
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the calculation of equilibrium thermodynamic properties from molecular dynamics
Peter V.
Coveney
* and
Shunzhou
Wan
Centre for Computational Science, Department of Chemistry, University College London, London WC1H 0AJ, UK. E-mail: p.v.coveney@ucl.ac.uk; Tel: +44 (0)2076791560
First published on 3rd May 2016
Abstract
The purpose of statistical mechanics is to provide a route to the calculation of macroscopic properties of matter from their constituent microscopic components. It is well known that the macrostates emerge as ensemble averages of microstates. However, this is more often stated than implemented in computer simulation studies. Here we consider foundational aspects of statistical mechanics which are overlooked in most textbooks and research articles that purport to compute macroscopic behaviour from microscopic descriptions based on classical mechanics and show how due attention to these issues leads in directions which have not been widely appreciated in the field of molecular dynamics simulation.
Introduction
There are a few fundamental questions in science. While the discussion of and attempted resolution of these questions are of widespread interest, not only to scientists but also to the general public, they rarely appear to affect the day to day work of jobbing scientists.One such question concerns the nature of time, in particular how to make sense of the fact that while the microscopic description of matter is time-reversal symmetric, macroscopic behaviour is temporally asymmetric.† Thermodynamic systems are observed to approach equilibrium; they do not retreat from it. Conventional attempts to explain this time asymmetry frequently proceed from the paradoxical assumption that the reversible microscopic world, which we cannot directly apprehend, is the true one; while the observable irreversible macroscopic world is an illusion, however persistent. That such an interpretation does not stand up to scientific scrutiny has been pointed out by numerous authors of scientific and philosophical works.1–7
In short, if one regards the concept of equilibrium as being as valid as the quantum mechanical or classical description of the molecules comprising the systems governed by thermodynamics, then we are obliged to reconcile these seemingly incompatible representations of matter.
Dynamical systems, ergodic theory and the approach to equilibrium
In this article, we shall only consider classical systems. Our aim is to discuss the use of classical molecular dynamics for the determination of macroscopic properties, in particular the Gibbs free energy for systems at equilibrium. While the formulation of classical mechanics typically presented in expositions of molecular dynamics found in textbooks of molecular dynamics is based on Newton's equations of motion (and their equivalent Hamiltonian and Lagrangian formulations), there is a further probabilistic representation to which relatively scant regard is paid. However, it is important to always keep this representation in mind, especially when considering statistical mechanics. Instead of working with Newton's equations of motion, in the probabilistic representation we employ the Liouville equation| ∂tρt = Lρt | (1) |
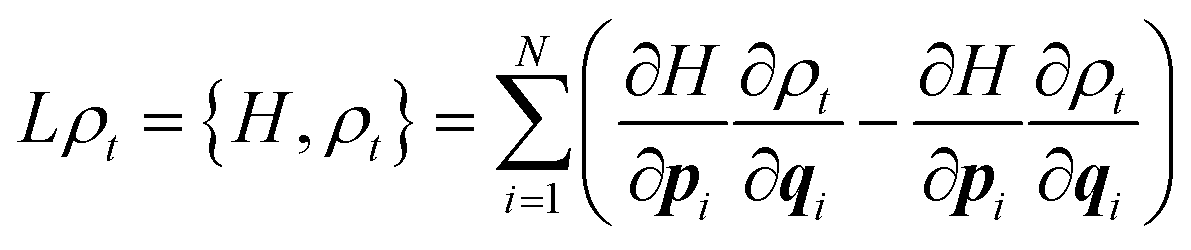 | (2) |
Macroscopic averages are given by ensemble averages of dynamical observables, G, denoted 〈G〉t. Thus
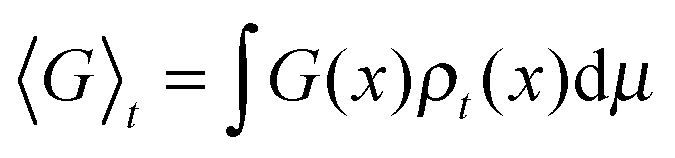 | (3) |
Despite the frequent assertion that the Newtonian, trajectory based, formulation and the probabilistic description are equivalent, the two representations of dynamics cannot always be used interchangeably. In particular, this is the case in discussing the approach to equilibrium and, indeed, the very concept of equilibrium itself. To see this, one can use the inverse Laplace transform to show that the solution of the Liouville equation may be written as
 | (4) |
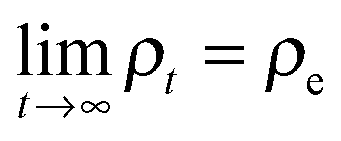 | (5) |
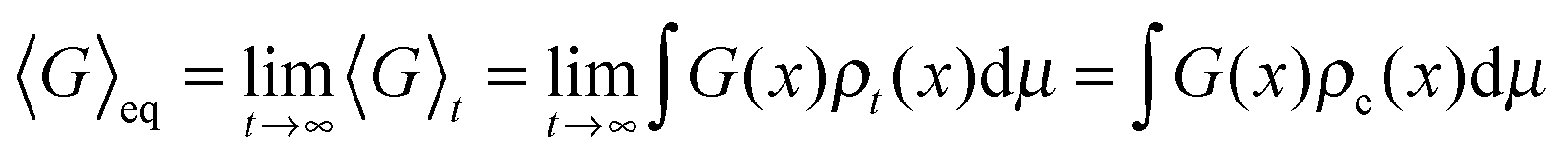 | (6) |
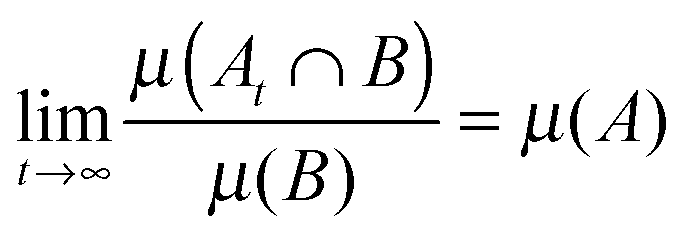 | (7) |
Mixing systems are one class of dynamical systems in the classification provided by ergodic theory8,9 rooted in the probabilistic description of dynamical systems governed by the Liouville equation. Ergodic theory has grown out of the relationship between the global properties of dynamical systems and statistical mechanics.10–12 Unfortunately, due to the level of mathematical rigour involved in the subject, it has become rather detached from workaday research in statistical mechanics and computer simulation. In a nutshell, the ergodic hierarchy embraces (i) ergodic; (ii) mixing; (iii) Kolmogorov; and (iv) Bernoulli dynamical systems, whose distinct properties are governed by the nature of the spectrum of L. These systems display an increasing level of dynamical instability, each implying the properties of the level before it, but not being implied by it.
In many textbooks on statistical mechanics and molecular simulation, attention is focused on ergodicity to provide the link between dynamics and thermodynamic equilibrium. Ergodic systems are ones that pass through every available point in phase space (given sufficient time), but ergodicity is not a sufficient condition: such systems do not display an approach to equilibrium. A greater degree of instability in the dynamics is required. As noted above, mixing systems are ergodic and they also display an approach to equilibrium. Kolmogorov and Bernoulli systems manifest even greater dynamical instabilities than mixing flows; they too display an approach to equilibrium and are, of course, also ergodic.
The key point is this. From ergodic theory, we know that if a system is going to exhibit an equilibrium state, it must be at least mixing. There is, however, an important additional requirement for these properties to be relevant to statistical mechanics. That is, in thermodynamics we are dealing with systems with very large numbers of atoms and molecules. In particular, statistical mechanics works because, in the thermodynamic limit (large N and large volume V, such that the density (N/V) is constant), ensemble averages converge to the same behaviour as the average properties of a single system, since the fluctuations become negligible.§
Classical molecular dynamics
As noted, many textbooks and research papers in the domain of interest here pay no attention to the foregoing discussion. Instead, they advocate the calculation of “statistical averages” from molecular dynamics by reference to the “ergodic theorem” (and the associated “ergodic hypothesis”). In short, they assume that it is possible to perform a sufficiently long single microscopic trajectory calculation such that, in the long time limit, one may replace the ensemble averages 〈G〉 which are fundamental in statistical mechanics by time averages: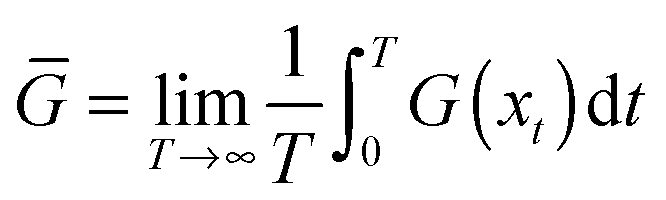 | (8) |
![[G with combining overline]](https://www.rsc.org/images/entities/i_char_0047_0305.gif) . Thus, for example, one reads in Understanding molecular simulation that molecular dynamics is concerned with time averaging.13 The problem with eqn (8), however, is obvious. The equation is valid in the infinite time limit; in practice this has been taken to mean that one runs a simulation for as long as is possible, computes its time averaged properties, and then claims that the values reported represent the macroscopic thermodynamic properties of the system. More fundamentally, such an approach to computing macroscopic averages lacks the generality one seeks in statistical mechanics. It is manifestly restricted to the case of systems in equilibrium; it cannot be employed for non-equilibrium systems where the system's properties depend explicitly on time.
. Thus, for example, one reads in Understanding molecular simulation that molecular dynamics is concerned with time averaging.13 The problem with eqn (8), however, is obvious. The equation is valid in the infinite time limit; in practice this has been taken to mean that one runs a simulation for as long as is possible, computes its time averaged properties, and then claims that the values reported represent the macroscopic thermodynamic properties of the system. More fundamentally, such an approach to computing macroscopic averages lacks the generality one seeks in statistical mechanics. It is manifestly restricted to the case of systems in equilibrium; it cannot be employed for non-equilibrium systems where the system's properties depend explicitly on time.
What is more or less universally ignored is the requirement, if an equilibrium state is to be reached, that the probabilistic dynamics must be at least mixing. Neighbouring trajectories in the “underlying” phase space diverge exponentially fast. A single trajectory will never capture such behaviour since on the Newtonian trajectory level of representation each instant in time is equivalent to every other. Indeed, in this representation the very concept of equilibrium has no meaning.
Rather than pursue such a tenuous approach to determine equilibrium properties based on single trajectories—no matter how “long” they be—we prefer to embrace the tenets of statistical mechanics and directly compute ensemble averages by running sufficiently large numbers of replicas of the system of interest.
None of the foregoing theory tells us anything about whether the systems under investigation in molecular dynamics simulation fulfil the mixing or higher instability properties in the ergodic hierarchy. Nor does that theory say anything about the rate at which equilibrium is approached, or the number of replicas one needs to include in an ensemble. As regards the first of these questions, it is known that rigorous proofs aiming to show that real systems reside in a specific class within the ergodic hierarchy are almost impossible to establish; but since these properties are the ones known to guarantee equilibrium behaviour, one must assume them to be at least mixing unless they are proved not to be so.¶ It is the central purpose of non-equilibrium statistical mechanics to calculate kinetic descriptions that describe the rate at which equilibrium is attained.11 However, the existing rigorous theories are again restricted to idealized and often abstract dynamical systems.14–16 Both the first and the second question therefore need to be investigated by computational means. And this is precisely the programme we have pursued.
The ensemble approach to free energy determination using molecular dynamics computer simulation
Although the use of ensemble simulations in molecular dynamics was first proposed some time ago,17,18 the approach was not systematically investigated. We focus here on the calculation of the Gibbs free energy of binding of a ligand to a protein in water. Assuredly these systems display experimentally observed equilibrium states, and one can measure the Gibbs free energy of binding. It follows from this that the system, if faithfully described by classical molecular dynamics, is at least a mixing system with trajectories that diverge exponentially in time, no matter how close they are initially.We start, therefore, from a suitably selected ensemble of initial conditions, working within the probabilistic interpretation of the dynamical behaviour. As noted earlier, statistical mechanical theory provides no guide as to how large such ensembles should be, while we must also keep an eye on the thermodynamic limit. Starting, for example, from an initial structure provided by X-ray diffraction, we build models with the ligand docked into the active site of the protein. Once the models are constructed and ready for molecular dynamics analysis, a sufficiently large number of replicas are created, which differ only by the random number seed that assigns them the differing velocities of all the atoms at the initial time when the simulations kick off. Two issues arise. First, how many replicas are required, and second what role is played by the thermodynamic limit? The answer to the first is established by numerical simulation; the thermodynamic limit is simply attained by the same process – a macroscopic system of this kind would just have of order one mole of ligand and protein present. Since there are no significant interactions between the individual ligand–protein complexes, we can access the thermodynamic limit directly by aggregating the properties of the replicas in the ensemble.
Our extensive studies of numerous protein–ligand systems show that such ensembles require no more than 4 nanosecond duration simulation and no fewer than 25 replicas in order to return errors on the free energy predictions of around 1 kcal mol−1 or less. The free energies are computed by two classes of method, known as ESMACS19,20 and TIES.20,21 One can control the errors in these free energy calculations through a combination of the duration in time of each molecular dynamics simulation and the number of replicas used. Errors at this level, which for one of our methods of free energy determination (TIES) are certainly less than experimental values, are also reproducible. Accuracy, precision and reproducibility of binding free energy predictions are all hallmarks of these ensemble based methods, while being notoriously elusive in the history of the use of molecular dynamics to predict these quantities.
What emerges from these studies is that the properties one computes from individual molecular dynamics trajectories are well described by Gaussian random processes. That is, they can be thought of as stochastic processes with mean and standard deviation conforming to a Gaussian distribution (Fig. 1a).9 This in turn makes it possible to reinterpret numerous equations and calculational routes in statistical mechanics as stochastic integrals (of which just one example are the equations for relative free energy changes in terms of thermodynamic integration over alchemically combined pairs of distinct molecular states), thereby allowing us to draw on the theory of stochastic calculus to compute relevant properties reliably.9
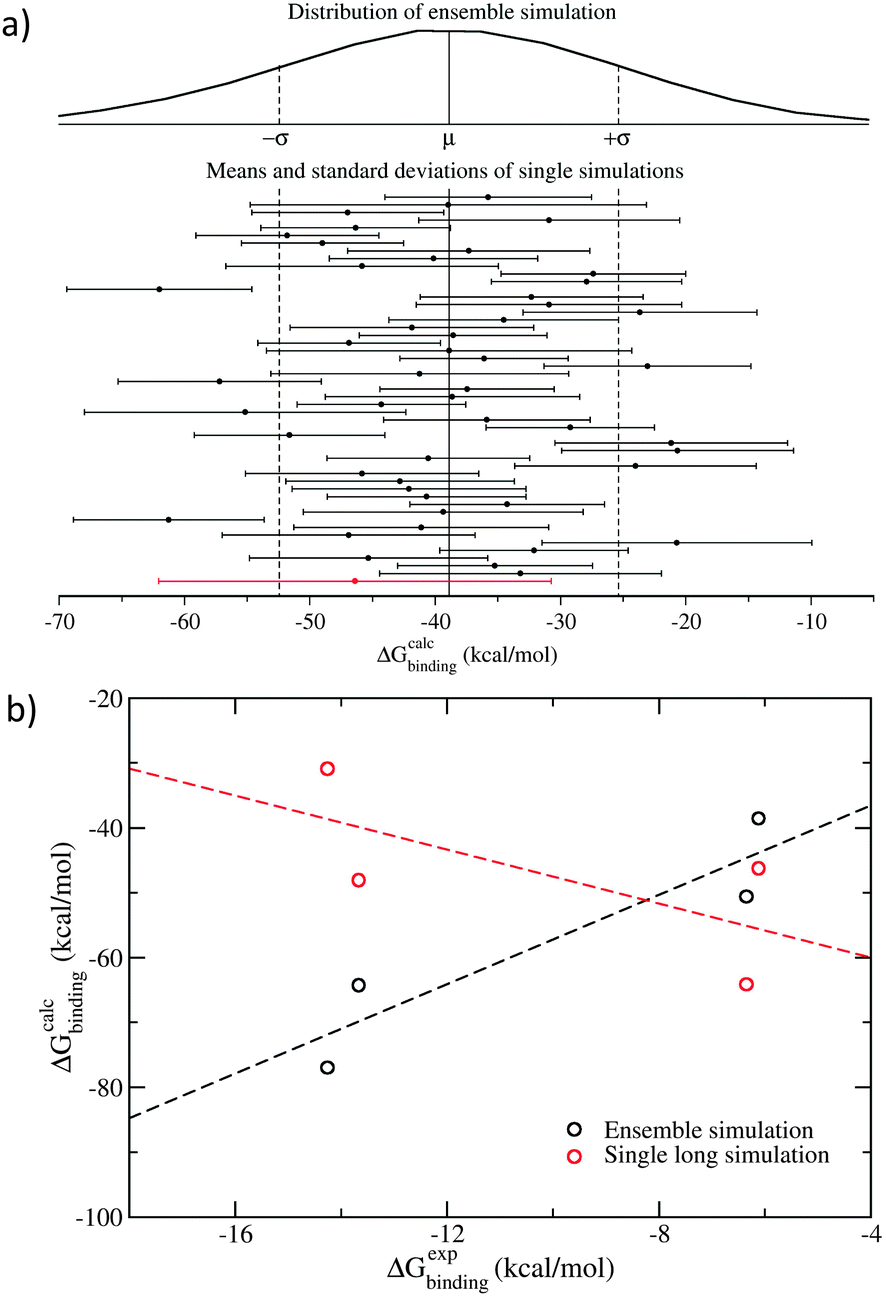 | ||
| Fig. 1 Comparison of ensemble and single trajectory simulations of four peptide–MHC complexes. Results for one peptide sequence, AAAKTPVIV (9 amino acid residues in single-character notation), are shown here as an example. All others exhibit similar behaviour. (a) The Gaussian distribution of free energies from ensemble simulation, comprising 50 independent replicas each of 4 ns duration, has a larger spread than from any individual replica; single microsecond trajectories (red line), five times longer than the aggregated length of all replicas in the ensemble, behave like 4 ns replicas.19 (b) The ensemble simulations correctly predict the ranking of the binding energies for the four peptides considered (black circles and dashed line), while single long trajectories produce rankings which are anti-correlated with those observed (red circles and dashed line). | ||
The use of these ensemble simulations can be compared to what happens when one performs a “long time duration” single trajectory simulation. Our work demonstrates compellingly that such long simulations (in cases we have studied, these have been run for up to a couple of microseconds or longer) produce predictions that are no better than those of a single replica taken from a 25-member ensemble, even though the former is several times longer in temporal duration than the entire ensemble simulation (Fig. 1).19,22 Our free energy studies show that binding affinities obtained from two independent simulations of the same molecular system, differing only in initially assigned atomic velocities, can vary by up to 10 kcal mol−1 in the small molecule–protein complexes,23,24 and by up to 43 kcal mol−1 in the peptide–MHC (major histocompatibility complex) systems.19 These variations are much larger than the experimentally observed maximum binding free energy difference of the inhibitors under investigation.
We use bootstrapping analysis to quantify convergence and deduce errors in these ensemble simulations for a given number of replicas and simulation duration.24 A very large number of bootstrap samples are generated, drawing with replacement from the population of the original data set. The standard deviation of the bootstrap samples provides an estimate of the error in the original data set. The magnitude of the estimated error decreases by increasing the number of replicas in an ensemble simulation, directly providing information on convergence of the results.
Thus, the success of such ensemble based simulation studies is due to a highly effective exploration of the relevant parts of phase space. One can of course envisage situations when there may be multiple minima pertaining to different ligand–protein bound states, or even simply a number of separate rotameric states of (say) the ligand, which may not be accessible from the initial ensemble within the timeframe of 4 ns. The absence of active–inactive transitions and the lack of sampling of inactive conformations do not make a significant contribution to the free energies of binding. If such transitions (translating into so-called “on” and “off” rates) and additional bound states are of interest, then one must ensure they are targeted by inclusion of ensembles of initial conditions that guarantee there is an appreciable probability for these processes to occur. One-off (single replica) simulations do not have any reliability owing to the random nature of molecular dynamics trajectories.
When the kinetics of conformational transitions and/or ligand binding is of interest, much longer molecular simulations, usually on the microsecond timescale,25,26 will be required than those recommended in our studies for equilibrium binding affinities. The kinetic information, as well as equilibrium properties, can be reconstructed from studies with so-called “enhanced sampling techniques” which are usually based on tempering the system, modifying the underlying potential energy surface, or a combination of both. Such enhanced sampling methods may be required to obtain relevant metastable states for computing equilibrium properties if barriers are sufficiently high between these states. However, attempts made to report kinetics from single simulations are likely to be just as error prone as single simulation based attempts to calculate free energies. Extreme caution must be exercised in seeking to make general inferences about a system's behaviour based on these, no matter how long the simulated time is reported to be.
Previous molecular dynamics studies have demonstrated that protein systems can get trapped in one or a few conformations even in a relatively long time duration simulation. The epidermal growth factor receptor (EGFR), for example, may25 or may not25,27 overcome the energy barrier between its active and inactive states in the course of molecular dynamics simulations on a time scale of 10 μs or longer. Even if the transition does occur, a probabilistic description cannot be meaningfully constructed because of the rare nature of the events concerned.
The increasing speed of modern computers is largely determined by the number of cores and accelerators available on them. This development in computer architectures is well aligned with the need articulated here to perform ensemble-based molecular dynamics studies since, in the elapsed time needed to perform one such simulation, we can do all of them if the machine is large enough. The ability to perform such calculations fast – in a few hours – and reliably will have an important impact on the use of such methods in areas such as drug discovery and clinical decision making.20,28
Conclusions
We started by recalling known properties of dynamical systems which approach and reside in equilibrium states. The central property, largely overlooked in discussions of statistical mechanics and molecular simulation methods designed to compute equilibrium properties from the microscopic level, is that of “mixing” in the ergodic hierarchy which classifies the types of global behaviours leading to thermodynamic equilibrium states under dynamical evolution.Basing the determination of equilibrium properties on ergodic theory in this manner ensures that we compute ensemble-averaged behaviour in a manner which is truly consistent with the methods of statistical mechanics. We indeed find that neighbouring trajectories diverge very rapidly in phase space. Properties derived from these show all the hallmarks of Gaussian random processes.
Calculating macroscopic properties in this manner from classical molecular dynamics provides the means to report reproducible results including errors intrinsic to the method. The errors are under the direct control of the person performing the calculations through the choice of number of replicas and duration of the molecular dynamics simulation time.
In several respects, this approach to molecular dynamics calculations is analogous to weather forecasting. To predict tomorrow's weather today, meteorologists do not run a single simulation. They recognise that they will never know exactly the initial conditions that are required to perform a single fluid dynamics based calculation, in a system of Navier–Stokes equations known to exhibit dynamical chaos. So they perform many simulations with slightly varying initial conditions—i.e. they compute the behaviour of ensembles—in order to make reliable probabilistic predictions of what the weather will look like the next day. Speed too is of the essence here as it is there. Just as the public wishes to know tomorrow's weather today, not in three weeks time, for the application of free energy calculations in drug development and clinically based personalised medicine28 we must be able to deliver accurate and reliable results within hours to drive decision making.
Acknowledgements
The authors thank EPSRC for its support via the 2020 Science Programme (EP/I017909/1), the EU Framework Programme 7 p-medicine (ICT-2009.5.3), the EU Horizon 2020 ComPat project (671564), the VPH-SHARE (FP7-ICT 269978) projects, the Qatar National Research Fund (7-1083-1-191), MRC for a Medical Bioinformatics Grant (MR/L016311/1), and funding from the UCL Provost. We made use of ARCHER to which access was provided through the EPSRC 2020 Science programme, and SuperMUC to which access was funded by the Gauss Centre for Supercomputing.Notes and references
- P. V. Coveney and R. Highfield, The arrow of time: a voyage through science to solve time's greatest mystery, W. H. Allen, London, 1990 Search PubMed.
- P. Baert, Time in contemporary intellectual thought, North Holland, Amsterdam, 1st edn, 2000 Search PubMed.
- S. Hawking, A brief history of time: from the big bang to black holes, Bantam Books, Toronto, 1988 Search PubMed.
- O. Penrose, Rep. Prog. Phys., 1979, 42, 1937–2006 CrossRef CAS.
- R. Penrose, The road to reality: a complete guide to the laws of the universe, Jonathan Cape, London, 2004 Search PubMed.
- I. Prigogine, From being to becoming: time and complexity in the physical sciences, W. H. Freeman, San Francisco, 1980 Search PubMed.
- H. Reichenbach, The direction of time, University of California Press, Berkeley, 1956 Search PubMed.
- R. Frigg, J. Berkovitz and F. Kronz, in The Stanford Encyclopedia of Philosophy, ed. E. N. Zalta, Summer 2014 edn, 2014 Search PubMed.
- A. Klenke, Probability theory: a comprehensive course, Springer, London, 2008 Search PubMed.
- V. I. Arnold and A. Avez, Ergodic problems of classical mechanics, Benjamin, New York, 1968 Search PubMed.
- R. Balescu, Equilibrium and nonequilibrium statistical mechanics, Wiley, New York, 1975 Search PubMed.
- D. Ruelle, Thermodynamic formalism: the mathematical structure of equilibrium statistical mechanics, Cambridge University Press, Cambridge, 2nd edn, 2004 Search PubMed.
- D. Frenkel and B. Smit, Understanding molecular simulation: from algorithms to applications, Academic Press, San Diego, 2nd edn, 2002, p. 17 Search PubMed.
- P. V. Coveney and O. Penrose, J. Phys. A: Math. Gen., 1992, 25, 4947–4966 CrossRef.
- O. Penrose and P. V. Coveney, Proc. R. Soc. A, 1994, 447, 631–646 CrossRef.
- P. V. Coveney and A. K. Evans, J. Stat. Phys., 1994, 77, 229–258 CrossRef.
- A. Elofsson and L. Nilsson, J. Mol. Biol., 1993, 233, 766–780 CrossRef CAS PubMed.
- L. S. D. Caves, J. D. Evanseck and M. Karplus, Protein Sci., 1998, 7, 649–666 CrossRef CAS PubMed.
- S. Wan, B. Knapp, D. W. Wright, C. M. Deane and P. V. Coveney, J. Chem. Theory Comput., 2015, 11, 3346–3356 CrossRef CAS PubMed.
- S. Wan, A. P. Bhati, S. J. Zasada and P. V. Coveney, Rapid, accurate, precise and reproducible ligand-protein binding free energy prediction, 2016 Search PubMed , submitted.
- T. D. Bunney, S. Wan, N. Thiyagarajan, L. Sutto, S. V. Williams, P. Ashford, H. Koss, M. A. Knowles, F. L. Gervasio, P. V. Coveney and M. Katan, EBioMedicine, 2015, 2, 194–204 CrossRef PubMed.
- S. K. Sadiq, D. W. Wright, O. A. Kenway and P. V. Coveney, J. Chem. Inf. Model., 2010, 50, 890–905 CrossRef CAS PubMed.
- S. Wan and P. V. Coveney, J. R. Soc., Interface, 2011, 8, 1114–1127 CrossRef CAS PubMed.
- D. W. Wright, B. A. Hall, O. A. Kenway, S. Jha and P. V. Coveney, J. Chem. Theory Comput., 2014, 10, 1228–1241 CrossRef CAS PubMed.
- Y. B. Shan, A. Arkhipov, E. T. Kim, A. C. Pan and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7270–7275 CrossRef CAS PubMed.
- A. C. Pan, D. W. Borhani, R. O. Dror and D. E. Shaw, Drug Discovery Today, 2013, 18, 667–673 CrossRef CAS PubMed.
- S. Wan, D. W. Wright and P. V. Coveney, Mol. Cancer Ther., 2012, 11, 2394–2400 CrossRef CAS PubMed.
- D. W. Wright, S. Wan, N. Shublaq, S. J. Zasada and P. V. Coveney, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2012, 4, 585–598 CrossRef CAS PubMed.
Footnotes |
| † We are excluding the strange decays of the long-lived kaon, which violate time-reversal symmetry. |
| ‡ The computable numbers form a set of zero measure so it is possible that computer generated molecular dynamics trajectories will behave pathologically. |
| § An important issue here is whether or not the limits t → ∞ and N → ∞ commute, which is assumed here. There is good reason to believe that they do not commute for some systems. One way in which the commutativity of these limits can break down is if the system becomes trapped in a “metastable” state and stays there for a very long time (cf. the later discussion on metastable states). |
| ¶ There exist dynamical systems that are neither ergodic nor integrable, the so-called non-integrable systems, to which the KAM theorem is applicable. However, the general view on these systems, to which we subscribe, is that the measure of phase space in which regular motion takes place tends to zero in the thermodynamic limit; at which point these systems are all members of the ergodic hierarchy with instabilities at least of the mixing variety. |
| This journal is © the Owner Societies 2016 |