
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic evidence for the formation of pentalene+ in the dissociative ionization of naphthalene†
Jordy
Bouwman
*a,
Arjen J.
de Haas
a and
Jos
Oomens
ab
aRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7c, NL-6525 ED Nijmegen, The Netherlands. E-mail: j.bouwman@science.ru.nl
bvan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, NL-1098 XH Amsterdam, The Netherlands
First published on 5th January 2016
Abstract
Although acetylene loss is well known to constitute the main breakdown pathway of polycyclic aromatic hydrocarbon (PAH) species, the molecular structure of the dissociation products remains only poorly characterized. For instance, the structure of the C8H6 product ion formed upon acetylene loss from the smallest PAH naphthalene (C10H8) has not been experimentally established. Several C8H6+ isomers are conceivable, including phenylacetylene, benzocyclobutadiene, pentalene as well as a number of a-cyclic products. Here we present infrared (IR) spectroscopic evidence for the formation of the (anti-aromatic) pentalene structure using a combination of tandem mass spectrometry and IR laser spectroscopy. The formation of pentalene is suggestive of facile 6- to 5-membered ring conversion, which possibly has implications for the PAH/fullerene interrelationship in energetic settings such as the interstellar medium and combustion environments.
Polyaromatic hydrocarbon (PAH) structures are perfused throughout chemistry and play a pivotal role in regions where energetic processing occurs, such as combustion chemistry and interstellar chemistry.1–4 The breakdown chemistry upon radiative or collisional activation has been subject of a large number of studies.5–12 For gaseous PAHs, various mass-spectrometric studies have shown that loss of one or more C2H2-units constitutes the major pathway for breakdown of the hexagonal carbon framework of catacondensed PAHs.9–12 However, these MS-based studies do not provide structural information on the fragmentation products, leaving mechanistic details of the breakdown chemistry largely unknown. We do not know whether the hexagonal framework remains largely intact with ethynyl group(s) on the periphery, or whether more substantial rearrangements take place. This study addresses this question for the smallest PAH naphthalene.
The dissociative ionization of naphthalene has received considerable attention in theoretical as well as experimental studies, as the unimolecular C2H2-loss is prototypical for PAH fission.9,10,13–17 In an attempt to elucidate the molecular structure of the C8H6+ product formed in the electron impact ionization of naphthalene, Schroeter et al.15 employed a variety of mass spectrometric methods. Based on their charge reversal (CR) data they dispute the formation of the phenylacetylene cation (PA+) and suggest that the benzocyclobutadiene cation (BCB+) is formed, as was also suggested by Ling et al.14 However, the possible formation of other structures is not excluded.15 To date, no unambiguous identification of these reaction products has been reported.
High-level quantum-chemical computations have been performed on the C10H8+ potential energy surface (PES).16–18 A summary of the PES reported by Dyakov et al.17 computed at the G3(MP2,CC)//B3LYP level of theory showing only the highest transition state barriers referenced to the naphthalene cation is shown in Fig. 1. Solano and Mayer19 determined product branching fractions for the dissociation of the naphthalene cation from ab initio calculations in conjunction with Rice–Ramsperger–Kassel–Marcus (RRKM) calculations and predicted that the pentalene cation (PE+) is the main C2H2-loss product at low internal energies of the naphthalene cation, followed by PA+ and BCB+. The contributions of PA+ and BCB+ grow as the internal energy of the naphthalene cation rises. These predictions await experimental verification.
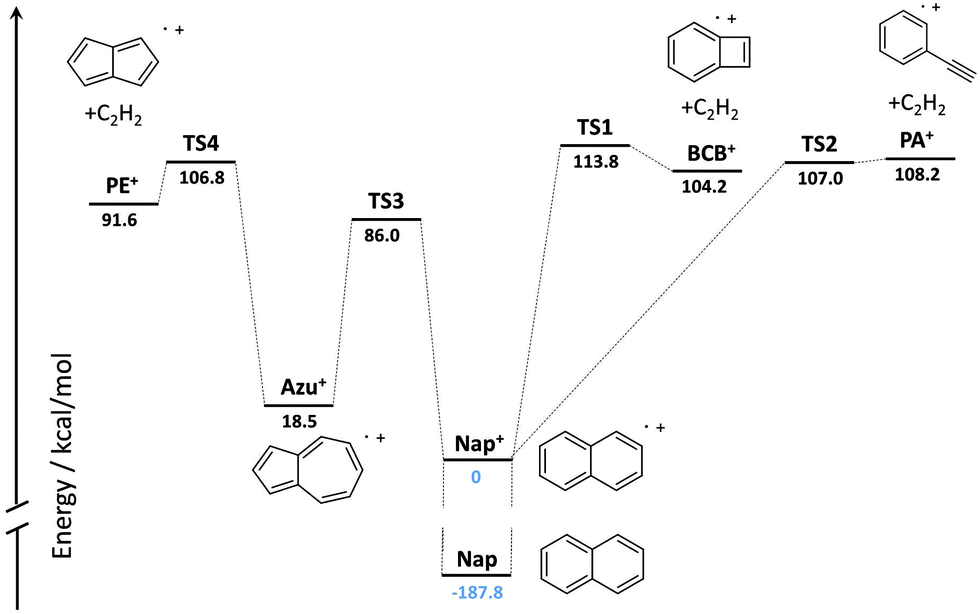 | ||
| Fig. 1 Summary of the possible C2H2 loss channels on the C10H8+ potential energy surface constructed from the data reported in Dyakov et al.17 | ||
West et al.12 studied the dissociation of naphthalene employing both tandem mass spectrometry and imaging photoelectron photoion coincidence spectrometry (iPEPICO). From their measurements they found that H-loss and C2H2-loss are the lowest energy dissociation channels. iPEPICO measurements have recently also been reported on quinoline and isoquinoline dissociation and it was suggested that either PA+, or BCB+ are the HCN-loss products that are formed from their dissociative ionization.20 An unambiguous identification of the C8H6+ isomer is however not possible from these experiments.
Here, we apply IR spectroscopy to identify the structure of the C8H6+ product that is formed in the dissociative ionization of naphthalene. The measurements have been performed in an ion trap mass spectrometer coupled to the free electron laser for infrared experiments (FELIX). A detailed description of the instrument is given in the ESI,† with the accompanying references.21–26 Briefly, the C8H6+ dissociation product is formed by two-photon dissociative ionization of naphthalene vapor at 193 nm (ArF wavelength) and mass isolated in a Paul type quadrupole ion trap by means of a tailored RF (SWIFT) pulse.24,23 The IR spectrum of the mass-isolated product ion is then recorded by infrared multiphoton dissociation (IRMPD) spectroscopy.27
Fig. 2 shows the mass spectrum of dissociatively ionized naphthalene (top), together with that of the subsequently mass-isolated m/z = 102 fragment ion (middle). The fragment signal is significantly enhanced when helium buffer gas is admitted to the trap at a pressure of 5 × 10−5 mbar. Furthermore, the SWIFT pulse that is used to isolate the fragment ion excites the secular frequency of the naphthalene cation, causing further collisional activation and an enhancement of the integrated fragment ion intensity by a factor of two. The mass spectrum of the C8H6+ fragment upon resonant IR irradiation at 1350 cm−1 is shown in the bottom panel. IR-induced dissociation yields weak but clearly observable signals at m/z = 76 and 50, corresponding to C6H4+ and C4H2+ fragment ions that are formed by C2H4 and C4H4-loss, respectively.
 | ||
| Fig. 2 Mass spectra of naphthalene dissociatively ionized at 193 nm (top trace), the SWIFT isolated fragment at m/z = 102 (middle trace) and the SWIFT isolated fragment dissociated with intense IR radiation at 1350 cm−1. | ||
The infrared spectrum of the isolated C8H6+ ion is constructed by plotting the fragmentation yield in the mass spectrum as a function of IR laser wavelength (see Fig. 3). The fragmentation yield is determined as the integrated fragment ion intensity divided by the total integrated ion intensity, i.e. the sum of the fragment and parent ion intensity. Also shown in Fig. 3 are B3LYP/6-311+G(d,p) computed infrared spectra of three C8H6+ isomers, PE, BCB and PA. Calculated frequencies are scaled by a factor of 0.9679 to correct for anharmonicities.26 The computed stick spectrum is convolved with a 30 cm−1 Full Width at Half Maximum (FWHM) Gaussian profile to facilitate comparison with the experimental spectrum.
 | ||
| Fig. 3 Infrared multiphoton dissociation spectrum of C8H6+ formed spectrum of C8H6+ formed from the dissociative ionization of naphthalene (top) shown together with the computed spectra of the C8H6+ isomers pentalene, benzocyclobutadiene and phenylacetylene. | ||
Predicted spectra of the three isomers that are possibly formed exhibit vibrational modes that are very diagnostic. Clearly, the computed spectrum of PE+ resembles the experimental spectrum closely. The main discrepancy is the computed absorption band at ∼1460 cm−1 that is observed only very weakly in the experimental spectrum. This is possibly due to the non-linear nature of the IRMPD technique, although an incorrect prediction of the band intensity is also possible. A weak absorption band is observed in the experimental spectrum between 1600 and 1700 cm−1 and is possibly caused by a small fraction of the product ions having the BCB+ isomeric structure, which is predicted to possess an absorption band in this wavelength range (C–C stretch of the aromatic ring), however, none of the other strong modes for BCB+ are observed. Hence, PE+ is identified as the dominant naphthalene dissociation product.
The detection of PE+ as a product of the dissociative ionization of naphthalene can be rationalized based on the summary of the C10H8+ PES shown in Fig. 1. The formation of PE+ occurs after isomerization of naphthalene+ to azulene+. The energy available for isomerization in the experiment reported here is 2 × 193 nm (295.8 kcal mol−1) less the ionization energy of naphthalene (187.8 kcal mol−1),28 which equals 108.0 kcal mol−1. This energy would be barely sufficient to cross the rate limiting transition state (TS4) that leads to PE+, but not yet sufficient to cross the barrier to form PA+. Collisions with helium induced by driving the secular frequency of the metastable naphthalene ion likely gives the ions just enough energy to dissociate. The rate limiting transition state to the formation of BCB+ is slightly higher than the barriers leading to PE+ and PA+. This hypothesis is supported by the RRKM model of Solano and Mayer,19 who predict PE+ to be the dominant product ion at low internal energies of the naphthalene cation. Future measurements using a F2 excimer laser (157 nm) or electron gun to induce ionization may raise the internal energy of the parent ion above the barriers leading to BCB+ and PA+ and will be used to investigate their formation.
The identification of PE+ as a product of the dissociative ionization strongly suggests that naphthalene undergoes isomerization to azulene prior to dissociation, thereby providing the first experimental evidence for 5/7 membered ring isomerization to occur upon energetic excitation. Such isomerization reactions may be prototypical for large catacondensed (irregularly-shaped) interstellar PAHs upon C2H2-loss. Similar rearrangements forming 5/7-membered ring defects in the 6-membered ring honeycomb structure, referred to as Stone–Wales isomerizations, have been suggested to occur upon energizing pericondensed PAHs.29,30 However, these isomerizations in pericondensed PAHs may be out-competed by H-loss reactions that typically exhibit a lower barrier.31 Experiments on larger PAHs exhibiting C2H2-loss are in progress to verify whether this mechanism can be established in these larger catacondensed PAH species as well.
JB acknowledges the Netherlands Organisation for Scientific Research (Nederlandse Organisatie voor Wetenschappelijk Onderzoek, NWO) for a VENI grant (grant number 722.013.014). JO acknowledges NWO for a VICI grant (grant number 724.011.002). This work was further supported by NWO Exacte Wetenschappen (Physical Sciences) for the use of the supercomputer facilities at SurfSara. The authors gratefully thank the FELIX laboratory staff for the beamtime and for their support in the construction, modification, and commissioning of the ion trap apparatus.
References
- A. Leger and J. L. Puget, Astron. Astrophys., 1984, 137, L5–L8 CAS.
- L. J. Allamandola, A. Tielens and J. R. Barker, Astrophys. J., 1985, 290, L25–L28 CrossRef CAS.
- H. Richter and J. B. Howard, Prog. Energy Combust. Sci., 2000, 26, 565–608 CrossRef CAS.
- A. Tielens, Interstellar polycyclic aromatic hydrocarbon molecules, Annual Reviews, Palo Alto, 2008, vol. 46, pp. 289–337 Search PubMed.
- S. J. Pachuta, H. I. Kenttamaa, T. M. Sack, R. L. Cerny, K. B. Tomer, M. L. Gross, R. R. Pachuta and R. G. Cooks, J. Am. Chem. Soc., 1988, 110, 657–665 CrossRef CAS.
- W. D. Cui, B. Hadas, B. P. Cao and C. Lifshitz, J. Phys. Chem. A, 2000, 104, 6339–6344 CrossRef CAS.
- M. J. Dibben, D. Kage, J. Szczepanski, J. R. Eyler and M. Vala, J. Phys. Chem. A, 2001, 105, 6024–6029 CrossRef CAS.
- J. Zhen, P. Castellanos, D. M. Paardekooper, H. Linnartz and A. G. G. M. Tielens, Astrophys. J., Lett., 2014, 797, L30 CrossRef CAS.
- H. W. Jochims, E. Ruhl, H. Baumgartel, S. Tobita and S. Leach, Astrophys. J., 1994, 420, 307–317 CrossRef CAS.
- S. P. Ekern, A. G. Marshall, J. Szczepanski and M. Vala, J. Phys. Chem. A, 1998, 102, 3498–3504 CrossRef CAS.
- H. A. B. Johansson, H. Zettergren, A. I. S. Holm, N. Haag, S. B. Nielsen, J. A. Wyer, M. B. S. Kirketerp, K. Stochkel, P. Hvelplund, H. T. Schmidt and H. Cederquist, J. Chem. Phys., 2011, 135, 084304 CrossRef CAS PubMed.
- B. West, C. Joblin, V. Blanchet, A. Bodi, B. Sztaray and P. M. Mayer, J. Phys. Chem. A, 2012, 116, 10999–11007 CrossRef CAS PubMed.
- G. Granucci, Y. Ellinger and P. Boissel, Chem. Phys., 1995, 191, 165–175 CrossRef CAS.
- Y. Ling, J. M. L. Martin and C. Lifshitz, J. Phys. Chem. A, 1997, 101, 219–226 CrossRef CAS.
- K. Schroeter, D. Schroder and C. Schwarz, J. Phys. Chem. A, 1999, 103, 4174–4181 CrossRef CAS.
- W. J. van der Hart, Int. J. Mass Spectrom., 2002, 214, 269–275 CrossRef CAS.
- Y. A. Dyakov, C. K. Ni, S. H. Lin, Y. T. Lee and A. M. Mebel, Phys. Chem. Chem. Phys., 2006, 8, 1404–1415 RSC.
- Y. A. Dyakov, C.-K. Ni, S. H. Lin, Y. T. Lee and A. M. Mebel, J. Phys. Chem. A, 2005, 109, 8774–8784 CrossRef CAS PubMed.
- E. A. Solano and P. M. Mayer, J. Chem. Phys., 2015, 143, 104305 CrossRef PubMed.
- J. Bouwman, B. Sztaray, J. Oomens, P. Hemberger and A. Bodi, J. Phys. Chem. A, 2015, 119, 1127–1136 CrossRef CAS PubMed.
- J. Oomens, A. J. A. van Roij, G. Meijer and G. von Helden, Astrophys. J., 2000, 542, 404–410 CrossRef CAS.
- W. Paul, Rev. Mod. Phys., 1990, 62, 531–540 CrossRef CAS.
- S. Guan and A. G. Marshall, Int. J. Mass Spectrom. Ion Processes, 1996, 157–158, 5–37 CrossRef.
- V. M. Doroshenko and R. J. Cotter, Rapid Commun. Mass Spectrom., 1996, 10, 65–73 CrossRef CAS PubMed.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CAS.
- M. P. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- N. C. Polfer and J. Oomens, Phys. Chem. Chem. Phys., 2007, 9, 3804–3817 RSC.
- NIST, NIST Chemistry WebBook, National Institute of Standards and Technology, Gaithersburg MD, 20899, 2009, vol. NIST Standard Reference Database Number 69.
- A. Ricca, C. W. Bauschlicher, Jr. and L. J. Allamandola, Astrophys. J., 2011, 729, 94 CrossRef.
- S. Öttl, S. E. Huber, S. Kimeswenger and M. Probst, Astron. Astrophys., 2014, 568, A95 CrossRef.
- A. I. S. Holm, H. A. B. Johansson, H. Cederquist and H. Zettergren, J. Chem. Phys., 2011, 134, 044301 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A detailed description of the experimental apparatus. See DOI: 10.1039/c5cc10090a |
| This journal is © The Royal Society of Chemistry 2016 |