenuation due to lifetime. This is unavoidable in our experiment because the 10 ns lifetime is shorter than the 20 ns duration of our ionization laser pulse.
 | ||
| Fig. 5 The 1 + 1′ REMPI spectrum (lower plot, in blue) of the à ← (410) transition of formaldehyde is compared with the LIF spectrum (upper plot, in red), at a similar rotational temperature. The LIF spectrum was acquired from a molecular jet expansion in the source chamber of our apparatus, with 0.5 bar He as backing gas. The fluorescence signal was integrated over a 100 ns window that started 45 ns after the laser excitation. The REMPI spectrum was acquired in our UHV chamber from a skimmed molecular beam, which was generated with 12 bar H2 as backing gas. The delay between the center of the two laser pulses in the REMPI spectrum was ∼3 ns. A simulated absorption spectrum at T = 8 K is shown in black with downward-directed peaks. The simulated line intensities (shown) are convolved with a Lorentzian lineshape with FWHM = 0.12 cm−1. | ||
The integrated line intensities from the 1 + 1′ REMPI spectrum of Fig. 5 are shown in a Boltzmann plot in Fig. 6. As with our simulated intensities, we calculate the partition functions of the non-interconverting ortho and para manifolds separately, and we use a room temperature population factor, P = 3 or 1 for ortho and para levels, respectively. The lower state energies on the x-axis are relative to the ground state energy of the respective manifolds. The resulting plot is linear, to within experimental uncertainty, and gives a best-fit rotational temperature of 8.7 ± 0.4 K. A table listing the line frequencies and integrated intensities is provided in Table S1 of the ESI.† We determine the absolute average error of our integrated line intensities to be 26.0%.
 | ||
| Fig. 6 A Boltzmann plot constructed from the 1 + 1′ REMPI spectrum shown in Fig. 5. Ortho transitions are plotted as blue squares and para transitions are plotted as red circles. In the y-axis label, S is the integrated line strength, μ is the relative rotational transition moment, and g is the degeneracy factor. Because of the non-interconversion of ortho and para levels in the molecular beam expansion, the partition function Q is calculated for the ortho and para manifolds separately, and a population factor P = 3 or 1 is applied. On the x-axis, the energy of the lower state in the transition is given relative to the ground rotational state of the respective manifold, E0 = 10.54 or 0 cm−1 for ortho or para levels, respectively. | ||
E. 1 + 1′ REMPI spectrum of a scattered molecular beam
As a proof of principle that the 1 + 1′ REMPI scheme can be used in molecular beam surface scattering experiments, we scattered the rotationally cold formaldehyde beam from a “dirty” Au(111) surface (i.e. a surface that had not been subject to the usual Ar+ ion sputtering and annealing procedure) and probed the rotationally excited scattering products by moving the resonant laser beam several millimeters above the axis of the incoming beam (where signal from the incoming molecular beam is negligible), at a distance of 7.2 mm from the surface. The surface temperature was maintained at 300 K. We used an uncleaned surface for this experiment because surface impurities promote sticking of the formaldehyde molecule to the surface, and ensure that trapping-desorption is the dominant scattering mechanism. Under these conditions, molecules are physisorbed to the surface and undergo thermal desorption. During this residence time, molecular degrees of freedom are thermalized to the surface temperature, and we expect a thermal rotational distribution with Trot ≈ 300 K. We may neglect the effects of detailed balance on the rotational state distribution of desorbing molecules.33,34 These effects are expected to be small because the characteristic rotational energy (0.026 eV) is much less than the binding energy (∼0.24 eV).35We obtain further evidence for the trapping-desorption mechanism by measuring the average kinetic energy of the incoming and scattered molecular beam. The average incoming kinetic energy (0.5 eV) was measured by comparing the arrival time of the molecular beam at several distances from the surface. From this data, the average arrival time at the surface can also be obtained. The average kinetic energy of the scattered molecular beam (0.04 eV) was then determined from the time-of-flight distribution of scattered molecules arriving 18 mm from the surface. The surface residence time is assumed to be negligible relative to the flight time. For time-of-flight measurements, we employ the single color 2 + 1 REMPI scheme of Liu et al.12 We probe with a tightly focused laser beam in the region of the unresolved (n,3px) ← (Ka′ = 1 ← Ka′′ = 1) transitions at a two-photon energy of 67730 cm−1. Since the kinetic energy of the scattered molecules corresponds to a kinetic energy loss much greater than the Baule limit for hard sphere collision of formaldehyde with gold—but approximately equal to the most probable velocity at room temperature—this provides evidence that trapping-desorption dynamics dominate under the conditions of our experiment.
A portion of the à ← 410 band, recorded via 1 + 1′ REMPI of the desorbed molecules is shown in Fig. 7, and is compared with the simulated 300 K absorption spectrum. The mean of the absolute value of the difference between the experimental and simulated 300 K spectrum is 3.1% of the peak intensity, which is only moderately larger than the noise level in our spectrum. Discrepancies between the observed and simulated spectrum are due mostly to the lineshape rather than to the integrated line intensities. We simulate the spectrum with Lorentzian lineshapes, but our experimentally observed lines are slightly broadened at the baseline relative to the power-broadened Lorentzian as a result of the spectral width of the dye laser pulse used to excite the à ← transition.
 | ||
| Fig. 7 The 1 + 1′ REMPI spectrum of a beam of formaldehyde molecules, scattered from an uncleaned Au(111) surface (upper spectrum, red), is compared with the simulated à ← 410 absorption spectrum at 300 K (lower spectrum, blue). The energy of the resonant laser pulse was 3.9 mJ, and the delay between the center of the two laser pulses was 5 ns. The simulated line intensities (shown in black) are convolved with a Lorentzian lineshape with FWHM = 0.15 cm−1. | ||
The relative intensities in the spectrum shown in Fig. 7 exhibit excellent agreement with simulated absorption intensities at 300 K. Because the relative intensities in the measured spectrum agree with the simulation over a wide spectral range, with values of J′′ ≤ 15 and Ka′′ ≤ 8, this is a good indication that the 157 nm ionization cross section appears to be approximately independent of the rotational quantum state that is probed. Furthermore, levels such as 110 that predissociate on a timescale similar to the 5 ns delay between the laser pulses (Fig. 4), appear to be rare, and do not affect the spectral intensities significantly at high temperature. This is consistent with the results of Shibuya and Lee,31 who investigate the single rovibrational level fluorescence quantum yields in the 41 level over a similar J′ and Ka′ range, and report an average lifetime of 91 ns. Finally, the absorption intensities obtained from the Watson Hamiltonian appear to be correct, indicating that the intensity of the à 1A2 ← 1A1 410 band is well described by a vibronic borrowing model, and that rotationally-dependent intensity borrowing effects are negligible over the observed range of J and Ka.
In the same region probed by our 1 + 1′ REMPI experiment (∼11.4 eV), synchrotron experiments15,36 show that the one-photon photoionization cross section of the ground state (1.3 × 10−17 cm2) is also independent of the photon energy. This is approximately the energy region in which the (n,3d) and (n,4p) Rydberg states are expected to appear,37 and it is possible that these states lend intensity to the photoionization cross section. However, the intensity appears to be spread over a broad bandwidth where auto-ionization occurs too rapidly for resonant features to be observed. This is consistent with our observation that the ionization step remains unsaturated up to 157 nm laser fluences of ∼800 J m−2, and that the absolute ionization efficiency appears to be within 1–2 orders of magnitude of the NO 1 + 1 REMPI efficiency via the A 2Σ+ ← X 2Π transition, for which the ionization cross section is independent of rotational quantum numbers and depends only moderately on vibration (see Section S1 of the ESI†).
IV. Conclusions and future directions
In this work, we have demonstrated a 1 + 1′ REMPI scheme for sensitive rotationally-resolved state-specific detection of formaldehyde, compatible with UHV surface scattering experiments. Previously reported tunable REMPI schemes for formaldehyde10–14 were not well suited for such applications because they did not provide the required resolution or sensitivity (they relied on resonances with short-lived predissociated states or involved higher-order multiphoton absorption steps). Our technique is not only much more sensitive than the LIF scheme to which we compare in Fig. 5, but also provides a more accurate measurement of relative populations, because the relative LIF intensities deviate from the relative absorption cross sections for levels that have short predissociative lifetimes.In future work, we are interested in extending our scheme to the detection of vibrationally excited formaldehyde molecules. Many low-lying vibrational levels of -state formaldehyde are expected to have favorable vibronic absorption intensity into either the 41 or the 40 level of the à state—depending on the vibrational symmetry of the lower-state in question—and can therefore be detected by such a scheme. However, although we have provided evidence in the current work that the ionization cross section of the à 41 level exhibits no strong rotational dependence, the vibrational dependence of the 157 nm ionization cross section in the à state is completely unknown and must be measured before 1 + 1′ REMPI via other Ã-state levels can be employed for quantitative measurements.
One potential concern is that the photon energy of our F2 laser lies only 780 cm−1 below the transition energy from the state to the (n,3py) Rydberg state, and might therefore excite accidental hot band resonances of the (n,3py) ← transition. This could lead to undesired background signal from 1′ + 1′ ionization, under conditions where the molecule is vibrationally excited. Our F2 laser operates on two molecular transitions at 157.52 and 157.63 nm, each with 10 pm bandwidth. These photon energies are insufficient to excite the vibrationless (n,3py) ← 0–0 transition, even at rotational temperatures up to 2000 K, so that accidental resonance is not a concern for formaldehyde in its ground vibrational state. However, the possible occurrence of such accidental resonances must be investigated and characterized if the technique is to be extended to vibrationally excited molecules.
Another area of future work involves refinement of the spectroscopic Hamiltonian of the low-lying vibrational levels of the à state. Although we are able to simulate the 300 K absorption spectrum very accurately, preliminary results from direct scattering experiments in which rotational temperatures up to 1000 K are reached reveal discrepancies between the calculated and observed line frequencies involving highly excited levels. These discrepancies arise because these lines were not previously available as input to the spectroscopic Hamiltonian fits.
Acknowledgements
The authors thank Dirk Schwarzer for building the nozzle used in this experiment. A. M. W. and G. B. P. acknowledge support from the Alexander von Humboldt Foundation.References
- D. W. Chandler and P. L. Houston, Two-dimensional imaging of state-selected photodissociation products detected by multiphoton ionization, J. Chem. Phys., 1987, 87(2), 1445–1447 CrossRef CAS.
- M. N. R. Ashfold, N. Hendrik Nahler, A. J. Orr-Ewing, O. P. J. Vieuxmaire, R. L. Toomes, T. N. Kitsopoulos, I. Anton Garcia, D. A. Chestakov, S.-M. Wu and D. H. Parker, Imaging the dynamics of gas phase reactions, Phys. Chem. Chem. Phys., 2006, 8, 26–53 RSC.
- A. M. Wodtke, D. Matsiev and D. J. Auerbach, Energy transfer and chemical dynamics at solid surfaces: the special role of charge transfer, Prog. Surf. Sci., 2008, 83(3), 167–214 CrossRef CAS.
- A. W. Kleyn and T. C. M. Horn, Rainbow scattering from solid surfaces, Phys. Rep., 1991, 199(4), 191–230 CrossRef CAS.
- B. D. Kay, T. D. Raymond and M. E. Coltrin, Observation of direct multiquantum vibrational excitation in gas-surface scattering: NH3 on Au(111), Phys. Rev. Lett., 1987, 59, 2792–2794 CrossRef CAS PubMed.
- C. T. Rettner, F. Fabre, J. Kimman and D. J. Auerbach, Observation of direct vibrational excitation in gas-surface collisions: NO on Ag(111), Phys. Rev. Lett., 1985, 55, 1904–1907 CrossRef CAS PubMed.
- D. J. Clouthier and D. A. Ramsay, The spectroscopy of formaldehyde and thioformaldehyde, Annu. Rev. Phys. Chem., 1983, 34, 31–58 CrossRef CAS.
- C. B. Moore and J. C. Weisshaar, Formaldehyde photochemistry, Annu. Rev. Phys. Chem., 1983, 34(1), 525–555 CrossRef CAS.
- D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, J. Rheinecker, L. B. Harding and J. M. Bowman, The roaming atom: straying from the reaction path in formaldehyde decomposition, Science, 2004, 306(5699), 1158–1161 CrossRef CAS PubMed.
- D. S. Bomse and S. Dougal, Multiphoton ionization of formaldehyde: observation of 3py and 3pz Rydberg states, Laser Chem., 1987, 7(1), 35–40 CrossRef CAS.
- S. Weizhong, X. Xingbin, L. Li and Z. Cunhao, Double resonance REMPI of formaldehyde 3A2 (n,3px) and 1A2 (n,3px) Rydberg states, Chin. J. Chem. Phys., 1989, 2, 184–192 Search PubMed.
- J. Liu, H.-T. Kim and S. L. Anderson, Multiphoton ionization and photoelectron spectroscopy of formaldehyde via its 3p Rydberg states, J. Chem. Phys., 2001, 114(22), 9797–9806 CrossRef CAS.
- M. Meisinger, A. M. Schulenburg, F. Merkt and P. P. Radi, Rotationally resolved spectroscopy and dynamics of the 3px1A2 Rydberg state of formaldehyde, Phys. Chem. Chem. Phys., 2010, 12, 15592–15599 RSC.
- L. Xiaonong, X. Xingbin, L. Li and Z. Cunhao, Rotationally resolved REMPI spectroscopy of the à 1A2 ← 1A1 (420) magnetic dipole transition, Chin. J. Chem. Phys., 1991, 4(4), 235–239 Search PubMed.
- J. E. Mentall, E. P. Gentieu, M. Krauss and D. Neumann, Photoionization and absorption spectrum of formaldehyde in the vacuum ultraviolet, J. Chem. Phys., 1971, 55(12), 5471–5479 CrossRef CAS.
- C. R. Lessard, D. C. Moule and S. Bell, The lower Rydberg states of formaldehyde, Chem. Phys. Lett., 1974, 29(4), 603–605 CrossRef CAS.
- C. R. Lessard and D. C. Moule, The 1A2 (n,3px) Rydberg state of formaldehyde, J. Mol. Spectrosc., 1976, 60(1–3), 343–347 CrossRef CAS.
- M. J. Weiss, C. E. Kuyatt and S. Mielczarek, Inelastic electron scattering from formaldehyde, J. Chem. Phys., 1971, 54(10), 4147–4150 CrossRef CAS.
- S. V. Andreyev, V. S. Antonov, I. N. Knyazev and V. S. Letokhov, Two-step photoionization of H2CO by radiation of N2 and H2 lasers and measurement of the lifetime of its 1A2 state, Chem. Phys. Lett., 1977, 45(1), 166–168 CrossRef.
- H. Zacharias, F. de Rougemont, T. F. Heinz and M. M. T. Loy, Ionization probabilities of A 2Σ+ (v′ = 0, 1, 2) and B 2Π (v′ = 0, 2) states of NO, J. Chem. Phys., 1996, 105(1), 111–117 CrossRef CAS.
- S. Wurm, P. Feulner and D. Menzel, Resonance-enhanced multiphoton ionization spectroscopy of X 1Σ+ and a 3Π carbon monoxide using electron stimulated desorption as a source for rovibronically excited species, J. Chem. Phys., 1996, 105(16), 6673–6687 CrossRef CAS.
- K.-D. Rinnen, M. A. Buntine, D. A. V. Kliner, R. N. Zare and W. M. Huo, Quantitative determination of H2, HD, and D2 internal-state distributions by (2 + 1) resonance-enhanced multiphoton ionization, J. Chem. Phys., 1991, 95(1), 214–225 CrossRef CAS.
- J. M. Beames, F. Liu and M. I. Lester, 1 + 1′ resonant multiphoton ionisation of OH radicals via the A 2Σ+ state: insights from direct comparison with A–X laser-induced fluorescence detection, Mol. Phys., 2014, 112(7), 897–903 CrossRef CAS.
- N. Bartels, K. Golibrzuch, C. Bartels, L. Chen, D. J. Auerbach, A. M. Wodtke and T. Schäfer, Dynamical steering in an electron transfer surface reaction: oriented NO (v = 3, 0.08 < Ei < 0.89 eV) relaxation in collisions with a Au(111) surface, J. Chem. Phys., 2014, 140(5), 054710 CrossRef PubMed.
- G. B. Park, B. C. Krüger, S. Meyer, D. Schwarzer and T. Schäfer, The ν6 fundamental frequency of the à state of formaldehyde and Coriolis perturbations in the 3ν4 level, J. Chem. Phys., 2016, 144, 194308 CrossRef PubMed.
- R. F. Curl, J. V. V. Kasper and K. S. Pitzer, Nuclear spin state equilibration through nonmagnetic collisions, J. Chem. Phys., 1967, 46(8), 3220–3228 CrossRef CAS.
- H. S. P. Müller, G. Winnewisser, J. Demaison, A. Perrin and A. Valentin, The ground state spectroscopic constants of formaldehyde, J. Mol. Spectrosc., 2000, 200(1), 143–144 CrossRef PubMed.
- D. T. Co, T. F. Hanisco, J. G. Anderson and F. N. Keutsch, Rotationally resolved absorption cross sections of formaldehyde in the 28100–28500 cm−1 (351–356 nm) spectral region: implications for in situ LIF measurements, J. Phys. Chem. A, 2005, 109(47), 10675–10682 CrossRef CAS PubMed.
- H. Zacharias, R. Schmiedl and K. H. Welge, State selective step-wise photoionization of NO with mass spectroscopic ion detection, Appl. Phys., 1980, 21(2), 127–133 CAS.
- J. C. Weisshaar and C. B. Moore, Collisionless nonradiative decay rates of single rotational levels of S1 formaldehyde, J. Chem. Phys., 1979, 70(11), 5135–5146 CrossRef CAS.
- K. Shibuya and E. K. C. Lee, Rotational dependence of the fluorescence quantum yields of H2CO and D2CO (Ã 1A2): single rovibronic level values and their average values for the 41 level, J. Chem. Phys., 1978, 69(12), 5558–5560 CrossRef CAS.
- W. E. Henke, H. L. Selzle, T. R. Hays and E. W. Schlag, Single rotational lifetimes of formaldehyde in a hypersonic jet, J. Chem. Phys., 1982, 76(3), 1327 CrossRef CAS.
- J. C. Polanyi and R. J. Wolf, Dynamics of simple gas–surface interaction. II. Rotationally inelastic collisions at rigid and moving surfaces, J. Chem. Phys., 1985, 82(3), 1555–1566 CrossRef CAS.
- R. R. Cavanagh and D. S. King, Rotational- and spin-state distributions: NO thermally desorbed from Ru(001), Phys. Rev. Lett., 1981, 47, 1829–1832 CrossRef CAS.
- W.-K. Chen, S.-H. Liu, M.-J. Cao, Q.-G. Yan and C.-H. Lu, Adsorption and dissociation of methanol on Au(111) surface: a first-principles periodic density functional study, J. Mol. Struct.: THEOCHEM, 2006, 770(1–3), 87–91, DOI:10.1016/j.theochem.2006.05.040.
- L. G. Dodson, L. Shen, H. D. Savee, N. C. Eddingsaas, O. Welz, C. A. Taatjes, D. L. Osborn, S. P. Sander and M. Okumura, VUV photoionization cross sections of HO2, H2O2, and H2CO, J. Phys. Chem. A, 2015, 119, 1279–1291 CrossRef CAS PubMed.
- C. M. Hadad, J. B. Foresman and K. B. Wiberg, Excited states of carbonyl compounds. 1. Formaldehyde and acetaldehyde, J. Phys. Chem., 1993, 97(17), 4293–4312 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of sensitivity to a benchmark system and tabulation of line frequencies and intensities. See DOI: 10.1039/c6cp03833f |
| This journal is © the Owner Societies 2016 |