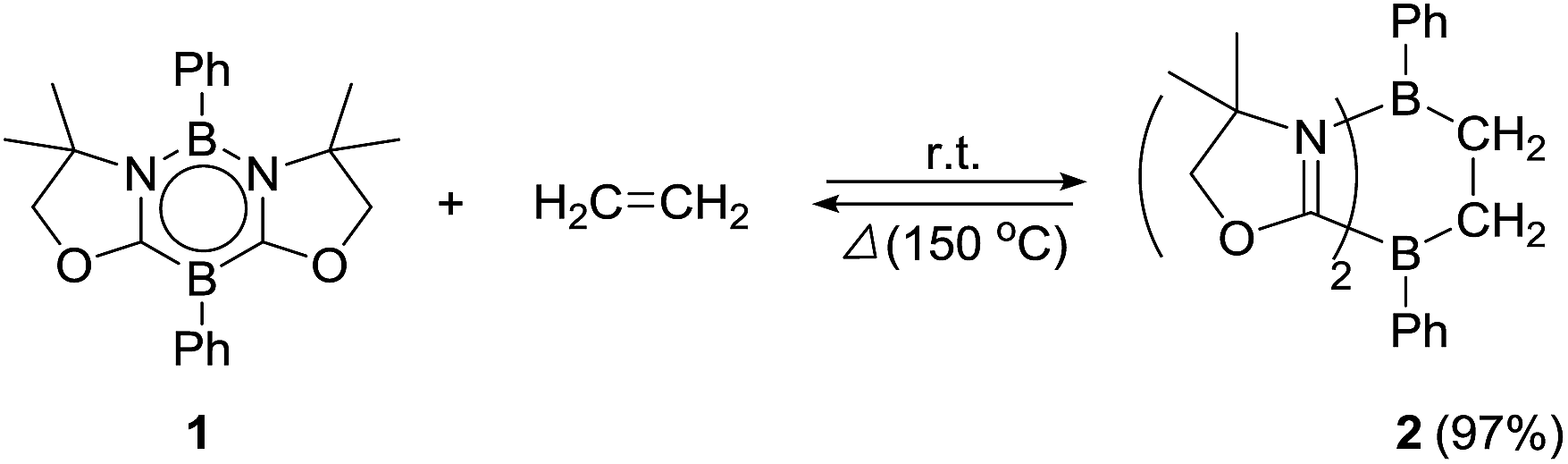DOI:
10.1039/C5SC03174E
(Edge Article)
Chem. Sci., 2015,
6, 7150-7155
Reversible [4 + 2] cycloaddition reaction of 1,3,2,5-diazadiborinine with ethylene†
Received
26th August 2015
, Accepted 14th September 2015
First published on 15th September 2015
Abstract
Under ambient conditions, a [4 + 2] cycloaddition reaction of 1,3,2,5-diazadiborinine 1 with ethylene afforded a bicyclo[2.2.2] derivative 2, which was structurally characterized. The cyclization process was found to be reversible, and thus retro-[4 + 2] cycloaddition reproduced 1 quantitatively, concomitant with the release of ethylene. Compound 1 reacted regio-selectively and stereo-selectively with styrene derivatives and norbornene, respectively, and these processes were found to be reversible too. Computational studies determined the reaction pathways which were consistent with the regio-selectivity observed in the reaction of styrene, and the reaction was suggested to be essentially concerted but highly asynchronous.
Introduction
Reversible coordination processes between a metal center and olefins are of paramount significance in transition metal catalysis.1 The labile yet attractive metal-olefin interactions can be accounted for by the Dewar–Chatt–Duncanson model;2 this model rationalizes, for example, the bonding mode in Zeise's salt [Cl3Pt(η2-C2H4)]K,3 in which the donation of π-electrons from ethylene to an unoccupied d-orbital of the metal and π-backdonation of d-electrons from the metal to the π*-orbital of ethylene occur concurrently.4 Such simultaneous donor–acceptor interactions give unique chemical properties to transition metal complexes.
In recent years, it has been amply demonstrated that main group elements can behave as transition metals,5e.g., in unique activation reactions of ethylene6 as well as other olefins7 where several systems were found to bind to olefins reversibly (Fig. 1). In 2009, Power et al. reported a cycloaddition reaction of distannyne Ia with two equivalents of ethylene, which afforded a bicycle[2.2.0] derivative Ib.8a Remarkably, Ib readily released ethylene molecules via thermal retro-cycloaddition under reduced pressure or upon standing at 25 °C, to reproduce Ia. Kato, Baceiredo, and co-workers demonstrated that ethylene could be incorporated onto phosphonium sila-ylide, giving rise to silirane IIb.9 The formation of IIb depended on the ethylene pressure, and indeed, IIb reverted to IIa after a decrease in ethylene pressure. Recently, Tuononen and Power et al. reported the reversible binding of ethylene to two-coordinate acyclic silylene IIIa under ambient conditions, which generated sililane IIIb with a tetra-coordinate silicon atom.10 Very recently, Tokitoh and Sasamori et al. reported that 1,2-digermacyclobutene IVa underwent sequestration of two ethylene molecules to produce IVb.11 Retro-conversion of IVb to IVa was observed by reducing the ethylene pressure. In these reactions, the formation of the cycloadducts is inferred to involve a [1 + 2] interaction between the heavier group 14 element center and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of ethylene in the initial step. With a group 13 compound, Jordan et al. reported that the cationic aluminum β-diketiminate derivative Va and ethylene underwent a [4 + 2] cycloaddition reaction, yielding the cycloadduct Vb, which was characterized only by NMR spectroscopy.12 Although Vb did not release ethylene even under vacuum, the addition of N,N-dimethylaniline to a solution of Vb allowed ethylene elimination concomitant with the formation of Vc at room temperature. Additive-free reversible [4 + 2] cyclization between a 6π-compound and ethylene still remains highly challenging.13
C bond of ethylene in the initial step. With a group 13 compound, Jordan et al. reported that the cationic aluminum β-diketiminate derivative Va and ethylene underwent a [4 + 2] cycloaddition reaction, yielding the cycloadduct Vb, which was characterized only by NMR spectroscopy.12 Although Vb did not release ethylene even under vacuum, the addition of N,N-dimethylaniline to a solution of Vb allowed ethylene elimination concomitant with the formation of Vc at room temperature. Additive-free reversible [4 + 2] cyclization between a 6π-compound and ethylene still remains highly challenging.13
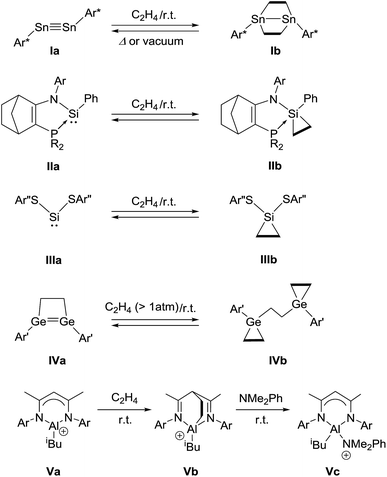 |
| | Fig. 1 Examples of reversible cycloaddition reactions between main group compounds and ethylene. | |
Recently, we reported the preparation of aromatic 1,3,2,5-diazadiborinine 1,14a which formally involves a neutral tricoordinate boron center surrounded by eight electrons.5a,14b–h Preliminary studies indicated the unique reactivity of 1 toward unsaturated compounds such as carbon dioxide, forming bicyclo[2.2.2] derivatives, which prompted us to investigate the reactions of 1 with ethylene and other non-activated olefins in detail. Herein, we report the reversible cycloaddition reactions of 1 with various alkenes. We also describe the single-crystal X-ray diffraction analysis of the cycloadducts, kinetic experiments, and computational studies.
Results and discussion, experimental
Reaction of 1 with ethylene
Ethylene was introduced into a benzene solution of 1 at 1 bar and the reaction mixture was stirred at ambient temperature for 3 h. After the removal of benzene and ethylene in vacuo, compound 2 was obtained as a white power in 97% yield (Scheme 1). In the 11B NMR spectrum of 2, two peaks were detected at −16.0 ppm and 1.6 ppm that are shifted upfield with respect to those (7.3 ppm and 24.9 ppm) for the corresponding boron atoms in 1.14 Recrystallization from a saturated benzene solution at room temperature afforded single crystals of 2, and the solid-state structure was confirmed by X-ray diffraction analysis (Fig. 2). Both of the boron centers are tetra-coordinated, indicating sp3 hybridization. The B1–C1 (1.653(5) Å) and B2–C2 (1.671(5) Å) distances are significantly longer than the typical B–C single bond (1.59 Å). The long C1–C2 (1.550(4) Å) distance is an indication of a C(sp3)–C(sp3) single bond. There is a lengthening of the B1–N1 (1.588(5) Å) and B2–C3 (1.593(5) Å) bonds and a shortening of the C3–N1 (1.302(4) Å) bond, compared to those of 1.14 Compound 2 is stable at room temperature, both in the solid state and in solution. However, upon heating a C6D6 solution of 2 at 150 °C, a clean ring opening occurred, giving rise to 1 quantitatively. Note that the Diels–Alder reaction between benzene and ethylene as well as the retro Diels–Alder reaction have scarcely been described thus far,15 despite the extensive application of the elementary reactions in organic synthesis.16
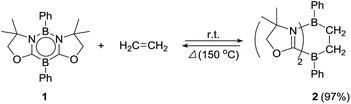 |
| | Scheme 1 Reversible [4 + 2] cycloaddition reaction between 1 and ethylene. | |
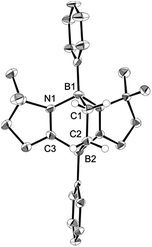 |
| | Fig. 2 Solid-state structure of 2. Thermal ellipsoids were set at the 50% probability level. Hydrogen atoms except for those on the C1 and C2 atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: B1–C1 1.653(5), B1–N1 1.588(5), B2–C2 1.671(5), B2–C3 1.593(5), C1–C2 1.550(4), C3–N1 1.302(4); N1–B1–C1 104.0(3), B1–C1–C2 110.4(3), C1–C2–B2 113.3(3), C2–B2–C3 100.7(3). | |
Reaction of 1 with styrene derivatives
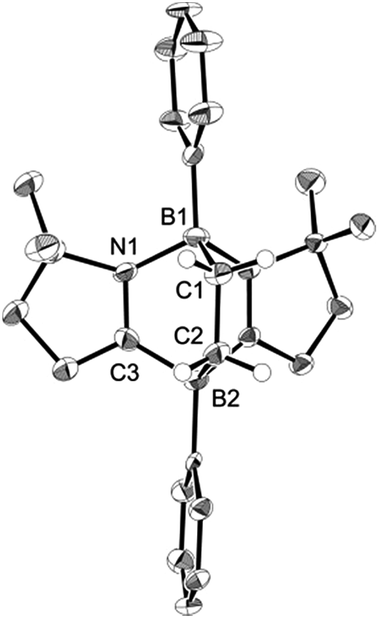
To examine the scope of the cycloaddition reaction, we employed styrene derivatives 3a–e as alkene substrates (Scheme 2). A slight excess (2.0 eq.) of styrene 3a was added to a C6D6 solution of 1 in a J. Young NMR tube, and the reaction was monitored by NMR spectroscopy. This reaction was drastically slower than that of ethylene, with full conversion of 1 observed only after 12 h at room temperature. After the workup, 4a was isolated as a white powder in 90% yield. In the 11B NMR spectrum of 4a, two singlets appeared at −14.3 ppm and 0.8 ppm, which are comparable to those (−16.0 ppm and 1.6 ppm) of 2. X-ray diffraction analysis revealed the bicyclo[2.2.2] structure of 4a and confirmed that the CH2 and the CHPh carbons of 3a selectively bind to the boron between two nitrogen atoms and the boron between two carbon atoms in 1, respectively (Fig. 3a). Remarkably, retro [4 + 2] cycloaddition of 4a was observed at ambient conditions. Thus, when isolated 4a was re-dissolved in C6D6 and the solution was kept at ambient temperature, partial regeneration of 1 and 3a was detected in the 1H NMR spectrum. Meanwhile, the full conversion of 4a into 1 and 3a was confirmed upon heating the solution at 110 °C. Cyclization reactions of 1 with para-substituted styrene derivatives 3b–e also proceeded cleanly at room temperature, and the corresponding [4 + 2] cycloadducts 4b–e were obtained. Analogous to 4a, the bicyclo[2.2.2] structures of 4b and 4d formed via regio-selective [4 + 2] cycloaddition were confirmed by X-ray diffraction studies (Fig. 3b and c). We also confirmed that 4b–e underwent retro [4 + 2] cycloaddition reactions at 110–150 °C, to afford 1 and 3b–e in 80–100% yields (see the ESI†).
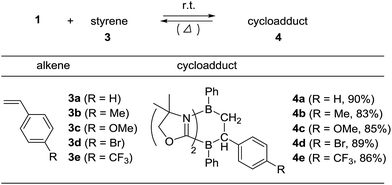 |
| | Scheme 2 Regio-selective [4 + 2] cycloaddition reactions between 1 and styrenes 3a–e. | |
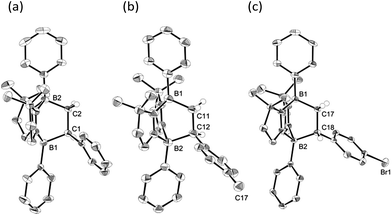 |
| | Fig. 3 Solid-state structures of 4a (a), 4b (b) and 4d (c). Thermal ellipsoids were set at the 50% probability level. Most of the hydrogen atoms are omitted for clarity. | |
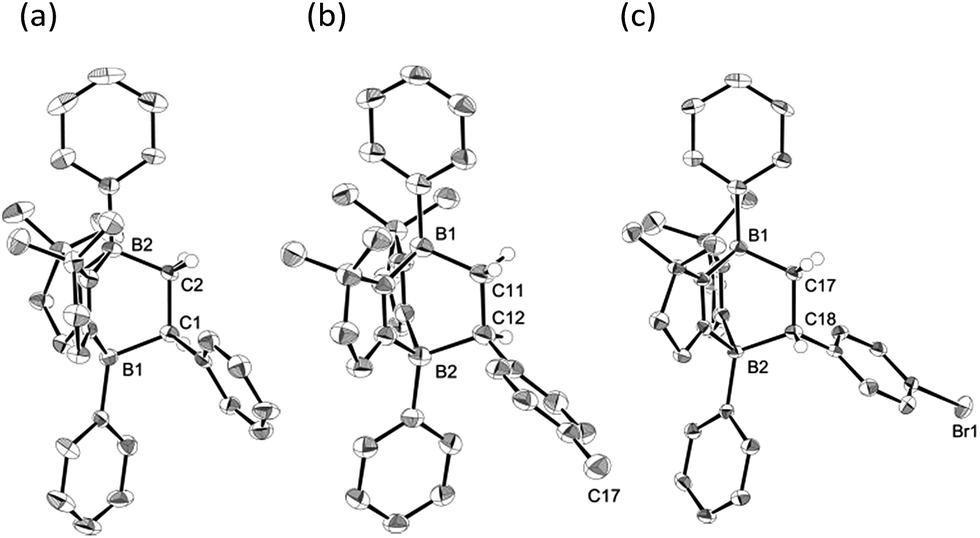
Interestingly, we noted that the cyclization reaction proceeded faster with styrenes bearing an electron-withdrawing group under the same reaction conditions. Thus, while the reactions with 3a–c required more than 12 h for the full conversion into the cycloadducts 4a–c, the reactions with 3d and 3e completed within 6 h and 3 h, respectively. Indeed, the equilibrium constant for the reversible cycloaddition highly depends on the para-substituent of the styrene derivative (Table 1). To evaluate the substituent effect on the cycloaddition reaction, we checked the Hammett correlation for the reaction of 1 with 3a–e.17 The rate constants k were measured based on the conversion rate of 1 using its integral values in the 1H NMR spectra (see the ESI†). Fig. 4 shows a linear correlation between log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k and the substituent constants with ρ of +1.43, which is in line with the sensitivity of the cycloaddition reaction to the inductive effect of the substituents.
k and the substituent constants with ρ of +1.43, which is in line with the sensitivity of the cycloaddition reaction to the inductive effect of the substituents.
Table 1 Equilibrium constants (K4–1) at room temperature
| Styrene |
3a
|
3b
|
3c
|
3d
|
3e
|
|
K
4–1
|
1.148 × 103 |
0.601 × 103 |
0.457 × 103 |
8.741 × 103 |
2.011 × 104 |
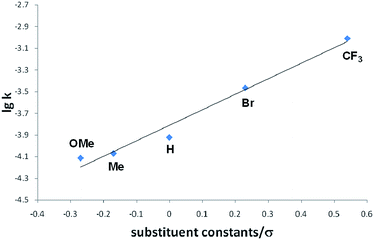 |
| | Fig. 4 Hammett plot for the reactions of 1 with para-substituted styrenes 3a–e in THF-d8 at room temperature. | |
Computational study on reaction pathway
To gain insight into the reaction mechanism, pathways for the cycloaddition reaction between 1 and styrene 3a were explored theoretically using DFT calculations at the M06-2X(SCRF, solvent = benzene)/def2-TZVP//M06-2X/6-31G* level of theory, where the M06-2X functional accounts for the dispersion effects.18 We attempted to obtain pathways for the concerted and stepwise mechanisms. However, only concerted pathways could be determined (Fig. 5a), which suggests that the reaction proceeds in a concerted fashion. We could determine two possible pathways for the concerted reaction, namely, a favored pathway (path A) leading to the experimentally observed product and a less favored pathway (path B). Stephan et al. demonstrated a stepwise activation of olefin with an intermolecular frustrated Lewis pair system.19 Thus, by employing a Lewis acidic borane tethered to a vinyl group through an alkyl chain, they experimentally confirmed that a unique borane–olefin van der Waals (vdW) complex was formed prior to nucleophilic attack by a Lewis base. In our system, a similar vdW interaction could not be observed at the stage of a reactant complex (RC). An important steric reason appears to be that the phenyl ring attached to the Lewis acidic boron atom in the N–B–N moiety is perpendicular to the diazadiborinine ring, and thus the phenyl ring hinders the full coordination of an olefin molecule onto the boron. Moreover, both Lewis acidic and Lewis basic boron centers are present in 1 and interact with each other in the 6π-system, which could allow the cooperative activation of olefin.
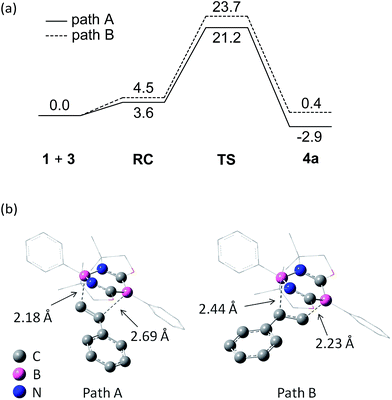 |
| | Fig. 5 (a) DFT-calculated free energy profiles (kcal mol−1) for the proposed mechanism of the [4 + 2] cycloaddition reaction between 1 and ethylene. The optimized structures were obtained at the M06-2X/6-31G* level. The ΔG values in C6H6 at 298 K were obtained from the M06-2X/6-31G* gas-phase harmonic frequencies and the energy in C6H6 calculated with the def2-TZVP basis set. (b) Transition-state geometries. | |
Consistent with the experimental observation, path A has a lower energy barrier. The computationally estimated activation parameters for the [4 + 2] cycloaddition process on path A (ΔH‡ = 6.6 kcal mol−1, ΔS‡ = −48.9 e.u., ΔG‡(298) = 21.2 kcal mol−1) agreed reasonably with the experimental values (ΔH‡ = 11.6 ± 0.9 kcal mol−1, ΔS‡ = −37.6 ± 3.0 e.u., ΔG‡(298) = 22.8 ± 1.8 kcal mol−1). The reaction on path A was calculated as slightly exergonic, consistent with the fact that heating is necessary for the retro-conversion (Scheme 2). It is interesting to note that although the reaction is essentially concerted, the transition state (TS) geometries shown in Fig. 5b suggest that the reaction is highly asynchronous. On path A, one of the two forming B–C bonds has a distance of 2.18 Å, whereas the other B–C bond is longer (2.69 Å). The same trend was also observed recently by Liu et al.20 The short distance of the former B–C bond reflects an efficient orbital interaction between the LUMO+3 of 1 and the HOMO of styrene 3a which has a large amplitude on the terminal carbon (Fig. 6). In addition to the orbital interaction, the effects of exchange and electrostatic interactions should play important roles in determining the regioselectivity. The exchange repulsion between the HOMOs of styrene 3a and 1 may be reduced in this asynchronous configuration, because the HOMO of 1 has a significant amplitude on the C–B–C moiety and the phenyl ring substituted on the boron atom. Furthermore, as shown in Fig. S4-2,† the boron atoms at the N–B–N and C–B–C moieties in 1 have positive (+0.35) and negative (−0.18) charges, respectively, whereas the terminal carbon (−0.39) in styrene 3a is more electronegative than the internal carbon (−0.12). Therefore, the TS for path B will feel a larger electrostatic repulsion. In terms of orbital interaction, path B should be favorable because it can form an efficient HOMO (1)–LUMO (3a) interaction (Fig. 6). However, the stabilization arising from this orbital interaction is outweighed by the above-mentioned repulsive effects. It is inferred that in addition to the repulsion at the reaction centers, the steric repulsion between the phenyl ring of 3a and the two dimethyl moieties (–CMe2–) of 1 in the TS of path B may also contribute to the distinct regio-selectivity in this reaction (Fig. 5b).
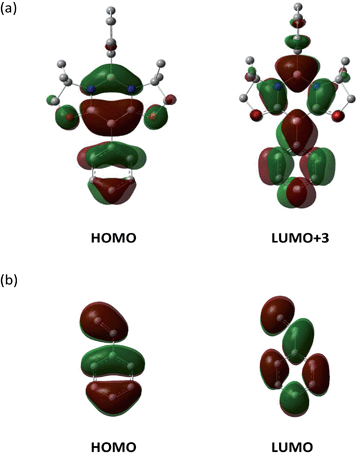 |
| | Fig. 6 Key frontier orbitals of 1 (a) and styrene 3a (b). | |
Reaction of 1 with norbornene
We also investigated the limitations of the substrate scope. Cyclization reactions of 1 with cyclic and acyclic internal alkenes such as 1,1-bis(trimethylsilyl)ethene, trans-diphenylethene, cyclohexene, cyclohexadiene did not proceed even under heating conditions (r.t. ∼150 °C), presumably because of steric hindrance. By contrast, the reaction of 1 with norbornene 5, which features a strained bicyclic skeleton, underwent stereo-selective [4 + 2] cycloaddition at 90 °C, and 6 was obtained as a solo product in 83% yield (Scheme 3). The stereoselectivity of the reaction originated probably from the fact that the steric hindrance of the CH2 moiety is smaller than that of the CH2CH2 part of 5. Retro-[4 + 2] cycloaddition of 6 proceeded thermally by heating at 150 °C, which cleanly reproduced 1 and 5.
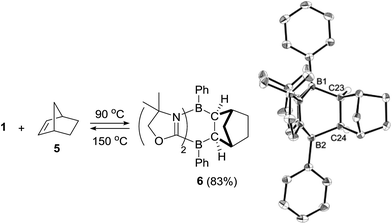 |
| | Scheme 3 Stereo-selective [4 + 2] cycloaddition reaction between 1 and norbornene 5 and its retro-conversion. | |
Conclusions
In summary, we have demonstrated a [4 + 2] cycloaddition reaction between 1 and ethylene which afforded the bicyclo[2.2.2] compound 2 at room temperature. We also showed regio-selective and stereo-selective [4 + 2] cycloadditions of 1 with styrene derivatives and norbornene, respectively. All of the cycloadducts underwent retro-[4 + 2] cycloaddition reactions that reproduced 1 and the corresponding alkenes quantitatively. Computational studies using DFT calculations showed that the transition state for the cycloaddition is concerted, but highly asynchronous, and provided explanations for the observed regioselectivity. Studies on the reversible cycloaddition of 1 with substrates featuring other unsaturated bonds, and its application20 are currently under investigation in our laboratory.
Acknowledgements
We are grateful to Nanyang Technological University (NTU) and PSF/A*STAR (SERC 1321202066) of Singapore, for financial support. H. H. thanks the High Performance Computing Centre of NTU for computer resources.
Notes and references
-
(a)
N. Winterton and G. J. Leigh, Modern Coordination Chemistry: The Legacy of Joseph Chatt, RSC, Cambridge, UK, 2002 Search PubMed;
(b) R. Jones, Chem. Rev., 1968, 68, 785–806 CrossRef CAS.
-
(a) J. Chatt, L. A. Duncanson and L. M. Venanzi, J. Chem. Soc., 1955, 4456–4460 RSC;
(b) J. Chatt and L. A. Duncanson, J. Chem. Soc., 1953, 2939–2947 RSC;
(c) M. Dewar, Bull. Soc. Chim. Fr., 1951, 18, C79 Search PubMed.
-
(a) W. C. Zeise, Poggendorff's Ann. Phys., 1837, 40, 234–252 CrossRef PubMed;
(b) W. C. Zeise, Poggendorff's Ann. Phys., 1831, 21, 497–541 CrossRef PubMed;
(c) W. C. Zeise, Poggendorff's Ann. Phys., 1827, 9, 632–633 Search PubMed;
(d) W. C. Zeise, Overs. K. Dan. Vidensk. Selsk. Forh., 1825, 13 Search PubMed.
-
(a) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed;
(b) G. Frenking and N. Fröhlich, Chem. Rev., 2000, 100, 717–774 CrossRef CAS PubMed;
(c) A. Dedieu, Chem. Rev., 2000, 100, 543–600 CrossRef CAS PubMed.
-
(a) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed;
(b) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed;
(c) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed;
(d) S. K. Mandal and H. W. Roesky, Acc. Chem. Res., 2012, 45, 298–307 CrossRef CAS PubMed;
(e) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC;
(f) S. Yao, Y. Xiong and M. Driess, Organometallics, 2011, 30, 1748–1767 CrossRef CAS;
(g) P. P. Power, Acc. Chem. Res., 2011, 44, 627–637 CrossRef CAS PubMed;
(h) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed;
(i) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
-
(a) T. J. Hadlington, J. Li, M. Hermann, A. Davey, G. Frenking and C. Jones, Organometallics, 2015, 34, 3175–3185 CrossRef CAS;
(b) C. A. Caputo, Z. Zhu, Z. D. Brown, J. C. Fettinger and P. P. Power, Chem. Commun., 2011, 47, 7506–7508 RSC;
(c) S. Ishida, T. Iwamoto and M. Kira, Heteroat. Chem., 2011, 22, 432–437 CrossRef CAS PubMed;
(d) J. S. Han, T. Sasamori, Y. Mizuhata and N. Tokitoh, J. Am. Chem. Soc., 2010, 132, 2546–2547 CrossRef CAS PubMed;
(e) R. Kinjo, M. Ichinohe, A. Sekiguchi, N. Takagi, M. Sumimoto and S. Nagase, J. Am. Chem. Soc., 2007, 129, 7766–7767 CrossRef CAS PubMed;
(f) J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968–4971 CrossRef CAS PubMed;
(g) N. Wiberg, S. K. Vasisht, G. Fischer and P. Z. Mayer, Z. Anorg. Allg. Chem., 2004, 630, 1823–1828 CrossRef CAS PubMed;
(h) T. Ohtaki and W. Ando, Organometallics, 1996, 15, 3103–3105 CrossRef CAS.
-
(a) B.-H. Xu, K. Bussmann, R. Fröhlich, C. G. Daniliuc, J. G. Brandenburg, S. Grimme, G. Kehr and G. Erker, Organometallics, 2013, 32, 6745–6752 CrossRef CAS;
(b) S. Schwendemann, S. Oishi, S. Saito, R. Fröhlich, G. Kehr and G. Erker, Chem–Asian J., 2013, 8, 212–217 CrossRef CAS PubMed;
(c) B.-H. Xu, R. A. A. Yanez, H. Nakatsuka, M. Kitamura, R. Fröhlich, G. Kehr and G. Erker, Chem–Asian J., 2012, 7, 1347–1356 CrossRef CAS PubMed;
(d) M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6594–6607 CrossRef CAS;
(e) T. Voss, C. Chen, G. Kehr, E. Nauha, G. Erker and D. W. Stephan, Chem.–Eur. J., 2010, 16, 3005–3008 CrossRef CAS PubMed;
(f) J.-B. Sortais, T. Voss, G. Kehr, R. Fröhlich and G. Erker, Chem. Commun., 2009, 7417–7418 RSC.
-
(a) Y. Peng, B. D. Ellis, X. Wang, J. C. Fettinger and P. P. Power, Science, 2009, 325, 1668–1670 CrossRef CAS PubMed . For reversible [1 + 2] cycloaddition between stannylene and alkyne, see:;
(b) L. R. Sita and R. D. Bickerstaff, J. Am. Chem. Soc., 1988, 110, 5208–5209 CrossRef CAS.
- R. Rodriguez, D. Gau, T. Kato, N. Saffon-Merceron, A. D. Cózar, F. P. Cossío and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 10414–10416 CrossRef CAS PubMed.
- F. Lips, J. C. Fettinger, A. Mansikkamäki, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2014, 136, 634–637 CrossRef CAS PubMed.
- T. Sasamori, T. Sugahara, T. Agou, K. Sugamata, J.-D. Guo, S. Nagase and N. Tokitoh, Chem. Sci., 2015, 6, 5526–5530 RSC.
- C. E. Radzewich, M. P. Coles and R. F. Jordan, J. Am. Chem. Soc., 1998, 120, 9384–9385 CrossRef CAS.
- For recent reports on reversible [4 + 2] reaction with activated olefins, see (references are therein):
(a) R. J. Burford, B. Li, M. Vasiliu, D. A. Dixon and S.-Y. Liu, Angew. Chem., Int. Ed., 2015, 54, 7823–7827 CrossRef CAS PubMed;
(b) A. K. H. Hirsch, P. Reutenauer, M. L. Moignan, S. Ulrich, P. J. Boul, J. M. Harrowfield, P. D. Jarowski and J.-M. Lehn, Chem.–Eur. J., 2014, 20, 1073–1080 CrossRef CAS PubMed;
(c) S. Kotha and S. Banerjee, RSC Adv., 2013, 3, 7642–7666 RSC;
(d) J. Chen, Z. Bajko, J. W. Kampf and A. J. Ashe, Organometallics, 2007, 26, 1563–1564 CrossRef CAS.
-
(a) D. Wu, L. Kong, Y. Li, R. Ganguly and R. Kinjo, Nat. Commun., 2015, 6, 7340 CrossRef CAS PubMed;
(b) J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2015, 54, 4469–4473 CrossRef PubMed;
(c) L. Kong, R. Ganguly, Y. Li and R. Kinjo, Chem. Sci., 2015, 6, 2893–2902 RSC;
(d) L. Kong, Y. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280–9283 CrossRef CAS PubMed;
(e) D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837–7839 RSC;
(f) M. A. Celik, R. Sure, S. Klein, R. Kinjo, G. Bertrand and G. Frenking, Chem.–Eur. J., 2012, 18, 5676–5692 CrossRef CAS PubMed;
(g) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed.
-
(a) A. Talbot, D. Devarajan, S. J. Gustafson, I. Fernández, F. M. Bickelhaupt and D. H. Ess, J. Org. Chem., 2015, 80, 548–558 CrossRef CAS PubMed;
(b) J. J. Serrano-Pérez, F. d. Vleeschouwer, F. d. Proft, D. Mendive-Tapia, M. J. Bearpark and M. A. Robb, J. Org. Chem., 2013, 78, 1874–1886 CrossRef PubMed;
(c) U. Streit and C. G. Bochet, Beilstein J. Org. Chem., 2011, 7, 525–542 CrossRef CAS PubMed;
(d) O. D. Lucchi and V. Lucchini, J. Chem. Soc., Chem. Commun., 1982, 7, 1105–1106 RSC;
(e) K. E. Wilzbach and L. Kaplan, J. Am. Chem. Soc., 1971, 93, 2073–2074 CrossRef CAS;
(f) C. A. Grob, H. Kny and A. Gagneux, Helv. Chim. Acta, 1957, 40, 130–140 CrossRef CAS PubMed.
-
(a)
S. Kobayashi and K. A. Jørgensen, Cycloaddition Reactions in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2001 CrossRef PubMed;
(b)
S. Ma, Handbook of Cyclization Reaction, vol. 1 and 2, Wiley-VCH, Weinheim, Germany, 2009 Search PubMed;
(c) M. Juhl and D. Tanner, Chem. Soc. Rev., 2009, 38, 2983–2992 RSC;
(d) K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed.
-
(a)
J. H. Espenson, Chemical Kinetics and Reaction Mechanisms, McGraw-Hill, New York, 1995, 2nd edn, pp. 214–228 Search PubMed;
(b) L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS;
(c) L. P. Hammett, Chem. Rev., 1935, 17, 125–136 CrossRef CAS.
- Details of the quantum chemical calculations are given in the ESI†.
-
(a) X. Zhao and D. W. Stephan, J. Am. Chem. Soc., 2011, 133, 12448–12450 CrossRef CAS PubMed , see also:;
(b) Y. Guo and S. Li, Eur. J. Inorg. Chem., 2008, 2008, 2501–2505 CrossRef PubMed;
(c) W. A. Herrebout and B. J. van der Veken, J. Am. Chem. Soc., 1997, 119, 10446–10454 CrossRef CAS;
(d) P. R. Jones, J. Org. Chem., 1972, 37, 1886–1889 CrossRef CAS.
- L. Liu, C. Chan, J. Zhu, C.-H. Cheng and Y. Zhao, J. Org. Chem., 2015, 80, 8790–8795 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and calculation details, and crystallographic information for 2, 4a, 4b, 4d, and 6. CCDC 1418724–1418728. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03174e |
|
| This journal is © The Royal Society of Chemistry 2015 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a