
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoupling of chromophores with exactly opposite luminescence behaviours in mesostructured organosilicas for high-efficiency multicolour emission†
Dongdong
Li‡
ab,
Yuping
Zhang‡
a,
Zhiying
Fan
a,
Jie
Chen
c and
Jihong
Yu
*a
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun, 130012, P. R. China. E-mail: jihong@jlu.edu.cn
bDepartment of Materials Science, Jilin University, Changchun, 130012, P. R. China
cKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China
First published on 20th July 2015
Abstract
Aggregation-induced emission (AIE) and aggregation-caused quenching (ACQ) materials are important for various fluorescence-based applications but cannot easily collaborate because of their opposite luminescence behaviours. Here, we demonstrate a strategy to integrate AIE and ACQ chromophores in periodic mesoporous organosilicas (PMOs) for high-efficiency multicolour emission. Tetraphenylethene (TPE)-bridged AIE-PMOs are prepared as hosts to encapsulate ACQ dyes (e.g. RhB), which enables fine-tuning of ACQ@AIE-PMO emissions over the entire visible spectrum in the solid and film states. Significantly, high-quality white light is achieved with CIE coordinates of (0.32, 0.33) and a quantum yield of up to 49.6%. Because of their high stability and solution processability, the ACQ@AIE-PMOs can be applied in solid-state lighting and bioimaging. This design concept opens up new perspectives for developing high-performance luminescent materials by the combination of a wide variety of AIE and ACQ chromophores.
Introduction
Conventional fluorescent chromophores such as fluorescein, rhodamine and cyanine derivatives have been widely used in biomedicine, sensing, and environmental detection because of their excellent photophysical properties.1 These chromophores typically display high quantum yields in dilute solutions, but their emissions are usually weakened or even completely quenched in concentrated solutions or in the solid state.2 This aggregation-caused quenching (ACQ) effect is disadvantageous to fluorescence efficiency, which leads to poor sensitivity in fluorescence sensory systems and prevents their application in solid-state lighting. Recently, a novel class of luminophores demonstrating aggregation-induced emission (AIE) has drawn increasing research interest as the opposite of ACQ systems, offering new potential for solid-state optical devices, sensors and bioprobes.3 In most cases, the enhanced emission in the solid state is attributed to the effective restriction of intramolecular rotations (RIR).4 Because of their exactly opposite luminescent behaviours, AIE and ACQ materials are not easily coupled to construct new efficient luminescent materials. Some attempts were made to combine AIE and ACQ molecules by covalent bonding,5 but they failed in realising the advantages of combining both classes of dye. The development of a feasible approach for achieving the effective collaboration of AIE and ACQ dyes and constructing highly efficient and tuneable multicolour-emission materials for practical applications remains an important challenge.Solid-state light-emitting materials, particularly white-lighting materials, have aroused considerable interest in recent years.6 Luminescent periodic mesoporous organosilicas (PMOs) are an interesting class of inorganic–organic hybrid materials with excellent thermal, mechanical and photochemical stabilities, in which the chromophores can be densely and covalently embedded within the framework forming the pore walls.7 So far, the reported fluorescent PMO materials are mainly based on ACQ bridging molecules.8 A high chromophore concentration in the pore walls results in relatively low fluorescence quantum yields compared to those of their precursors because they tend to form nonemissive aggregates.9 Thus, the design of PMO materials with highly efficient emission suitable for solid-state lighting applications is required. Recently, we developed AIE-luminogen-functionalised mesoporous materials using post-grafting methods;10 their emission intensities were enhanced with increased loading amounts of the AIE-active unit. It is believed that the introduction of AIE chromophores instead of conventional dyes as the bridging groups is an efficient approach to construct high-performance luminescent PMOs. Significantly, the ordered channels of AIE-unit-embedded PMOs can also host a variety of chromophores and stabilize their optical properties, which is advantageous for the fabrication of new donor–acceptor systems that allow the coupling of AIE and ACQ dyes on the basis of efficient fluorescence resonance energy transfer (FRET). In addition, the pore size of PMOs located in the working distances of photoinduced electron transfer and/or energy transfer can dramatically enhance the electron transfer efficiency.11 We expect that such unique features of mesostructured organosilicas will not only afford great advantages for enhancing the luminescence of the AIE chromophores in the mesoporous framework but also lead to multicolour emission, including the emission of pure white light, upon encapsulation of ACQ dyes in the mesochannels.
Here, we introduce an efficient strategy to achieve the coupling of AIE and ACQ chromophores in PMOs via FRET for the construction of high-efficiency and tuneable multicolour-emission materials (Fig. 1). Blue-emitting TPE-bridged organosilane is used as a precursor for preparing high-performance luminescent PMOs, which avoid the typical fluorescence quenching suffered by ACQ dyes; these AIE-PMOs are employed as energy donors. Conventional ACQ dyes, such as rhodamine B (RhB) and rhodamine 6G (R6G), are encapsulated in the pores of the AIE-PMOs and used as energy acceptors. The resulting ACQ@AIE-PMO composites show multicolour emission over a wide range of the visible spectrum with high quantum yields. Significantly, highly pure solid-state white-light emission can be obtained with a quantum yield as high as 49.6%, which is superior to the level of some reported inorganic–organic hybrid materials.6a–c,11d This design concept provides new perspectives for developing high-performance luminescent materials by the combination of a wide variety of AIE and ACQ chromophores for fluorescence-based applications in solid-state lighting, biomedicine and other areas.
 | ||
| Fig. 1 Schematic illustration of the construction of ACQ@AIE-PMO with tuneable multicolour emission. AIE-PMO nanospheres are prepared by using AIE-active TPE-Si as a precursor, where TPE units are covalently embedded within the framework forming the pore walls; ACQ molecules (such as RhB) are then encapsulated in the mesoporous channels of the AIE-PMO; tuneable multicolour emission of PMO is achieved on the basis of FRET from the AIE-PMO donor to the ACQ acceptor. Note that the TPE-Si precursor is nonemissive in solution but luminesces intensively upon molecular aggregation, while RhB dye is emissive in solution but suffers from aggregation-caused fluorescence quenching. | ||
Results and discussion
Synthesis and characterisation of TPE-bridged PMOs
The AIE-luminogen-containing organosilica (TPE-Si) precursor was prepared by reacting 3-aminopropyltriethoxysilane (APTS) with 1,2-bis[4-(bromomethyl)phenyl]-1,2-diphenylethene (BTPE) without further purification. The presence of the TPE-Si precursor was proved by mass spectroscopic (MS) data. It emits strong blue luminescence in a THF/H2O mixture with 90% water under UV light illumination (365 nm), showing a typical AIE character (Fig. S1†). AIE-PMOs with different amounts of organosilica were then synthesised by basic hydrolysis and polycondensation of TPE-Si with TEOS in the presence of cetyltrimethylammonium bromide (CTAB) surfactant. The concentrations of the TPE unit introduced into the materials were approximately 0.12, 0.17 and 0.21 mmol g−1, as determined by elemental analysis (Table S1†); the resulting materials are denoted as AIE-PMO1, AIE-PMO2 and AIE-PMO3, respectively. The ordered 2D hexagonal mesostructures of the AIE-PMO nanoparticles were confirmed by small-angle powder X-ray diffraction (XRD) (Fig. 2a), scanning electron microscopy (SEM) (Fig. 2b and S2†), nitrogen adsorption–desorption isotherms (Fig. 2c) and transmission electron microscopy (TEM) (Fig. 2d and S3†). When the amount of TPE-Si unit embedded in the PMOs was increased, the d values of the AIE-PMOs slightly increased, perhaps because the introduction of more TPE molecules partially reduced the pore size, as shown in Fig. 2a and c. On the other hand, as TPE-Si is a hydrophobic molecule, the CTAB surfactant micelles will be changed with the introduction of more TPE-Si, which may further affect their morphology even to the point of forming a spherical morphology.12 The bands at 1400–1600 cm−1 in the Fourier transform-infrared (FT-IR) spectra attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration of aromatic rings confirm that the AIE-active TPE molecules were covalently embedded within the framework forming the pore walls (Fig. S4†).13
C stretching vibration of aromatic rings confirm that the AIE-active TPE molecules were covalently embedded within the framework forming the pore walls (Fig. S4†).13
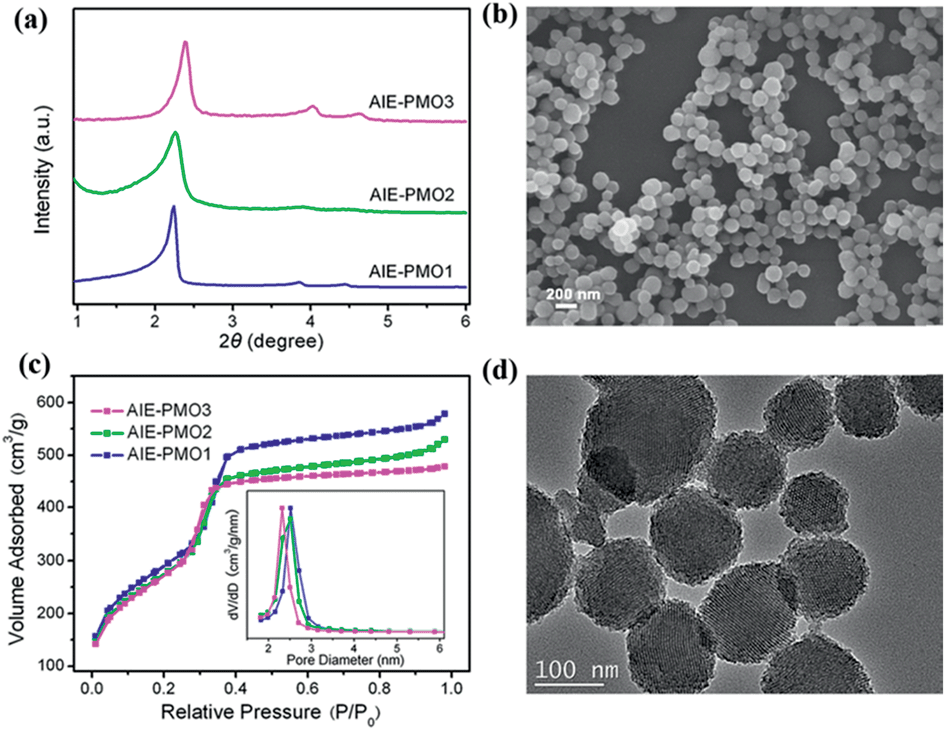 | ||
| Fig. 2 (a) Powder XRD patterns of mesostructured AIE-PMOs. (b) SEM image of AIE-PMO3. (c) N2 adsorption–desorption isotherms of AIE-PMOs and their corresponding pore size distributions. (d) TEM image of AIE-PMO3. | ||
All of the AIE-PMO materials exhibited strong fluorescence centred at approximately 480 nm (Fig. 3a). The emission behaviour is attributed primarily to the ability of the rigid framework of the mesostructured materials to greatly restrict the intramolecular rotations of the embedded TPE molecules by covalent bonding, thus blocking the nonradiative relaxation channel, and to populate radiative decay to the ground state, making the materials highly emissive.4b Furthermore, the photoluminescence (PL) intensity was noticeably enhanced with increased embedding amount of the TPE chromophores, and their quantum yields are 18.1%, 20.0% and 26.9% for AIE-PMO1, AIE-PMO2 and AIE-PMO3, respectively.
 | ||
| Fig. 3 (a) Fluorescence spectra of AIE-PMOs in the solid state. (b) Fluorescence spectra of RhB@AIE-PMO2 powders with different RhB contents. (c) Corresponding emission colours of RhB@AIE-PMO2 powders (marked by the dots) in the CIE 1931 chromaticity diagram. (d) Fluorescence decay profiles of RhB@AIE-PMO2 with different RhB contents at λex = 365 nm and λem = 480 nm. | ||
Tuneable multicolour emission of RhB-doped AIE-PMOs
A new donor–acceptor system of ACQ@AIE-PMO was constructed by using the blue-emitting AIE-PMOs as energy donors and the encapsulated ACQ dyes as energy acceptors. As a model system for demonstrating the utility and validity of this approach, a red-emitting RhB dye was selected because of the sufficient spectral overlap of its absorption band with the emission bands of the AIE-PMOs (Fig. S5†). Owing to the similar structure and emission property of the AIE-PMOs, AIE-PMO2 was chosen as a host for the encapsulation of RhB. Powder samples of AIE-PMO2 were first immersed in aqueous solutions with different RhB concentrations. After 24 h, the soaked samples were removed from solution and thoroughly washed with water. As the concentrations of the encapsulated RhB dye increased, the emission of AIE-PMO2 at approximately 480 nm gradually decreased, and an emission band at approximately 576 nm attributed to the RhB appeared and increased in intensity under excitation at 365 nm (Fig. 3b). Meanwhile, the average emission lifetime value of the TPE unit at 480 nm in AIE-PMO2 was gradually shortened from 4.63 ns to 3.84 ns to 1.91 ns, with the increased doping amount of RhB from 0 mol% to 0.36 mol% to 1.71 mol% (with respect to the amount of TPE unit) (Fig. 3d and Table S2†). These results suggest an energy transfer from the AIE-PMO framework to the encapsulated RhB dyes. The emission colour of the RhB@AIE-PMO2 powders clearly changed from blue to red via white light under UV irradiation. The Commission International de I'Eclairage (CIE) coordinates were (0.17, 0.28), (0.32, 0.33) and (0.50, 0.42) for an RhB content of 0 mol%, 0.36 mol% and 1.71 mol%, respectively (Fig. 3c). The quantum yield of RhB@AIE-PMO2 under excitation at 365 nm increased with increasing RhB content from 20.0% (blue) to 49.6% (white) to 80.1% (red). The enhancement of the quantum yield shows that energy transfer can occur directly from the AIE unit in the framework to the RhB dye in the channels without a radiation–reabsorption process. Significantly, the resulting white light was very close to pure white light with CIE coordinates of (0.33, 0.33), and RhB@AIE-PMO2 exhibited a high quantum yield compared with the white-light-emitting inorganic–organic hybrid materials reported to date, such as MOFs, PMOs, nanomaterials and semiconductor bulk materials (Table S3†).6a–c,11d Other mesoporous silica, such as SBA-15, can also be used as a host for coupling the AIE and ACQ dyes (Fig. S6†).The optical stability of RhB@AIE-PMO2 was investigated. After continuous irradiation of the solid sample containing 0.36 mol% RhB with 365 nm UV light for 45 min, the emission spectral profiles remained approximately identical (Fig. S7†). Meanwhile, the time-dependent emission spectra of RhB@AIE-PMO2 powder dispersed in water also showed no obvious change (Fig. S8†). After 24 hours, less than 0.2% RhB was detected with UV-vis spectroscopic measurement due to the slight desorption from RhB@AIE-PMO2 in aqueous solution (Fig. S9†). These results indicate the excellent stability of RhB@AIE-PMO materials in the solid state and in solution.
The bioimaging abilities of the RhB@AIE-PMO nanoparticles were evaluated with HeLa cells for in vitro cellular imaging. After the cells were incubated with RhB@AIE-PMO2 samples with different RhB concentrations (0 mol%, blue; 0.36 mol%, white; 1.71 mol%, red) for 4 h at 37 °C, strong blue, white and red light emissions, respectively, were observed under the confocal microscope (Fig. 4). The bright-field and fluorescent images of the HeLa cells clearly demonstrate that these luminescent nanoparticles were successfully internalized into the cells, indicating their potential applications in biomedicine.
 | ||
| Fig. 4 (Left) Bright-field and (right) fluorescent images of HeLa cells after incubation with RhB@AIE-PMO2 with different RhB concentrations: (a and b) 0 mol%, (c and d) 0.36 mol%, and (e and f) 1.71 mol%. | ||
Tuneable multicolour emission of RhB-doped AIE-PMO films
We further fabricated PMO composite films with different doping amounts of RhB dye by an acidic sol–gel one-step polycondensation via evaporation-induced self-assembly processes in the presence of P123 surfactant (Fig. S10†). The mesoporous structure of the prepared AIE-PMO film was confirmed by XRD (Fig. S11†) and TEM analysis (Fig. S12†). Doping of RhB dye into the mesostructured AIE-PMOs enabled flexible tuning of the fluorescence emission colour of the films within a wide range of the visible spectrum. As shown in Fig. 5a, the emission at approximately 574 nm, which was attributed to the doped RhB dye, appeared and increased in intensity under excitation at 365 nm. In addition, the emission intensity of TPE at approximately 474 nm decreased when the RhB content was increased from 0 to 5.0 mol%, indicating efficient energy transfer from the AIE-PMO framework to the guest RhB dye. The emission colour of the PMO composite films changed from blue, with CIE coordinates of (0.16, 0.24), to yellow-red (0.42, 0.41) (Fig. 5b). More importantly, the RhB@AIE-PMO film containing 3.2 mol% RhB exhibited a high-quality white-light emission with CIE coordinates of (0.31, 0.33). Remarkably, the RhB@AIE-PMO films were highly transparent, which is crucial for avoiding the loss of incident light via scattering. Moreover, the machinability of the sol solution for the preparation of PMOs further facilitated drop-casting onto various substrates, as typically used for various assemblies. Fig. 5c shows a blue-emitting transparent thin film of AIE-PMO fabricated on a large-area glass substrate, and a white-light emitting film of RhB@AIE-PMO deposited directly onto a quartz plate. The high solution-processability of ACQ@AIE-PMOs suggests their potential for practical solid-state lighting applications. | ||
| Fig. 5 (a) Fluorescence spectra of RhB@AIE-PMO films with different RhB content. (b) Corresponding emission colours of the films (marked by the dots) in the CIE 1931 chromaticity diagram. (c) PMO composite films coated on 10 × 10 cm glass under sunlight (left), blue film on 10 × 10 cm glass (middle) and white-light film on 5 × 5 cm quartz (right) under 365 nm UV irradiation. | ||
Tuneable multicolour emission of R6G-doped AIE-PMOs
To estimate the generality and versatility of our design concept for achieving the collaboration of AIE and ACQ dyes and constructing multicolour emission luminescent materials, R6G dye was also selected as an encapsulate. The PL spectra of AIE-PMO2 allow for partial spectral overlap with the absorption of R6G (Fig. S13†). This overlap suggests the possibility of FRET with R6G molecules as the acceptor and AIE-PMO2 as the donor. The energy transfer process from the AIE-PMO2 framework to the R6G dye inside the pore in the solid state was characterised by fluorescence spectroscopy (Fig. S14a†). With increasing R6G content, the donor blue-light emission of AIE-PMO2 at 480 nm was quenched dramatically, whereas the acceptor emission at 560 nm of R6G increased gradually. The perceived emission colour of R6G-doped AIE-PMO2 varied from blue to yellow, as plotted in the CIE 1931 chromaticity diagram shown in Fig. S14b†. The fluorescence quantum yield increased from 20.0% to 38.0% and 46.9%, when the R6G content was 0 mol%, 0.23 mol% and 0.68 mol%, respectively. These results strongly suggest that the excitation energy of the TPE units in the silica framework can also transfer to the R6G moieties in the solid state.Conclusions
In summary, we have successfully demonstrated a facile strategy to achieve the effective coupling of AIE and ACQ dyes with exactly opposite photoluminescence behaviours by taking advantage of the unique structure of PMOs. The emission of the resulting ACQ@AIE-PMO composites can be fine-tuned over the entire visible spectrum by adjusting the content of encapsulated ACQ dyes. Significantly, highly pure solid-state white light can be achieved with a quantum yield as high as 49.6%. Furthermore, white-emitting ACQ@AIE-PMO transparent films can be fabricated on a large scale through sol–gel polycondensation. The considerable flexibility of the rational combination of a great number of AIE chromophores and a wide range of ACQ dyes may facilitate the design of a variety of high-efficiency and tuneable multicolour emission PMOs. This work may create a new perspective for the exploration of novel high-performance solid luminescent materials that are easy-to-fabricate for various fluorescence-based applications in solid-state lighting, biomedicine and others.Acknowledgements
This work is supported by the State Basic Research Project of China (Grant No. 2011CB808703) and the National Natural Science Foundation of China (Grant No. 91122029 and 21320102001).Notes and references
- (a) R. Weissleder, C. H. Tung, U. Mahmood and A. Bogdanov, Nat. Biotechnol., 1999, 17, 375 CrossRef CAS PubMed; (b) Y. Urano, D. Asanuma, Y. Hama, Y. Koyama, T. Barrett, M. Kamiya, T. Nagano, T. Watanabe, A. Hasegawa, P. L. Choyke and H. Kobayashi, Nat. Med., 2009, 15, 104 CrossRef CAS PubMed; (c) H. N. Kim, Z. Q. Guo, W. H. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79 RSC; (d) A. Razgulin, N. Ma and J. H. Rao, Chem. Soc. Rev., 2011, 40, 4186 RSC; (e) J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973 CrossRef CAS PubMed.
- S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef PubMed.
- (a) J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (c) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441 CrossRef CAS PubMed; (d) Y. P. Zhang, D. D. Li, Y. Li and J. H. Yu, Chem. Sci., 2014, 5, 2710 RSC; (e) C. X. Shi, Z. Q. Guo, Y. L. Yan, S. Q. Zhu, Y. S. Xie, Y. S. Zhao, W. H. Zhu and H. Tian, ACS Appl. Mater. Interfaces, 2013, 5, 192 CrossRef CAS PubMed; (f) A. D. Shao, Y. S. Xie, S. J. Zhu, Z. Q. Guo, S. Q. Zhu, J. Guo, P. Shi, T. D. James, H. Tian and W. H. Zhu, Angew. Chem., Int. Ed., 2015, 54, 7275 CrossRef CAS PubMed.
- (a) J. Huang, N. Sun, Y. Q. Dong, R. L. Tang, P. Lu, P. Cai, Q. Q. Li, D. G. Ma, J. G. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329 CrossRef CAS PubMed; (b) Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- (a) W. Z. Yuan, P. Lu, S. M. Chen, J. W. Y. Lam, Z. M. Wang, Y. Liu, H. S. Kwok, Y. G. Ma and B. Z. Tang, Adv. Mater., 2010, 22, 2159 CrossRef CAS PubMed; (b) Q. L. Zhao, X. A. Zhang, Q. Wei, J. Wang, X. Y. Shen, A. J. Qin, J. Z. Sun and B. Z. Tang, Chem. Commun., 2012, 48, 11671 RSC; (c) A. Ozawa, A. Shimizu, R. Nishiyabu and Y. Kubo, Chem. Commun., 2015, 51, 118 RSC; (d) G. Chen, W. B. Li, T. R. Zhou, Q. Peng, D. Zhai, H. X. Li, W. Z. Yuan, Y. M. Zhang and B. Z. Tang, Adv. Mater., DOI:10.1002/adma.201501981.
- (a) M. Roushan, X. Zhang and J. Li, Angew. Chem., Int. Ed., 2012, 51, 436 CrossRef CAS PubMed; (b) T. Wang, V. Chirmanov, W. H. M. Chiu and P. V. Radovanovic, J. Am. Chem. Soc., 2013, 135, 14520 CrossRef CAS PubMed; (c) C. Y. Sun, X. L. Wang, X. Zhang, C. Qin, P. Li, Z. M. Su, D. X. Zhu, G. G. Shan, K. Z. Shao, H. Wu and J. Li, Nat. Commun., 2013, 4, 2717 Search PubMed; (d) M. M. Shang, C. X. Li and J. Lin, Chem. Soc. Rev., 2014, 43, 1372 RSC; (e) E. R. Dohner, E. T. Hoke and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 1718 CrossRef CAS PubMed.
- (a) P. N. Minoofar, B. S. Dunn and J. I. Zink, J. Am. Chem. Soc., 2005, 127, 2656 CrossRef CAS PubMed; (b) F. Hoffmann and M. Fröba, Chem. Soc. Rev., 2011, 40, 608 RSC; (c) A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328 CrossRef CAS PubMed; (d) Y. Yamamoto, H. Takeda, T. Yui, Y. Ueda, K. Koike, S. Inagaki and O. Ishitani, Chem. Sci., 2014, 5, 639 RSC; (e) T. Tani, N. Mizoshita and S. Inagaki, J. Mater. Chem., 2009, 19, 4451 RSC.
- (a) N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789 RSC; (b) S. H. Cheng, C. C. Hsiehc, N. T. Chen, C. H. Chu, C. M. Huang, P. T. Chou, F. G. Tseng, C. S. Yang, C. Y. Mou and L. W. Lo, Nano Today, 2011, 6, 552 CrossRef CAS PubMed; (c) Y. Li, F. Auras, F. Löbermann, M. Döblinger, J. Schuster, L. Peter, D. Trauner and T. Bein, J. Am. Chem. Soc., 2013, 135, 18513 CrossRef CAS PubMed.
- Y. Goto, N. Mizoshita, O. Ohtani, T. Okada, T. Shimada, T. Tani and S. Inagaki, Chem. Mater., 2008, 20, 4495 CrossRef CAS.
- (a) D. D. Li, J. H. Yu and R. R. Xu, Chem. Commun., 2011, 47, 11077 RSC; (b) D. D. Li, J. Z. Liu, R. T. K. Kwok, Z. Q. Liang, B. Z. Tang and J. H. Yu, Chem. Commun., 2012, 48, 7167 RSC.
- (a) M. Waki, Y. Maegawa, K. Hara, Y. Goto, S. Shirai, Y. Yamada, N. Mizoshita, T. Tani, W. J. Chun, S. Muratsugu, M. Tada, A. Fukuoka and S. Inagaki, J. Am. Chem. Soc., 2014, 136, 4003 CrossRef CAS PubMed; (b) K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562 CrossRef CAS PubMed; (c) B. J. Scott, M. H. Bartl, G. Wirnsberger and G. D. Stucky, J. Phys. Chem. A, 2003, 107, 5499 CrossRef CAS; (d) N. Mizoshita, Y. Goto, Y. Maegawa, T. Tani and S. Inagaki, Chem. Mater., 2010, 22, 2548 CrossRef CAS.
- (a) G. W. Zhou, Y. J. Chen, J. H. Yang and S. H. Yang, J. Mater. Chem., 2007, 17, 2839 RSC; (b) Y. Guillemin, J. Ghanbaja, E. Aubert, M. Etienne and A. Walcarius, Chem. Mater., 2014, 26, 1848 CrossRef CAS.
- E. Fuente, J. A. Menéndez, M. A. Díez, D. Suárez and M. Montes-Morán, J. Phys. Chem. B, 2003, 107, 6350 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis, TEM, SEM, XRD patterns, FTIR spectra, absorption and PL spectra. See DOI: 10.1039/c5sc02044a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |