
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of extracellular DNA in uranium precipitation and biomineralisation†
Joseph
Hufton
a,
John H.
Harding
b and
Maria E.
Romero-González
 *a
*a
aDepartment of Geography, The University of Sheffield, Sheffield, S10 2TN, UK. E-mail: m.e.romero-gonzalez@sheffield.ac.uk
bDepartment of Materials Science and Engineering, Sir Robert Hadfield Building, University of Sheffield, Mappin Street, Sheffield, S1 3JD, UK
First published on 27th September 2016
Abstract
Bacterial extra polymeric substances (EPS) have been associated with the extracellular precipitation of uranium. Here we report findings on the biomineralisation of uranium, with extracellular DNA (eDNA) used as a model biomolecule representative of EPS. The complexation and precipitation of eDNA with uranium were investigated as a function of pH, ionic strength and varying concentrations of reactants. The role of phosphate moieties in the biomineralisation mechanism was studied by enzymatically releasing phosphate (ePO4) from eDNA compared to abiotic phosphate (aPO4). The eDNA–uranium precipitates and uranium minerals obtained were characterised by Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FT-IR) spectroscopy, Scanning Electron Microscopy-Energy Dispersive X-Ray analysis (SEM-EDX), X-Ray Powder Diffraction (XRD) and X-Ray Photoelectron Spectroscopy (XPS). ATR-FT-IR showed that at pH 5, the eDNA–uranium precipitation mechanism was predominantly mediated by interactions with phosphate moieties from eDNA. At pH 2, the uranium interactions with eDNA occur mainly through phosphate. The solubility equilibrium was dependent on pH with the formation of precipitate reduced as the pH increased. The XRD data confirmed the formation of a uranium phosphate precipitate when synthesised using ePO4. XPS and SEM-EDX studies showed the incorporation of carbon and nitrogen groups from the enzymatic orthophosphate hydrolysis on the obtained precipitated. These results suggested that the removal of uranium from solution occurs via two mechanisms: complexation by eDNA molecules and precipitation of a uranium phosphate mineral of the type (UO2HPO4)·xH2O by enzymatic orthophosphate hydrolysis. This demonstrated that eDNA from bacterial EPS is a key contributor to uranium biomineralisation.
1 Introduction
The use of nuclear power may present a solution to the global problem of energy supply, since uranium fission can be used for energy production. However the release of radionuclides, either through large-scale accidents (such as the Chernobyl nuclear disaster) or through leakage from underground stored radioactive waste, is an environmental hazard. Therefore, the safe removal, storage and detoxification of these contaminants are of utmost importance. The use of microorganisms may provide a cheap alternative to the material-heavy and expensive physiochemical treatment processes currently used.1–4 However, the biochemical mechanisms and the impact of solution chemistry involved in bioaccumulation and biomineralisation of radiounuclides are not fully understood, which makes their use and application difficult for remediation, extraction and reuse.The complexation of soluble radionuclide species, such as a highly mobile uranyl ion (UO22+), has been associated with the constituents of bacterial extra polymeric substances (EPS).2,5–8 Bacterial EPS consist of large quantities of biopolymers, such as phosphorylated peptides, polysaccharides, lipids and nucleic acids that have either been secreted by bacteria or released into the surrounding environment from the lysis of dead bacterial cells.9,10 Due to the complexity of bacterial EPS, it is difficult to identify detailed mechanisms for extracellular uranium biomineralisation. Merroun et al. (2011) suggested that phosphate containing and phosphorylated biopolymers are responsible for sequestering uranium through biosorption to negatively charged functional groups, or through acid phosphatase activity that liberates inorganic phosphate from biological molecules to mineralise uranium.
Extracellular DNA (eDNA) is a major constituent of EPS and for some species it could represent up to 50% of their EPS components.11 Elevated levels of eDNA production have been reported following the investigations of metal interactions with bacterial biofilms.12 In certain species, such as Pseudomonas putida and Bacillus cereus, eDNA is fundamental to biofilm formation.11,13 The main source of eDNA is likely to be the lysis of dead bacterial cells.14–17 However, additional eDNA can be supplied via bacterial extrusion (as a survival or enrichment mechanism). Deinococcus radiodurans secretes damaged portions of genomic DNA into its EPS to be replaced by de novo synthesis18 while other genera such as Bacillus and Pseudomonas release eDNA into liquid media during bacterial growth.19 We hypothesise that the complexation of uranium within EPS is facilitated by eDNA under acidic conditions through phosphate interactions, due to large amounts of phosphate within DNA in comparison to that found in sugars or lipids.
Acid phosphatase activity has been associated with the biomineralisation of uranium phosphate mineral-like phases20–23 since this enzyme hydrolyses organic phosphate esters from biological molecules.24 It has been suggested that uranium biomineralisation due to microbial acid phosphatase activity does not depend on the microbial structure, be it of Gram positive or Gram-negative origin, but instead, depends on pH.2 Enzymatic uranium biomineralisation occurs under mildly acidic to circumneutral conditions producing uranium phosphate mineral-like phases, such as meta-autunite, HUO2PO4 and NaUO2PO4, both inside and outside the cell wall. By contrast, bioprecipitation with phosphate groups in biomolecules more commonly occurs under mild to acidic conditions.2,20,25,26 This suggested that free phosphate of biogenic origin plays an important role in the mineralisation of uranium. It has been previously demonstrated that phosphate from DNA released via phosphatase activity may be the sole source of phosphorus involved in the precipitation of metals.27
This study aims to reveal the mechanism of formation of uranium-phosphate minerals in the presence of eDNA as a key contributor to the biomineralisation of uranium in microbial systems. We have quantified the reaction mechanism for the complexation and precipitation of eDNA with uranium and the formation of uranium phosphate minerals by enzymatically liberated phosphate (ePO4). We hypothesised that enzymatically liberated phosphate would bind to a highly mobile U(VI) species under acidic conditions.
The interactions of uranium with eDNA and ePO4 were studied using Fourier Transform-Infrared spectroscopy (FT-IR), Scanning Electron Microscopy-Energy Dispersive X-ray (SEM-EDX) analysis, X-Ray Difrraction (XRD) and X-ray Photoelectron Spectroscopy (XPS). FT-IR is a suitable tool to identify the functional groups involved in binding since there are well-defined changes in the peak position and intensity upon uranium binding to biomass. XRD is used to determine the mineral structures of any uranium precipitates formed following accumulation.7,23,28,29 We used eDNA as a model biomolecule to represent uranium biomineralisation within complex bacterial EPS for two reasons: (i) it contains phosphate moieties bound to organic compounds, which allow us to quantify the efficiency of organic phosphate to precipitate uranium, and (ii) DNA–organic phosphate can be enzymatically released as phosphate (ePO4) that could be used to form uranium minerals.
The novelty of this work consists in the fact that, whereas previous uranium biomineralisation experiments resorted to investigating whole bacterial strains and their corresponding EPS, this study aims to quantify the process by using representative biomolecules (purified DNA and ePO4) to investigate whether phosphate-containing biomolecules are responsible for uranium biomineralisation within EPS. Understanding the mechanistic processes involved in uranium binding to eDNA provides insight into how a bacterial EPS interacts with uranium.
2 Experimental procedures
2.1 Materials and stock solutions
The composition of eDNA obtained from bacteria was comparable to the composition of DNA from other organisms.27 In order to simplify the experiments, lyophilised DNA from salmon sperm was obtained from Sigma-Aldrich (D1626) since it was readily available. DNA stock solutions of 2 mg mL−1 (referred to from here on as eDNA) were prepared using DNase/RNase-free water. Purified acid phosphatase (6.3 units mg−1 solid) was obtained from Sigma-Aldrich (P1146) and solutions were prepared directly before use with DNase/RNase-free water. U(VI) solutions were prepared using 0.1 M uranyl nitrate hexahydrate (UO2(NO3)2·6H2O) in 1% HNO3 (Fluka Analytical: 94270). The pH of the solutions was adjusted by the addition of HCl or NaOH while the ionic strength was controlled by the addition of NaCl.2.2 Uranium–eDNA complexation and precipitation
Uranium–eDNA complexation and precipitation experiments were designed to determine the amount of uranium removed from solution by a given quantity of extracellular DNA (eDNA). 1 mg mL−1 eDNA was incubated with 0.5 mM U(VI) in 0.1 M NaCl (pH 5). All experiments were carried out at room temperature for 1 hour. The uranium concentration used represented and exceeded a typical amount of uranium in contaminated water.30,31 To investigate the effects of pH, ionic strength and reactant concentrations on the precipitation process, the pH range studied was 2–7, ionic strength was varied between 1000 mM and 1 mM NaCl, DNA concentrations ranged from 2.0 to 0.25 mg mL−1 and U(VI) concentrations ranged from 0.05 to 2 mM.To quantify the free uranium remaining in solution, the residual DNA was removed from solution. The eDNA–uranium complexes and subsequent precipitates were removed from solution by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 minutes (4 °C), lyophilised and stored for spectral analysis. 50 μL of 3 M sodium acetate buffer (pH 5.0) and 550 μL of 2-propanol were added sequentially to 500 μL of the supernatant to precipitate any remaining DNA from solution. Samples were stored on ice for 15 minutes and centrifuged at full speed for a further 15 minutes (4 °C). Supernatants were acidified using 1% HNO3 and uranium was quantified by Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) (ELAN DRC II, Perkin Elmer).
000 × g for 10 minutes (4 °C), lyophilised and stored for spectral analysis. 50 μL of 3 M sodium acetate buffer (pH 5.0) and 550 μL of 2-propanol were added sequentially to 500 μL of the supernatant to precipitate any remaining DNA from solution. Samples were stored on ice for 15 minutes and centrifuged at full speed for a further 15 minutes (4 °C). Supernatants were acidified using 1% HNO3 and uranium was quantified by Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) (ELAN DRC II, Perkin Elmer).
The Quant-iT™ PicoGreen® dsDNA reagent kit from Life Technologies (P11496) was used to quantify free eDNA in solution following the removal of the DNA–uranium precipitate. Free DNA was diluted using 1× TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.5) to below 100 μg mL−1 while the Quant-iT™ PicoGreen® dsDNA reagent (Component A) was diluted 50 fold using 1× TE buffer for analysis. 10 μL of free DNA was added to 200 μL of the reagent and analysed using the FLUOstar OPTIMA fluorescent microplate reader with samples excited at 485 nm and the fluorescence emission intensity recorded at 545 nm. DNA was quantified by comparison to known standards and relative fluorescence units to obtain a linear calibration.
2.3 ATR-FT-IR
For the spectroscopic analysis of uranium precipitates and minerals, samples were centrifuged at 10000 × g for 15 minutes (4 °C), the supernatant was removed and the pellet was lyophilised (Alpha 1-2 LD Plus freeze dryer). Measurements were performed using Attenuated Total Reflectance (ATR)-FT-IR. These were obtained using a Silver Gate Evolution ATR accessory, consisting of a germanium crystal, coupled to a Perkin Elmer Spectrum One FT-IR spectrometer. A total of 30 scans were performed on each sample within the scanning range 2000–800 cm−1. An average spectrum was obtained from 3 replicates and was normalised to 1.5 arbitrary units using the 1063 cm−1 absorption band from the control eDNA spectra, corresponding to the vibrational stretching of phosphate groups. All data acquisition and processing were performed using the PerkinElmer Spectrum version 3.3.
2.4 Uranium biomineralisation using enzymatically generated phosphate
Enzymatic phosphate from eDNA (ePO4) was hydrolysed from 1 mg mL−1 DNA using 2 units mL−1 acid phosphatase in the presence of 0.1 M NaCl (pH 5) for 72 hours at 37 °C. Solutions were centrifuged and membrane filtered (0.45 μm PES) to remove the acid phosphatase and any other particulates. The supernatant was quantified for enzymatic inorganic orthophosphate (ePO4) using a modified molybdenum blue method.32 To 80 μL of the sample, 100 μL of 0.1 M ascorbic acid containing 0.5 M trichloroacetic acid (TCA), 50 μL of 0.01 M ammonium molybdate and 50 μL of 0.1 M sodium citrate containing 0.2 M sodium arsenite were added sequentially. Samples were incubated at room temperature for 30 minutes with gentle agitation and the absorbance of molybdenum blue was measured at 850 nm.ePO4 was incubated with uranium at varying molar ratios (pH 5) for 24 hours at 37 °C. Minerals were removed by centrifugation and stored for further analysis. The supernatant was divided into 2 aliquots: one aliquot was used to determine the remaining phosphate in solution, and an acidified aliquot was used to quantify uranium.
2.5 Scanning electron microscopy (SEM)
Scanning electron microscopy was carried out using an FEI Quanta 650 Environmental Scanning Electron Microscope (ESEM) with a tungsten thermionic emission gun equipped with an Oxford Instruments Aztec EDX system. Lyophilised uranium precipitate powders were immobilised on PELCO™ carbon conductive tabs (12 mm) and samples were carbon coated to a 10 nm approximate thickness using a Quorum Industries K150 carbon coating unit before analysis.2.6 X-ray powder diffraction (XRD) analysis
XRD patterns of lyophilised eDNA–uranium precipitates and uranium minerals were recorded using a STOE STADI P diffractometer with a molybdenum radiation source (λ = 0.71) over the range of 3–40 (2θ) with a step length of 0.01°. Before data acquisition, samples were flattened between 2 acetate sheets using a hydraulic press (SPECAC) and placed in the sample holder. The molecular structures of the eDNA–uranium precipitates and uranium minerals were identified through comparison with standard powder diffraction files (PDF-4 + International Centre for Diffraction Data's (ICDD) database).2.7 X-ray photoelectron spectroscopy (XPS)
For XPS analysis, uranium minerals formed from the reaction of ePO4 with uranium were immobilised on a double-sided carbon adhesive tape and mounted onto sample studs. XPS analysis was conducted using a KRATOS AXIS 165 Ultra Photoelectron spectrometer and an Al Kα X-ray source (1486.6 eV). Each sample was analysed by a wide survey scan (pass energy 160 eV, 1.0 eV step size) and a high resolution scan (pass energy 20 eV, 0.1 eV step size) for carbon, oxygen, phosphorus and uranium. Binding energies were determined using the CasaXPS software (Version 2.3.16). The fitting of the C1s peak was set at 285.0 eV and the full width at half maximum was kept constant for subsequent peak calibration.3 Results and discussion
3.1 Uranium precipitation using eDNA
Uranium and eDNA precipitated from solution following their interaction, and were quantified by ICP-MS and PicoGreen fluorescence, respectively. The percentage removals (eqn (1)) and maximum accumulation capacities were calculated for a variety of pH values, ionic strengths and varying concentrations of reactants in solution.| % Removal = ((C0 − CI)/C0) × 100 | (1) |
Fig. 1 shows the percentage of uranium and eDNA removed from solution as a function of the parameters studied. Changes in pH affect the adsorption of uranium to eDNA from aqueous solution, with an increase in the removal of both reactants as the pH decreases to as low as pH 2, Fig. 1A. Within a low pH environment, almost all the eDNA (1 mg mL−1) is removed from solution by 0.5 mM uranium. 99.9 ± 0.244% of eDNA and 95.3 ± 0.4% of uranium were removed at pH 2, decreasing to 92.3 ± 2.6% eDNA and 88.2 ± 0.7% uranium removed at pH 4. As the pH increases to 5, the amount of uranium precipitation with eDNA decreased substantially and 31.7 ± 13.9% of eDNA and 42.6 ± 3.3% of uranium were removed from solution.
 | ||
| Fig. 1 Uranium removal by eDNA as a function of pH, NaCl concentration, initial DNA concentration and initial uranium concentration. | ||
DNA has a net negative charge in acidic solutions, due to its negatively charged deoxyribose phosphate backbone.33,34 The highly mobile positively charged U(VI) ion, UO22+, is the dominant uranium(VI) species in acidic environments. UO22+ interacted strongly with eDNA at low pH. The results indicated the almost complete removal of both reactants between pH 2 and pH 4. At pH 6 and 7 in the absence of any strong anion, U(VI) was found in the form of larger species such as (UO2)3(OH)5+ (UO2)4(OH)7+. The difference in size and net charge of these U(VI) species, in comparison to UO22+, may have hindered interactions with functional groups within the eDNA structure. At circumneutral pH, uranium reached its solubility equilibrium and was not present in solution. However, between 10 and 20% of the DNA was removed, possibly concomitant with the insoluble U(VI) species.
The amount of eDNA interacting with U(VI) species decreased as the solution pH increased. Although uranium biosorption with EPS has been reported to take place over a wide range of pH,8,35 both precipitation and sorption may be involved in the process resulting in the higher removal of uranium compared to our results. EPS are composed of various biomolecules, including polysaccharides, nucleic acids, peptides and lipids.9,10 Polysaccharides have previously been shown to bind preferentially to uranium at closer to neutral pH compared to other biomass present within bacterial EPS.36 At low pH, uranium would interact more strongly with components such as eDNA that have an overall net negative charge at low pH. As the pH increases, the net positive charge of eDNA increases; hence uranium prefers to bind to other biopolymers. The carboxyl groups of polysaccharides are deprotonated at pH higher than 5, and therefore, are more likely to bind positively charged U(VI) species from solution. These results indicated that there is preferential binding to different biomasses present within EPS as a function of pH.
Changes in ionic strength, obtained by varying the concentration of sodium chloride with 0.5 mM U(VI) (pH 5), affected the precipitation of eDNA–uranium complexes from solution, Fig. 1B. As the concentration of sodium chloride decreased in solution, there was a reduction in the formation of eDNA–uranium precipitates. At 1 M sodium chloride and pH 5, the amounts of 1 mg mL−1 eDNA and 0.5 mM uranium precipitated from solution were 36.8 ± 6.9% and 56.3 ± 2.2%, respectively. This precipitation decreased to 13.7 ± 4.1% eDNA and 10.6 ± 7.6% uranium when no sodium chloride was present in the solution. The increase in uranium precipitating with eDNA was due to the increased ionic strength. As the ionic strength increased there was an overall increase in the net negative charge of the eDNA structure, facilitating the interaction. This finding agrees with other studies that have investigated the effect of ionic strength on the adsorption of protons to biomass of bacterial origin.37,38
The uranium speciation also changed as a function of NaCl concentration (Visual MINTEQ). In the presence of 1 M NaCl, the abundance of U(VI) species was 16.3% UO22+ and 40.9% (UO2)3(OH)5+. As the NaCl concentration decreased to zero, the relative abundances changed to 7.3% and 62.1%, respectively, making (UO2)3(OH)5+ the dominant species. The speciation of uranium hence controlled the reactivity of uranium with eDNA.
Changes in the initial concentration of reactants to determine the stoichiometry of the reaction suggested that uranium precipitation with eDNA at pH 5 is not favourable. At 0.5 mM uranium initial concentration and 2 mg mL−1 eDNA in solution, a maximum of 64.5 ± 6.9% of uranium is removed by 16.0 ± 15.3% of the eDNA. As this initial eDNA concentration decreased, the amount of uranium removed from solution (by 68.0 ± 7.2% of 0.25 mg mL−1 eDNA) decreased to 37.2 ± 4.3%, Fig. 1C. The changes in the initial ratio of concentrations of both reactants changed the efficiency of removal by changing the number of available reaction sites.
The increase of uranium concentration in solution resulted in a higher percentage removal of eDNA, Fig. 1D. When the initial concentration of uranium was 0.25 mM, 40.0 ± 6.0% was removed from solution by 16.3 ± 3.4% of eDNA in solution. This removal of both reactants increased with initial uranium concentration. 75.0 ± 4.2% of uranium was removed by 96.2 ± 1.8% of eDNA for an initial concentration of 2 mM. We did not observe uranium precipitates on the control samples, indicating that all uranium was soluble in solution in the studied concentration range. Up to an initial concentration of 1 mM uranium in solution, the removal of 1 mg mL−1 eDNA was higher than the percentage of uranium removal from solution. At 2 mM uranium there was a higher percentage of DNA removed than uranium. This could be due to changes in the structural conformation. Increasing concentrations of uranium and the size of U(VI) species could lead to the deformation of any secondary structures and breaking of the double helix structure. This change in the structure could then lead to an increase in the bioavailability of functional groups (that were not located on the surface of the eDNA molecule in its original conformation) for uranium binding.39 At low concentrations, the quantity of U(VI) species in solution would result in mostly electrostatic interactions at the surface of the molecule.34
From the quantification data obtained, we suggest that at low pH, there is a higher percentage removal of both reactants from solution due to the electrostatic interactions between the positively charged uranyl ion and the negatively charged biomolecule. This removal is further enhanced by high ionic strength, where ions in solution change the accessibility of certain functional groups due to conformational changes of the eDNA structure, leading to higher uranium binding and removal from solution.
3.2 Characterisation of eDNA–uranium interactions using ATR-FT-IR
FT-IR spectra were collected in triplicate between 2000 and 800 cm−1 in order to identify changes in the functional groups of eDNA involved in precipitating uranium from solution. The shifts, and changes in shape and intensity of absorption bands can be attributed to a number of different functional groups in eDNA binding to uranium.Table 1 summarises the absorption band assignments for the control eDNA as well as eDNA–uranium precipitates prepared by interacting eDNA with 0.5 mM uranium solution at pH values 2–5. The changes in eDNA molecular vibrations following their interaction with increasing uranium concentrations (up to 2 mM U(VI) at pH 5) were also investigated (Table S1, ESI†). The full spectral region for both investigations is shown in Fig. S1 and S2 (ESI†).
| Control | pH 2 | pH 3 | pH 4 | pH 5 | Allocationa |
|---|---|---|---|---|---|
| a Functional group allocation is based on ref. 21 and 39–43 | |||||
| 1700 sh | 1689 s | 1700 sh | 1700 sh | 1700 sh | ν(C–NH2) bases; guanine/adenine |
| 1649 m | 1632 sh | 1646 w | 1647 w | 1649 m |
ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
| 1606 w | 1599 sh | 1600 w | 1602 w | 1606 w | — |
| 1579 vw | 1579 vw | Adenine/cytosine | |||
| 1529 w | 1534 vw | 1531 vw | 1529 vw | 1529 w |
δ(NH)/ν(CO), cytosine/guanine |
| 1486 m | 1478 w | 1482 w | 1483 w | 1490 m | C–NH2 scissoring |
| 1419 w | 1419 vw | 1419 vw | 1419 w | — | |
| 1369 w | 1366 vw | 1365 vw | 1365 vw | 1367 vw |
ν(CNH) nitrogenous bases |
| 1230 s | 1209 s | 1212 s | 1214 s | 1218 s | ν as(PO2) |
| 1063 vs | 1059 vs | 1061 vs | 1064 vs | 1060 vs | ν s(PO2), ν(C–OH, CO–C, C–C) |
| 965 s | 970 m | 970 m | 970 m | 970 m | ν(P–O) |
| 928 s | 927 s | 930 s | 918 s | ν(U–Oligand) | |
An increase in shape and a shift in the absorption band position, corresponding to changes in the vibrational stretching of C–NH2 groups of nitrogenous bases,34,40 were observed for the eDNA–uranium precipitates at pH 2. The shift from 1700 cm−1 to 1689 cm−1 suggested binding to nitrogenous bases at very acidic pH.
Shifts in the absorption band position corresponding to the vibrational stretching of the CO bond were observed as the pH increased from 2 to 5. At pH 2, stretching is observed at 1632 cm−1. As the pH increases to 3, 4 and 5, the absorption band position shifts to 1646, 1647 and 1649 cm−1, respectively. The vibrational stretching of CO groups within the eDNA control spectra was observed at 1649 cm−1; therefore it was suggested that the binding of uranium to carbonyl groups in nitrogenous bases was pH-dependent.41 Similarly, the precipitation depended on the concentration of uranium. At pH 5, there was a shift of the C–NH2 band from 1649 cm−1 to 1647 cm−1 as the uranium concentration increased to 2 mM. Changes in the region between 1550 and 1350 cm−1 corresponding to the binding of U(VI) species to the nitrogenous bases of eDNA were also observed. A shift occurred in the absorption band, corresponding to the changes in the scissoring mode of C–NH2 groups following uranium interactions with eDNA at pH 2. This absorption band shifted from 1486 cm−1 to 1478 cm−1. This change indicates higher binding affinity at low pH. This is further supported by changes in the absorption bands at 1529 cm−1 (shifting to 1534 cm−1 as the pH decreases) and at 1369 cm−1 (shifting to 1366 cm−1), corresponding to δN–H, and νCNH, respectively.34,40,41 Furthermore, as the concentration of uranium increases in solution at pH 5, the absorption bands at 1529 cm−1, 1486 cm−1 and 1369 cm−1 shift to 1525 cm−1, 1490 cm−1 and 1365 cm−1, respectively, further supporting the binding of uranium to nitrogenous bases at higher concentrations.
Fig. 2 shows characteristic absorption bands in the spectral region between 1300 and 900 cm−1 as a function of pH and uranium concentration. Fig. 2A shows the effect of pH on the precipitates formed when precipitating eDNA with 0.5 mM uranium solution. Fig. 2B shows the effect of the precipitates formed when interacting 1 mg mL−1 eDNA with increasing uranium concentrations at pH 5.
 | ||
| Fig. 2 1300–900 cm−1 FTIR region of uranium binding to eDNA as a function of pH [A] and initial uranium concentration [B]. | ||
At 1230 cm−1, vibrational asymmetric stretching of phosphate is observed within the eDNA control spectra. A strong shift in the absorption band position was observed in the presence of uranium at different pH values (Fig. 2A). At pH 5 the absorption band is observed at 1218 cm−1 compared to pH 2 where it has changed to 1209 cm−1. A similar shift was observed in the complexes formed at increasing uranium concentration, Fig. 2B. In the presence of 0.25 mM uranium at pH 5, this absorption band shifted strongly to 1218 cm−1 and to 1206 cm−1 as the concentration increased to 2 mM. This change in the absorption band position was attributed to the weakening of the P–O bond due to uranium accumulation,28,42 indicating uranium binding to phosphate groups at relatively low concentrations of uranium. This was due to the electrostatic interactions between positively charged uranyl ions and negatively charged phosphate groups located on the surface of the eDNA. These results are in agreement with molecular dynamics simulation studies investigating the sensitivity of the asymmetric stretching of phosphate, where a red shift was observed for the vibrational frequency of the (PO2) groups.43,44 Similar shifts were also observed in the 2D infrared spectra of dioleoylphosphatidylcholine (DOPC) reverse micelles,43 supporting the hypothesis that uranyl ions are complexed with eDNA via electrostatic interactions.
A shift in the 1063 cm−1 absorption band of the eDNA control spectra, corresponding to the vibrational symmetric stretching of PO2 and the vibrations of C–OH, C–O–C, and C–C, was observed following uranium interactions with eDNA. This absorption band position shifts to 1060 cm−1 when the solution pH of the eDNA–uranium interaction experiment increases to 5,34Fig. 2A. This smaller shift in the absorption band position was most likely due to the lack of uranium interactions with C–OH, C–O–C and C–C from deoxyribose sugars.41
Changes in the absorption band position within the eDNA control spectra were observed at 965 cm−1 following uranium interactions with eDNA at pH 2–5. In the presence of uranium, the absorption bands shifted to 970 cm−1 for all pH values investigated.34,42 This change in the absorption band position was explained as U(VI) species binding to phosphate through the changes in the vibrational frequency of the P–O single bond. A shift in the absorption band position of this functional group suggested preferential binding to phosphate groups over a wide range of acidic pH. As the pH decreased, the highly mobile UO22+ U(VI) species would further bind to other functional groups within the eDNA, such as those present within the nitrogenous bases. Additional absorption bands were observed within the region of 928–918 cm−1 as the pH increased from 2 to 5 corresponding to the vibrations of the U–O ligand.45,46
3.3 The proposed precipitation of uranium on eDNA
Overall, FT-IR was found to be a suitable technique to investigate the interactions between eDNA and uranium as a function of pH and initial uranium concentration. The results suggest that at very low pH, coordination of the U(VI) species with eDNA occurs predominantly through interactions with phosphate groups and the nitrogenous bases. As the pH increased to 5, phosphate groups alone mediated U(VI) coordination at a starting concentration of 0.5 mM uranium. At low U(VI) concentration, i.e. 0.25 mM, coordination with eDNA occurred through the negatively charged groups of the deoxyribose-phosphate backbone. Rare earth metal recovery using salmon milt DNA indicated adsorption of dysprosium and lutetium through phosphate groups, as determined using EXAFS.33Coordination with other chemical moieties such as the N–H, CO and C–N of nitrogenous bases occurred more frequently as the uranium concentration increased, suggesting a preferential binding to phosphate groups until the bioavailability of these groups decreased due to uranium saturation. Thereafter, uranium accumulation would occur through less preferred functional groups. This has been demonstrated when investigating the binding of cadmium to DNA to elucidate the toxic effects of the metallic species. At low concentrations Cd2+ primarily interacted with phosphate groups, with further coordination with guanine and adenine as the Cd2+ concentration increased.34 Similar interactions were observed following single and double stranded DNA interactions with transition metal species. Transition metal species, including Mn2+ and Co2+, readily interacted with the N7 atom of guanine in dsDNA and phosphate groups, N1 and N7 atoms of purines and N3 atoms of pyrimidines of ssDNA, as well as other metal specific interactive sites.47 Furthermore, chelation of heavy metal species between phosphate groups and N7 atoms of purines has been proposed in the application of DNA-based biosensors for the determination of heavy metal species.39
The predominant interaction of U(VI) species with eDNA phosphate groups would depend on the net charge of the U(VI) species. The proposed U(VI) interactions with eDNA phosphate groups as a function of U(VI) species are shown in eqn (2). w is the overall net charge of the U(VI) species, y is the number of uranyl ions in the U(VI) molecule and Lz is the number of additional ligands, e.g. OH groups present in U(VI) species such as (UO2)3(OH)5+.
| w(eDNA–PO4−) + (UO2)y(Lz)w+ ⇌ w(eDNA–PO4)(UO2)y(Lz) | (2) |
U(VI) species with an overall net charge of +1 would interact with one phosphate group in the eDNA structure (Fig. 3A). U(VI) species with an overall net charge of +2 can interact with eDNA phosphate groups in more than one way; binding with two phosphate groups of multiple eDNA molecules (Fig. 3B) or with 2 phosphate groups of one eDNA molecule (Fig. 3C).
 | ||
| Fig. 3 U(VI) interactions with eDNA phosphate groups under acidic conditions. | ||
3.4 Uranium biomineralisation using enzymatically released phosphate
The formation of uranium phosphate minerals was identified as the primary mechanism for the removal of uranium from solution, due to the low solubility of uranium phosphate complexes. The stoichiometry of the reaction was studied using enzymatically-released phosphate from eDNA (ePO4) and uranyl ion at different molar ratios (Fig. 4). The experimental parameters were optimised for the maximum orthophosphate release for subsequent interactions with uranium, Fig. S3 (ESI†). Enzymatic phosphate (ePO4) was hydrolysed from 1 mg mL−1 eDNA using 2 units mL−1 acid phosphatase (0.1 M NaCl, pH 5) for 72 hours and interacted with uranium (pH 5) at various molar ratios. The ePO4 obtained using this method was comparable to the organically bound phosphate released by bacteria using indigenous acid phosphatase for the biomineralisation of uranium.21 | ||
| Fig. 4 Uranium and phosphate removal as a function of the H2PO4−:(UO2)3(OH)5+ molar ratio and phosphate origin. | ||
After 72 hours ∼2000 μM ePO4 was released from eDNA. The stoichiometry of the biomineralisation reaction using ePO4 and uranium was studied using varying molar ratios of both reactants. ePO4 and uranium were quantified to determine the amount remaining in solution and, by mass balance, the amount accumulated in the precipitates formed, Fig. 4. (UO2)3(OH)5+ was used as the predominant uranium species as calculated by Visual MINTEQ.48 The fraction of (UO2)3(OH)5+ was estimated from the total uranium quantified by ICP and the stoichiometry of the reaction was calculated assuming that the complexation reaction took place primarily with this species. These findings were compared with the formation of an abiotic precipitate (aPO4) in which the phosphate in solution originated from 2000 μM sodium phosphate (NaH2PO4). At pH 5, the dominant U(VI) species present within solution was (UO2)3(OH)5+ at concentrations higher than 0.25 mM U(VI). The dominant phosphate species at pH 5 was determined to be H2PO4− at approximately 86%, according to Visual MINTEQ calculations.48 The molar ratios of reactants were calculated accordingly using these dominant species, Fig. 4.
At a molar ratio of 1.8:1 H2PO4−:(UO2)3(OH)5+, uranium was the limiting reactant, and hence it was completely removed from solution and converted into a precipitate using both ePO4 and aPO4. At this molar ratio, 72% of the uranium was precipitated from solution using ePO4 and approximately 28% of unreacted uranium remained in solution, Fig. 4A. At a molar ratio of 3.8:1 H2PO4−:(UO2)3(OH)5+, almost all of both reactants were completely removed from solution, with approximately 6.5% of H2PO4− remaining in solution. This suggested that a molar ratio of approximately 3.5:1 (H2PO4−:(UO2)3(OH)5+) could precipitate all reactants from solution.
There were noticeable differences in the H2PO4− removal depending on the molar ratio and the H2PO4− source, Fig. 4B. At a molar ratio of 1.8:1 H2PO4−:(UO2)3(OH)5+, there were no ePO4 and aPO4 remaining in solution. In the presence of an excess of H2PO4− in solution, for example at a molar ratio of 37.6:1 H2PO4−:(UO2)3(OH)5+, only 45% of ePO4 was removed from solution. No differences in orthophosphate remaining in solution were observed when the concentration of uranium decreased in the starting solution. On the other hand, there was a relative decrease in the aPO4 removal up to a molar ratio of 150.4:1 H2PO4−:(UO2)3(OH)5+ where 96% aPO4 remained in solution following the biomineralisation of uranium. The results suggest that all the uranium is incorporated into the minerals formed, with an excess of orthophosphate liberated from eDNA for uranium biomineralisation and subsequent remediation, thus suggesting that uranium was the limiting factor in this reaction. Differences in phosphate precipitation between ePO4 and aPO4 suggested the presence of other particulates from the enzymatic hydrolysis of eDNA which may interact with reactants removing excess quantifiable phosphate.
3.5 Characterisation of uranium minerals
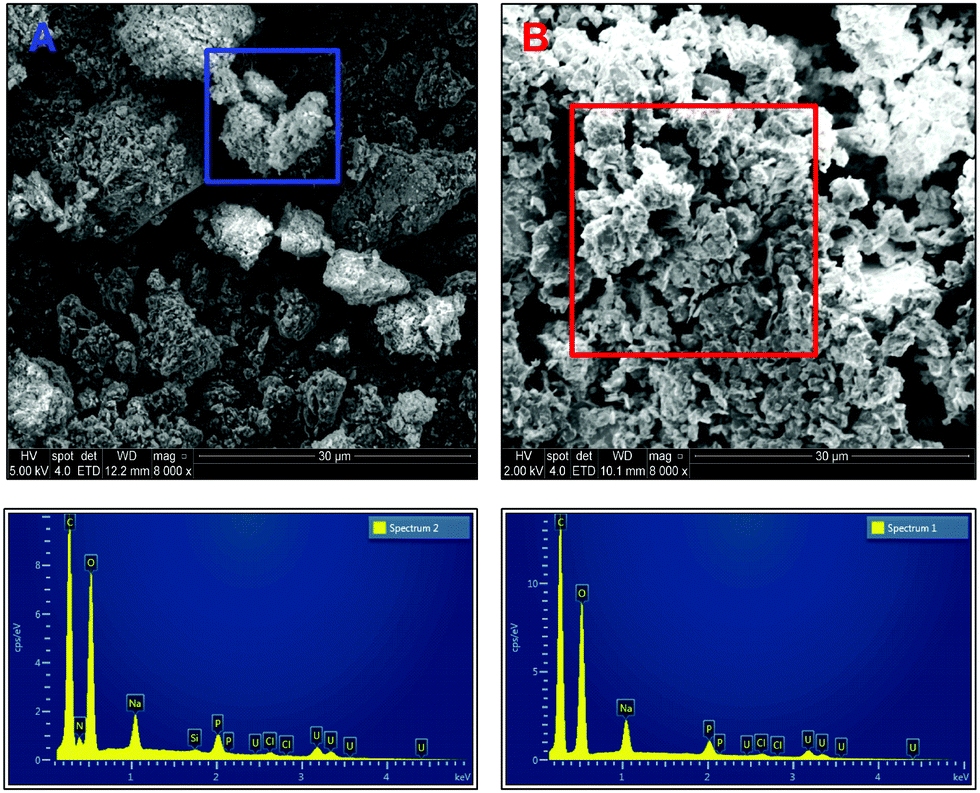 | ||
| Fig. 5 Scanning electron micrographs of carbon coated uranium precipitates using ePO4 [A] and aPO4 [B]. | ||
EDX analysis of particles and aggregates of both samples indicates similar chemical composition of the precipitates formed but with one noticeable difference, the presence of nitrogen within the precipitates formed with ePO4. This peak is absent in the EDX spectrum of the aPO4 precipitate. The presence of nitrogen in the ePO4 precipitate was due to impurities in the enzymatic phosphate following enzymatic hydrolysis of the eDNA in which the residual soluble components of the eDNA remain in solution. This could explain the differences in particle size, shape and aggregation between the two precipitates. Other impurities could include carbon from the enzymatic phosphate hydrolysis of eDNA. However, it was impossible to distinguish this incorporation due to the adherence of the samples to the carbon tape and subsequent coating.
:(UO2)3(OH)5+ were analysed by XRD (Fig. 6). The XRD patterns of all samples showed the same peaks when overlaying the patterns. This result revealed that they were the same uranium mineral phase at varying levels of crystallinity. At a molar ratio of 7.5:1, ePO4:uranium, the diffraction pattern showed poorly defined and broad peaks, suggesting an amorphous phase or a poorly crystalline material. As the initial concentration of uranium increased to a molar ratio of 1.8:1, the diffraction peaks were better defined with a greater intensity. This suggested an increased structural ordering and therefore a more crystalline material.49 The differences in crystallinity between the uranium precipitates formed using ePO4 or aPO4 could have been due to the incorporation of impurities from the ePO4 synthesis, such as nitrogen- and carbon-containing compounds arising from the enzymatic hydrolysis of eDNA.
 | ||
| Fig. 6 XRD patterns of uranium precipitates formed using aPO4 and ePO4 at varying molar ratios of H2PO4:(UO2)3(OH)5+. | ||
The XRD pattern of the aPO4 samples showed an increase in the intensity of the peaks. This suggested the formation of the same mineral phase, but with a higher degree of crystallinity. Similar results were obtained for the low uranium concentration samples where poor crystallinity shown by the poorly defined diffraction peaks was observed (Fig. S4, ESI†).
The XRD patterns and the corresponding d-spacing values obtained are comparable to those from minerals such as meta-autunite (Ca(UO2)2(PO4)2), (ICDD PDF 00-039-1351 and 04-012-5106) and other uranium phosphate hydrated minerals (ICDD PDF 00-037-0373 and 00-064-0815). However, in the absence of calcium in the sample, the calculated d-spacing values of 8.45, 4.91, 3.47 and 2.20 may be attributed to a uranium-phosphate mineral.
A high-resolution scan of the C1s region showed additional carbon-related peaks in the ePO4 minerals (Fig. 7) that were absent within the aPO4 minerals (Fig. S5, ESI†). Since there was no carbon present in the reagents for the abiotic reaction using uranium and aPO4, the carbon XPS peaks at 285, 286.49 and 288.88 eV (Table 2) were assigned to the carbon tape. This was confirmed by the spectral bands observed in carbon tape control (Fig. S6, ESI†). The C1s peak at 287.91 eV observed in the ePO4 mineral corresponded to a carbon oxygen double bond. Additional carbon satellite peaks were present at 293.24 and 295.95 eV. The peaks corresponding to carbon-related compounds in the ePO4 sample are from residual impurities remaining after the ePO4 synthesis.
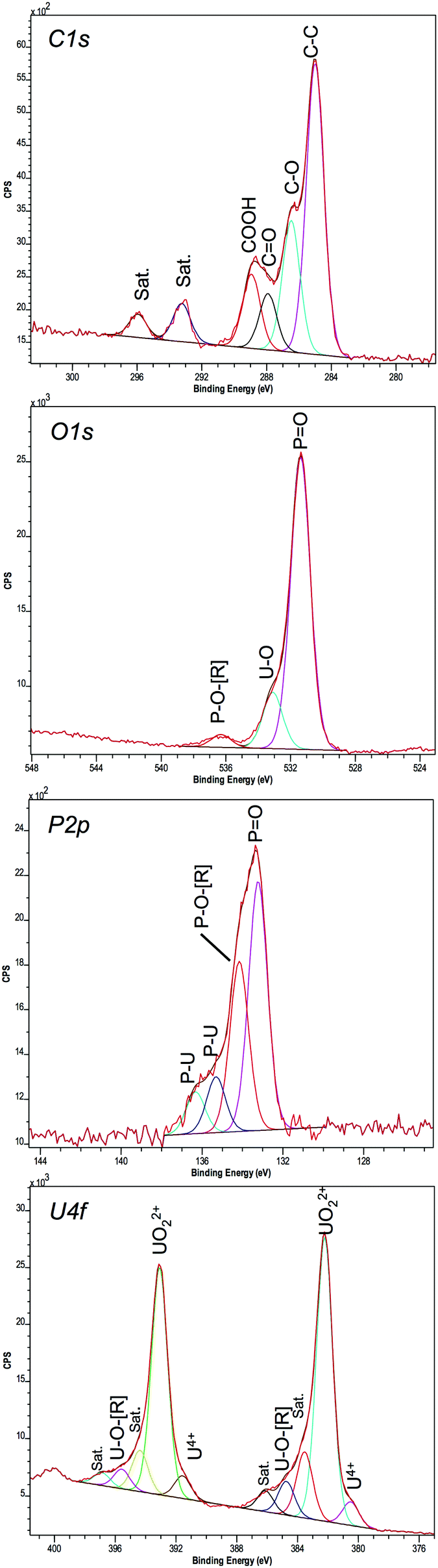 | ||
| Fig. 7 XPS high resolution spectra of uranium minerals synthesised using ePO4. | ||
| Binding energy (eV) | % area | Assignment | ||||
|---|---|---|---|---|---|---|
| ePO4 | aPO4 | U(VI) | ePO4 | aPO4 | U(VI) | |
| a Carbon tape associated peaks. | ||||||
| C 1s | ||||||
| 285 | 285 | 285 | 52.67 | 69.66 | 69.45 | C–C, Ha |
| 286.48 | 286.49 | 286.67 | 23.78 | 16.44 | 20.57 | C–Oa C–N |
| 287.91 | 10.26 | CO, O–C–O, COOR |
||||
| 288.93 | 288.88 | 289.23 | 13.28 | 13.9 | 9.98 | COOHa, O–CO |
| 293.24 | N/A | Satellite | ||||
| 295.95 | N/A | Satellite | ||||
| O 1s | ||||||
| 531.38 | 531.37 | 80.86 | 83.78 | PO, CO |
||
| 533.1 | 533.13 | 533.17 | 15.59 | 12.07 | 100 | U–O, P/C—OH |
| 536.29 | 536.35 | 3.56 | 4.15 | P–O–[R], U–[R] | ||
| P 2p | ||||||
| 133.23 | 133.3 | 48.26 | 64.55 | PO, P–OH |
||
| 134.16 | 134.19 | 32.86 | 35.45 | P–O–[R] | ||
| 135.31 | 10.8 | [R]–P–O–[R] | ||||
| 136.32 | 8.08 | [R]–P–O–[R] | ||||
| U 4f | ||||||
| 380.5 | 380.56 | 3.16 | 1.93 | U4+ | ||
| 382.21 | 382.25 | 382.99 | 37.86 | 42.47 | 42.61 | UO22+, U–O |
| 383.53 | 383.58 | 384.89 | 8.8 | 6.14 | 8.43 | U–O–[R], satellite |
| 384.73 | 384.81 | 386.7 | 4.49 | 3.73 | 5.83 | U–O–[R], satellite |
| 386.1 | 386.07 | 2.79 | 2.46 | Satellite | ||
| 391.56 | 391.77 | 3.01 | 2.66 | U4+ | ||
| 393.08 | 393.12 | 393.84 | 30.16 | 31.95 | 32.41 | UO22+, U–O |
| 394.37 | 394.38 | 395.68 | 5.47 | 4.43 | 6.27 | U–O–[R], satellite |
| 395.58 | 395.62 | 397.52 | 2.57 | 2.65 | 4.45 | U–O–[R], satellite |
| 396.87 | 396.89 | 1.7 | 1.57 | Satellite | ||
The U4f XPS spectra of uranium minerals synthesised using both ePO4 and aPO4 showed a peak separation of 10.87 eV between 4f7/2 and 4f5/2. This peak separation was in agreement with that of UO2(NO3)2·6H2O (Table 2) and that reported in the previous literature.50,51 Aside from the main XPS peaks, there were additional peaks 1.35–1.71 eV lower than and also up to 3.89 eV higher than the main 4f7/2 and 4f5/2 peaks. The additional peaks at higher energy are satellite peaks and arise from the interactions of photoelectrons with valence electrons during the photo-excitation process. The satellite binding energy is much more sensitive to the oxidation state of the uranium than that of the main U4f peaks.50,52
The U4f XPS region of UO2(NO3)2·6H2O, a U(VI) species, showed two additional peaks for both 4f7/2 and 4f5/2 peaks (Fig. S5, ESI†). Within both uranium minerals there are three additional peaks at higher energy for both 4f7/2 and 4f5/2 peaks. The highest additional peak (∼3.7 eV) is also a satellite peak indicating that the majority of the uranium in the mineral is in the oxidation state VI.53 The other two higher energy peaks (at approximately +1.3 and +2.5 eV higher than the binding energies of the two most intense peaks) could be either satellite peaks or due to the interaction of uranium with the phosphate. The latter hypothesis can be further supported by the additional XPS peaks within the spectrum of the P2p region for both samples, with further peaks observed in the ePO4 samples due to impurities.
Interestingly, the XPS results showed evidence that not all the uranium minerals formed using ePO4 and aPO4 contained the U(VI) oxidation state alone. The peaks at 380.56 and 391.6 eV do correspond to U(VI).53,54 However, using the U4f7/2 peak, the percentage of U as U(VI) (UO22+) is 92.3% and 7.7% as U(IV) (U4+) for ePO4 precipitates. These percentages change to 95.7% and 4.3% for the aPO4 precipitates. These peaks were not observed in the UO2(NO3)2·6H2O control, where only peaks attributable to U(VI) were present.
Overall, the XPS data of uranium minerals formed at a molar ratio of 1.8:1 H2PO4−:(UO2)3(OH)5+ using ePO4 and aPO4 suggested slight differences in the chemical nature. The presence of impurities of carbon and nitrogen on the ePO4 changed the mineral structure compared to the aPO4 formed mineral. Uranium minerals formed by bacteria outside and within the cell wall have been reported as meta-autunite-like minerals. For example, calcium- and sodium-bearing autunite minerals have been reported for certain strains of bacteria.8,20–22 Meta-autunite-like precipitates were identified for the marine bacterium Idiomarina loihiensis MAH1.35 The observation of nitrogen on uranium minerals formed by Bacillus thuringiensis was reported as nano-uramphite ((NH4)(UO2)PO4).55 A clear identification of these minerals was not possible due to the presence of impurities; however the mineral formed can be identified as a simple uranium phosphate mineral such as UO2HPO4. Further spectroscopic characterisation of the precipitates was investigated using ATR-FT-IR (Fig. S7, S8 and Table S2, ESI†) that confirms the formation of a uranium phosphate mineral of similar structure using enzymatic and abiotic phosphate.
3.6 The proposed interaction of ePO4 with U(VI)
Acid phosphatase (APase) hydrolyses phosphate esters from biological molecules.24 Between pH 5 and 7, approximately 2 mM ePO4 was hydrolysed from 1 mg mL−1 using 2 units mL−1 acid phosphatase. No ePO4 was released from eDNA at pH 4 and below due to a decrease in the acid phosphatase activity. This is similar to that of microbial acid phosphatase activity in which uranium biomineralisation does not occur at pH as low as 2 to 3.21,56 The proposed hydrolysis of ePO4 (predominantly in the form of H2PO4− at pH 5) from eDNA using acid phosphatase is shown in eqn (3)–(5).| (APase) + eDNA–PO4− + H2O ⇌ (APase)–eDNA–PO4− + H2O | (3) |
| (APase)–eDNA–PO4− + H2O ⇌ (APase)–PO3− + eDNA–O + H2O | (4) |
| (APase)–PO3− + eDNA–O + H2O ⇌ (APase) + eDNA–O + H2PO4− | (5) |
The stoichiometric data obtained from the interactions of ePO4 with uranium at varying molar ratios indicated that approximately a 3.5:1 H2PO4−:(UO2)3(OH)5+ molar ratio was responsible for the removal of reactants from solution. All of the uranium (in the form of (UO2)3(OH)5+) was precipitated using approximately 94% of ePO4 at an initial molar ratio of 3.8:1 in the starting solution, Fig. 4. XRD analysis of the uranium minerals formed indicated that they were of uranium phosphate origin. The uranium phosphate hydrated mineral (UO2HPO4)·xH2O (ICDD PDF# 00-037-0373) displayed similar diffraction patterns to both ePO4 and aPO4 minerals. Therefore, we propose that the biomineralisation of uranium with ePO4 occurs as described in eqn (6). This proposed biomineralisation reaction is based on a molar ratio of 3:1 H2PO4−:(UO2)3(OH)5+. The difference in the proposed molar ratio and the recorded molar ratio (3.5:1) is probably due to an overestimation of the ePO4 removed by uranium from solution. Phosphate is also removed from solution by impurities from the organic material and nitrogen-containing compounds from the enzymatic hydrolysis of eDNA which can complex with the phosphate and remove it from solution.
| 3H2PO4− + (UO2)3(OH)5+ + 2H+ ⇌ 3UO2HPO4·1.67H2O | (6) |
Enzymatic phosphate released from microbial acid phosphatase has been associated with uranium biomineralisation through the formation of uranium phosphate bearing minerals (Martinez et al., 2007). These include the precipitation of simple crystalline uranium phosphate compounds such as (UO2)3(PO4)2·H2O,28 chernikovite (H2(UO2)2(PO4)2)57 and HUO2PO4·4H2O.20 The incorporation of other elements into uranium phosphate minerals has been documented for numerous strains of bacteria in the precipitation of minerals such as meta-autunite (Ca(UO2)2(PO4)2)20,21 and nano-uramphite ((NH4)(UO2)PO4).55
4 Conclusions
The present work reveals the mechanism whereby extracellular DNA (i) complexes uranium and (ii) mineralises uranium from solution. The complexation and precipitation of eDNA with uranium are mainly mediated by negatively charged phosphate groups within the eDNA structure, with a higher percentage of eDNA complexed with uranium at very low pH. This was due to an increased availability of phosphate groups on the eDNA and the reactivity of uranium species. The reaction depends on both pH and uranium concentration. Acid phosphatase hydrolysed phosphate from eDNA making it available for uranium biomineralisation. The biominerals formed during this study were of phosphate nature and were similar to their abiotic proxies but contained a fraction of organic components that made them less crystalline in their structure. At circumneutral pH, the enzymatic release of phosphate from acid phosphatase activity resulted in the formation of uranium phosphate-like mineral phases in which the uranium oxidation state was mainly VI. The structure of the mineral obtained was similar to that of uranium phosphate-bearing minerals such as (UO2HPO4)·xH2O. The stoichiometry of the reaction was 3:1 H2PO4−:(UO2)3(OH)5+ and the reaction occurred under mild acidic conditions (pH 5). The results demonstrated that DNA can mineralize uranium from solution via complexation and precipitation after DNA-phosphate hydrolysis. Therefore DNA could be considered a major contributor to the biomineralisation of uranium within bacterial EPS. The results presented here demonstrate that phosphate-rich bio-macromolecules affect the behaviour of uranium in the natural environment.
Acknowledgements
The authors thank the EPSRC consortium “Hard-soft matter interfaces: from understanding to engineering” (EP/I001514/1) for financial support during the experimental study. Further thanks go to Dr Deborah Hammond and Dr Mathew Pringle (The University of Sheffield) for help with XPS data acquisition and interpretation.References
- J. R. Lloyd and J. C. Renshaw, Curr. Opin. Biotechnol., 2005, 16, 254–260 CrossRef CAS PubMed.
- M. L. Merroun and S. Selenska-Pobell, J. Contam. Hydrol., 2008, 102, 285–295 CrossRef CAS PubMed.
- L. Newsome, K. Morris and J. R. Lloyd, Chem. Geol., 2014, 363, 164–184 CrossRef CAS.
- S. Choudhary and P. Sar, Rev. Environ. Sci. Bio/Technol., 2015, 14, 347–355 CrossRef CAS.
- E. Krawczyk-Baersch, K. Grossmann, T. Arnold, S. Hofmann and A. Wobus, Geochim. Cosmochim. Acta, 2008, 72, 5251–5265 CrossRef CAS.
- J. A. Finlay, V. J. M. Allan, A. Conner, M. E. Callow, G. Basnakova and L. E. Macaskie, Biotechnol. Bioeng., 1999, 63, 87–97 CrossRef CAS PubMed.
- M. L. Merroun, C. Hennig, A. Rossberg, T. Reich and S. Selenska-Pobell, Radiochim. Acta, 2003, 91, 583–591 CrossRef CAS.
- F. Jroundi, M. L. Merroun, J. M. Arias, A. Rossberg, S. Selenska-Pobell and M. T. Gonzalez-Munoz, Geomicrobiol. J., 2007, 24, 441–449 CrossRef CAS.
- M. Marvasi, P. T. Visscher and L. C. Martinez, FEMS Microbiol., 2010, 313, 1–9 CrossRef CAS PubMed.
- J. Wingender, T. R. Neu and H. C. Flemming, Microbial Extracellular Polymeric Substances: Characterization, Structure, and Function, Springer, Berlin, 1999 Search PubMed.
- R. E. Steinberger and P. A. Holden, Appl. Environ. Microbiol., 2005, 71, 5404–5410 CrossRef CAS PubMed.
- J. H. Priester, S. G. Olson, S. M. Webb, M. P. Neu, L. E. Hersman and P. A. Holden, Appl. Environ. Microbiol., 2006, 72, 1988–1996 CrossRef CAS PubMed.
- S. Vilain, J. M. Pretorius, J. Theron and V. S. Brozel, Appl. Environ. Microbiol., 2009, 75, 2861–2868 CrossRef CAS PubMed.
- K. M. Nielsen, P. J. Johnsen, D. Bensasson and D. Daffonchio, Environ. Biosaf. Res., 2007, 6, 37–53 CrossRef CAS PubMed.
- D. J. Levy-Booth, R. G. Campbell, R. H. Gulden, M. M. Hart, J. R. Powell, J. N. Klironomos, K. P. Pauls, C. J. Swanton, J. T. Trevors and K. E. Dunfield, Soil Biol. Biochem., 2007, 39, 2977–2991 CrossRef CAS.
- M. Allesen-Holm, K. B. Barken, L. Yang, M. Klausen, J. S. Webb, S. Kjelleberg, S. Molin, M. Givskov and T. Tolker-Nielsen, Mol. Microbiol., 2006, 59, 1114–1128 CrossRef CAS PubMed.
- T. Das, S. Sehar and M. Manefield, Environ. Microbiol. Rep., 2013, 5, 778–786 CrossRef CAS PubMed.
- J. R. Battista, Annu. Rev. Microbiol., 1997, 51, 203–224 CrossRef CAS PubMed.
- K. Matsui, N. Ishii and Z. Kawabata, Appl. Environ. Microbiol., 2003, 69, 2399–2404 CrossRef CAS PubMed.
- L. E. Macaskie, K. M. Bonthrone, P. Yong and D. T. Goddard, Microbiology, 2000, 146, 1855–1867 CrossRef CAS PubMed.
- M. L. Merroun, M. Nedelkova, J. J. Ojeda, T. Reitz, M. Lopez Fernandez, J. M. Arias, M. Romero-Gonzalez and S. Selenska-Pobell, J. Hazard. Mater., 2011, 197, 1–10 CrossRef CAS PubMed.
- R. J. Martinez, M. J. Beazley, M. Taillefert, A. K. Arakaki, J. Skolnick and P. A. Sobecky, Environ. Microbiol., 2007, 12, 9 Search PubMed.
- T. Sousa, A. P. Chung, A. Pereira, A. P. Piedade and P. V. Morais, Metallomics, 2013, 5, 390–397 RSC.
- J. B. Vincent, M. W. Crowder and B. A. Averill, Trends Biochem. Sci., 1992, 17, 105–110 CrossRef CAS PubMed.
- L. E. Macaskie, R. M. Empson, A. K. Cheetham, C. P. Grey and A. J. Skarnulis, Science, 1992, 257, 782–784 CAS.
- A. Koban, G. Geipel, A. Rossberg and G. Bernhard, Radiochim. Acta, 2004, 92, 903–908 CrossRef CAS.
- G. E. Pinchuk, C. Ammons, D. E. Culley, S. W. Li, J. S. Mclean, M. F. Romine, K. H. Nealson, J. K. Fredrickson and A. S. Beliaev, Applied and environmental microbiology, 2008, 74, 1198–1208 CrossRef CAS PubMed.
- S. Choudhary and P. Sar, J. Hazard. Mater., 2011, 186, 336–343 CrossRef CAS PubMed.
- X. Lu, X.-J. Zhou and T.-S. Wang, J. Hazard. Mater., 2013, 262, 297–303 CrossRef CAS PubMed.
- M. Alrakabi, G. Singh, A. Bhalla, S. Kumar, A. Srivastava, B. Rai, N. Singh, J. S. Shahi and D. Mehta, J. Radioanal. Nucl. Chem., 2012, 294, 221–227 CrossRef CAS.
- K. Belitz, M. S. Fram and T. D. Johnson, Environ. Sci. Technol., 2015, 49, 8330–8338 CrossRef CAS PubMed.
- Z. Q. He and C. W. Honeycutt, Commun. Soil Sci. Plant Anal., 2005, 36, 1373–1383 CrossRef CAS.
- Y. Takahashi, K. Kondo, A. Miyaji, Y. Watanabe, Q. Fan, T. Honma and K. Tanaka, PLoS One, 2014, 9, e114858 Search PubMed.
- S. Alex and P. Dupuis, Inorg. Chim. Acta, 1989, 157, 271–281 CrossRef CAS.
- F. Morcillo, M. T. Gonzalez-Munoz, T. Reitz, M. E. Romero-Gonzalez, J. M. Arias and M. L. Merroun, PLoS One, 2014, 9, e91305 Search PubMed.
- S. K. Kazy, P. Sar and S. F. D'Souza, Biorem. J., 2008, 12, 47–57 CrossRef CAS.
- A. C. C. Plette, W. H. Vanriemsdijk, M. F. Benedetti and A. Vanderwal, J. Colloid Interface Sci., 1995, 173, 354–363 CrossRef CAS.
- P. G. Burnett, H. Heinrich, D. Peak, P. J. Bremer, A. J. McQuillan and C. J. Daughney, Geochim. Cosmochim. Acta, 2006, 70, 1914–1927 CrossRef CAS.
- S. S. Babkina and N. A. Ulakhovich, Anal. Chem., 2005, 77, 5678–5685 CrossRef CAS PubMed.
- A. Cecal, D. Humelnicu, V. Rudic, L. Cepoi, D. Ganju and A. Cojocari, Bioresour. Technol., 2012, 118, 19–23 CrossRef CAS PubMed.
- S. J. Parikh and J. Chorover, Langmuir, 2006, 22, 8492–8500 CrossRef CAS PubMed.
- A. A. Kamnev, L. P. Antonyuk, L. Y. Matora, O. B. Serebrennikova, M. V. Sumaroka, M. Colina, M. F. Renou-Gonnord and V. V. Ignatov, J. Mol. Struct., 1999, 481, 387–393 CrossRef.
- R. Costard, I. A. Heisler and T. Elsaesser, J. Phys. Chem. Lett., 2014, 5, 506–511 CrossRef CAS PubMed.
- D. J. Floisand and S. A. Corcelli, J. Phys. Chem. Lett., 2015, 6, 4012–4017 CrossRef CAS PubMed.
- G. J. Lumetta, B. K. McNamara, B. M. Rapko and J. E. Hutchison, Inorg. Chim. Acta, 1999, 293, 195–205 CrossRef CAS.
- S. K. Kazy, S. F. D'Souza and P. Sar, J. Hazard. Mater., 2009, 163, 65–72 CrossRef CAS PubMed.
- J. G. Duguid, V. A. Bloomfield, J. M. Benevides and G. J. Thomas, Biophys. J., 1995, 69, 2623–2641 CrossRef CAS PubMed.
- J. P. Gustafsson, Visual MINTEQ (v3.1) Search PubMed, http://vminteq.lwr.kth.se.
- S. Handley-Sidhu, J. A. Hriljac, M. O. Cuthbert, J. C. Renshaw, R. A. D. Pattrick, J. M. Charnock, B. Stolpe, J. R. Lead, S. Baker and L. E. Macaskie, Environ. Sci. Technol., 2014, 48, 6891–6898 CrossRef CAS PubMed.
- R. Asuvathrarnan, K. I. Gnanasekar, P. C. Clinsha, T. R. Ravindran and K. V. G. Kutty, Ceram. Int., 2015, 41, 3731–3739 CrossRef.
- J. H. Liu, S. Van den Berghe and M. J. Konstantinovic, J. Solid State Chem., 2009, 182, 1105–1108 CrossRef CAS.
- S. Kushwaha, B. Sreedhar and P. Padmaja, Langmuir, 2012, 28, 16038–16048 CrossRef CAS PubMed.
- P. Benard, D. Louer, N. Dacheux, V. Brandel and M. Genet, Chem. Mater., 1994, 6, 1049–1058 CrossRef CAS.
- G. C. Allen, P. M. Tucker and J. W. Tyler, J. Phys. Chem., 1982, 86, 224–228 CrossRef CAS.
- X. H. Pan, Z. Chen, F. B. Chen, Y. J. Cheng, Z. Lin and X. Guan, J. Hazard. Mater., 2015, 297, 313–319 CrossRef CAS PubMed.
- T. Reitz, A. Rossberg, A. Barkleit, S. Selenska-Pobell and M. L. Merroun, PLoS One, 2014, 9, e102447 Search PubMed.
- M. J. Beazley, R. J. Martinez, S. M. Webb, P. A. Sobecky and M. Taillefert, Geochim. Cosmochim. Acta, 2011, 75, 5648–5663 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional ATR-FT-IR spectra, enzymatic orthophosphate hydrolysis results, XRD patterns, XPS spectra and raw data. See DOI: 10.1039/c6cp03239g |
| This journal is © the Owner Societies 2016 |