
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base mediated 1,3-dipolar cycloaddition of α-substituted vinyl phosphonates with diazo compounds for synthesis of 3-pyrazolylphosphonates and 5-pyrazolcarboxylates†
Nataliya S.
Goulioukina
 *ab,
Nikolay N.
Makukhin
a,
Egor D.
Shinkarev
a,
Yuri K.
Grishin
a,
Vitaly A.
Roznyatovsky
a and
Irina P.
Beletskaya
*ab
*ab,
Nikolay N.
Makukhin
a,
Egor D.
Shinkarev
a,
Yuri K.
Grishin
a,
Vitaly A.
Roznyatovsky
a and
Irina P.
Beletskaya
*ab
aDepartment of Chemistry, Moscow State University, Leninskie Gory, GSP-1, 119991 Moscow, Russian Federation. E-mail: goulioukina@org.chem.msu.ru
bFrumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninsky Pr. 31, 119071 Moscow, Russian Federation
First published on 26th September 2016
Abstract
5-Aryl-substituted pyrazol-3-ylphosphonates have been conveniently synthesized by 1,3-dipolar cycloaddition of 1-formamidovinylphosphonates and aryldiazomethanes under K2CO3/MeOH conditions at room temperature. These pyrazoles are formed in one pot via spontaneous elimination of formamide. Basic conditions prevent competitive formation of cyclopropylphosphonates. 3-Aryl substituted pyrazol-5-carboxylates can be synthesized by the same methodology from 1-arylvinylphosphonates and ethyl diazoacetate, although a stronger base NaH is necessary to ensure the success of the aromatization stage with the elimination of the diethoxylphosphoryl moiety.
Introduction
A pyrazole moiety is regarded as a privileged heterocyclic skeleton with multifarious applications. Pyrazoles represent the core structure of numerous naturally occurring molecules (e.g., pyrazol-3(5)-carboxylic acid from the sponge Tedania anhelans, (S)-β-pyrazolylalanine from Citrullus vulgaris, and alkaloid Withasomnine from Indian medicinal plant Withania somnifera),1 pharmaceuticals (e.g., the marketed drugs Celebrex and Viagra), and crop protection agents.2 Pyrazoles are also efficient coordinating ligands in catalysis3 and supramolecular chemistry.4 Among various functionalized pyrazoles, pyrazolylphosphonates have received considerable attention over the past few decades due to their remarkable bioactivity profiles5 and possible applications as organic precursors.6 Increasing interest in pyrazolylphosphonates has been reflected by the development of various synthetic methodologies allowing straightforward access to these structural motifs, however, the main focus had been on 4-pyrazolylphosphonates,7 while 3(5)-pyrazolylphosphonates8 received much less attention. A remarkable progress had been initiated by the pioneering work of Namboothiri et al. who proposed to employ the Bestmann–Ohira reagent as a 1,3-dipolar precursor in base-mediated cycloaddition reactions with conjugated nitroalkenes.9 This methodology was further extended on a variety of dipolarophiles and nowadays presents the most developed synthetic route to 3(5)-pyrazolylphosphonates.8,10,11 By contrast, an alternative approach utilizing α,β-unsaturated phosphonates as cycloaddition partners has been scarcely reported, is frequently complicated by competitive formation of cyclopropylphosphonates, and thus remains a challenging task.Recently we have reported an efficient approach to the synthesis of 2-substituted 1-aminocyclopropylphosphonates based on the regioselective 1,3-dipolar cycloaddition reaction of diazo compounds with dimethyl 1-formamidovinylphosphonate (1) (Scheme 1).12,13 The reaction pathway involves the formation of 1-pyrazoline. The subsequent elimination of a nitrogen molecule affords the cyclopropane ring. However in the case of (4-diethoxyphosphorylphenyl)diazomethane containing a strong electron-acceptor substituent in the ring,14 the major reaction product is 2-pyrazoline, which is resistant to nitrogen extrusion. It was also shown that the aromatization of 1- and 2-pyrazolines in acidic media is accompanied by the elimination of formamide to give 3-pyrazolylphosphonates.
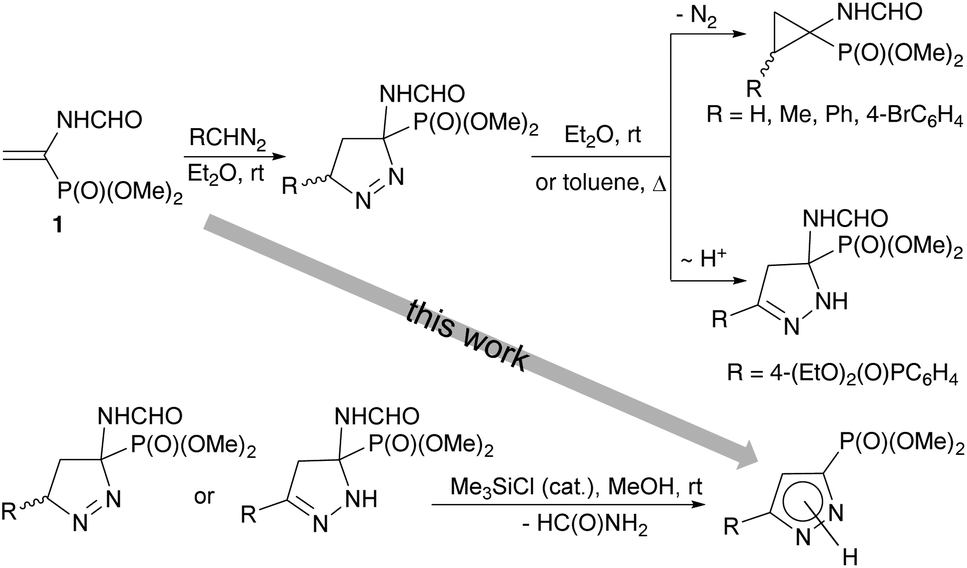 | ||
| Scheme 1 Summary of our previous results on 1,3-dipolar cycloaddition reaction of vinylphosphonate 1 with diazo compounds. | ||
In the present work, the possibility to give a new turn to the general reaction scheme and to adapt it for the target one-pot synthesis of 3(5)-phosphonylated pyrazoles was demonstrated. All one has to do is provide conditions for the fast isomerization of the initially formed 1-pyrazoline to 2-pyrazoline (thus suppressing the possibility of nitrogen extrusion with cyclopropane formation) and the elimination of formamide. Both steps would be expected to be catalyzed by base.15,16 Therefore the preliminary optimization of the reaction conditions included the variation of bases and solvents.
Results and discussion
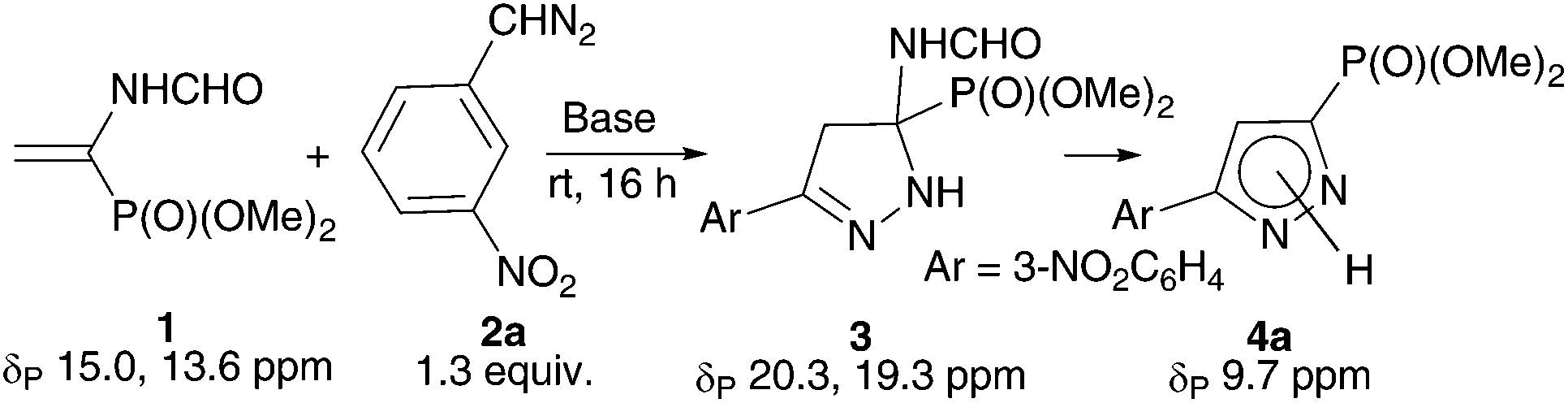
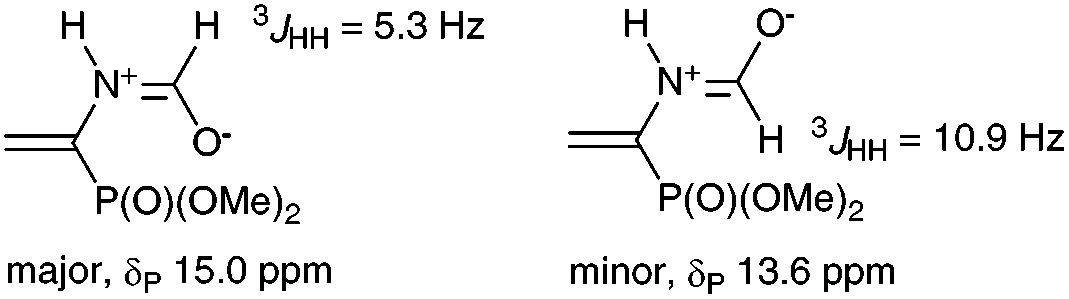
Reasonably stable and, hence, easy to handle (3-nitrophenyl)diazomethane (2a) was chosen as a model diazo compound. The reaction course was monitored using the 31P NMR method by the disappearance of signals from two rotamers of vinylphosphonate 1 at δP 15.0 and 13.6 ppm (ref. 17) and accumulation of signals from the products. The obtained results presented in Table 1 show that the reaction of 1 with 1.3 equiv. of 2a in ether in the presence of 10 mol% K2CO3 completes in 16 h and affords almost exclusively 2-pyrazoline 3 (two rotamers in a ratio of 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14; δP 20.3 and 19.3 ppm), which precipitates (entry 1). The yield of pyrazolylphosphonate 4a (δP 9.7 ppm) was only 3% and increased to 25% when the experiment was repeated in CH2Cl2 in which all reaction products are soluble (entry 2). The use of MeOH turned out to be most productive: in this case, the yield of the target pyrazole 4a was 89% (entry 3). Note that in all experiments the conversion of vinylphosphonate 1 was close to quantitative, which is consistent with the known fact that the rate of 1,3-dipolar cycloaddition is almost insensitive to the polarity of the medium.18 In all experiments the formation of dimethyl 1-formamido-2-(3-nitrophenyl)cyclopropylphosphonates (δP 23.3 and 22.7 ppm for the cis-isomer and δP 25.7 and 24.9 ppm for the trans-isomer) was detected, but their total yield did not exceed 2%.
a
:14; δP 20.3 and 19.3 ppm), which precipitates (entry 1). The yield of pyrazolylphosphonate 4a (δP 9.7 ppm) was only 3% and increased to 25% when the experiment was repeated in CH2Cl2 in which all reaction products are soluble (entry 2). The use of MeOH turned out to be most productive: in this case, the yield of the target pyrazole 4a was 89% (entry 3). Note that in all experiments the conversion of vinylphosphonate 1 was close to quantitative, which is consistent with the known fact that the rate of 1,3-dipolar cycloaddition is almost insensitive to the polarity of the medium.18 In all experiments the formation of dimethyl 1-formamido-2-(3-nitrophenyl)cyclopropylphosphonates (δP 23.3 and 22.7 ppm for the cis-isomer and δP 25.7 and 24.9 ppm for the trans-isomer) was detected, but their total yield did not exceed 2%.
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | Conversion of 1b (%) |
Yieldb,c (%) | |
| 3 | 4a | ||||
| a Reaction conditions: 1 (0.25 mmol), 2a (0.325 mmol), base (10 mol%), solvent (0.65 ml), rt, 16 h. b Determined by 31P NMR analysis of the crude reaction mixture. c Isolated yields are given in the parentheses. | |||||
| 1 | Et2O | K2CO3 | 96 | 87 (79) | 3 |
| 2 | CH2Cl2 | K2CO3 | 98 | 68 | 25 |
| 3 | MeOH | K2CO3 | 98 | 1 | 89(76) |
| 4 | MeOH | Cs2CO3 | 99 | 5 | 84 |
| 5 | MeOH | K3PO4 | 99 | 5 | 86 |
| 6 | MeOH | MeONa | 99 | 3 | 89 |
The variation of bases showed that the replacement of K2CO3 by Cs2CO3 (entry 4), K3PO4 (entry 5), or MeONa (entry 6) exerted almost no effect on the yield of the target product 4a and, therefore, cheaper potash was used in further experiments.
The absence of a noticeable effect of the nature of the base and a rather strong solvent effect is consistent with the proposed reaction mechanism (Scheme 2), according to which the rate determining stage of 1,3-dipolar cycloaddition leads to 1-pyrazoline formation. The base initiates the fast isomerization of 1-pyrazoline to thermodynamically more stable conjugated 2-pyrazoline. The role of alcohol is most probably the protonation of the leaving group in the next step of formamide elimination rather than increasing the solubility of an inorganic base.19
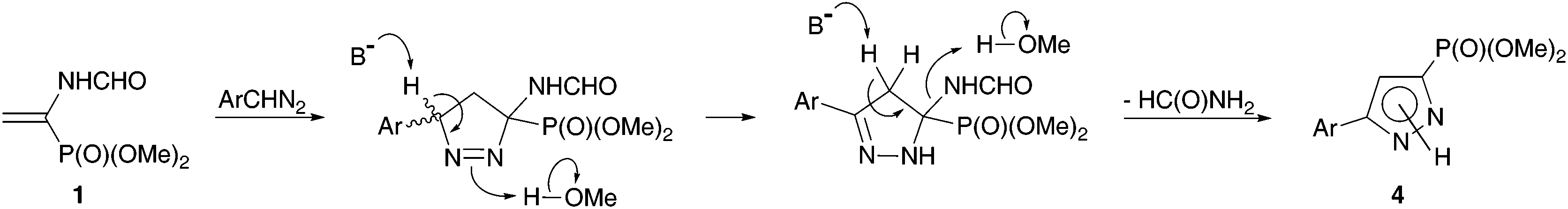 | ||
| Scheme 2 Proposed pathway for pyrazolylphosphonate 4 formation. | ||
Under the optimal conditions found, a large series of aryldiazomethanes 2a–p containing both acceptor and donor substituents in the ring were introduced into the reaction with vinylphosphonate 1, and the corresponding 5-aryl-substituted dimethyl (1H-pyrazol-3-yl)phosphonates 4a–p were obtained in high yields after chromatographic purification (Table 2). Commenting on the obtained results, two points should be mentioned. First, the reaction time, required for quantitative conversion of vinylphosphonate 1, is noticeably longer in the case of aryldiazomethanes containing strong electron-acceptor substituents in the ring, especially 2b.20 The same concerns the sterically hindered aryldiazomethanes 2e,f containing ortho-substituents. These two factors are combined in (2,4-dinitrophenyl)diazomethane, which turned out to be completely inert under the reaction conditions. The second point is the necessity to use a substantial excess of aryldiazomethanes with electron-donor substituents in the ring (e.g., 4-EtO (2p), 4-MeO (2o), and 4-i-Pr (2n)), since these diazo compounds are noticeably decomposed during the reaction to form substituted cis- and trans-stilbenes and dibenzylidenehydrazines.21 Not unexpectedly, the amount of cyclopropanes increases to 9–10% of total yield, since the donor substituents should impede the proton transfer and isomerization of 1-pyrazoline to 2-pyrazoline.
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 (equiv.) | Ar | Product | Reaction timeb (h) | Yield of 4c (%) |
| a Reaction conditions: 1 (0.25 mmol), 2 (equiv., 0.5 M solution in MeOH), K2CO3 (10 mol%), rt. b Time to completion, estimated by 31P NMR. c Isolated yield. d The yield in the reaction performed on a 2.5 mmol scale is given in the parentheses. | |||||
| 1 | 2a (1.3) | 3-NO2C6H4 | 4a | 16 | 76 |
| 2 | 2b (1.3) | 4-NO2C6H4 | 4b | 30 | 72 |
| 3 | 2c (1.5) | 4-(EtO)2P(O)C6H4 | 4c | 24 | 79 |
| 4 | 2d (1.5) | 4-MeO2CC6H4 | 4d | 18 | 78 |
| 5 | 2e (1.4) | 2-ClC6H4 | 4e | 17 | 63 |
| 6 | 2f (1.3) | 2-BrC6H4 | 4f | 16 | 54 |
| 7 | 2g (1.3) | 3-ClC6H4 | 4g | 12 | 78 |
| 8 | 2h (1.3) | 4-ClC6H4 | 4h | 4 | 87 |
| 9 | 2i (1.4) | 4-BrC6H4 | 4i | 3 | 87 |
| 10 | 2j (3) | 4-MeSC6H4 | 4j | 3 | 86 |
| 11 | 2k (3) | 3-MeOC6H4 | 4k | 3 | 71 |
| 12 | 2l (1.3) | Ph | 4l | 3 | 85 (91)d |
| 13 | 2m (1.5) | 4-FC6H4 | 4m | 3 | 67 |
| 14 | 2n (4) | 4-i-PrC6H4 | 4n | 3 | 80 |
| 15 | 2o (2) | 4-MeOC6H4 | 4o | 3 | 76 |
| 16 | 2p (1.6) | 4-EtOC6H4 | 4p | 2 | 63 |
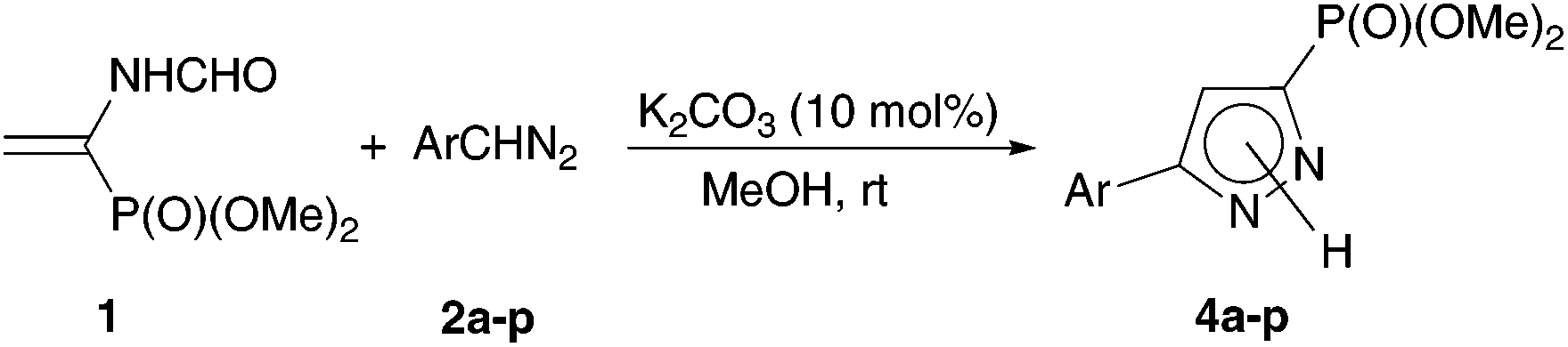
The structure of pyrazolylphosphonate 4c was unambiguously proved by the X-ray diffraction analysis data (Fig. 1).13,22 In the 1H NMR spectra of pyrazolylphosphonates 4 the doublet of the C(4)H proton of the pyrazole ring at δH 6.8–7.3 ppm (3JPH = 1.8–2.4 Hz) is characteristic. The analysis of the 13C NMR spectra is complicated by the fact that the signals of the carbon atoms of the pyrazole ring and the ipso-carbon of the aryl fragment broadened, which is explained by prototropic ring tautomerism in the solution.11b,c,15,23 The detection of a doublet of the quaternary carbon atom bound to phosphorus and lying at δC 132.7–138.0 ppm (1JPC = 220–229 Hz) was particularly difficult. The unambiguous assignment was made on the basis of 2D NMR spectroscopy HMBC experiments, which makes it possible to observe cross peaks arising from the 2JCH coupling constants between the C(4)H proton and the carbon atoms C(3) and C(5).
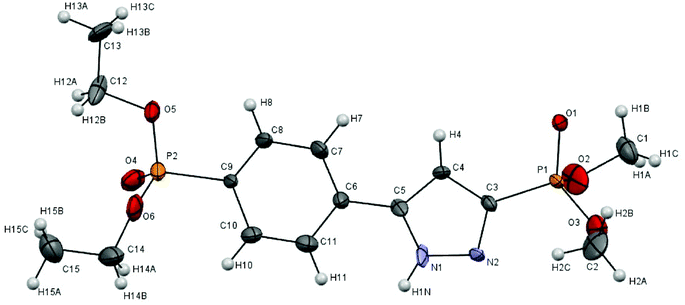 | ||
| Fig. 1 Single X-ray crystal structure of 4c (CCDC 959780). Thermal ellipsoids are drawn at a 50% probability. | ||
Ethyl diazoacetate reacts with vinylphosphonate 1 only at elevated temperatures. At 80 °C without solvent, the main reaction product was 2-pyrazoline 5, which was isolated chromatographically in 80% yield (Table 3, entry 1). The temperature decrease to 50 °C results in a decrease in the reaction rate (entry 2), and the use of the solvent (EtOH in the presence of 10 mol% K2CO3 (entry 3) or PhMe (entry 4)) induces a dramatic decrease in the selectivity of the process.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | T (°C) | Conversion of 1b (%) |
Yield of 5b,c (%) |
| a Reaction conditions: 1 (0.28 mmol), N2CHCO2Et (0.56 mmol), 4 h. b Determined by 31P NMR analysis of the crude reaction mixture. c Isolated yield after column chromatography is given in the parentheses. d In the presence of K2CO3 (10 mol%). | ||||
| 1 | None | 80 | 100 | 85 (80) |
| 2 | None | 50 | 64 | 59 |
| 3 | EtOHd | 80 | 66 | 20 |
| 4 | PhMe | 80 | 59 | 12 |
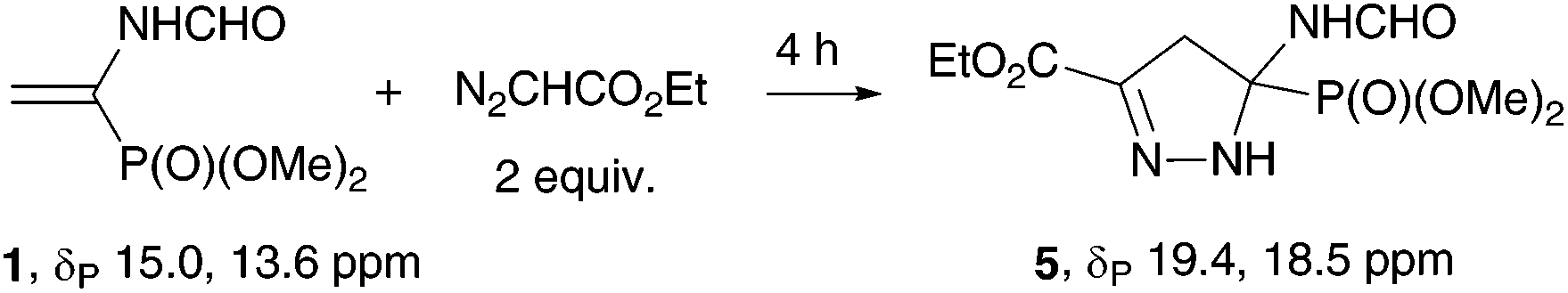
An attempt to aromatize 2-pyrazoline 5 under the conditions used in the synthesis of pyrazolylphosphonates 4 (MeOH, K2CO3, 20 °C) was unsuccessful: the reaction proceeded very slowly and was accompanied by transesterification of the ester fragment. Pyrazolylphosphonate 65c was obtained in high yield when the reaction was carried out in methanol with addition of a catalytic amount of Me3SiCl (Scheme 3).
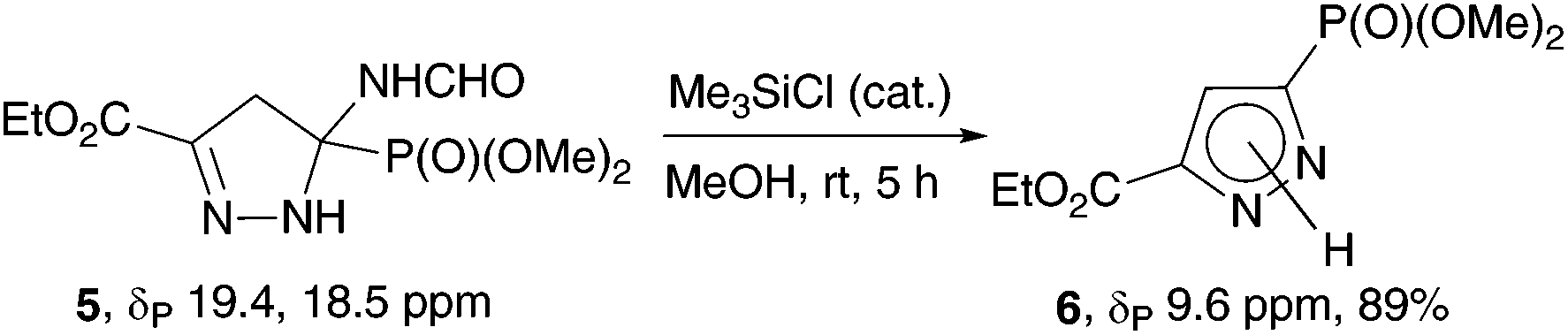 | ||
| Scheme 3 Aromatization of 2-pyrazoline 5 with pyrazolylphosphonate 6 formation. | ||
It is worthy of note that no elimination of diethyl phosphite was observed in the reactions of vinyl phosphonate 1 with diazo compounds. Meanwhile, rare examples for the aromatization of phosphonylated pyrazolines with the formation of pyrazole due to the C–P bond cleavage are briefly mentioned in the literature.24 We employed this possibility in the synthesis of 3-aryl-substituted 1H-pyrazole-5-carboxylates,25,26viz., carboxyl analogs of pyrazolylphosphonates 4. Diethyl 1-arylvinylphosphonates 7 available by the Conant reaction served as the starting dipolarophiles.27
The synthesis conditions were optimized for the model diethyl 1-phenylvinylphosphonate (7a). The reaction course was monitored by 31P and 1H NMR spectroscopy. On heating at 50 °C a solution of vinylphosphonate 7a and ethyl diazoacetate (2 equiv.) in THF in the presence of NaH (2.5 equiv.), the reaction took 18 h for completion. After neutralization of the reaction mixture with a solution of HCl in ether followed by treatment with triethylamine, the target pyrazolecarboxylate 9a was isolated chromatographically in 82% yield (Table 4, entry 1).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | Base | T (°C) | Reaction time (h) | Conv. of 7ab (%) |
Yield (%) | |
|
8b,c |
9a | ||||||
| a Reaction conditions: 7a (0.4 mmol), N2CHCO2Et (0.8 mmol), base, solvent (2 ml). b Determined by 31P NMR analysis of the crude reaction mixture. c Isolated yield is given in the parentheses. d Isolated yield after column chromatography. e Formation of tetraethyl (1-phenylethane-1,2-diyl)bis(phosphonate) (11%; δP 27.7 and 29.2 ppm (3JP–P = 82.9 Hz)) was also detected by NMR analysis of the crude reaction mixture. f Determined by 1H NMR analysis of the crude reaction mixture; doublets due to ortho-protons are observed at 7.51, 7.45, and 7.72 ppm for compounds 7a, 8, and 9a, respectively. | |||||||
| 1 | THF | NaH (2.5 equiv.) | 50 | 18 | 97 | 0 | 82d,e |
| 2 | THF | K2CO3 (5 mol%) | 50 | 48 | 97 | 92(43) | 0f |
| 3 | EtOH | K2CO3 (5 mol%) | 60 | 24 | 92 | 86 | 0f |
| 4 | Dioxane | K2CO3 (5 mol%) | 80 | 20 | 92 | 63 | 0f |

The reaction mechanism (Scheme 4), assuming the deprotonation of 1-pyrazoline formed at the first step, followed by the elimination of the diethyl phosphite anion via the E1cB mechanism, and repeated deprotonation to form an aromatic structure, explains the necessity to use two equivalents of sodium hydride.
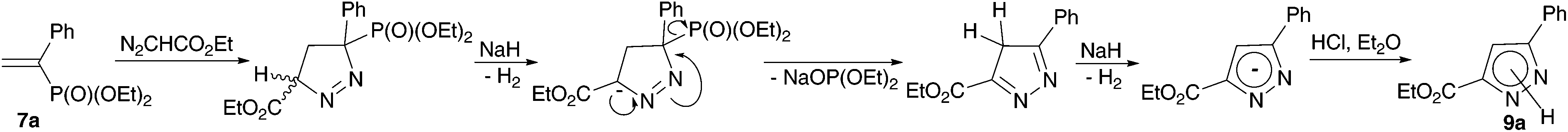 | ||
| Scheme 4 Proposed pathway for pyrazolcarboxylate 9a formation. | ||
The replacement of NaH by K2CO3 results in the termination of the reaction at the step of formation of 2-pyrazoline 8 when using THF (entry 2) or ethanol (entry 3) as a solvent. No formation of target pyrazolecarboxylate 9a was observed even on increasing the reaction temperature to 80 °C (in dioxane), but the selectivity of the process decreased noticeably because of the formation of by-products (entry 4).
2-Pyrazoline 8 was isolated and characterized. The aromatization of 2-pyrazoline 8 occurs smoothly in the presence of 2 equiv. NaH in THF. An increase of temperature from ambient to 50 °C shortens the reaction time from 24 to 5 h. The aromatization can be carried out both for individual pyrazoline 8 and in situ, adding 2 equiv. NaH to the reaction mixture in THF after the reaction of vinylphosphonate 7a with ethyl diazoacetate in the presence of K2CO3 has ceased. In both cases, pyrazolecarboxylate 9a was isolated in a yield of 66%.
A series of vinylphosphonates 7b–e containing 4-chlorophenyl, 4-isobutylphenyl, 4-diphenyl, and 2-naphthyl substituents in the α-position were introduced into the reaction with ethyl diazoacetate to extend the scope of the method. Since the yields of product 9a were comparable when using both the one-step (method A) and two-step (method B) protocols, we checked both procedures for each substrate of 7b–e. The obtained results (Table 5) showed that both protocols made it possible to obtain products 9b–e in good yields. The use of the longer method B is justified in the case of substrates 7b,d because it gave better yields of the corresponding products 9b,d.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 7 | Ar | Product | Isolated yield (%) | |
| Method Aa | Method Bb | ||||
| a Reaction conditions: 7 (0.4 mmol), N2CHCO2Et (0.8 mmol), NaH (1.0 mmol), THF (2 ml), 50 °C, 18 h. b Reaction conditions: (1) 7 (0.4 mmol), N2CHCO2Et (0.6–0.8 mmol), K2CO3 (5 mol%), THF (2 ml), 50 °C, 48–55 h (31P NMR monitoring); (2) NaH (0.8 mmol), 50 °C, 5 h. | |||||
| 1 | 7a | Ph | 9a | 82 | 66 |
| 2 | 7b | 4-ClC6H4 | 9b | 57 | 62 |
| 3 | 7c | 4-i-BuC6H4 | 9c | 60 | 56 |
| 4 | 7d | 4-PhC6H4 | 9d | 76 | 83 |
| 5 | 7e | 2-Napth | 9e | 74 | 68 |
Products 9 were characterized by all relevant methods. The spectral characteristics of substrates 9a,b,e are consistent with those described in the literature.25d,l Note that the published X-ray diffraction data for the 4-methoxyphenyl analog of products 9 show that in the crystalline state pyrazolecarboxylates 9, unlike pyrazolylphosphonates 4, exist as an alternative tautomer ethyl 3-aryl-1H-pyrazole-5-carboxylate.25d
Conclusions
In conclusion, 1,3-dipolar cycloaddition of aryldiazomethanes with dimethyl 1-formamidovinylphosphonate (1) exhibits a condition-controlled product-selectivity. Under K2CO3/MeOH conditions nitrogen extrusion with cyclopropane ring formation is suppressed and the reaction affords 5-aryl substituted dimethyl (1H-pyrazol-3-yl)phosphonates 4 in high yields. Mild reaction conditions and broad functional group tolerance make this strategy synthetically useful. In much the same way, 1,3-dipolar cycloaddition of ethyl diazoacetate with diethyl 1-arylvinylphosphonates 7 under NaH/THF conditions provides regioisomerically pure 3-aryl substituted ethyl 1H-pyrazol-5-carboxylates 9, the reaction being accompanied by the elimination of the diethoxylphosphoryl moiety.Experimental
Reactions were routinely performed under dry conditions in oven-dried glassware and under an air atmosphere unless otherwise specified. Methanol was distilled from magnesium methoxide, and THF and Et2O were distilled from sodium benzophenon ketyl prior to use. The starting material vinylphosphonate 1 was prepared according to the known method.17,28 Vinylphosphonates 7 were obtained as previously reported.27 Ethyl diazoacetate was purchased from Aldrich and used as received. Aryldiazomethanes 2 were generated from sodium salts of tosylhydrazones of the corresponding aromatic aldehydes by the vacuum pyrolysis method29 (for 2d,o,p) or by the pyrolysis in the ethylene glycol procedure30 (for 2a–c,e–n). (Caution! Although we did not experience any problems in handling aryldiazomethanes, full safety precautions should be taken due to their potentially explosive nature.)NMR data were recorded on Bruker Avance-300, 400 or Agilent 400-MR spectrometers at ambient temperature. 13C and 31P NMR spectra were 1H decoupled. Chemical shifts are reported on the δ-scale in parts per million relative to the solvent (CDCl3: δC 77.0; Acetone-d6: δC 29.8; CD3OD: δC 49.0; DMSO-d6: δC 39.5) or the residual solvent peak (CHCl3: δH 7.25; Acetone-d6: δH 2.04; CD3OD: δH 3.30) as internal standards, or to external 85% H3PO4 (δP 0). The IR spectra were taken on a SPECORD 75 IR instrument using KBr pellets. High resolution mass spectra (HRMS) were measured on a Bruker maXis spectrometer using electrospray ionization (ESI). Elemental analyses were carried out on an Elementar Vario MICRO Cube analyzer. Melting points were measured with Electrothermal 9100 apparatus and are uncorrected. Column chromatography was carried out using Macherey-Nagel silica gel 60 (0.015–0.04 mm). Preparative thin-layer chromatography (TLS) was performed using 20 × 20 cm pre-coated glass plates SIL G-100 UV254 (Macherey-Nagel) and visualized by UV (254 nm).
Dimethyl [5-(formamido)-3-(3-nitrophenyl)-4,5-dihydro-1H-pyrazol-5-yl]phosphonate (3)
A foil-covered 4 ml vial was charged with vinylphosphonate 1 (45 mg, 0.25 mmol), 0.5 M solution of the diazo compound 2a in Et2O (0.65 ml, 0.325 mmol), and K2CO3 (3.5 mg, 0.025 mmol). The reaction mixture was stirred at rt for 16 h. The resultant precipitate was collected by filtration, washed with Et2O, acetone, and water and dried in a vacuum desiccator over P2O5 to give 2-pyrazoline 3 as a yellowish solid (68 mg) in 79% yield; a 86:14 mixture of two rotamers in CDCl3. 31P NMR (162 MHz, CDCl3): δ 20.3 (major), 19.3 (minor). 1H NMR (400 MHz, CDCl3) (only for the major rotamer): δ 3.53–3.67 m (2H, CH2), 3.91 (d, 3JH–P = 10.6 Hz, 3H, OCH3), 3.93 (d, 3JH–P = 10.4 Hz, 3H, OCH3), 6.36 (br. s, 1H, NHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 7.07 (br. s, 1H, NNH), 7.56 (dd, 3JH–H = 3JH–H = 8.0 Hz, 1H, ArH), 8.04 (m, 3JH–H = 8.0 Hz, 1H, ArH), 8.18 (s, 1H, CHO), 8.20 (m, 3JH–H = 8.0 Hz, 1H, ArH), 8.40 (dd, 4JH–H = 4JH–H = 1.7 Hz, 1H, ArH). 13C NMR (101 MHz, CDCl3) (only for the major rotamer): δ 42.2 (d, 2JC–P = 3.1 Hz, CH2), 54.3 (d, 2JC–P = 7.5 Hz, OCH3), 55.2 (d, 2JC–P = 6.7 Hz, OCH3), 76.1 (d, 1JC–P = 197.7 Hz, PC), 121.0 (CH), 123.8 (CH), 129.7 (CH), 131.7 (CH), 133.4 (CAr), 148.5 (d, JC–P = 14.4 Hz, CN), 148.7 (CNO2), 161.1 (d, 3JC–P = 5.2 Hz, CO). Anal. calcd for C12H15N3O5P·2H2O: C, 38.10; H, 5.06; N, 14.81. Found: C, 38.61; H, 4.55; N, 14.41.
O), 7.07 (br. s, 1H, NNH), 7.56 (dd, 3JH–H = 3JH–H = 8.0 Hz, 1H, ArH), 8.04 (m, 3JH–H = 8.0 Hz, 1H, ArH), 8.18 (s, 1H, CHO), 8.20 (m, 3JH–H = 8.0 Hz, 1H, ArH), 8.40 (dd, 4JH–H = 4JH–H = 1.7 Hz, 1H, ArH). 13C NMR (101 MHz, CDCl3) (only for the major rotamer): δ 42.2 (d, 2JC–P = 3.1 Hz, CH2), 54.3 (d, 2JC–P = 7.5 Hz, OCH3), 55.2 (d, 2JC–P = 6.7 Hz, OCH3), 76.1 (d, 1JC–P = 197.7 Hz, PC), 121.0 (CH), 123.8 (CH), 129.7 (CH), 131.7 (CH), 133.4 (CAr), 148.5 (d, JC–P = 14.4 Hz, CN), 148.7 (CNO2), 161.1 (d, 3JC–P = 5.2 Hz, CO). Anal. calcd for C12H15N3O5P·2H2O: C, 38.10; H, 5.06; N, 14.81. Found: C, 38.61; H, 4.55; N, 14.41.
Representative synthesis: dimethyl [5-(3-nitrophenyl)-1H-pyrazol-3-yl]phosphonate (4a)
A foil-covered 4 ml vial was charged with vinylphosphonate 1 (45 mg, 0.25 mmol), 0.5 M solution of the diazo compound 2a in MeOH (0.65 ml, 0.325 mmol), and K2CO3 (3.5 mg, 0.025 mmol). The reaction mixture was stirred at rt for 16 h. Volatile components were removed on a rotary evaporator and the residue was purified by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.4) to afford pyrazolylphosphonate 4a as a colorless solid (56.5 mg) in 76% yield. Mp 185 °C. 31P NMR (162 MHz, CDCl3): δ 9.7. 1H NMR (400 MHz, CDCl3): δ 3.87 (d, 3JH–P = 11.6 Hz, 6H, OCH3), 7.09 (d, 3JH–P = 2.4 Hz, 1H, C(4)H), 7.61 (dd, 3JH–H = 3JH–H = 8.0 Hz, 1H, ArH), 8.17–8.22 (m, 2H, ArH), 8.67 (dd, 4JH–H = 4JH–H = 1.9 Hz, 1H, ArH), 12.86 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.6 (d, 2JC–P = 5.5 Hz, OCH3), 109.1 (d, 2JC–P = 19.1 Hz, C(4)), 120.7 (CH), 122.9 (CH), 129.8 (CH), 131.6 (CH), 132.7 (d, 1JC–P = 223 Hz, CP), 133.6 (CAr), 148.7 (CNO2), 149.3 (d, 3JC–P = 14.3 Hz, C(5)). IR (KBr): ν 3400, 1550, 1529, 1344, 1248, 1184, 1063, 1032, 1003, 843, 781, 741 cm−1. Anal. calcd for C11H12N3O5P: C, 44.45; H, 4.07; N, 14.14. Found: C, 44.40; H, 4.31; N, 13.84.Dimethyl [5-(4-nitrophenyl)-1H-pyrazol-3-yl]phosphonate (4b)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2b in MeOH (0.65 ml, 0.325 mmol); the reaction time was 30 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.3) gave pyrazolylphosphonate 4b as a white solid (53.5 mg) in 72% yield. Mp 198 °C. 31P NMR (162 MHz, acetone-d6): δ 7.4 (major tautomer), 11.7 (minor tautomer) in 83:17 ratio. 1H NMR (400 MHz, acetone-d6): δ 3.80 (d, 3JH–P = 11.4 Hz, 6H, OCH3), 7.38 (d, 3JH–P = 1.9 Hz, 1H, C(4)H), 8.18 (br. d, 3JH–H = 8.8 Hz, 2H, ArH), 8.31 (br. d, 2H, ArH), 13.59 (br. s, 1H, NH). 13C NMR (101 MHz, acetone-d6): δ 53.5 (d, 2JC–P = 5.6 Hz, OCH3), 111.2 (d, 2JC–P = 20.1 Hz, C(4)), 124.9 (2CH), 127.2 (2CH), 139.0 (br., C(5)), 148.3 (CNO2). IR (KBr): ν 3420, 1518, 1346, 1236, 1184, 1 061, 1030, 854, 600 cm−1. Anal. calcd for C11H12N3O5P: C, 44.45; H, 4.07; N, 14.14. Found: C, 44.29; H, 4.12; N, 13.
Dimethyl [5-(4-(diethoxyphosphoryl)phenyl)-1H-pyrazol-3-yl]phosphonate (4c)
The reaction was carried out as described for 4a using 0.5 M solution of diazo compound 2c in MeOH (0.75 ml, 0.375 mmol); the reaction time was 24 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.17) gave pyrazolylphosphonate 4c as a pale yellow oil (77 mg) in 79% yield. 31P NMR (121 MHz, CD3OD): δ 11.6, 18.4. 1H NMR (300 MHz, CD3OD): δ 1.33 (dt, 3JH–H = 7.1 Hz, 4JH–P = 0.5 Hz, 6H, CCH3), 3.84 (d, 3JH–P = 11.4 Hz, 6H, OCH3), 4.13 (m, 4H, OCH2), 7.24 (d, 3JH–P = 1.9 Hz, 1H, C(4)H), 7.85 (dd, 3JH–P = 12.9 Hz, 3JH–H = 8.6 Hz, 2H, ArH), 7.96 (dd, 4JH–P = 4.0 Hz, 3JH–H = 8.6 Hz, 2H, ArH). 13C NMR (75 MHz, CD3OD): δ 16.5 (d, 3JC–P = 6.2 Hz, CCH3), 54.1 (d, 2JC–P = 5.9 Hz, OCH3), 64.0 (d, 2JC–P = 5.9 Hz, CH2), 111.0 (d, 2JC–P = 22.3 Hz, C(4)), 127.0 (d, 3JC–P = 15.3 Hz, 2CH), 128.7 (d, 1JC–P = 191.5 Hz, CP), 133.4 (d, 2JC–P = 10.3 Hz, 2CH), 135.5 (d, 4JC–P = 3.3 Hz, C), 138.0 (d, 1JC–P = 228.3 Hz, CP), 147.8 (dd, 3JC–P = 13.3 Hz, 5JC–P = 1.0 Hz, C(5)). IR (KBr): ν 3433, 1480, 1238, 1186, 1130, 1051, 1028, 970 cm−1. HRMS (ESI): m/z [M + H]+ calcd for C15H24N2O6P2: 389.1025. Found 389.1020.Dimethyl [5-(4-(methoxycarbonyl)phenyl)-1H-pyrazol-3-yl]phosphonate (4d)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2d in MeOH (0.75 ml, 0.375 mmol); the reaction time was 18 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.19) gave pyrazolylphosphonate 4d as a pale yellow solid (61 mg) in 78% yield. Mp 184 °C. 31P NMR (162 MHz, CDCl3): δ 10.6. 1H NMR (400 MHz, CDCl3): δ 3.84 (d, 3JH–P = 11.5 Hz, 6H, POCH3), 3.92 (s, 3H, COCH3), 7.06 (d, 3JH–P = 2.1 Hz, 1H, C(4)H), 7.88 (pseudo d, 3JH–H = 8.4 Hz, 2H, ArH), 8.09 (pseudo d, 3JH–H = 8.4 Hz, 2H, ArH), 13.04 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 52.1 (COCH3), 53.5 (d, 2JC–P = 5.5 Hz, POCH3), 109.4 (d, 2JC–P = 19.8 Hz, C(4)), 125.6 (2CH), 129.8, 130.2 (2CH), 133.7 (d, 1JC–P = 223.4 Hz, CP), 135.5 (CAr), 149.5 (br., C(5)), 166.7 (CO). IR (KBr): ν 3400, 1724, 1457, 1284, 1240, 1188, 1161, 1103, 1057, 1026, 787 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C13H15N2O5PNa: 333.0611. Found 333.0602.
Dimethyl [5-(2-chlorophenyl)-1H-pyrazol-3-yl]phosphonate (4e)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2e in MeOH (0.70 ml, 0.35 mmol); the reaction time was 17 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.21) gave pyrazolylphosphonate 4e as a pale yellow solid (45 mg) in 63% yield. Mp 89 °C. 31P NMR (162 MHz, CDCl3): δ 11.7. 1H NMR (400 MHz, CDCl3): δ 3.82 (d, 3JH–P = 11.5 Hz, 6H, OCH3), 7.15 (d, 3JH–P = 1.8 Hz, 1H, C(4)H), 7.25–7.31 (m, 2H, ArH), 7.44 (m, 1H, ArH), 7.71 (m, 1H, ArH), 8.94 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.3 (d, 2JC–P = 5.5 Hz, OCH3), 112.2 (d, 2JC–P = 21.5 Hz, C(4)), 127.2 (CH), 129.2, 129.7 (CH), 130.48 (CH), 130.53 (CH), 131.9 (CCl), 135.4 (d, 1JC–P = 226.4 Hz, CP), 145.6 (d, 3JC–P = 14.3 Hz, C(5)). IR (KBr): ν 3420, 1479, 1244, 1188, 1030, 837, 787, 758 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C11H12ClN2O3PNa: 309.0166. Found 309.0166.Dimethyl [5-(2-bromophenyl)-1H-pyrazol-3-yl]phosphonate (4f)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2f in MeOH (0.65 ml, 0.325 mmol); the reaction time was 16 h. Purification by preparative TLC (EtOAc/hexane: 20/1, Rf 0.21) using EtOAc/hexane as the eluent gave pyrazolylphosphonate 4f as a white solid (45 mg) in 54% yield. Mp 125 °C. 31P NMR (162 MHz, CDCl3): δ 11.3. 1H NMR (400 MHz, CDCl3): δ 3.83 (d, 3JH–P = 11.5 Hz, 6H, OCH3), 7.13 (d, 3JH–P = 1.9 Hz, 1H, C(4)H), 7.20 (ddd, 3JH–H = 8.0 Hz, 3JH–H = 7.5 Hz, 4JH–H = 1.6 Hz, 1H, ArH), 7.35 (ddd, 3JH–H = 7.6 Hz, 3JH–H = 7.5 Hz, 4JH–H = 1.2 Hz, 1H, ArH), 7.63 (dd, 3JH–H = 7.6 Hz, 4JH–H = 1.6 Hz, 1H, ArH), 7.65 (dd, 3JH–H = 8.0 Hz, 4JH–H = 1.2 Hz, 1H, ArH), 12.77 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.4 (d, 2JC–P = 5.6 Hz, OCH3), 112.5 (d, 2JC–P = 21.2 Hz, C(4)), 121.8 (CBr), 127.6 (CH), 129.9 (CH), 131.1 (CH), 131.7, 133.7 (CH), 134.7 (d, 1JC–P = 224 Hz, CP), 147.3 (br., C(5)). IR (KBr): ν 3471, 1475, 1242, 1180, 1157, 1055, 1030, 987, 958, 860, 845, 835, 766 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C11H12BrN2O3PNa: 352.9661. Found 352.9659.Dimethyl [5-(3-chlorophenyl)-1H-pyrazol-3-yl]phosphonate (4g)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2g in MeOH (0.65 ml, 0.325 mmol); the reaction time was 12 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.29) gave pyrazolylphosphonate 4g as a white solid (56 mg) in 78% yield. Mp 81 °C. 31P NMR (162 MHz, CDCl3): δ 11.4. 1H NMR (400 MHz, CDCl3): δ 3.81 (d, 3JH–P = 11.5 Hz, 6H, OCH3), 6.97 (d, 3JH–P = 2.0 Hz, 1H, C(4)H), 7.28 (m, 1H, ArH), 7.31 (m, 1H, ArH), 7.67 (m, 1H, ArH), 7.82 (m, 1H, ArH), 10.26 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.4 (d, 2JC–P = 5.5 Hz, OCH3), 108.8 (d, 2JC–P = 20.2 Hz, C(4)), 123.9 (CH), 125.9 (CH), 128.4 (CH), 130.1 (CH), 132.8, 134.8 (CCl), 134.1 (d, 1JC–P = 223.4 Hz, CP), 148.6 (d, 3JC–P = 14.4 Hz, C(5)). IR (KBr): ν 3369, 1479, 1246, 1182, 1032, 839, 787, 775 cm−1. Anal. calcd for C11H12N2ClO3P: C, 46.09; H, 4.22; N 9.77. Found: C, 46.07; H, 4.64; N, 9.34.Dimethyl [5-(4-chlorophenyl)-1H-pyrazol-3-yl]phosphonate (4h)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2h in MeOH (0.65 ml, 0.325 mmol); the reaction time was 4 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.2) gave pyrazolylphosphonate 4h as a white solid (62 mg) in 87% yield. Mp 143 °C. 31P NMR (162 MHz, CDCl3): δ 11.0. 1H NMR (400 MHz, CDCl3): δ 3.84 (d, 3JH–P = 11.5 Hz, 6H, OCH3), 6.98 (d, 3JH–P = 2.1 Hz, 1H, C(4)H), 7.38 (pseudo d, 3JH–H = 8.5 Hz, 2H, ArH), 7.74 (pseudo d, 3JH–H = 8.5 Hz, 2H, ArH), 13.22 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.4 (d, 2JC–P = 5.5 Hz, OCH3), 108.8 (d, 2JC–P = 20.4 Hz, C(4)), 127.1 (2CH), 129.0 (2CH), 129.6, 134.2 (CCl), 134.1 (d, 1JC–P = 223.3 Hz, CP), 148.8 (d, 3JC–P = 14.4 Hz, C(5)). IR (KBr): ν 3388, 1495, 1246, 1178, 1059, 1018, 833, 771 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C11H12ClN2O3PNa: 309.0166. Found 309.0167.Dimethyl [5-(4-bromophenyl)-1H-pyrazol-3-yl]phosphonate (4i)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2i in MeOH (0.70 ml, 0.35 mmol); the reaction time was 3 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.2) gave pyrazolylphosphonate 4i as a white solid (72 mg) in 87% yield. Mp 159 °C. 31P NMR (162 MHz, CDCl3): δ 10.75. 1H NMR (400 MHz, CDCl3): δ 3.84 (d, 3JH–P = 11.5 Hz, 6H, OCH3), 6.98 (d, 3JH–P = 2.1 Hz, 1H, C(4)H), 7.54 (pseudo d, 3JH–H = 8.5 Hz, 2H, ArH), 7.68 (pseudo d, 3JH–H = 8.5 Hz, 2H, ArH), 11.90 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.5 (d, 2JC–P = 5.5 Hz, OCH3), 108.9 (d, 2JC–P = 19.9 Hz, C(4)), 122.4 (CBr), 127.4 (2CH), 130.2, 132.0 (2CH), 133.6 (d, 1JC–P = 222.6 Hz, CP), 149.4 (d, 3JC–P = 14.8 Hz, C(5)). IR (Nujol): ν 1492, 1385, 1240, 1175, 1080, 1050, 998, 845, 795 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C11H12BrN2O3PNa: 352.9661. Found 352.9660.Dimethyl [5-(4-(methylthio)phenyl)-1H-pyrazol-3-yl]phosphonate (4j)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2j in MeOH (1.5 ml, 0.75 mmol); the reaction time was 3 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.17) gave pyrazolylphosphonate 4j as a white solid (64 mg) in 86% yield. Mp 138 °C. 31P NMR (162 MHz, CDCl3): δ 11.6. 1H NMR (400 MHz, CDCl3): δ 2.49 (s, 3H, SCH3), 3.84 (d, 3JH–P = 11.5 Hz, 6H, OCH3), 6.98 (d, 3JH–P = 1.9 Hz, 1H, C(4)H), 7.28 (pseudo d, 3JH–H = 8.3 Hz, 2H, ArH), 7.69 (pseudo d, 3JH–H = 8.3 Hz, 2H, ArH), 13.04 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 15.6 (SCH3), 53.3 (d, 2JC–P = 5.5 Hz, OCH3), 108.3 (d, 2JC–P = 20.8 Hz, C(4)), 126.1 (2CH), 126.7 (2CH), 127.4, 139.2 (CSMe), 135.0 (d, 1JC–P = 225.6 Hz, CP), 148.8 (d, 3JC–P = 13.2 Hz, C(5)). IR (KBr): ν 3383, 1242, 1493, 1171, 1068, 1038, 985, 841, 779 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C12H15N2O3PSNa: 321.0433. Found 321.0429.Dimethyl [5-(3-methoxyphenyl)-1H-pyrazol-3-yl]phosphonate (4k)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2k in MeOH (1.5 ml, 0.75 mmol); the reaction time was 3 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.25) gave pyrazolylphosphonate 4k as a white solid (50 mg) in 71% yield. Mp 113 °C. 31P NMR (162 MHz, CDCl3): δ 11.7. 1H NMR (400 MHz, CDCl3): δ 3.83 (d, 3JH–P = 11.5 Hz, 6H, POCH3), 3.85 (s, 3H, COCH3), 6.99 (d, 3JH–P = 2.0 Hz, 1H, C(4)H), 6.90 (m, 1H, ArH), 7.30–7.38 (m, 3H, ArH), 12.69 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.3 (d, 2JC–P = 5.5 Hz, POCH3), 55.3 (OMe), 108.8 (d, 2JC–P = 20.8 Hz, C(4)), 111.1 (CH), 114.4 (CH), 118.3 (CH), 130.0 (CH), 132.0, 135.0 (d, 1JC–P = 222.0 Hz, CP), 149.2 (d, 3JC–P = 14.4 Hz, C(5)), 160.0 (COMe). IR (KBr): ν 3369, 1493, 1248, 1188, 1034, 841, 787 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C12H15N2O4PNa: 305.0662. Found 305.0654.Dimethyl (5-phenyl-1H-pyrazol-3-yl)phosphonate (4l)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2l in MeOH (0.65 ml, 0.325 mmol); the reaction time was 3 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.24) gave pyrazolylphosphonate 4l as a white solid (54 mg) in 85% yield. Mp 79 °C. 31P NMR (162 MHz, CDCl3): δ 11.85. 1H NMR (400 MHz, CDCl3): δ 3.83 (d, 3JH–P = 11.5 Hz, 6H, OCH3), 7.01 (d, 3JH–P = 2.0 Hz, 1H, C(4)H), 7.33 (t, 3JHH = 7.4 Hz, 1H, ArH), 7.41 (dd, 3JHH = 3JHH = 7.4 Hz, 2H, ArH), 7.78 (d, 3JHH = 7.4 Hz, 2H, ArH), 11.52 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.3 (d, 2JC–P = 5.5 Hz, OCH3), 108.6 (d, 2JC–P = 21.2 Hz, C(4)), 125.8 (2CH), 128.5 (CH), 128.9 (2CH), 130.5, 135.4 (d, 1JC–P = 225.6 Hz, CP), 148.7 (d, 3JC–P = 13.8 Hz, C(5)). IR (KBr): ν 3406, 1495, 1250, 1180, 1038, 787 cm−1. Anal. calcd for C11H13N2O3P: C, 52.39; H, 5.20; N, 11.11. Found: C, 52.03; H, 5.36; N, 11.20.This reaction was also repeated using vinylphosphonate 1 (448 mg, 2.5 mmol), 0.5 M solution of the diazo compound 2l in MeOH (6.5 ml, 3.25 mmol), and K2CO3 (35 mg, 0.25 mmol). The product 4l was isolated by column chromatography in 91% yield (574 mg).
Dimethyl [5-(4-fluorophenyl)-1H-pyrazol-3-yl]phosphonate (4m)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2m in MeOH (0.75 ml, 0.375 mmol); the reaction time was 3 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.38) gave pyrazolylphosphonate 4m as a colorless solid (45 mg) in 67% yield. Mp 117 °C. 31P NMR (162 MHz, CDCl3): δ 11.5. 1H NMR (400 MHz, CDCl3): δ 3.83 (d, 3JH–P = 11.5 Hz, 6H, OCH3), 6.96 (d, 3JH–P = 2.0 Hz, 1H, C(4)H), 7.09 (dd, 3JH–H = 8.7 Hz, 3JH–F = 8.7 Hz, 2H, ArH), 7.76 (dd, 3JH–H = 8.7 Hz, 4JH–F = 5.3 Hz, 2H, ArH), 13.16 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.4 (d, 2JC–P = 5.5 Hz, OCH3), 108.7 (d, 2JC–P = 20.4 Hz, C(4)), 115.8 (d, 2JC–F = 21.8 Hz, 2CH), 127.3 (d, 4JC–F = 2.7 Hz), 127.6 (d, 3JC–F = 8.2 Hz, 2CH), 134.1 (d, 1JC–P = 223.1 Hz, CP), 149.0 (d, 3JC–P = 14.2 Hz, C(5)), 162.8 (d, 1JC–F = 248.0 Hz, CF). IR (KBr): ν 3400, 1508, 1246, 1178, 1066, 1034, 837, 785 cm−1. Anal. calcd for C11H12FN2O3P: C, 48.90; H, 4.48; N, 10.37. Found: C, 48.98; H, 4.70; N, 10.07.Dimethyl [5-(4-isopropylphenyl)-1H-pyrazol-3-yl]phosphonate (4n)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2n in MeOH (2 ml, 1 mmol); the reaction time was 3 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.3) gave pyrazolylphosphonate 4n as a white solid (59 mg) in 80% yield. Mp 142 °C. 31P NMR (162 MHz, CDCl3): δ 11.9. 1H NMR (400 MHz, CDCl3): δ 1.26 (d, 3JH–H = 6.9 Hz, 6H, CCH3), 2.92 (septet, 3JH–H = 6.9 Hz, 1H, CHiPr), 3.83 (d, 3JH–P = 11.4 Hz, 6H, OCH3), 6.96 (d, 3JH–P = 1.9 Hz, 1H, C(4)H), 7.28 (pseudo d, 3JH–H = 8.2 Hz, 2H, ArH), 7.66 (pseudo d, 3JH–H = 8.2 Hz, 2H, ArH), 12.97 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 23.8 (CCH3), 33.9 (CHiPr), 53.3 (d, 2JC–P = 5.6 Hz, OCH3), 108.6 (d, 2JC–P = 21.5 Hz, C(4)), 125.8 (2CH), 127.0 (2CH), 128.0, 135.7 (d, 1JC–P = 226.4 Hz, CP), 148.5 (d, 3JC–P = 12.9 Hz, C(5)), 149.4 (CPri). IR (KBr): ν 3400, 1502, 1236, 1178, 1043, 787 cm−1. Anal. calcd for C14H19N2O3P: C, 57.14; H, 6.51; N, 9.52. Found: C, 57.35; H, 6.80; N, 9.20.Dimethyl [5-(4-methoxyphenyl)-1H-pyrazol-3-yl]phosphonate (4o)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2o in MeOH (1 ml, 0.5 mmol); the reaction time was 3 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.24) gave pyrazolylphosphonate 4o as a white solid (64 mg) in 86% yield. Mp 184 °C. 31P NMR (162 MHz, CDCl3): δ 12.0. 1H NMR (400 MHz, CDCl3): δ 3.80 (s, 3H, COCH3), 3.82 (d, 3JH–P = 11.5 Hz, 6H, POCH3), 6.91 (d, 3JH–P = 1.8 Hz, 1H, C(4)H), 6.92 (pseudo d, 3JH–H = 8.7 Hz, 2H, ArH), 7.68 (pseudo d, 3JH–H = 8.7 Hz, 2H, ArH), 10.72 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 53.3 (d, 2JC–P = 5.5 Hz, POCH3), 55.3 (COCH3), 108.0 (d, 2JC–P = 21.4 Hz, C(4)), 114.3 (2CH), 123.1, 127.1 (2CH), 135.5 (d, 1JC–P = 223.8 Hz, CP), 148.4 (d, 3JC–P = 11.5 Hz, C(5)), 159.9 (COMe). IR (KBr): ν 3452, 1618, 1510, 1252, 1230, 1178, 1049, 1028, 841, 827, 785 cm−1. Anal. calcd for C12H15N2O4P: C, 51.07; H, 5.36; N, 9.93. Found: C, 50.81; H, 5.33; N, 9.79.Dimethyl [5-(4-ethoxyphenyl)-1H-pyrazol-3-yl]phosphonate (4p)
The reaction was carried out as described for 4a using 0.5 M solution of the diazo compound 2p in MeOH (0.8 ml, 0.4 mmol); the reaction time was 2 h. Purification by preparative TLC (CH2Cl2/MeOH: 20/1, Rf 0.26) gave pyrazolylphosphonate 4p as a yellowish solid (47 mg) in 63% yield. Mp 165 °C. 31P NMR (162 MHz, CDCl3): δ 12.5. 1H NMR (400 MHz, CDCl3): 1.40 (t, 3JH–H = 7.0 Hz, 3H, CCH3), 3.80 (d, 3JH–P = 11.4 Hz, 6H, OCH3), 4.02 (q, 3JH–H = 7.0 Hz, 2H, CH2), 6.88 (d, 3JH–P = 1.8 Hz, 1H, C(4)H), 6.90 (pseudo d, 3JH–H = 8.8 Hz, 2H, ArH), 7.66 (pseudo d, 3JH–H = 8.8 Hz, 2H, ArH), 12.98 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 14.8 (CCH3), 53.3 (d, 2JC–P = 5.5 Hz, OCH3), 63.5 (CH2), 107.8 (d, 2JC–P = 21.6 Hz, C(4)), 114.8 (2CH), 122.8, 127.1 (2CH), 135.9 (d, 1JC–P = 229.1 Hz, CP), 148.2 (br., C(5)), 159.3 (COEt). IR (KBr): ν 3369, 1508, 1250, 1178, 1030, 985, 841, 781 cm−1. Anal. calcd for C13H17N2O4P: C, 52.70; H, 5.78; N, 9.46. Found: C, 52.17; H, 5.64; N, 8.94.Ethyl 5-(dimethoxyphosphoryl)-5-(formamido)-4,5-dihydro-1H-pyrazole-3-carboxylate (5)
A foil-covered 4 ml vial was charged with vinylphosphonate 1 (50 mg, 0.28 mmol) and ethyl diazoacetate (60 μl, 0.56 mmol). The reaction mixture was stirred at 80 °C for 4 h. Purification by column chromatography (CH2Cl2/MeOH: 20/1, Rf 0.32) afforded 2-pyrazoline 5 as a colorless oil (66 mg) in 80% yield; a 84:16 mixture of two rotamers in CDCl3. 31P NMR (162 MHz, CDCl3): δ 19.4 (major), 18.5 (minor). 1H NMR (400 MHz, CDCl3) (only for the major rotamer): δ 1.29 (t, 3JH–H = 7.1 Hz, 3H, CCH3), 3.31 (dd, 3JH–P = 18.2 Hz, 2JH–H = 18.2 Hz, 1H, C(4)H), 3.43 (dd, 3JH–P = 7.2 Hz, 2JH–H = 18.2 Hz, 1H, C(4)H), 3.80 (d, 3JH–P = 10.6 Hz, 3H, OCH3), 3.83 (d, 3JH–P = 10.4 Hz, 3H, OCH3), 4.25 (q, 3JH–H = 7.1 Hz, 2H, OCH2) 7.77 (br. s, 1H, NHCO), 7.93 (br. s, 1H, NNH), 8.09 (s, 1H, CHO). 13C NMR (101 MHz, CDCl3): δ 14.1 (CCH3, major), 14.0 (CCH3, minor), 41.1 (2JC–P = 3.2 Hz, C(4), major), 41.8 (2JC–P = 2.9 Hz, C(4), minor), 54.0 (2JC–P = 7.3 Hz, OCH3, minor), 54.2 (2JC–P = 7.3 Hz, OCH3, major), 55.1 (2JC–P = 6.6 Hz, OCH3, major), 55.3 (2JC–P = 6.3 Hz, OCH3, minor), 61.5 (OCH2, major), 61.6 (OCH2, minor), 75.1 (1JC–P = 198.1 Hz, CP, minor), 76.2 (1JC–P = 199.8 Hz, CP, major), 141.5 (3JC–P = 12.2 Hz, C(3), minor), 142.3 (3JC–P = 11.4 Hz, C(3), major), 161.7 (CHO, minor), 161.8 (3JC–P = 3.1 Hz, CHO, major), 163.4 (C(O)O, minor), 163.6 (C(O), major). IR (KBr): ν 3192, 1697, 1666, 1460, 1379, 1263, 1213, 1178, 1037, 1022 cm−1. HRMS (ESI): m/z [M + Na]+ calcd for C9H16N3O6PNa: 316.0669. Found 316.0672.
Ethyl 3-(dimethoxyphosphoryl)-1H-pyrazole-5-carboxylate (6)
To a solution of 2-pyrazoline 5 (52 mg, 0.18 mmol) in anhydrous methanol (2 mL) two drops of Me3SiCl were added. After stirring at rt for 4 h under an argon atmosphere, the mixture was evaporated under vacuum 2 mmHg. The residue was suspended in CH2Cl2 and filtered. The filtrate was evaporated under vacuum to provide the pure pyrazole 6 as a colorless solid (39 mg) in 89% yield. Mp 109 °C. 31P NMR (162 MHz, CDCl3): δ 9.6. 1H NMR (400 MHz, CDCl3): δ 1.36 (t, 3JH–H = 7.1 Hz, 3H, CCH3), 3.79 (d, 3JH–P = 11.5 Hz, 6H, 2OCH3), 4.38 (q, 3JH–H = 7.1 Hz, 2H, CH2), 7.21 (d, 3JH–P = 11.5 Hz, 1H, C(4)H). 13C NMR (101 MHz, CDCl3): δ 14.17 (CCH3), 53.5 (d, 2JC–P = 5.6 Hz, OCH3), 61.4 (CH2), 114.6 (d, 2JC–P = 19.9 Hz, C(4)), 134.6 (1JPC = 224.2 Hz, CP), 141.5 (d, 3JPC = 13.6 Hz, C(5)), 160.8 (CO). IR (KBr): ν 3442, 1734, 1730, 1462, 1257, 1242, 1176, 1047 cm−1. Anal. calcd for C8H13N2O5P: C, 38.72; H, 5.28; N, 11.29. Found: C, 38.87; H, 5.40; N, 10.99.Ethyl 5-(diethoxyphosphoryl)-5-phenyl-4,5-dihydro-1H-pyrazole-3-carboxylate (8)
To a solution of vinylphosphonate 7a (96 mg, 0.4 mmol) in THF (2 ml) ethyl diazoacetate (63 μl, 0.6 mmol) and K2CO3 (3 mg, 0.02 mmol) were added. After stirring at 50 °C for 48 h, the mixture was evaporated under vacuum. The viscous residue was solidified by vigorous stirring with hexane. The resultant precipitate was collected by filtration, washed with Et2O, hexane and dried under vacuum to give 2-pyrazoline 8 as a yellow solid (61 mg) in 43% yield. 31P NMR (162 MHz, CDCl3): δ 21.5. 1H NMR (400 MHz, CDCl3): δ 1.17 (t, 3JH–H = 7.1 Hz, 3H, CH3), 1.23 (t, 3JH–H = 7.1 Hz, 3H, CH3), 1.32 (t, 3JH–H = 7.1 Hz, 3H, CH3), 3.33 (dd, 3JH–P = 24.2 Hz, 2JH–H = 17.6 Hz, 1H, C(4)H), 3.81 (dd, 3JH–P = 26.2 Hz, 2JH–H = 17.6 Hz, 1H, C(4)H), 3.86–4.09 M (m, 4H, POCH2), 4.28 (q, 3JH–H = 7.1 Hz, 2H, COCH2), 6.98 (br. d, 3JH–P = 4.5 Hz, 1H, NH), 7.30 (m, 1H, ArH), 7.37 (m, 2H, ArH), 7.45 (m, 2H, ArH). 13C NMR (101 MHz, CDCl3): δ 14.2 (COCH2CH3), 16.27 (POCH2CH3), 16.31 (POCH2CH3), 41.7 (C(4)), 61.3 (COCH2), 63.4 (d, 2JC–P = 7.4 Hz, POCH2), 64.0 (d, 2JC–P = 6.8 Hz, POCH2), 70.5 (d, 1JC–P = 152.6 Hz, CP), 126.6 (d, 3JC–P = 4.0 Hz, 2CH), 128.1 (CH), 128.6 (2CH), 138.7, 142.1 (C(3)), 162.0 (CO). Anal. calcd for C16H23N2O5P: C, 54.23; H, 6.54; N, 7.91. Found: C, 53.99; H, 6.31; N, 7.75.
Representative synthesis: ethyl 3-phenyl-1H-pyrazole-5-carboxylate (9a)
Method A. To a solution of vinylphosphonate 7a (96 mg, 0.4 mmol) and ethyl diazoacetate (84 μl, 0.8 mmol) in THF (2 ml) sodium hydride (60% dispersion in mineral oil, Aldrich; 40 mg, 1 mmol) was added in an argon atmosphere. The reaction mixture was stirred at 50 °C for 18 h, cooled to rt, and neutralized by the slow dropwise addition of the satd. ethereal solution of HCl (300 μl), followed by the addition of NEt3 (130 μl). Volatile components were removed on a rotary evaporator and the residue was purified by column chromatography (EtOAc/hexane: 1/2, Rf 0.8) afforded pyrazolecarboxylate 9a as a white solid (71 mg) in 82% yield.Method B. A mixture of vinylphosphonate 7a (96 mg, 0.4 mmol), ethyl diazoacetate (84 μl, 0.8 mmol), K2CO3 (3 mg, 0.8 mmol), and THF (2 ml) was stirred at 50 °C for 48 h and cooled to rt in an argon atmosphere. After the addition of sodium hydride (60% dispersion in mineral oil, Aldrich; 32 mg, 0.8 mmol), the reaction mixture was further stirred at 50 °C for 5 h, cooled, and worked up as described in Method A. Column chromatography afforded pyrazolecarboxylate 9a (57 mg) in 66% yield.
Mp 185 °C. 1H NMR (400 MHz, CDCl3): δ 1.26 (t, 3JH–H = 7.1 Hz, 3H, CH3), 4.25 (q, 3JH–H = 7.1 Hz, 2H, CH2), 7.02 (s, 1H, C(4)H), 7.32 (t, 3JH–H = 7.4 Hz, 1H, ArH), 7.38 (dd, 3JH–H = 3JH–H = 7.4 Hz, 2H, ArH), 7.72 (d, 3JH–H = 7.4 Hz, 2H, ArH), 11.40 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 14.0 (CH3), 61.0 (CH2), 105.1 (C(4)), 125.6 (2CH), 128.5 (CH), 128.8 (2CH), 130.3, 140.1 (br., C(5)), 148.1 (br., C(3)), 160.9 (CO). IR (KBr): ν 3435, 1726, 1493, 1241, 1194, 1138, 762 cm−1. Anal. calcd for C12H12N2O2: C, 66.65; H, 5.59; N, 12.96. Found: C, 66.56; H, 5.78; N, 12.85.
Ethyl 3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (9b)
Method A. The reaction was carried out as described for 9a using vinylphosphonate 7b (110 mg, 0.4 mmol). Purification by column chromatography (EtOAc/hexane: 1/2, Rf 0.8) gave pyrazolecarboxylate 9b as a yellow solid (57 mg) in 57% yield.Method B. The reaction was carried out as described for 9a using vinylphosphonate 7b (110 mg, 0.4 mmol). Column chromatography afforded pyrazolecarboxylate 9b (62 mg) in 62% yield.
Mp 141 °C. 1H NMR (400 MHz, CDCl3): δ 1.32 (t, 3JH–H = 7.1 Hz, 3H, CH3), 4.35 (q, 3JH–H = 7.1 Hz, 2H, CH2), 7.04 (s, 1H, C(4)H), 7.37 (pseudo d, 3JH–H = 8.5 Hz, 2H, ArH), 7.69 (pseudo d, 3JH–H = 8.5 Hz, 2H, ArH), 10.24 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 14.2 (CH3), 61.4 (CH2), 105.4 (C(4)), 126.9 (2CH), 129.0 (2CH), 129.5, 134.3 (CCl), 138.4 (br., C(5)), 148.8 (br., C(3)), 160.3 (CO). IR (KBr): ν 3292, 1697, 1473, 1444, 1304, 1277, 1192, 1093, 1028, 957, 833, 773 cm−1. Anal. calcd for C12H11ClN2O2: C, 57.49; H, 4.42; N, 11.17. Found: C, 57.62; H, 4.57; N, 11.01.
Ethyl 3-(4-isobutylphenyl)-1H-pyrazole-5-carboxylate (9c)
Method A. The reaction was carried out as described for 9a using vinylphosphonate 7c (119 mg, 0.4 mmol). Purification by column chromatography (EtOAc/hexane: 1/2, Rf 0.5) gave pyrazolecarboxylate 9c as a yellowish solid (65 mg) in 60% yield.Method B. The reaction was carried out as described for 9a using vinylphosphonate 7c (110 mg, 0.4 mmol); the reaction time at the first stage was 53 h. Column chromatography afforded pyrazolecarboxylate 9c (61 mg) in 56% yield.
Mp 168 °C. 1H NMR (400 MHz, CDCl3): δ 0.90 (d, 3JH–H = 6.6 Hz, 6H, CH3 iBu), 1.23 (t, 3JH–H = 7.1 Hz, 3H, OCH2CH3), 1.86 (m, 1H, CHiBu), 2.47 (d, 3JH–H = 7.2 Hz, 2H, CH2 iBu), 4.21 (q, 3JH–H = 7.1 Hz, 2H, OCH2), 6.98 (s, 1H, C(4)H), 7.15 (pseudo d, 3JH–H = 8.1 Hz, 2H, ArH), 7.61 (pseudo d, 3JH–H = 8.1 Hz, 2H, ArH), 11.81 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 14.1 (OCH2CH3), 22.3 (CH3 iBu), 30.2 (CH iBu), 45.2 (CH2 iBu), 61.0 (OCH2), 105.0 (C(4)), 125.4 (2CH), 127.8, 129.6 (2CH), 140.2 (br., C(5)), 142.3 (CBui), 148.4 (br., C(3)), 161.0 (CO). IR (KBr): ν 3159, 1730, 1464, 1379, 1242, 1130, 985 cm−1. Anal. calcd for C16H20N2O2: C, 70.56; H, 7.40; N, 10.29. C16H20N2O2. Found: C, 70.34; H, 7.34; N, 10.19.
Ethyl 3-(biphenyl-4-yl)-1H-pyrazole-5-carboxylate (9d)
Method A. The reaction was carried out as described for 9a using vinylphosphonate 7d (127 mg, 0.4 mmol). Purification by column chromatography (EtOAc/hexane: 1/2, Rf 0.7) gave pyrazolecarboxylate 9d as a yellowish solid (89 mg) in 76% yield.Method B. The reaction was carried out as described for 9a using vinylphosphonate 7d (127 mg, 0.4 mmol); the reaction time at the first stage was 55 h. Column chromatography afforded pyrazolecarboxylate 9d (93 mg) in 83% yield.
Mp 192 °C. 1H NMR (400 MHz, CDCl3): δ 1.42 (t, 3JH–H = 7.1 Hz, 3H, CH3), 3.51 (br. s, 1H, NH), 4.42 (q, 3JH–H = 7.1 Hz, 2H, CH2), 7.15 (s, 1H, C(4)H), 7.36 (t, 3JH–H = 7.3 Hz, 1H, ArH), 7.45 (dd, 3JH–H = 7.3 Hz, 3JH–H = 7.9 Hz, 2H, ArH), 7.63 (d, 3JH–H = 7.9 Hz, 2H, ArH), 7.67 (d, 3JH–H = 8.3 Hz, 2H, ArH), 7.84 (d, 3JH–H = 8.3 Hz, 2H, ArH). 13C NMR (101 MHz, DMSO-d6): δ 14.2 (CH3), 60.5 (CH2), 105.3 (C(4)), 125.9 (2CH), 126.6 (2CH), 127.1 (2CH), 127.7 (CH), 129.0 (2CH), 129.3, 139.4, 139.9, 141.6 (br., C(5)), 146.6 (br., C(3)), 160.8 (CO). IR (KBr): ν 3288, 1697, 1472, 1442, 1267, 1186, 1024, 835, 762 cm−1. Anal. calcd for C16H20N2O2: C, 73.95; H, 5.52; N, 9.58. C18H16N2O2. Found: C, 74.14; H, 5.86; N, 9.41.
Ethyl 3-(2-naphthyl)-1H-pyrazole-5-carboxylate (9e)
Method A. The reaction was carried out as described for 9a using vinylphosphonate 7e (116 mg, 0.4 mmol). Purification by column chromatography (EtOAc/hexane: 1/2, Rf 0.75) gave pyrazolecarboxylate 9e as a yellowish solid (79 mg) in 74% yield.Method B. The reaction was carried out as described for 9a using vinylphosphonate 7e (116 mg, 0.4 mmol); the reaction time at the first stage was 53 h. Column chromatography afforded pyrazolecarboxylate 9e (73 mg) in 68% yield.
Mp 187 °C. 1H NMR (400 MHz, CDCl3): δ 1.39 (t, 3JH–H = 7.1 Hz, 3H, CH3), 4.38 (q, 3JH–H = 7.1 Hz, 2H, CH2), 7.23 (s, 1H, C(4)H), 7.46–7.51 (m, 2H, ArH), 7.82–7.92 (m, 4H, ArH), 8.23 (s, 1H), 11.70 (br. s, 1H, NH). 13C NMR (101 MHz, CDCl3): δ 14.2 (CH3), 61.4 (CH2), 105.8 (C(4)), 123.6 (CH), 124.6 (CH), 126.4 (CH), 126.5 (CH), 127.7 (CH), 128.22 (CH), 128.27, 128.7 (CH), 133.3, 133.4, 138.9 (br., C(5)), 149.8 (br., C(3)), 160.6 (CO). IR (KBr): ν 3261, 1711, 1469, 1257, 1184, 1144, 1024, 820, 779 cm−1. Anal. calcd for C16H14N2O22: C, 72.16; H, 5.30; N, 10.52. C18H16N2O2. Found: C, 72.29; H, 5.41; N, 10.29.
Acknowledgements
Financial support of this work by the Russian Foundation for Basic Research (Grant No. 15-03-04594-a) and the M. V. Lomonosov Moscow State University Program of Development is gratefully acknowledged.Notes and references
- V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2013, 69, 735 CrossRef CAS PubMed
.
- C. Lamberth, Heterocycles, 2007, 71, 1467 CrossRef CAS
-
(a) J. García-Antón, R. Bofill, L. Escriche, A. Llobet and X. Sala, Eur. J. Inorg. Chem., 2012, 4775 CrossRef
-
(a) J. Pérez and L. Riera, Eur. J. Inorg. Chem., 2009, 4913 CrossRef
-
(a) M. Lilley, B. Mambwe, M. J. Thompson, R. F. Jackson and R. Muimo, Chem. Commun., 2015, 51, 7305 RSC
-
(a) J. Modranka, R. Jakubowski, M. Rozalski, U. Krajewska, A. Janecka, K. Gach, D. Pomorska and T. Janecki, Eur. J. Med. Chem., 2015, 92, 565 CrossRef CAS PubMed
-
(a) T. E. Ali and S. M. Abdel-Kariem, Heterocycles, 2012, 85, 2073 CrossRef CAS
-
(a) N. S. Goulioukina, N. N. Makukhin and I. P. Beletskaya, Russ. Chem. Rev., 2016, 85, 667, DOI:10.1070/RCR4579
- R. Muruganantham, S. M. Mobin and I. N. N. Namboothiri, Org. Lett., 2007, 9, 1125 CrossRef CAS PubMed
- S. Mohapatra, C. Bhanja, S. Jena, S. Chakroborty and S. Nayak, Synth. Commun., 2013, 43, 1993 CrossRef CAS
- For some
recent papers on 3(5)-pyrazolylphosphonates by different research groups, see:
(a) R. Kumar, D. Nair and I. N. N. Namboothiri, Tetrahedron, 2014, 70, 1794 CrossRef CAS
- N. S. Goulioukina, N. N. Makukhin and I. P. Beletskaya, Tetrahedron, 2011, 67, 9535 CrossRef CAS
- N. N. Makukhin, N. S. Goulioukina, A. G. Bessmertnykh-Lemeune, S. Brandès, R. Guilard and I. P. Beletskaya, Synthesis, 2015, 279 CAS
-
(a) H. Duddeck and R. Lecht, Phosphorus, Sulfur Silicon Relat. Elem., 1987, 29, 169 CrossRef CAS
-
L. C. Behr, R. Fusco and C. H. Jarboe, The Chemistry of Heterocyclic Compounds: Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles and Condensed Rings, ed. R. H. Wiley, John Wiley & Sons, New York, 1967, vol. 22, 888 p Search PubMed
- D. B. Reddy, M. R. Sarma, A. Padmaja and V. Padmavathi, Phosphorus, Sulfur Silicon Relat. Elem., 2000, 164, 23 CrossRef
- Vinylphosphonate 1 had been synthesized for the first time by U. Schollkopf et al. [ U. Schöllkopf, I. Hoppe and A. Thiele, Liebigs Ann. Chem., 1985, 555–559 CrossRef CAS
. - J. Geittner, R. Huisgen and H. U. Reissig, Heterocycles, 1978, 11, 109 CrossRef CAS
- A. Yu. Platonov, A. N. Evdokimov, A. V. Kurzin and H. D. Maiyorova, J. Chem. Eng. Data, 2002, 47, 1175 CrossRef CAS
-
G. Maas, in Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, John Wiley & Sons, Inc., New York, 2002, pp. 539–621 Search PubMed
- W. R. Bamford and T. S. Stevens, J. Chem. Soc., 1952, 4735 RSC
- Crystallographic data for the structure 4c (deposition number CCDC 959780).
-
(a)
L. Yet, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, pp. 1–141 Search PubMed
-
(a) T. Minami, S. Tokumasu, R. Mimasu and I. Hirao, Chem. Lett., 1985, 1099 CrossRef CAS
- For some recent papers on 3(5)-pyrazolecarboxylate synthesis, see:
(a) K. Rikimaru, T. Wakabayashi, H. Abe, H. Imoto, T. Maekawa, O. Ujikawa, K. Murase, T. Matsuo, M. Matsumoto, C. Nomura, H. Tsuge, N. Arimura, K. Kawakami, J. Sakamoto, M. Funami, C. D. Mol, G. P. Snell, K. A. Bragstad, B. C. Sang, D. R. Dougan, T. Tanaka, N. Katayama, Y. Horiguchi and Y. Momose, Bioorg. Med. Chem., 2012, 20, 714 CrossRef CAS PubMed
- For some examples of practically important 3(5)-pyrazole carboxylic acid derivatives, see:
(a) F. Reviriego, A. Sanz, P. Navarro, J. Latorre, E. García-España and M. Liu-Gonzalez, Org. Biomol. Chem., 2009, 7, 3212 RSC
-
(a) J. B. Conant, A. D. MacDonald and A. M. Kinney, J. Am. Chem. Soc., 1921, 43, 1928 CrossRef CAS
-
F. Gu, US Pat., US20090229489A1, 2009 Search PubMed
- H. Tomioka, N. Kobayashi, S. Murata and Y. Ohtawa, J. Am. Chem. Soc., 1991, 113, 8771 CrossRef CAS
- X. Creary, Org. Synth., 1986, 64, 207 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all compounds. See DOI: 10.1039/c6ob01780k |
| This journal is © The Royal Society of Chemistry 2016 |