
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DNA based multi-copper ions assembly using combined pyrazole and salen ligandosides†
Meng
Su
,
María
Tomás-Gamasa
and
Thomas
Carell
*
Department of Chemistry, Ludwig-Maximilians-University Munich, Butenandtstraße 5-13, 81377, Munich, Germany. E-mail: thomas.carell@cup.uni-muenchen.de
First published on 11th August 2014
Abstract
The DNA structure is an ideal building block for the construction of functional nano-objects. In this direction, metal coordinating base pairs (ligandosides) are an appealing tool for the future specific functionalization of such nano-objects. We present here a study, in which we combine the metal ion coordinating pyrazole ligandoside with the interstrand crosslinking salen ligandoside system. We show that both ligandosides, when combined, are able to create stable multi-copper ion complexing DNA double helix structures in a cooperative fashion.
Introduction
The Watson–Crick base pairing of DNA is a phenomenon that is more and more exploited for the construction of complex two and three dimensional self-assembled nano-objects.1–3 Nowadays, sophisticated technologies provide scientists almost total control of the self-assembly process so that basically any desired nano-object can be created using DNA as the building block.4–6 The next phase in the field is the functionalization of these objects, thus facilitating access to materials of relevance, e.g. “intelligent” drug delivery devices7 or building blocks useful in nano-machines.8 One approach for the construction of alternative and functional DNA structures was based on the design of bases showing novel base-pairing schemes,9–11 including those held together by hydrogen bonding patterns different from those of the natural base pairs12 or by simply hydrophobic interactions.13,14 The development of metal-mediated base pairs (ligandosides) represents a research direction of particular interest in respect to the desired functionalization of DNA.15–17 They can add properties to DNA, such as conductivity or magnetism,18–20 or they can be used to construct DNA able to perform logic operations.21,22Metal base pairs comprise natural or artificial nucleobases that are able to coordinate a central metal ion. The first group involves the conversion of a natural mismatched base pair into a metal coordinated pair. Examples include the coordination of Hg2+ and Ag+ by T–T, U–U or C–A mismatches which goes in hand with the formation of stable metal base pairs.23–26
Regarding the artificial base pairs, a plethora of different metal modified double helixes were generated depending on the choice of the synthesized ligandoside. Diverse systems, including monodentate,27,28 bidentate29–34 as well as homo tridentate ligandosides,35–38 which can complex a variety of metal ions (e.g., Cu2+, Ni2+, Pd2+) have been reported to date. Fig. S1† summarizes current reported structures. Last examples comprise new bimetal base pairs,39,40 which in principle would allow to construct DNA structures with heavy metal ion loading.
In 2005, we introduced the salen concept for the construction of a new metal base pair.41–45 This ligandoside involves formation of a covalent linkage between the two strands via a bridge established by ethylenediamine, as depicted in Fig. 1a. Due to the formed interstrand crosslink, the salen base pair was proven to be extremely stable after coordination of a copper ion.
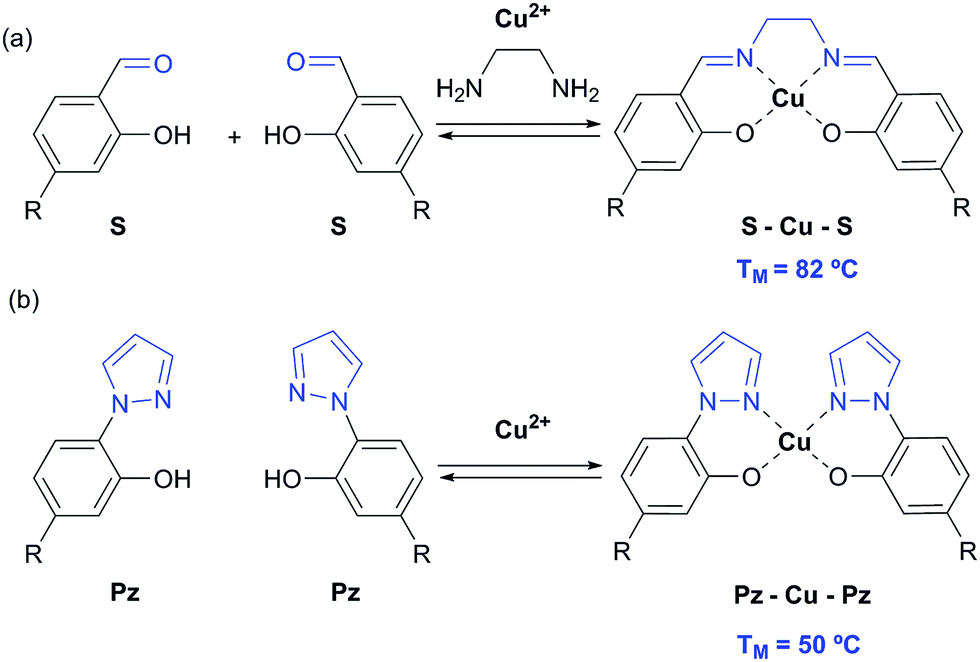 | ||
| Fig. 1 Depiction of (a) the reversible salen (S) self-base pair; (b) the pyrazole (Pz) self base pair. R = deoxyribose. | ||
In order to broaden this concept and to move away from the linking character of the salen system, we recently reported the pyrazole base pair (Pz) displayed in Fig. 1b.46 This ligandoside construction lacks the bridge but still retains the same metal coordination geometry.
Here we present stability and selectivity studies with the Pz ligandoside. We report that by combining the Pz and the salen ligandosides the generation of stable multi-Cu2+ complexing DNA duplexes becomes possible.
Results and discussion
We synthesized the pyrazole ligandosides Pz and Pm according to our published procedure.46Table 1 summarizes the modified DNA single strands prepared for this work.|
|
|||
|---|---|---|---|
| No. | 5′ ------ 3′a | Mass (calc.) | Massb (exp.) |
| a X = Pz, M = Pm, S = salen base. b Data from MALDI-TOF mass spectrometry. | |||
| 0a | CAC ATT AGT GTT GTA | 4579.8 | 4576.5 |
| 0b | CAC ATT ACT GTT GTA | 4557.8 | 4553.7 |
| 1a | CAC ATT AXT GTT GTA | 4588.8 | 4585.0 |
| 1b | TAC AAC AXT AAT GTG | 4606.8 | 4602.9 |
| 2a | CAC ATT AMT GTT GTA | 4602.8 | 4597.9 |
| 2b | TAC AAC AMT AAT GTG | 4620.9 | 4616.3 |
| 3a | CAC ATT XXT GTT GTA | 4613.8 | 4611.6 |
| 3b | TAC AAC AXX AAT GTG | 4640.9 | 4636.1 |
| 4a | GCGCC XXXXXXXXXX GGCCG | 6449.2 | 6452.4 |
| 4b | CGGCC XXXXXXXXXX CGCGC | 6369.2 | 6370.8 |
| 5a | CAC STT AXT GTS GTA | 4571.8 | 4570.0 |
| 5b | TAC SAC AXT AAS GTG | 4589.8 | 4586.8 |
| 6a | CAC STT AMT GTS GTA | 4585.8 | 4586.8 |
| 6b | TAC SAC AMT AAS GTG | 4603.8 | 4604.2 |
| 7a | CATGSTXGSAXCSTXCSTGCA | 6451.1 | 6450.7 |
| 7b | TGCASGXASGXTSCXASCATG | 6500.1 | 6500.1 |
| 8a | GCGCG XXXXX GGCCG | 4758.9 | 4757.6 |
| 8b | CGGCC XXXXX CGCGC | 4678.9 | 4676.6 |
Complexation properties
To determine the thermal stability of DNA duplexes containing the ligands Pz and Pm, melting points were measured in the absence and in the presence of copper ions.For duplexes 1a/1b and 2a/2b having one artificial base pair inserted, a decrease of the melting point value from 49 °C (TM of the reference duplex 0a/0b containing a GC base pair) to 41 °C and 42 °C was observed, respectively. Upon metal complexation, the duplex with the Pz–Cu–Pz complex furnished a melting temperature of 50 °C, slightly above the original TM of the duplex 0a/0b. In contrast, no increase of the TM value was detected for duplex 2a/2b (42 °C). This result shows that the presence of the phenolic hydroxyl groups and in particular, their ability to become deprotonated, are critical for metal complexation, which confirms the coordination geometry proposed in Fig. 1b.
In order to investigate in more detail the need for deprotonation, a study of the relationship between duplex thermostability and the pH value was carried out, using duplexes 1a/1b and 2a/2b. The data are compiled in Fig. 2, S2 and S3 and Table S1.† At pH 6.0, no substantial differences between the presence and absence of copper ions were found. The TM is approximately 8 °C lower than the TM of the canonical duplex 0a/0b. At this pH, the phenolic hydroxyl groups are obviously not deprotonated, which blocks Cu2+ complexation. Interestingly, at physiological pH 7.4, a slight difference (51 °C to 47 °C) regarding the melting point values between 1a/1b without and with Cu2+ was observed. We speculate that, at this pH, half deprotonation takes place, which leads, in the absence of Cu2+, to the formation of a stabilizing H-bonding bridge between the Pz–Pz base pair. Upon addition of Cu2+, the copper base pair is formed and is as stable as the H-bonded Pz–Pz self pair. At pH 9.0, the metal-free duplex is less stable, likely because of phenoxide formation followed by charge repulsion. Addition of Cu2+ stabilizes the system, in agreement with formation of the Pz–Cu–Pz base pair.
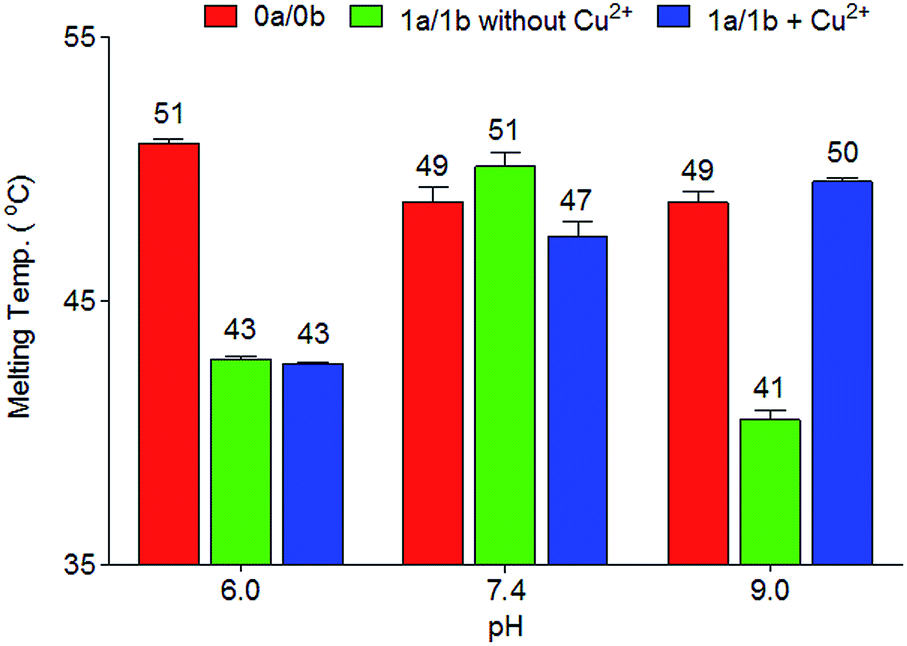 | ||
| Fig. 2 Melting temperatures of duplexes 0a/0b and 1a/1b at different pH values. Conditions: 150 mM NaCl, 10 mM Na2HPO4/NaH2PO4 buffer pH 6.0/7.4 or CHES buffer pH 9.0, 1 μM oligonucleotide, with or without 1 μM Cu2+, final volume of 200 μL. | ||
The overall duplex conformation was further investigated by circular dichroism spectroscopy. All measured CD spectra of DNA containing the Pz ligandoside in the absence and presence of Cu2+ support the presence of a B-form helix (Fig. S4 and S5†).
Selectivity of the Pz base
Next, the selectivity of the new Pz ligandoside self pair was studied. Melting profiles of a Pz base containing strand 1a with a complementary strand with a canonical base at the opposite position were measured at pH 9.0 (Fig. 3). The results showed that the Pz–C situation destabilizes the duplex dramatically while Pz–A/G/T arrangements give duplexes with lower but similar stability. In the presence of copper ions, the Pz–Cu–Pz metal base pair is by far the most stable structure which is the basis for selective Cu2+-complexation.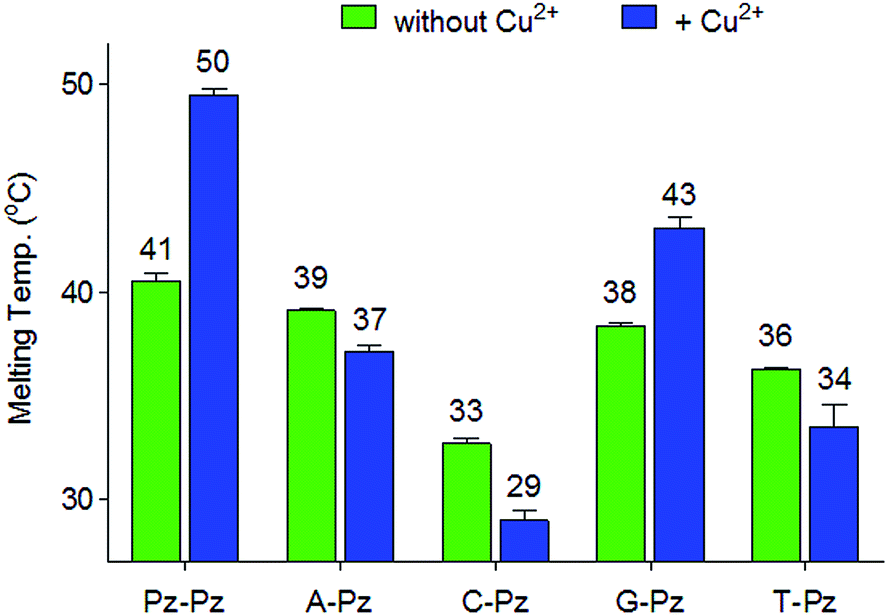 | ||
| Fig. 3 Melting temperatures of 1a combined with different counter strands having A, G, C, T or Pz as the counter base. Conditions: 150 mM NaCl, 10 mM CHES buffer pH 9.0, 1 μM oligonucleotide, with or without 1 μM Cu2+, final volume of 200 μL. | ||
Multiple metal ion binding by (Pz–Pz)n–DNA
To answer the question of whether the Pz ligandoside allows construction of DNA structures containing more than one metal ion stacking on top of each other we prepared oligonucleotides with several consecutive Pz ligandosides (Table 1, duplexes 3a/3b and 4a/4b).As a first approach, two complementary DNA single strands, each containing two neighbouring Pz pyrazole nucleosides, were hybridized to form the duplex 3a/3b. Titration melting profiles are depicted in Fig. 4. In the absence of copper ions, the TM of the duplex is decreased by about 11 °C (TM = 38 °C, Fig. 4, black line) with respect to the double strand 0a/0b and by 3 °C with respect to the duplex 1a/1b with only one artificial Pz base pair.
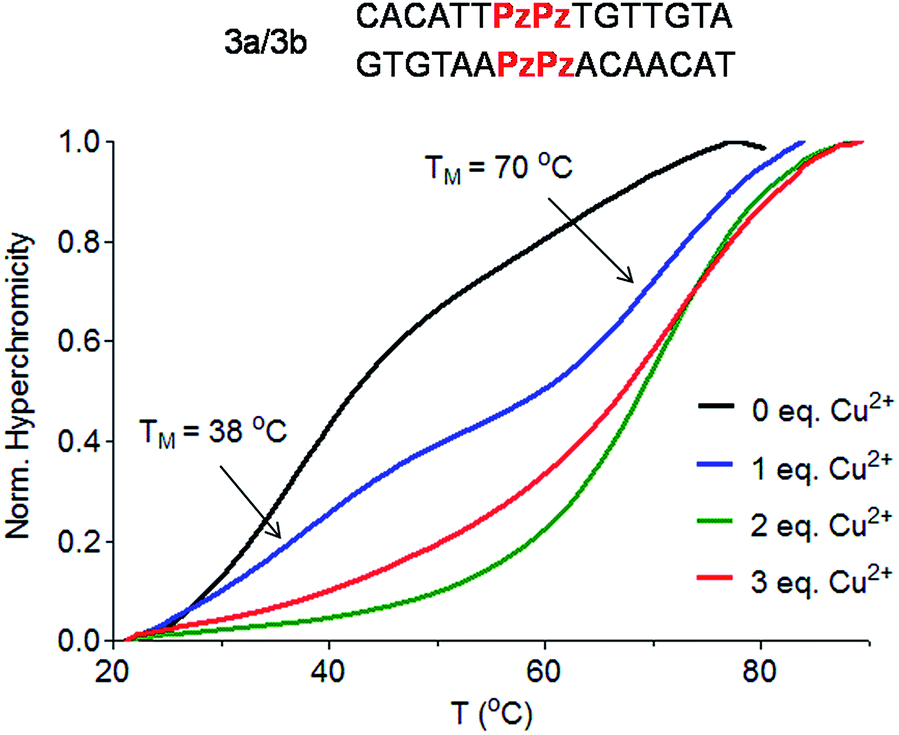 | ||
| Fig. 4 Schematic depiction of the investigated duplex 3a/3b and Cu2+ titration melting profiles of duplex 3a/3b (Pz = Pyrazole). Conditions: 150 mM NaCl, 10 mM CHES buffer pH 9.0, 1 μM oligonucleotide, final volume of 200 μL. | ||
When one equivalent of Cu2+ was added, the system showed two sigmoidal transitions (Fig. 4, blue line). One transition was detectable at TM = 38 °C (corresponding to the TM for the duplex without Cu2+) while the second transition appeared at a TM of 70 °C (see also Fig. S6†).
These data indicate the presence of two well-defined species in solution. With one equivalent of Cu2+ ions in solution, half of the duplexes contains two metal ions (TM = 70 °C), while the other duplexes feature no metal ion (TM = 38 °C), arguing that Cu2+ complexation by the Pz ligandoside is a cooperative effect. This is in agreement with the observation of isosbestic points in the UV and CD titration experiments (Fig. S7 and S8†). When two or more equivalents of Cu2+ are present, the melting curves exhibit only the transition at 70 °C (Fig. 4, green and red lines), showing full saturation of all Cu2+ binding sites.
In order to investigate if more than two Cu2+ ions can be arranged in a line, we next studied a duplex with ten consecutive Pz ligandosides, where GC rich DNA sequences were used as the terminus (Table 1, duplex 4a/4b).
After hybridization in the presence of Cu2+, the duplex 4a/4b was again subjected to thermal UV analysis. However, due to its high stability, the melting point could not be accurately measured any more under the tested conditions. Therefore, CD titrations (Fig. 5, see also Fig. S9† for titrations at 240 nm) and UV titrations (Fig. S10†) at the same concentration were employed to monitor the Cu2+ assembling process. The overlaid CD spectra revealed that the structure of the duplex changes significantly upon increasing Cu2+ complexation. A true isosbestic point is not present any more. Plotting of the ellipticity at 300 nm against the equivalents of Cu2+ ions displayed, however, a linearly decrease until a [Cu2+]/[duplex] ratio of about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was reached, in agreement with the expected complexation stoichiometry. From the experiments we conclude that the assembly of 10 × (Pz–Cu–Pz) is indeed possible. However, the duplex forms a distorted structure that appears to be tolerated by the flexible Pz-self base pair. The complexation process is certainly not a cooperative event, which explains the absence of clear isosbestic point. We also observe at 240 nm structural changes beyond the titration of 10 eq. of Cu2+. We believe that these changes are due to additional association of Cu2+ with the multiple Pz-structure. These additional structural changes are not observable at longer wavelength. We therefore examined next how the presence of stiff, while crosslinking, salen ligandosides (S, Fig. 1a) would influence the copper coordination process.
:1 was reached, in agreement with the expected complexation stoichiometry. From the experiments we conclude that the assembly of 10 × (Pz–Cu–Pz) is indeed possible. However, the duplex forms a distorted structure that appears to be tolerated by the flexible Pz-self base pair. The complexation process is certainly not a cooperative event, which explains the absence of clear isosbestic point. We also observe at 240 nm structural changes beyond the titration of 10 eq. of Cu2+. We believe that these changes are due to additional association of Cu2+ with the multiple Pz-structure. These additional structural changes are not observable at longer wavelength. We therefore examined next how the presence of stiff, while crosslinking, salen ligandosides (S, Fig. 1a) would influence the copper coordination process.
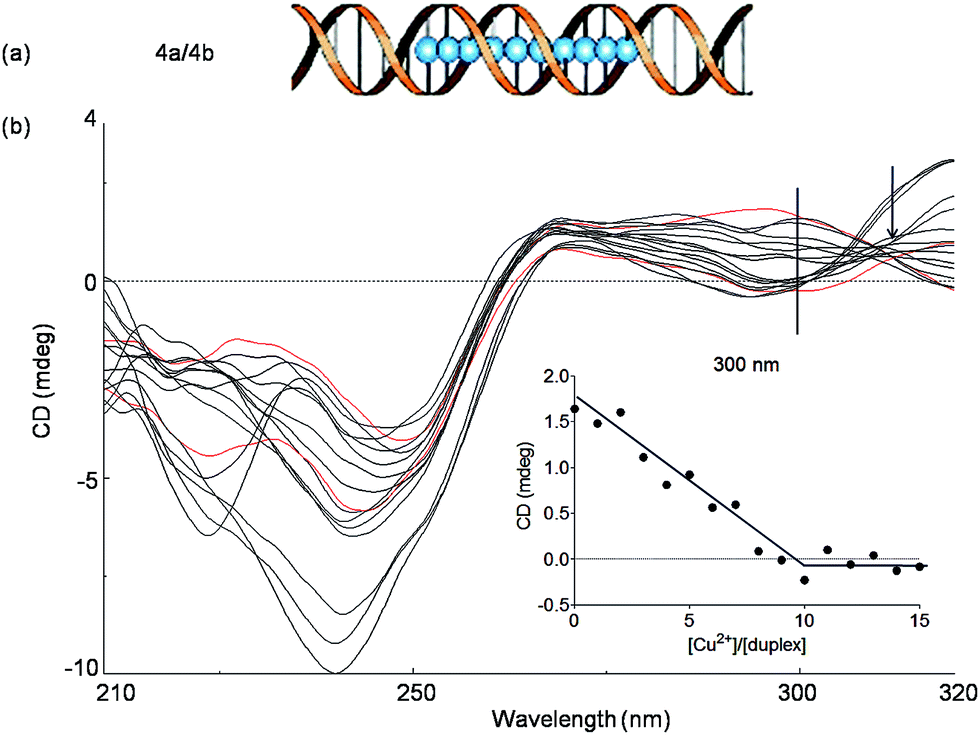 | ||
| Fig. 5 (a) Schematic depiction of the assembly of 10 Cu2+ pyrazole ligandosides inside a DNA duplex 4a/4b; (b) CD spectral changes of the duplex 4a/4b at various concentrations of Cu2+ (from 0 to 15 eq., step of 1 eq.), spectra of 0 eq. and 10 eq. are showed in red. Inset: plot of circular dichroic changes at 300 nm against the ratio of [Cu2+]/[4a/4b]. Conditions: 150 mM NaCl, 10 mM CHES buffer pH 9.0, 1 μM oligonucleotide, final volume of 200 μL. | ||
Multiple metal ion binding by hybrid Pz/S containing DNA
To this end mixed strands were prepared. For an initial study, oligonucleotides containing two salen nucleobases (S) and one additional Pz nucleobase in the middle were synthesized. In principle, this duplex can complex three metal ions (Table 1, duplex 5a/5b).The characteristic changes in the UV/Vis and CD spectrum of duplex 5a/5b that occur upon titration with Cu2+ ions are presented in Fig. 6a, S11 and S12.† The overlaid curves show now isosbestic points again at λ = 340 and 396 nm. A plot of the absorbance at 360 nm against the equivalents of Cu2+ ions is depicted in Fig. 6b. Initially, when one equivalent of Cu2+ was added, a value of 0.025 for the absorbance was measured. With increasing amounts of Cu2+ (up to three equivalents), the absorbance raised to 0.055, which is a typical behaviour for copper complexation by a salen ligand.42 Further titration did not affect the absorbance any more. A similar trend was observed at 235 nm (Fig. S13†). These results confirm the complexation of three metal ions and the data reflect that the first metal ion is complexed by the Pz–Pz pair, followed by complexation of the other two ions by the two salen ligandosides. It is clear that the complexation process by both ligandosides follows a different kinetic scheme.
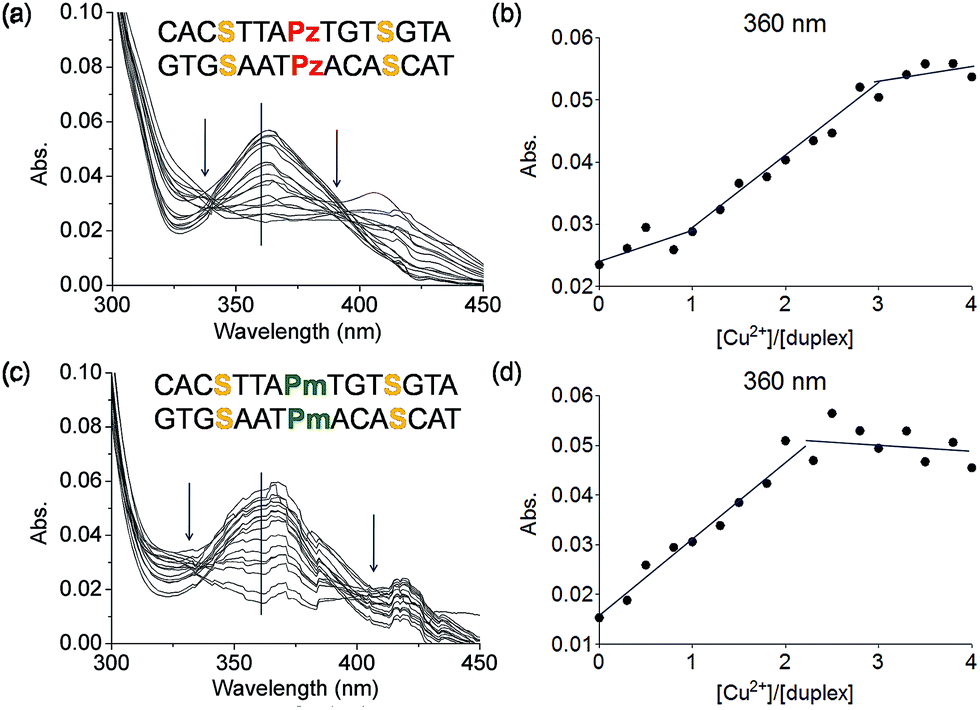 | ||
| Fig. 6 (a) Overlaid UV spectrum obtained at different concentrations of Cu2+ (from 0 to 4 eq.) of the duplex 5a/5b and (b) plot of UV absorbance at 360 nm against the ration of [Cu2+]/[5a/5b]; (c) overlaid UV spectrum at different concentrations of Cu2+ (from 0 to 4 eq.) of the duplex 6a/6b and (d) plot of UV absorbance at 360 nm against the ration of [Cu2+]/[6a/6b]. Conditions: 150 mM NaCl, 10 mM CHES buffer pH 9.0, 3 μM oligonucleotide, 30 eq. ethylenediamine, final volume of 200 μL. | ||
More experimental support for this hypothesis was obtained when the Pz nucleoside was replaced by methylated Pz (Pm), which is unable to coordinate copper ions. UV titration of duplex 6a/6b containing now the Pm instead of the Pz ligandosides showed that the absorbance at 360 nm increased up to a value of 0.050 after addition of 2 equivalents of Cu2+, characteristic for salen copper complexes (Fig. 6c/d). Further addition of metal ions didn't change the absorbance. As Pm fails to coordinate copper, the ions go directly into the two salen base pairs. Because the increase at 360 nm for 6a/6b is similar to those for 5a/5b, we conclude that two equivalents of Cu2+ are complexed in this case. These observations support the hypothesis, that the Pz–Pz base pair is the first ligandoside loaded with a metal ion.
For the characterization of these duplexes, ESI-Mass measurements were performed. Double strand 1a/1b, hybridized with copper, provided always two main signals corresponding to the two single strands. We were unable to detect the desired mass peaks of the duplex containing a copper ion inside, confirming that complexation and decomplexation of Pz–Pz ⇆ Pz–Cu–Pz is fast. In contrast, the same experiment performed with the duplex 5a/5b with an excess of ethylenediamine and 3 equivalents of Cu2+ showed in the mass spectrum quantitative formation of the duplex containing two copper complexes (Fig. 7 and S14†). The third copper ion could not be detected in agreement with fast Pz–Cu–Pz decomplexation. Obviously, and in agreement with earlier data,42 in the MS experiment, only the Cu–salen complexes are stable enough to survive the ESI conditions.
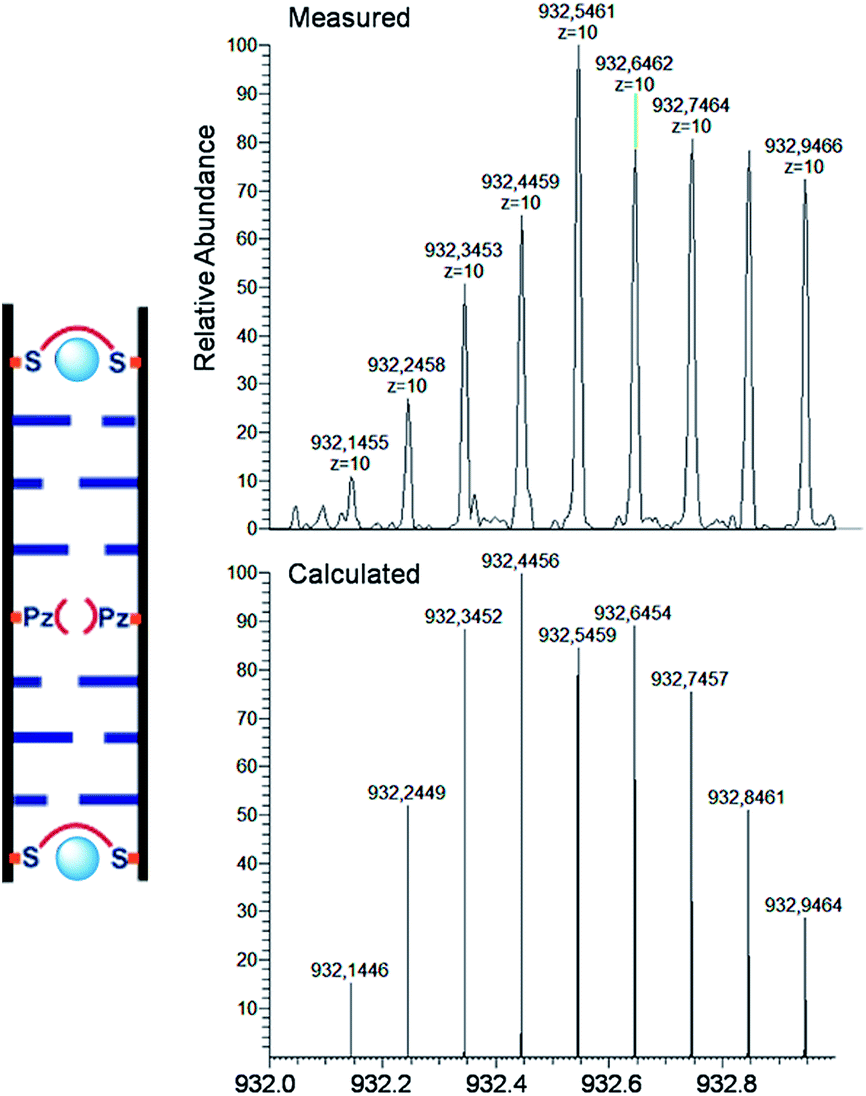 | ||
| Fig. 7 ESI-Mass spectrum and comparison of experimental data with calculated molecular weights of duplex 6a/6b with 2 eq. Cu2+, molecular formula C316H384O176N96P28Cu2. Peaks contain 10 charges (top) compared with calculation (bottom). Conditions: 150 mM NH4OAc, 30 μM oligonucleotide, 30 eq. ethylenediamine. | ||
A more complicated scenario was found when four Pz pairs were mixed with three salen pairs in duplex 7a/7b. The complexation of copper ions in this new situation was confirmed by CD titration. The CD data prove complexation of 7 equivalents of Cu2+ ions within the duplex (Fig. 8a/b; see also Fig. S15† for overlaid UV spectrum). The overlaid CD spectrum shows, despite the complex ligandoside composition of the duplex, two isosbestic points at 246 nm and 265 nm arguing for cooperative metal complexation. A plot of the UV absorbance at 405 nm during metal ion titration is displayed in Fig. 8d (see also Fig. S16† for absorbance at 360 nm). The UV data reveal two different trends which correspond to the two different coordination events established by the Pz and the salen base. Addition of three equivalents of Cu2+ led to a first rather sharp decrease in the absorbance at 405 nm, which corresponds to the first coordination process. With 3 Pz ligandosides we assume again initial complexation of Cu2+ by this ligandoside system. Thereafter, addition of another four equivalents of Cu2+ gave a further change of the UV data. Now, the reduction of the absorbance is slower. With all these evidences in hand, and accordingly to the previous hypothesis and results, we conclude that the Pz pairs complex the copper ions first while the 4 salen ligandosides are filled in the second process.
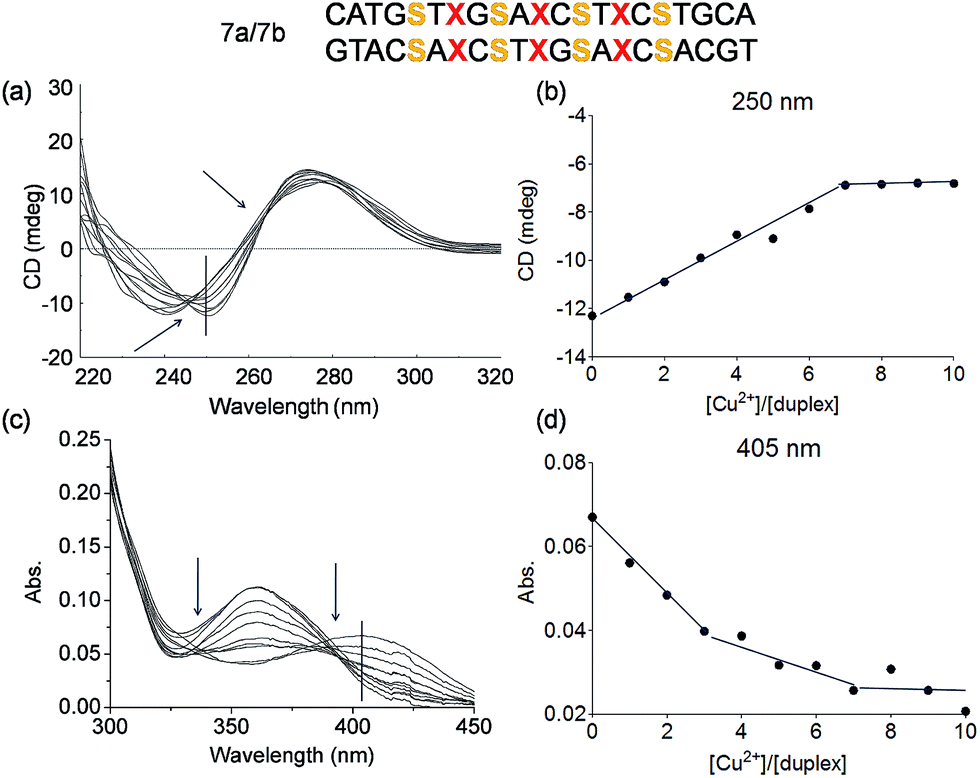 | ||
| Fig. 8 Schematic depiction of the investigated duplex, S = salen, X = pyrazole. (a) Overlaid CD spectrum at different concentrations of Cu2+ (from 0 to 10 eq.) of the duplex 7a/7b; (b) plot of circular dichroic changes at 250 nm against the ration of [Cu2+]/[7a/7b]; (c) overlaid UV spectrum under the same conditions as in (a); (d) plot of UV absorbance at 405 nm against the ration of [Cu2+]/[7a/7b]. Conditions: 150 mM NaCl, 10 mM CHES buffer pH 9.0, 3 μM oligonucleotide, 30 eq. ethylenediamine, final volume of 200 μL. | ||
Catalysis of a Diels–Alder reaction by (Pz–Cu2+–Pz)n–DNA
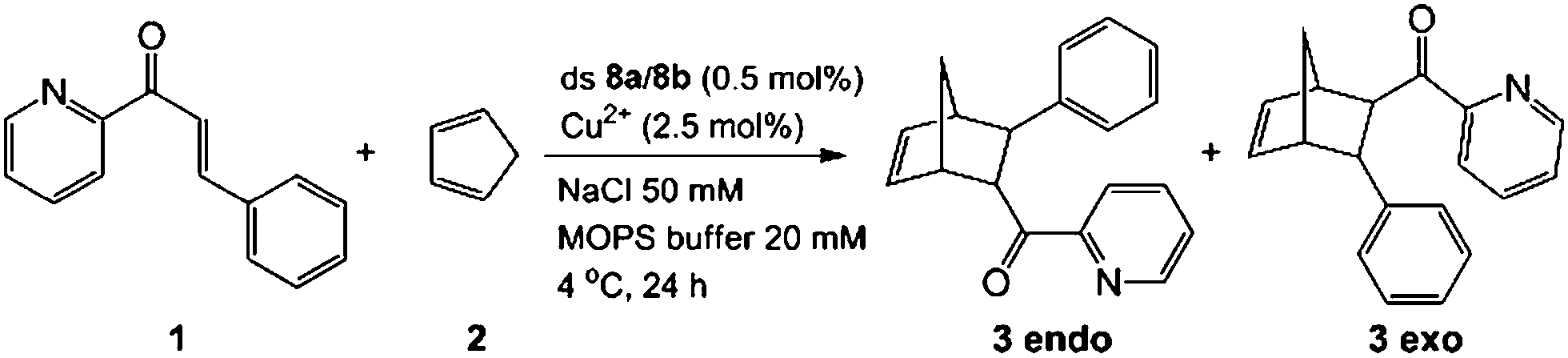
In order to investigate if the multiple Cu-containing DNA strands are able to exhibit some function, e.g. as catalyst, we started to investigate their ability to catalyze a Diels–Alder reaction. For other enantioselective Diels Alder reactions based on Cu2+–ligand–DNA see ref. 47 and 48 and for Cu2+-G-quadruplex see ref. 49.The D–A reaction between aza-chalcone (1) and cyclopentadiene (2), using duplex 8a/8b (system with five Cu2+ ions complexed up in Pz containing DNA) as a catalyst was selected for this initial study and the results are summarized in Table 2. For us a central question was if the duplexes, which contain the metal ions in the middle are able to perform the catalysis and whether enantioselectivity can be obtained. We noted that both single and double strands with the Pz-ligands catalyze the reaction in the presence of Cu2+ and in both cases some chirality transfer is observed. At the same time, the complex accelerates the conversion. Copper counter ion and surrounding pH are also playing a role in the catalytic process. The result shows that the Pz-containing DNA strands can be turned into catalyst. Now, their efficacy and the ee-values obtained for the reaction need to be improved.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | [Cu] | pH | ee (%) | Endo/exo | Conv. (%) |
| a See ESI for reaction details. All data are averaged over two experiments. OTf = trifluoromethanesulfonate. | ||||||
| 1 | 8a/8b | Cu(NO3)2 | 6.5 | 39 | 93 | 97 |
| 2 | — | Cu(NO3)2 | 6.5 | 1 | 87 | 76 |
| 3 | ss 8a | Cu(NO3)2 | 6.5 | 29 | 93 | 96 |
| 4 | 8a/8b | Cu(NO3)2 | 7.4 | 36 | 86 | 79 |
| 5 | 8a/8b | Cu(NO3)2 | 9.0 | 23 | 80 | 47 |
| 6 | 8a/8b | CuSO4 | 6.5 | 32 | 87 | 52 |
| 7 | 8a/8b | Cu(OTf)2 | 6.5 | 29 | 84 | 46 |
| 8 | 8a/8b | — | 6.5 | 3 | 76 | 19 |
Conclusion
The metal base pair, pyrazole ligandoside, was synthesized and incorporated into oligonucleotides. The ligandoside system shows a pH-dependent complexation behaviour due to the need for deprotonation of the phenolic groups. When the OH groups are replaced by a methoxy moiety as in the Pm ligandoside, no metal ion coordination is observed. Cu2+ ions stabilize a Pz–Pz containing duplex when compared to a canonical G–C pair, at pH 9.0.The Pz–Pz ligandoside base pair allows complexation of up to 10 Cu2+ ions within a duplex. Better complexation of multiple Cu2+ ions is, however, observed when the unbridged Pz ligandoside is combined with the bridged salen (S) system which seems to add so much integrity and duplex stiffness that the metal ion complexation process is dominated by cooperative effects. The copper ions show a kinetic preference for complexing first into the Pz–Pz base pair while the salen complex is loaded in a second independent step. This stepwise complexation enables in principle the design of logic gates. The described complexation properties are indeed now the basis for the construction of defined metal ions clusters within oligonucleotides, with the goal to construct multi-metal ion based catalysts and logic gates.
Acknowledgements
We thank the Alexander von Humboldt Foundation for a post-doctoral fellowship to M. T.-G. Funding for this research was obtained from the Volkswagen Foundation and the DFG (SFB749, TP4 and SFB1032, TPA5).Notes and references
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Hogberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- M. Endo, K. Hidaka, T. Kato, K. Namba and H. Sugiyama, J. Am. Chem. Soc., 2009, 131, 15570–15571 CrossRef CAS PubMed.
- S. H. Ko, M. Su, C. Zhang, A. E. Ribbe, W. Jiang and C. Mao, Nat. Chem., 2010, 2, 1050–1055 CrossRef CAS PubMed.
- X. Bai, T. G. Martin, S. H. W. Scheres and H. Dietz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 20012–20017 CrossRef CAS PubMed.
- Z.-S. Wu, Z. Shen, K. Tram and Y. Li, Nat. Commun., 2014, 5 CAS.
- E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. P. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73–76 CrossRef CAS PubMed.
- H. Pei, L. Liang, G. Yao, J. Li, Q. Huang and C. Fan, Angew. Chem., Int. Ed., 2012, 51, 9020–9024 CrossRef CAS PubMed.
- S. A. Benner, Acc. Chem. Res., 2004, 37, 784–797 CrossRef CAS PubMed.
- F. Wojciechowski and C. J. Leumann, Chem. Soc. Rev., 2011, 40, 5669–5679 RSC.
- I. Hirao, M. Kimoto and R. Yamashige, Acc. Chem. Res., 2012, 45, 2055–2065 CrossRef CAS PubMed.
- Z. Yang, F. Chen, J. B. Alvarado and S. A. Benner, J. Am. Chem. Soc., 2011, 133, 15105–15112 CrossRef CAS PubMed.
- A. A. Henry and F. E. Romesberg, Curr. Opin. Chem. Biol., 2003, 7, 727–733 CrossRef CAS PubMed.
- T. Lavergne, M. Degardin, D. A. Malyshev, H. T. Quach, K. Dhami, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2013, 135, 5408–5419 CrossRef CAS PubMed.
- G. H. Clever and M. Shionoya, Coord. Chem. Rev., 2010, 254, 2391–2402 CrossRef CAS PubMed.
- Y. Takezawa and M. Shionoya, Acc. Chem. Res., 2012, 45, 2066–2076 CrossRef CAS PubMed.
- P. Scharf and J. Müller, ChemPlusChem, 2013, 78, 20–34 CrossRef CAS.
- K. Tanaka, A. Tengeiji, T. Kato, N. Toyama and M. Shionoya, Science, 2003, 299, 1212–1213 CrossRef CAS PubMed.
- G. H. Clever, S. J. Reitmeier, T. Carell and O. Schiemann, Angew. Chem., Int. Ed., 2010, 49, 4927–4929 CrossRef CAS PubMed.
- S. Liu, G. H. Clever, Y. Takezawa, M. Kaneko, K. Tanaka, X. Guo and M. Shionoya, Angew. Chem., Int. Ed., 2011, 50, 8886–8890 CrossRef CAS PubMed.
- R. Freeman, T. Finder and I. Willner, Angew. Chem., Int. Ed., 2009, 48, 7818–7821 CrossRef CAS PubMed.
- T. Carell, Nature, 2011, 469, 45–46 CrossRef CAS PubMed.
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, J. Am. Chem. Soc., 2006, 128, 2172–2173 CrossRef CAS PubMed.
- S. Johannsen, S. Paulus, N. Düpre, J. Müller and R. K. O. Sigel, J. Inorg. Biochem., 2008, 102, 1141–1151 CrossRef CAS PubMed.
- A. Ono, S. Cao, H. Togashi, M. Tashiro, T. Fujimoto, T. Machinami, S. Oda, Y. Miyake, I. Okamoto and Y. Tanaka, Chem. Commun., 2008, 44, 4825–4827 RSC.
- T. Funai, Y. Miyazaki, M. Aotani, E. Yamaguchi, O. Nakagawa, S.-i. Wada, H. Torigoe, A. Ono and H. Urata, Angew. Chem., Int. Ed., 2012, 51, 6464–6466 CrossRef CAS PubMed.
- K. Tanaka, Y. Yamada and M. Shionoya, J. Am. Chem. Soc., 2002, 124, 8802–8803 CrossRef CAS PubMed.
- D. Böhme, N. Düpre, D. A. Megger and J. Müller, Inorg. Chem., 2007, 46, 10114–10119 CrossRef PubMed.
- K. Tanaka and M. Shionoya, J. Org. Chem., 1999, 64, 5002–5003 CrossRef CAS.
- H. Weizman and Y. Tor, J. Am. Chem. Soc., 2001, 123, 3375–3376 CrossRef CAS.
- K. Tanaka, A. Tengeiji, T. Kato, N. Toyama, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2002, 124, 12494–12498 CrossRef CAS PubMed.
- L. Zhang and E. Meggers, J. Am. Chem. Soc., 2004, 127, 74–75 CrossRef PubMed.
- C. Switzer and D. Shin, Chem. Commun., 2005, 41, 1342–1344 RSC.
- C. Switzer, S. Sinha, P. H. Kim and B. D. Heuberger, Angew. Chem., Int. Ed., 2005, 44, 1529–1532 CrossRef CAS PubMed.
- E. Meggers, P. L. Holland, W. B. Tolman, F. E. Romesberg and P. G. Schultz, J. Am. Chem. Soc., 2000, 122, 10714–10715 CrossRef CAS.
- N. Zimmermann, E. Meggers and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 13684–13685 CrossRef CAS PubMed.
- D. Shin and C. Switzer, Chem. Commun., 2007, 43, 4401–4403 RSC.
- B. D. Heuberger, D. Shin and C. Switzer, Org. Lett., 2008, 10, 1091–1094 CrossRef CAS PubMed.
- D. A. Megger, C. Fonseca Guerra, J. Hoffmann, B. Brutschy, F. M. Bickelhaupt and J. Müller, Chem.–Eur. J., 2011, 17, 6533–6544 CrossRef CAS PubMed.
- H. Mei, I. Röhl and F. Seela, J. Org. Chem., 2013, 78, 9457–9463 CrossRef CAS PubMed.
- G. H. Clever, K. Polborn and T. Carell, Angew. Chem., Int. Ed., 2005, 44, 7204–7208 CrossRef CAS PubMed.
- G. H. Clever, Y. Söltl, H. Burks, W. Spahl and T. Carell, Chem.–Eur. J., 2006, 12, 8708–8718 CrossRef CAS PubMed.
- K. Tanaka, G. H. Clever, Y. Takezawa, Y. Yamada, C. Kaul, M. Shionoya and T. Carell, Nat. Nanotechnol., 2006, 1, 190–194 CrossRef CAS PubMed.
- G. H. Clever and T. Carell, Angew. Chem., Int. Ed., 2007, 46, 250–253 CrossRef CAS PubMed.
- C. Kaul, M. Müller, M. Wagner, S. Schneider and T. Carell, Nat. Chem., 2011, 3, 794–800 CrossRef CAS PubMed.
- M. Su, M. Tomas-Gamasa, S. Serdjukow, P. Mayer and T. Carell, Chem. Commun., 2014, 50, 409–411 RSC.
- G. Roelfes and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 3230–3232 CrossRef CAS PubMed.
- J. Wang, E. Benedetti, L. Bethge, S. Vonhoff, S. Klussmann, J.-J. Vasseur, J. Cossy, M. Smietana and S. Arseniyadis, Angew. Chem., Int. Ed., 2013, 52, 11546–11549 CrossRef CAS PubMed.
- C. Wang, G. Jia, J. Zhou, Y. Li, Y. Liu, S. Lu and C. Li, Angew. Chem., Int. Ed., 2012, 51, 9352–9355 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc01567c |
| This journal is © The Royal Society of Chemistry 2015 |