
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge screening in RNA: an integral route for dynamical enhancements†
Joon Ho
Roh
*ab,
Madhu
Tyagi
cd,
Pulakesh
Aich
a,
Kimoon
Kim
aef,
R. M.
Briber
d and
Sarah A.
Woodson
g
aCenter for Self-assembly and Complexity (CSC), Institute for Basic Science (IBS), Pohang 37673, South Korea. E-mail: jhroh@ibs.re.kr
bBiomolecular Science, University of Science and Technology, Daejeon 34113, South Korea
cNIST Center for Neutron Research, National Institute of Standards and Techonology, Gaithersburg, Maryland 20899, USA
dDepartment of Materials Science and Engineering, University of Maryland, College Park, Maryland 20742, USA
eDepartment of Chemistry, Pohang University of Science and Technology, Pohang 37673, South Korea
fDivision of Advanced Materials Science, Pohang University of Science and Technology, Pohang 37673, South Korea
gT. C. Jenkins Department of Biophysics, Johns Hopkins University, Baltimore, Maryland 21218, USA
First published on 24th September 2015
Abstract
Electrostatic interactions of RNA are at the center of determining the dynamical flexibility and structural stability. By analysing neutron scattering spectroscopy, we show that fast dynamics of hydrated tRNA on ps to ns timescales increases with stronger charge screening, while its structural stability either increases or remains largely unchanged. An unprecedented electrostatic threshold for the onset of additional flexibility is induced from the correlation between the charge-screening density of counterions and the promoted dynamical properties. The results demonstrate that the enhanced dynamical flexibility of tRNA originates from local conformational relaxation coupled with stabilized charge screening rather than governed by fluctuation of hydrated counterions. The present study casts light on the specificity of electrostatic interactions in the thermodynamic balance between the dynamical flexibility and structural stability of RNA.
Biological macromolecules have evolved to serve their functions in accord with gene-commanded responses to endo- and exo-cellular environments. Dynamical interactions of biological macromolecules (in particular, RNAs and proteins) with solvents and hydrated solutes are an integral part of their functional adaptation in a variety of biological events, including enzymatic reaction1 and folding nucleation.2 However, understanding the conformational dynamics of biological macromolecules is non-trivial since surrounding environments lead to a complex contribution to it through electrostatic, hydration, or/and structure stabilizing interactions (hydrophobic for proteins and sequence-specific tertiary for RNA).
According to previous neutron scattering and simulation studies, the conformational dynamics of proteins is strongly coupled with diffusive motions of hydrating water molecules.3,4 More recently, our neutron scattering and dielectric studies on the dynamics of hydrated proteins and RNAs have shown that the coupled motions are attributed to the mutual response between the chemical structures of biopolymers and their interactions with water rather than controlled by the dynamics of hydrating water.4b,5
However, as compared with neutral and weakly charged proteins, understanding the dynamics of RNA remains more challenging in that the electrostatic neutralization of the negatively charged nucleic acids stabilizes the compact folded structure of RNA, while helping to increase the molecular mobility of the biological polyelectrolytes.1c,d Therefore, the dynamics of RNA must be described in the context of the specificity of charge screening to the thermodynamic link between dynamical electrostatic interactions and structural stabilization.6 A more satisfactory understanding of the charge-screening mediated connection between dynamical flexibility and structural stability requires a detailed examination of the effects of the strength of charge screening on the dynamical enhancements of hierarchically structured RNA samples.
This paper presents that pico- to nano-second conformational mobility of hydrated tRNA that translates into a dynamic energy factor is enhanced by larger charge-screening density of hydrated counterions, while it stabilizes RNA secondary or tertiary structures. This result suggests that the additional conformational mobility of tRNA is coupled with stabilized charge screening rather than the fluctuation of hydrated counterions. We also found a new turnover for the onset of additional conformational dynamics, which is ascribed to an electrostatic threshold evaluated from the correlation between the charge-screening density of counterions and the increased dynamic energy factors.
Wheat germ tRNA samples were used for this study. Details about the sample preparation including the deuteration of labile protons in tRNA, the addition of salts (MgCl2, NaCl, KCl), and hydration are described in the ESI.† The mole ratio (m) of each salt to the phosphate groups of the tRNA was 30. The equimolar amount was chosen among the salts to explore the concomitant variation of dynamics with (1) the strength of charge screening, (2) the charge density of hydrated counterions, (3) equivalent hydration, and (4) hierarchical structural stabilization. The charge density of hydrated counterions reflects the relative difference in the total electrostatic screening, referred to as the charge-screening density. Structural studies employing small-angle X-ray scattering spectroscopy verified that the addition of 30 M Mg2+ enables tRNA to fold into compact native structures with the formation of tertiary interactions, while it retains extended secondary conformations with only a monovalent counterion Na+ or K+.6,7 The hydration of tRNA samples was conducted under 100% D2O humidity in a desiccator. The extent of hydration for wet samples was 0.65h (h: the weight of hydrating water in grams/1 g tRNA), which corresponds to a single hydration layer. The difference in the total amount of hydrating water caused by the presence of additional 30 M Cl− for MgCl2 is estimated to be <0.02h. The dry sample contains a small amount of water corresponding to ∼0.05h after 48 hours of lyophilization under 0.3 mTorr. Elastic scattering scans were carried out on HFBS (NG2 at the NIST Center for Neutron Research) at a rate of 0.7 K min−1 between 50 and 320 K. Dynamic scattering spectra were collected in the energy window of 17 μeV and the resolution of 0.8 μeV, which cover the time range from 40 ps to 2 ns.
Fig. 1 shows the temperature variations of the mean-squared displacements, 〈r2(T)〉, of hydrogen atoms in dry and wet tRNA samples with or without counterions. It is estimated using the following equation: 〈r2(T)〉 = −3Q−2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln[Iel(Q, T)/Iel(Q, 4 K)], where Iel is the elastic neutron scattering intensity. 〈r2(T)〉 provides information about the damped local motions faster than the resolution ∼2 ns including segmental, secondary dynamics, and the collective vibrations.
ln[Iel(Q, T)/Iel(Q, 4 K)], where Iel is the elastic neutron scattering intensity. 〈r2(T)〉 provides information about the damped local motions faster than the resolution ∼2 ns including segmental, secondary dynamics, and the collective vibrations.
 | ||
| Fig. 1 Mean-squared displacements, 〈r2(T)〉, of H-atoms in tRNA samples. Error bars represent standard deviations. | ||
The temperature dependence of 〈r2(T)〉 reveals that all wet tRNA samples exhibit a dynamic transition for the onset of anharmonic motions, defined as the sudden rise of 〈r2(T)〉, at T ∼ 210 K, while it is absent in the dry sample. Given that the onset of the conformational relaxation is coupled with the dynamics of hydrating water,3 the similarity of dynamic transition temperatures (Td) suggests that the extent of hydration-induced dynamical enhancements changes little regardless of the addition of the salts. The Td values would otherwise shift to higher temperatures for less hydration as reported previously.5
The thermal scanning of the UV-absorption at 260 nm shows that the Mg2+-tRNA sample folds into 3D structures (Fig. S1, ESI†). In contrast, the Na+- and K+-tRNA samples hold mostly secondary structures with a lack of long-range tertiary interactions since the monovalent cations offer cationic charges as half as that of the Mg2+ (Fig. S1, ESI†).6 The largest 〈r2(T)〉 in Mg2+-tRNA reflects the greatest flexibility, while it has the most compact folded structures.7 Interestingly, the flexibility of the other tRNA samples becomes larger in the order of Na+ > K+ > dialyzed (Fig. 1), while the structural stability is slightly increased in the presence of the monovalent cations, but remains largely unchanged between them. The above results show that stronger electrostatic interactions attributed to greater charge density of hydrated counterions (Table 1) are a key factor in enhancing the dynamic collective motions of tRNA upon charge screening. Larger scale motions are probably coupled with the spatial ranges of stabilized charge screening rather than governed by the entropic fluctuation of hydrated cations in which case relatively loosely bound K+ would result in greater flexibility of tRNA like plasticizers: more flexibility of RNA with more strongly bound counterions upon hydration. It is worth noting that larger 〈r2(T)〉 of Na+-tRNA than that of K+-tRNA despite their similar structures evidences that the dependence of fast dynamics on charge-screening density is stronger than that of structural interactions. This is consistent with our earlier results that reported similar flexibility between RNA and synthetic polyelectrolytes.6
 at each temperature
at each temperature
| Cation | ζ [e Å−3] | T [K] |
|
|---|---|---|---|
| a ζ = 2e/(4π/3)R3, where R is the radius of the hydrated cation. | |||
| Mg2+ | 0.055 | 260 | 10.71 ± 0.08 |
| Na+ | 0.017 | 280 | 9.07 ± 0.07 |
| K+ | 0.012 | 300 | 7.57 ± 0.08 |

Since sole analysis of 〈r2(T)〉 is not able to discriminate among the different motions, we measure the energy-resolved spectra covering 40 ps to 2 ns. Fig. 2 shows dynamic structure factors summed over all Q. Consistent with the results of 〈r2(T)〉, the broadening of Quasi-Elastic Neutron Scattering (QENS) becomes larger following the same order as Mg2+ > Na+ > and K+ > dialyzed at all three temperatures. This again confirms that Mg2+-induced charge screening results in greater conformational flexibility of the RNA accessed in the time window (Fig. 2). The QENS spectra were fit to a function representing the deconvolution of the total scattering spectrum into an elastic and a quasielastic functions which correspond respectively to the scattering of immobile H-atoms and the relaxation motions of the mobile fraction: S(Q, E) = DW(Q)[EISF(Q)δ(E) + QISF(Q)L(Γ, E)] ⊗ R(E), where ⊗ signifies convolution, and L(Γ, E) = π−1[Γ/(E2 + Γ2)]. DW, L, Γ, and R(E) are the Debye–Waller factor, a Q-dependent Lorentzian function, the half-width at half-maximum of the QENS peak, and a resolution function, respectively. EISF(Q) and QISF(Q) denote the elastic and quasi-elastic incoherent scattering factors, respectively.
 | ||
| Fig. 2 S(Q, E) summed over all Q ranges of wet tRNA samples at T = (a) 260 K, (b) 280 K, and (c) 300 K. Resolution function is Na+-tRNA at 10 K. | ||
The relaxation rates are assessed with a single Lorentzian function (an example of the QENS analysis with a single Lorentzian function is shown in Fig. S2, ESI†). For the rationale behind the approximation of the relaxation spectrum as the Lorentzian function, please see ref. 6 and 8. In Fig. 3A the temperature variations of the relaxation times show slight non-Arrhenius behaviour indicating cooperative conformational motions. The average relaxation rate of tRNA becomes faster with hydrated counterions of lager charge-screening density at temperatures between 260 and 300 K.
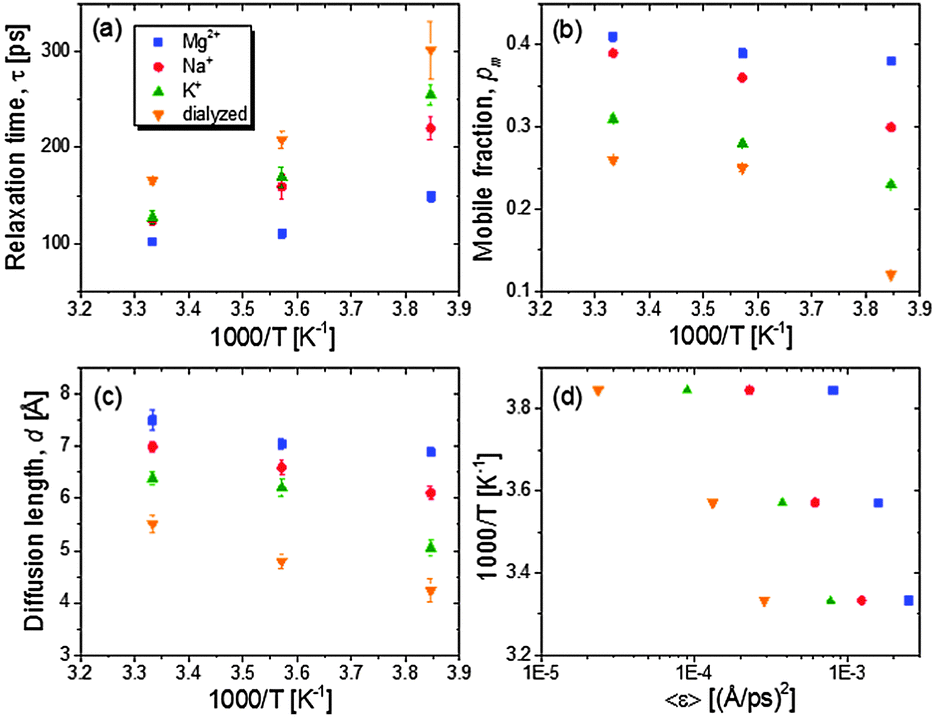 | ||
| Fig. 3 Dynamic parameters obtained from the analysis of quasielastic dynamic structure factor: (a) τ; (b) pm; and (c) d. The τ values are calculated from Γ values of quasielastic Lorentzian relaxation functions and averaged over Q ranges between 0.87 Å−1 and 1.68 Å−1, where no Q-dependence of τ is observed. (d) shows the variation of fractional dynamic energy factors with inverse temperatures. | ||
The partitioning of the hydrogen atoms into conformational relaxation and their diffusive distance are characterized from the Q-dependent fit of a model representing diffusion in a sphere: EISF(Q) = 1 − pm + pm[3j1(Qd)/(Qd)]2, where pm and d are mobile fraction and the radius of the sphere (Fig. S3, ESI†). Dialyzed tRNA exhibits a second decay in the EISF(Q) at higher Q ∼ 1.3 Å−1 (∼4 Å), which does not appear for all counterion-mediated tRNA samples. Though beyond the scope of this paper, this implies that more localized motions also exist distinctively in the unscreened structures, probably originating from local structures loosely constrained by weaker electrostatic repulsion formed by sparse surrounding negative charges. The present study focuses on the analysis of larger scale motions emerging at lower Q, the nature of which is comparable to the relaxation observed in salted tRNA samples, and presumably more likely to be affected by the electrostatic interactions.
The resulting values of pm and d are plotted against inverse temperatures (Fig. 3B and C). The pm value of Mg2+-tRNA turns out to be larger than that of Na+- and K+-tRNA, ranging from 0.38 at 260 K to 0.41 at 300 K. Since the accessed timescales are too limited to characterize the full distribution of relaxation, the estimated pm does not represent the total fraction of mobile hydrogen atoms. A strong increase of temperature-dependent pm is observed in Na+-tRNA, reaching 0.39 at 300 K, which is very similar to the pm of Mg2+-tRNA. The relaxation length scale, d, of Mg+-tRNA is also greater than that of other counterion-mediated tRNAs in the temperature range between 260 and 300 K. The d value of the folded Mg2+-tRNA is ∼7.5 Å at 300 K, which reflects the relaxation of its localized structures involving ∼40% of total hydrogen atoms (pm ∼ 0.4). The spatial ranges of the localized motions are about 50% and 20% of the size of its helical unit and global structure.5
MD simulation and NMR studies revealed that hinge motions emerge from the bulges or helix junctions in HIV transactivation response (TAR) RNA on nanosecond timescales.9 Similar to the hinge motions, the fast dynamics of tRNA traced by QENS is believed to originate from the average localized motions related to inter-secondary structures between the acceptor stem and the TψC loop stem, or between the anticodon stem and the D loop stem.5
As it shows lower 〈r2(T)〉 at T above Td, the K+-tRNA has the local conformational motions of smaller pm and d than Na+-tRNA at all temperatures. For dialyzed tRNA, the most remarkable result is the decrease of pm down to ∼0.1 at 260 K while it remains larger than 0.2 for salted samples. This indicates that the electrostatic repulsion drastically reduces the thermally mobile segments as temperature is near Td ∼ 210 K.
The energetic assessment of dynamical flexibility pertaining to complicated molecular motions of biopolymers is not trivial and requires understanding of several dynamical parameters.5 To follow the energetic variation associated with the fast conformational relaxation of tRNA, we introduce an averaged fractional dynamic energy factor, 〈ε〉, expressed by combining the three dynamic parameters: d, τ, and pm:  . (Note: the term “relative” describes the weighting of pm, which assesses the part of total energy). The presentation of the comprehensive dynamic properties 〈ε〉 is attempted to evaluate the relative kinetic energy of the locally diffusive motions of H-atoms in the tRNA samples.
. (Note: the term “relative” describes the weighting of pm, which assesses the part of total energy). The presentation of the comprehensive dynamic properties 〈ε〉 is attempted to evaluate the relative kinetic energy of the locally diffusive motions of H-atoms in the tRNA samples.
Fig. 3D shows that the calculated 〈ε〉 values nonlinearly vary with temperature (for an ideal case, thermal and dynamic energies are linearly dependent on a pre-factor 2/3), reflecting the cooperative segmental relaxation. The shift of temperature variation of 〈ε〉 to larger values with greater charge-screening density indicates that the dynamical flexibility pertaining to conformational relaxation of tRNA is enhanced by stronger charge screening.
The greater flexibility induced by ionizing salts was reported for halophilic proteins such as malate dehydrogenase and 2Fe–2S ferredoxin from Haloarcula marismorti, which also consist of highly negative surface charge densities.10 However, the structural stability of the proteins varies with different types of salts because of the thermodynamic difference in their interactions with water and the subsequent solvated structures: salting-in salts (Kosmotropic) stabilize the proteins, but salting-out (Chaotropic) destabilize at molar concentrations. Since the thermodynamic interplays become more complex for biologically important intermediate salts including NaCl and KCl, the specificity of electrostatic interactions to the balance between dynamic flexibility and structural stability has not been straightforwardly evaluated for the halophilic proteins.10
Likewise, the role of charge screening in the inextricable connection for RNA remains poorly assessed. We have been able to examine it in tRNA as we show below that the dynamical flexibility is strongly correlated with the charge-screening density engendered by hydrated counterions. The additional flexibility of tRNA driven by charge screening alone is examined in terms of Δ〈ε〉, which is obtained by subtracting the 〈ε〉 value of dialyzed tRNA from the 〈ε〉 value of salted tRNA samples (assuming there is no significant change in hydration-enhanced flexibility between the dialyzed and salted samples as long as they are hydrated with similar amount of water molecules, which is rationalized by the similar Td). Interestingly, Δε/T turns out to be directly proportional to the logarithmic of charge-screening density with unique ζ values extrapolated at the zero-point dynamic energy  at each temperature (Fig. 4 and Fig. S4, ESI,† and Table 1). Accordingly, the empirical correlation can be expressed as
at each temperature (Fig. 4 and Fig. S4, ESI,† and Table 1). Accordingly, the empirical correlation can be expressed as  , which manifests that the change of dynamic energy factor is reflected by the extent of charge-screening density of hydrated counterions. The presence of nonzero
, which manifests that the change of dynamic energy factor is reflected by the extent of charge-screening density of hydrated counterions. The presence of nonzero  implies that there is a threshold of charge screening sufficient to trigger the onset of additional conformational exploration on sub-ns timescales. Above the threshold, the relevant dynamic energy probably outcompetes localized structural interactions bridged by hydrated counterions. At 300 K, the electrostatic threshold is about 63% of the charge-screening density of hydrated K+. Weaker electrostatic interactions at higher temperatures account for the decreased
implies that there is a threshold of charge screening sufficient to trigger the onset of additional conformational exploration on sub-ns timescales. Above the threshold, the relevant dynamic energy probably outcompetes localized structural interactions bridged by hydrated counterions. At 300 K, the electrostatic threshold is about 63% of the charge-screening density of hydrated K+. Weaker electrostatic interactions at higher temperatures account for the decreased  with increasing temperature.
with increasing temperature.
 | ||
| Fig. 4 Change in dynamic energy factor, Δ〈ε〉, corresponding to additional conformational motions of wet tRNA driven by charge screening vs. logarithmic charge-screening density of hydrated counterions at 260, 280, and 300 K. | ||
In conclusion, we report that more and denser charge screening (i.e. larger charge-screening density around RNA) engendered by hydrated counterions serves to enhance the dynamic flexibility of tRNA, while also inducing structural stabilization. The increased mobility is concomitantly attributed to more mobile fractions of faster conformational motions with a larger mobile length scale. A threshold of charge screening that triggers the onset of the additional flexibility is estimated from the correlation between the charge-screening density of the hydrated counterions and the enhanced comprehensive dynamics of tRNA. This implies that larger flexibility of tRNA driven by the greater charge-screening density arises from local conformational relaxation coupled with stabilized charge screening rather than governed by the fluctuation of hydrated counterions.
Acknowledgements
J.H.R., P.A., and K.K. acknowledge support from the Institute for Basic Science (IBS) [IBS-R007-D1]. This work utilized facilities (NCNR High Flux Backscattering Spectrometer) supported in part by the National Science Foundation under Agreement No. DMR-0944772.Notes and references
- (a) B. F. Rasmussen, A. M. Stock, D. Ringe and G. A. Petsko, Nature, 1992, 357, 423 CrossRef CAS PubMed; (b) J. A. Rupley and G. Careri, Adv. Protein Chem., 1991, 41, 37 CrossRef CAS; (c) E. Westhof, Annu. Rev. Biophys. Biophys. Chem., 1988, 17, 125 CrossRef CAS PubMed; (d) J. Yoon, J.-C. Lin, C. Hyeon and D. Thirumalai, J. Phys. Chem. B, 2014, 118, 7910 CrossRef CAS PubMed.
- (a) Z. Bu, J. Cook and D. J. E. Callaway, J. Mol. Biol., 2001, 312, 865 CrossRef CAS PubMed; (b) V. Receveur, P. Calmettes, J. C. Smith, M. Desmadril, G. Coddens and D. Durand, Proteins: Struct., Funct., Genet., 1997, 28, 380 CrossRef CAS; (c) E. Mamontov, H. O'Neill and Q. Zhang, J. Biol. Phys., 2010, 36, 291 CrossRef CAS PubMed.
- (a) P. W. Fenimore, H. Frauenfelder, B. H. McMahon and F. G. Parak, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16047 CrossRef CAS PubMed; (b) A. L. Tournier, J. C. Xu and J. C. Smith, Biophys. J., 2003, 85, 1871 CrossRef CAS; (c) K. Wood, A. Frolich, A. Paciaroni, M. Moulin, M. Hartlein, G. Zaccai, D. J. Tobias and M. Weik, J. Am. Chem. Soc., 2008, 130, 4586 CrossRef CAS PubMed; (d) S. Khodadadi and A. P. Sokolov, Soft Matter, 2015, 11, 4984 RSC.
- (a) G. Zaccai, Philos. Trans. R. Soc. London, Ser. B, 2004, 359, 1269 CrossRef CAS PubMed; (b) S. Khodadadi, J. H. Roh, A. Kisliuk, E. Mamontov, M. Tyagi, S. A. Woodson, R. M. Briber and A. P. Sokolov, Biophys. J., 2010, 98, 1321 CrossRef CAS PubMed; (c) K. Wood, M. Plazanet, F. Gabel, B. Kessler, D. Oesterhel, D. J. Tobias, G. Zaccai and M. Weik, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18049 CrossRef CAS PubMed; (d) X. Li, M. Zamponi, K. Hong, L. Porcar, C.-Y. Shew, T. Jenkins, E. Liu, G. S. Smith, K. W. Herwig, Y. Liu and W.-R. Chen, Soft Matter, 2011, 7, 618 RSC.
- J. H. Roh, R. M. Briber, A. Damjanovic, D. Thirumalai, S. A. Woodson and A. P. Sokolov, Biophys. J., 2009, 96, 2755 CrossRef CAS PubMed.
- J. H. Roh, M. Tyagi, R. M. Briber, S. A. Woodson and A. P. Sokolov, J. Am. Chem. Soc., 2011, 133, 16406 CrossRef CAS PubMed.
- (a) D. E. Draper, D. Grilley and A. M. Soto, Annu. Rev. Biophys. Biomol. Struct., 2005, 34, 221 CrossRef CAS PubMed; (b) S. Moghaddam, G. Caliskan, S. Chauhan, C. Hyeon, R. M. Briber, D. Thirumalai and S. A. Woodson, J. Mol. Biol., 2009, 393, 753 CrossRef CAS PubMed.
- (a) M. Kofu, T. Someya, S. Tatsumi, K. Ueno, T. Ueki, M. Watanabe, T. Matsunaga, M. Shibayama, V. Garcia Sakai and M. Tyagi, Soft Matter, 2012, 8, 7888 RSC; (b) S. Khodadadi, S. Pawlus, J. H. Roh, V. G. Sakai, E. Mamontov and A. P. Sokolov, J. Chem. Phys., 2008, 128, 195106 CrossRef CAS PubMed.
- (a) Y. Mu and G. Stock, Biophys. J., 2006, 90, 391 CrossRef CAS PubMed; (b) Q. Zhang, X. Sun, E. D. Watt and H. M. Al-Hashimi, Science, 2006, 311, 653 CrossRef CAS PubMed.
- (a) M. Tehei, D. Madern, C. Pfister and G. Zaccai, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14356 CrossRef CAS PubMed; (b) M. Mevarech, F. Frolow and L. M. Gloss, Biophys. Chem., 2000, 86, 155 CrossRef CAS; (c) M. T. Record, W. Zhang and C. F. Anderson, Adv. Protein Chem., 1998, 51, 281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sm02084k |
| This journal is © The Royal Society of Chemistry 2015 |