
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceα-Methylphenacyl thioesters as convenient thioacid precursors†
Toru
Hatanaka
a,
Ryosuke
Yuki
a,
Ryota
Saito
ab and
Kaname
Sasaki
*a
aDepartment of Chemistry, Toho University, 2-2-1 Miyama, Funabashi 274-8510, Japan. E-mail: kaname.sasaki@sci.toho-u.ac.jp
bResearch Center for Materials with Integrated Properties, Toho University, 2-2-1 Miyama, Funabashi, Chiba 274-8510, Japan
First published on 25th October 2016
Abstract
α-Methylphenacyl (Mpa) thioesters are described as precursors of thioacids. Mpa thioesters are accessible via the condensation of carboxylic acids and phenacyl thiol, which is easily prepared without column chromatography. The Mpa thioesters are selectively deprotected by reduction with zinc dust in the presence of conventional thioacid protecting groups. In addition, the Mpa group exhibits orthogonal reactivity to the Boc group. These features are expected to facilitate the preparation of complex thioacids, including those in peptides.
The thioacid functional group, due to its unique reactivity, can be an expedient handle by which to convergently and chemoselectively install modification modules, particularly in peptides.1 For example, thioacid-based amide ligation reactions relying on unprotected aziridines,2 electron-deficient azides,3 electron-deficient arylsulfonamides,4 isonitriles,5 and isocyanates6 have been reported. Furthermore, condensation reactions between thioacids and amines have also been described recently, employing the Sanger and Mukaiyama reagents,7 HOBt,8 organonitrite,9 Cu(II),10 HOBt-Cu(OAc)2,11 and N,O-bis(trimethylsilyl)acetamide as activators.12
A promising approach to preparing peptide thioacids is to carry thioesters through the peptide elongation sequence and then release the thioacids by reliable and simple reactions. Currently, several thioesters are available as thioacid precursors, including 2,4,6-trimethoxybenzyl (Tmob),3 9-fluorenylmethyl (Fm),13 trityl (Trt),14 and 2-cyanoethyl,15 which release thioacids under either basic or acidic conditions. As for C-terminal peptide thioacids, other approaches were reported, which utilize N–S acyl transfer16–18 or acyl hydrazines.19 We were interested in a different type of precursor that would permit a wider range of applications for thioacid chemistry, and herein, report a novel thioacid precursor that has, notably, reactivity orthogonal to the Boc group. As thioesters are not generally stable to nucleophilic amines such as piperidine, Boc-based elongation would be the first choice for preparing thioacid-bearing peptides. Therefore, thioacid precursors with reactivity orthogonal to the Boc group could be useful for the preparation of peptide thioacids. In this context, we focused on phenacyl groups. As a protective group for carboxylic acids, phenacyl esters have been used due to their stability under acidic conditions and removal by one-electron reduction in mildly acidic media.20 However, phenacyl esters are known to be relatively electrophilic and susceptible to undesired transesterification. Therefore, we prepared phenacyl thiols 3a–c, which vary in bulkiness (Scheme 1), to examine the stabilities of their thioesters. Each variant was prepared from the commercially available phenacyl bromides 1a–c by a two-step sequence of substitution with thioacetate to give 2a–c, followed by deacetylation. These are all easily scalable spot-to-spot reactions, and afford phenacylthiol 3a, α-methylphenacylthiol 3b, and α,α-dimethylphenacylthiol 3c quantitatively, without the need for column chromatography.
 | ||
| Scheme 1 Preparation of thiols 3a–c and thioesters 4a–c. | ||
Each thiol was efficiently coupled with Boc-protected tryptophan using standard coupling procedures in the presence of DMAP.21 The lower yield in the preparation of 4a is due to the partial loss of 3a observed during silica gel column chromatography. Thioesters 4a–c were subjected to reducing conditions using zinc dust in aqueous acetic acid as a benchmark (Table 1). The efficiency in liberating the thioacid was evaluated after conversion to the corresponding benzyl thioester 5, because thioacids generally have two tautomeric forms (O- and S-acids) that complicate spectroscopic analysis.22 All three substrates were deprotected to give the corresponding thioacid. The reaction rate clearly depended on the bulkiness of the phenacyl moiety. The bulkier thioester 4c required a longer reaction time than 4a and 4b, even though the intermediate phenacyl radical of 4c would presumably be more stable than those of the others. Elevating the reaction temperature shortened the reaction time and improved the yield. We considered the α-methylphenacyl group, abbreviated as Mpa, to be a good entry as a precursor for thioacids.23

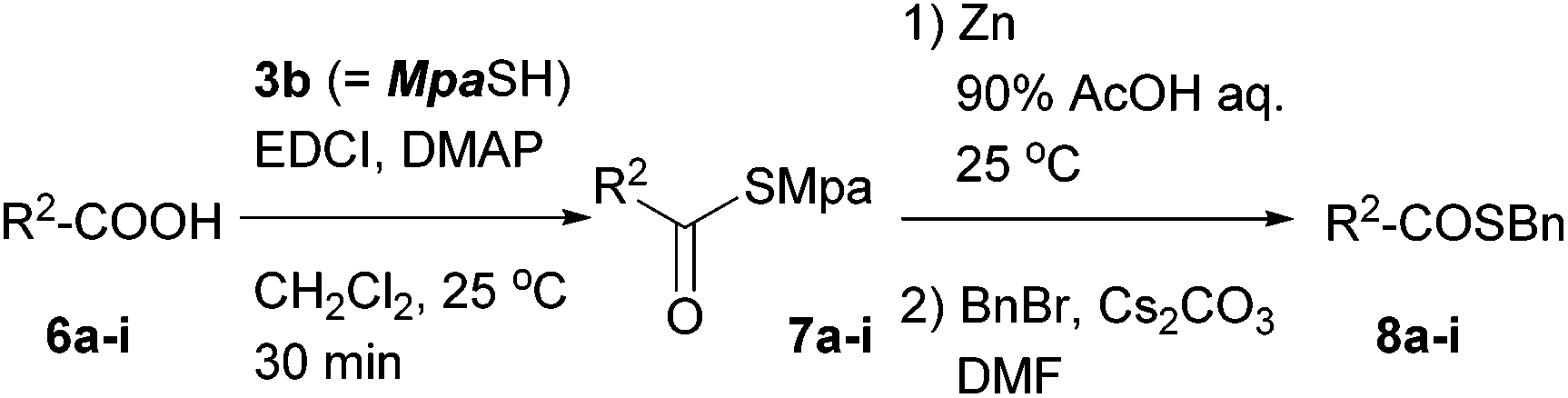
Table 2 summarizes the scope of this thioacid preparation procedure. Each of the aromatic, aliphatic, and amino acids 6a–i was converted to the Mpa thioester 7a–i using EDCI and DMAP in excellent isolated yields. These precursors afforded the corresponding thioacids smoothly when treated with zinc at ambient temperature. Excess zinc dust was removed by filtration through a silica gel pad, and then the solvent was evaporated to effectively give the thioacid, according to the yield after benzylation. It is noteworthy that no diastereomeric benzyl thioester of 8f was observed, which excludes the possibility of racemization in the reaction sequence. The present transformation was also applicable to Fmoc-protected amino acid 6g. Furthermore, carboxylic acids having additional thioester groups such as 6h and 6i could be converted to the bisthioesters, and the Mpa group could be removed in the presence of either an Fm- or Tmob-thioester. The thioacids could also be captured by 3-bromopropionitrile instead of benzyl bromide to give the 2-cyanoethyl thioester 8i′.
|
|
|||
|---|---|---|---|
| Entry | R2-COOH | Yield of 7a–i![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/% a/% |
Yield of 8a–ia/% |
| a Isolated yields. b 3-Bromopropionitrile was used instead of BnBr, which afforded 2-cyanoethyl thioester 8i′. | |||
| 1 | PhCOOH (6a) | 90 | 80 |
| 2 | n C9H19COOH (6b) | 92 | 91 |
| 3 | Boc-Phe-OH (6c) | 94 | 83 |
| 4 | Boc-Gly-OH (6d) | 95 | 83 |
| 5 | Boc-Ser(OBn)-OH (6e) | 98 | 78 |
| 6 | Boc-Thr(OBn)-OH (6f) | 94 | 73 |
| 7 | Fmoc-Trp-OH (6g) | 98 | 85 |
| 8 | Boc-Glu(SFm)-OH (6h) | 74 | 78 |
| 9 | Fmoc-Glu(STmob)-OH (6i) | 77 | 81 |
| 10 | 6i | — | 77b |
Next, we examined the orthogonality of the Mpa thioesters and Boc groups. As shown in Scheme 2, Boc-protected thioglycinate 7d was employed as the C-terminus, and peptide elongation based on Boc-TFA chemistry was conducted. In the presence of the Mpa thioester, the Boc group was successfully deprotected in 40% TFA/CH2Cl2. The subsequent coupling reaction afforded dipeptidyl thioester 9, where NMM performed better than DIPEA as a base. Another deprotection–coupling sequence was performed with lysine having a Boc group at the ω-amine to give tripeptide 10 without any problems. In the presence of the ω-Boc group, Mpa was cleanly removed using zinc at 40 °C to afford the peptidyl thioacid in good yield, which was confirmed by derivatization to the benzyl thioester 11.
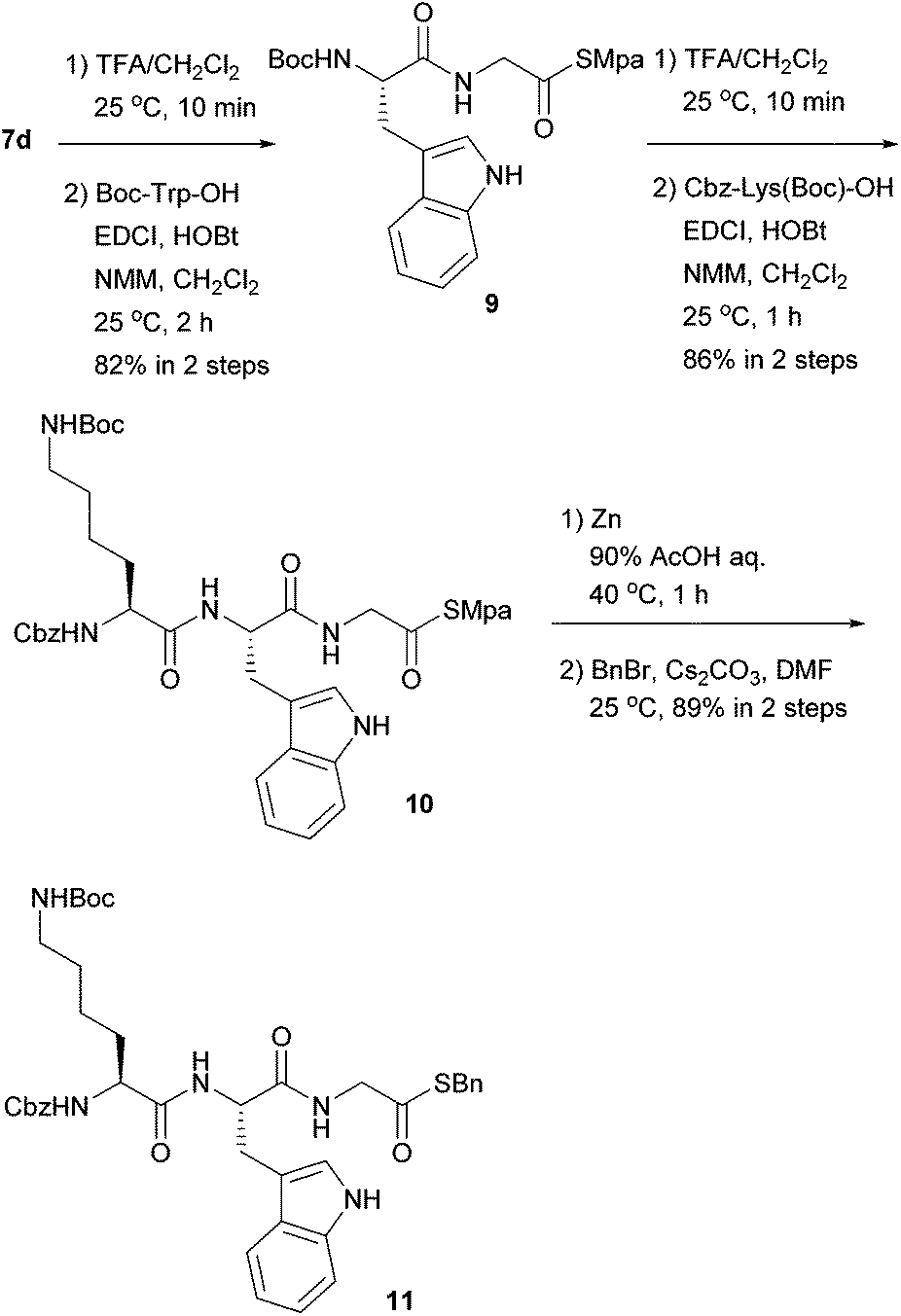 | ||
| Scheme 2 Synthesis of oligopeptide α-thioacid. | ||
Finally, as shown in Scheme 3, we prepared ω-Mpa thioglutamate 16 from the known glutamate 12 by protecting group manipulation, i.e., allylation of the α-carboxylic acid, deprotection of the ω-9-fluorenylmethyl ester, thioesterification using MpaSH, and deallylation using a Pd catalyst. This glutamate residue could be incorporated into a peptide by Boc-TFA chemistry without loss of the Mpa group to afford tripeptide 18. Again, the Mpa group could be removed in the presence of the Boc group, and benzyl thioester 19 was obtained after treatment with benzyl bromide. This orthogonality permits the preparation of peptidyl thioacids at either the C-terminus or the side-chain.
 | ||
| Scheme 3 Synthesis of oligopeptide having thioacid on its side-chain. | ||
Conclusions
α-Methylphenacyl (Mpa) thioesters were developed as precursors for thioacids. MpaSH is easily accessible from commercially available, low-cost materials and readily introduced to carboxylic acids. Mpa thioesters can facilitate the preparation of peptides bearing a thioacid either at the C-terminus or on the side chain. The orthogonal reactivity to the Boc-group and chemoselectivity over Tmob- and Fm-thioesters are expected to enable the preparation of complex thioacids such as peptide thioacids. We are now applying this chemistry to the preparation of oligo- and polypeptides.Acknowledgements
This work was financially in part supported by a JSPS Grant-in-Aid for Young Scientists B (JP26810062) to KS and the MEXT-Supported Program for the Strategic Research Foundation at Private Universities (2012–2016) to RS. We also thank Prof. Hidemasa Hikawa, Toho University, for help with FAB-HRMS experiments.Notes and references
- K. Sasaki, S. Aubry and D. Crich, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 1005–1018 CrossRef CAS.
- N. Assem, A. Natarajan and A. K. Yudin, J. Am. Chem. Soc., 2010, 132, 10986–10987 CrossRef CAS PubMed.
- N. Shangguan, S. Katukojvala, R. Greenberg and L. J. Williams, J. Am. Chem. Soc., 2003, 125, 7754–7755 CrossRef CAS PubMed.
- D. Crich, K. Sasaki, M. Y. Rahaman and A. A. Bowers, J. Org. Chem., 2009, 74, 3886–3893 CrossRef CAS PubMed.
- Y. Rao, X. Li and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 12924–12926 CrossRef CAS PubMed.
- D. Crich and K. Sasaki, Org. Lett., 2009, 11, 3514–3517 CrossRef CAS PubMed.
- D. Crich and I. Sharma, Angew. Chem., Int. Ed., 2009, 48, 2355–2358 CrossRef CAS PubMed.
- P. Wang and S. J. Danishefsky, J. Am. Chem. Soc., 2010, 132, 17045–17051 CrossRef CAS PubMed.
- J. Pan, N. O. Devarie-Baez and M. Xian, Org. Lett., 2011, 13, 1092–1094 CrossRef CAS PubMed.
- S. M. Mali, S. V. Jadhav and H. N. Gopi, Chem. Commun., 2012, 48, 7085–7087 RSC.
- R. Joseph, F. B. Dyer and P. Garner, Org. Lett., 2013, 15, 732–735 CrossRef CAS PubMed.
- W. Wu, Z. Zhang and L. S. Liebeskind, J. Am. Chem. Soc., 2011, 133, 14256–14259 CrossRef CAS PubMed.
- D. Crich, K. Sana and S. Guo, Org. Lett., 2007, 9, 4423–4426 CrossRef CAS PubMed.
- D. Crich and I. Sharma, Angew. Chem., Int. Ed., 2009, 48, 7591–7594 CrossRef CAS PubMed.
- R. Raz and J. Rademann, Org. Lett., 2012, 14, 5038–5041 CrossRef CAS PubMed.
- A. Shigenaga, Y. Sumikawa, S. Tsuda, K. Sato and A. Otaka, Tetrahedron, 2010, 66, 3290–3296 CrossRef CAS.
- S. L. Pira, E. Boll and O. Melnyk, Org. Lett., 2013, 15, 5346–5349 CrossRef CAS PubMed.
- T. Shimizu, R. Miyajima, K. Sato, K. Sakamoto, N. Naruse, M. Kita, A. Shigenaga and A. Otaka, Tetrahedron, 2016, 72, 992–998 CrossRef CAS.
- C. Chen, Y. Huang, L. Xu, Y. Zheng, H. Xu, Q. Guo, C. Tian, Y. Li and J. Shi, Org. Biomol. Chem., 2014, 12, 9413–9418 CAS.
- P. G. M. Wuts and T. W. Greene, in Protective groups in organic synthesis, John Wiley & Sons, Hoboken, New Jersey, 4th edn, 2007, pp. 568–569 Search PubMed.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- S. Kato, Y. Kawahara, H. Kageyama, R. Yamada, O. Niyomura, T. Murai and T. Kanda, J. Am. Chem. Soc., 1996, 118, 1262–1267 CrossRef CAS.
- In the reaction of 4b, conversion to the corresponding thioacid was verified by ESI-HRMS; calcd for C16H19N2O3S− [M − H]− 319.1122, found 319.1103.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob02256a |
| This journal is © The Royal Society of Chemistry 2016 |