
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Valorization of lignin waste from hydrothermal treatment of biomass: towards porous carbonaceous composites for continuous hydrogenation†
Gianpaolo
Chieffi
,
Nina
Fechler
and
Davide
Esposito
*
Max-Planck-Institute of Colloids and Interfaces, 14424 Potsdam, Germany. E-mail: davide.esposito@mpikg.mpg.de
First published on 20th July 2015
Abstract
Alkali lignin has been accumulated as a by-product mixed with barium salts during the hydrothermal treatment of rye straw with Ba(OH)2. Direct heat treatment followed by acid washing of such mineralized lignin were performed in order to obtain a porous material that was further exploited for the synthesis of a carbonaceous supported FeNi nanoparticle composite as active catalysts for continuous hydrogenation.
Introduction
Lignin is one of the most abundant organic polymers on our planet and the third major component of lignocellulosic biomass.1 Commercially, lignin is mostly obtained as the side product of cellulose pulp mills.2 Different pre-treatment processes that enable the isolation of the lignin fraction from the biomass have been reported.3 These are usually classified in accordance to the technology used and, among others, solvent fractionation (e.g. organosolv process), physical treatment (e.g. ball milling), biological treatment or chemical pre-treatment (oxidative, acidic or alkaline) have received great attention. Basic reagents such as sodium, calcium or ammonium hydroxide have been used for the effective chemical delignification of biomass.4 Such treatments favor to a good extent the cleavage of α- and β-ether linkages, producing a so-called alkali lignin mainly composed of oligomers in the range of 1–5 kDa.Lignin has been recently used for the production of activated carbon (AC) by thermal treatment in the presence of alkali metal hydroxides.5,6 For instance, Fierro et al. studied the optimization of AC manufacture from Kraft lignin (KL) using NaOH or KOH as the activating agents.7 Wang et al. used a two-step procedure involving the pre-carbonization of the lignin, which is successively mixed with KOH before undergoing an additional heat-treatment. Final washing with 1 N HCl provided porous carbon with very high surface area (∼3000 m2 g−1), which was successfully tested as an efficient Ni(II) adsorbent from wastewater.8
Metal loaded lignin has been also used as precursor for the preparation of supported nanoparticles (NPs). Mesoporous activated carbons, obtained by chemical activation of KL with H3PO4, were employed as supports for the synthesis of a carbon-based Pd material. The composites displayed activity for a broad range of transformations including the oxidation of toluene and xylene,9 selected example of hydrogenations and the Suzuki–Miyaura cross-coupling.10
Besides the use of rare and expensive metals, composites based on iron, the fourth most abundant element in the lithosphere, have also been prepared. In this regard, Rodriguez et al. used FeCl3 both as the activating agent for lignin isolated from alkaline-pulping black liquors as well as precursor for the introduction of stable Fe nanoparticles in the carbonaceous matrix. This system was used as catalyst in wet peroxide oxidation of phenol.11 Interestingly, carbonaceous supported FeNi NPs have also proved to be an efficient hydrogenation catalyst for a variety of substrates.12–14
Recently, our group developed an efficient alkaline hydrothermal treatment of biomass e.g., rye straw, which affords the platform chemical lactic acid in high yields.15,16 The method relies on the use of barium hydroxide excess and generates alkali lignin as a by-product. The latter can be recovered in the form of a mixture with barium inorganic salts.
In order to further increase the value chain and inspired by other groups that reported on the use of carbonaceous residues from biomass for the preparation of heterogeneous catalysts or chromatographic supports,17–19 we here utilized the waste stream of the alkaline hydrothermal digestion of biomass, i.e. mineralized lignin, as direct “all-in-one” starting material for the preparation of porous carbonaceous material where the inherently included barium salts are used as porogen.20–23
The obtained materials were then used as support for iron-nickel metal nanoparticles which exhibited remarkable catalytic activities for the continuous flow hydrogenation of nitrobenzene and phenylacetylene, commonly reduced using scarce and expensive noble metal supported catalysts.24
Result and discussion
The first step of our process was the hydrothermal treatment of biomass (rye straw) using Ba(OH)2, which despite its toxicity compared to other alkaline compounds, is known to promote the formation of lactic acid in high yields,15 and led to the recovery of a precipitate containing lignin-derived oligomers (see the ESI†). In this study, we focused on the valorization of this otherwise waste crude by-product consisting in a lignin/barium salt mixture (ML: mineralized lignin) as “all-in-one” precursor for porous carbonaceous materials. Initially, ML, which precipitates after the hydrothermal process, could be easily obtained by filtration (Scheme 1, step 1).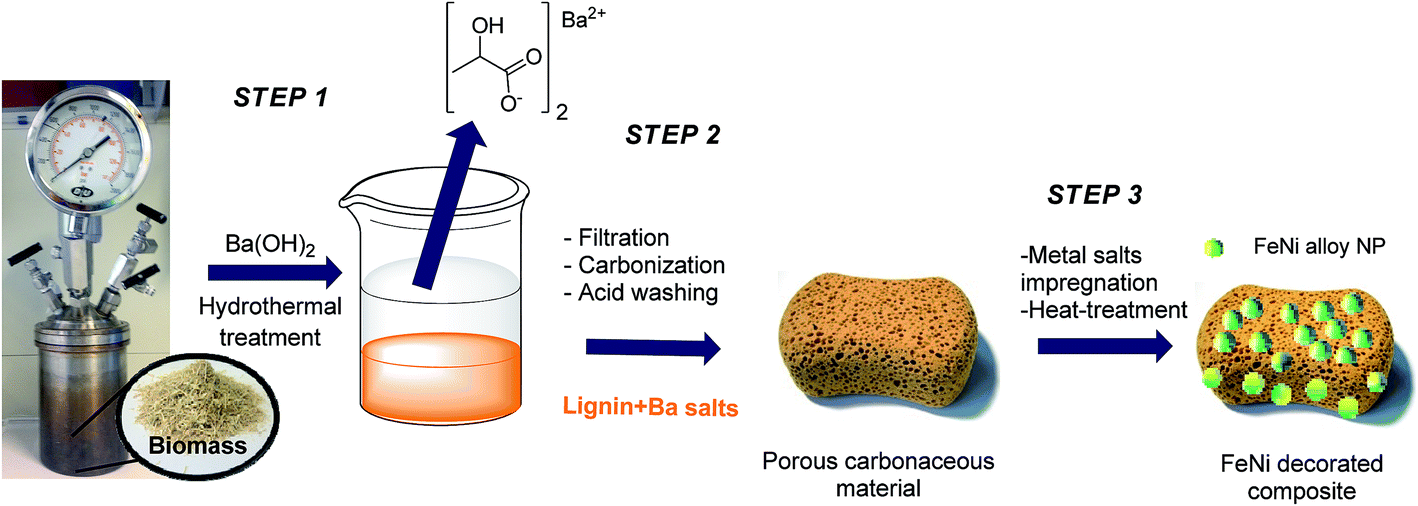 | ||
| Scheme 1 Process chain for the synthesis of porous lignin derived carbonaceous material and the respective supported FeNi NPs composite. | ||
The X-ray diffraction (XRD) pattern of the crude filter cake revealed the typical diffraction peaks of barium carbonate (Fig. 1a). Carbonates are classical by-products of alkali hydrothermal treatments of biomass.25 The crystallite size of the salt was calculated by Scherrer equation to be approx. 26 nm in diameter. The presence of this compound was additionally confirmed by comparison of the IR spectrum of the ML (Fig. 2a) with the one of the pure BaCO3 (Fig. 2b). The absorption bands at 400–1800 cm−1, also visible in ML, were attributed to vibrations of CO32− and reflect the asymmetric C–O stretching (1427 cm−1), out of plane bending (856 cm−1) and in plane bending vibration (692 cm−1).26
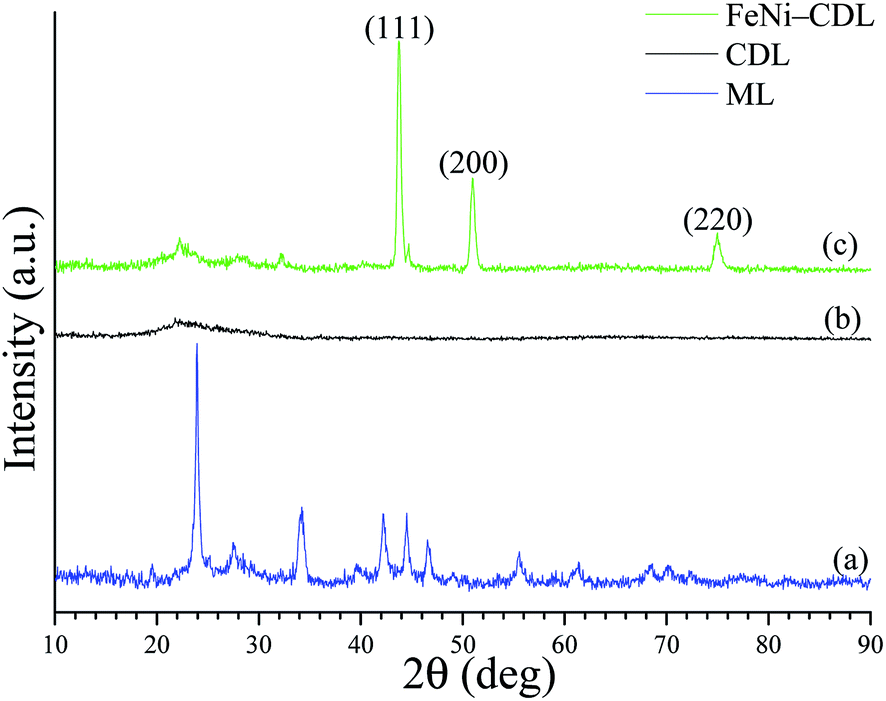 | ||
| Fig. 1 XRD patterns of the mineralized lignin (a), of the carbonized/demineralized lignin (b) and of the carbonized/demineralized lignin impregnated with the salt precursors of Fe and Ni and then heat-treated at 800 °C for 2 h under N2 (c). | ||
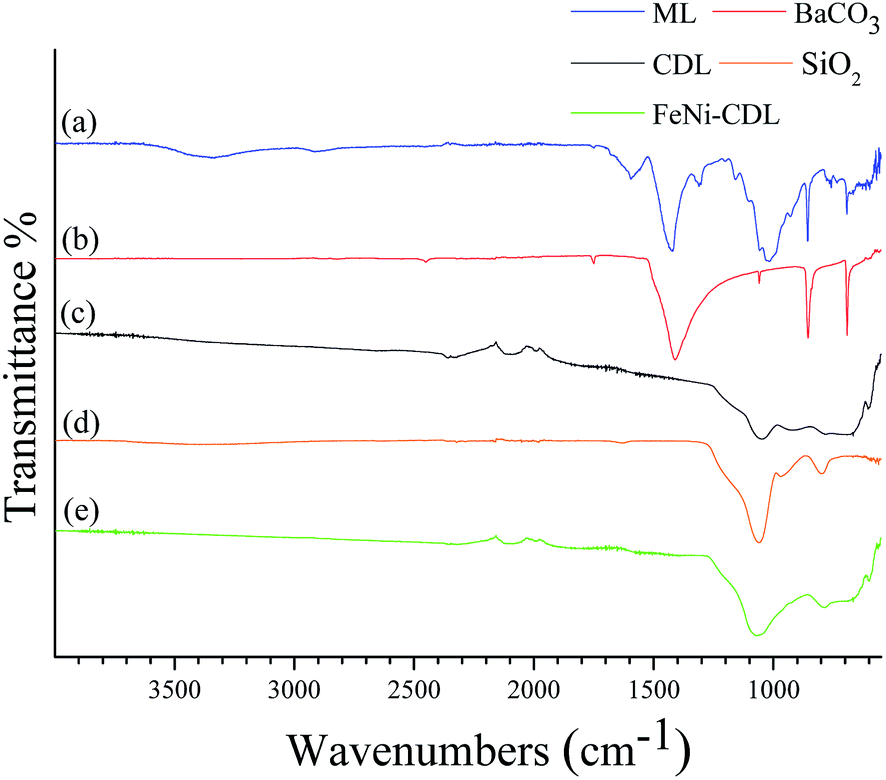 | ||
| Fig. 2 IR spectra of the mineralized lignin (a), of BaCO3 (b), of the carbonized/demineralized lignin (c), of SiO2 (d), and of the carbonized/demineralized lignin impregnated with the salt precursors of Fe and Ni and then heat-treated at 800 °C for 2 h under N2 (e). | ||
Besides BaCO3, the IR spectrum of ML also confirmed the presence of some characteristic infrared bands attributed to lignin: a broad band at 3340 cm−1 corresponding to the O–H of alcoholic and phenolic groups stretching vibrations, and the shoulder at 1644 cm−1 caused by the stretching of carbonyl groups conjugated with aromatic rings. Aromatic skeleton vibration was also visible and appeared at 1594 cm−1.27
Elemental analysis of the ML (see Table S1†) revealed a residual carbon content of ∼17 wt% and the relative TGA analysis under inert atmosphere (see Fig. S1†) showed a weight loss of 38% at 1000 °C associated with release of volatiles.28
At this stage, the organic–inorganic solid ML was directly used as carbonaceous precursor and heat-treated in a nitrogen oven at 800 °C for 2 h where BaCO3 served as the in situ activation and porogen agent. The carbonized product was further washed with diluted hydrochloric acid in order to remove the inorganic content and free the pores of the resulting carbonaceous material (CDL: carbonized demineralized lignin), which was isolated as a black powder in 16 wt% yield after simple vacuum drying (Scheme 1, step 2). The removal of crystalline components was confirmed by XRD (Fig. 1b). However, IR revealed the presence of an adsorption band centered at 1060 cm−1, which could be ascribed to the asymmetric bending modes of SiO2, pointing to residual amounts of silica (see Fig. 2c and d) commonly found in straw ashes.29,30 Scanning electron microscopy (SEM) revealed a different morphological structure for crude ML (Fig. 3a and a′) compared to CDL (Fig. 3b and b′).
 | ||
| Fig. 3 Low magnification (a–c) and high magnification (a′–c′) SEM images of the ML (a and a′), CDL (b and b′) and FeNi–CDL (c and c′). | ||
The crude ML material possessed a flake-like morphology with extended thick sheets over several microns. In contrast, CDL showed much finer structures which suggested the successful removal of the barium salts thus giving access to the actual carbonaceous material. In order to assess the porosity in more detail, nitrogen sorption measurements were performed (Fig. 4a and b).
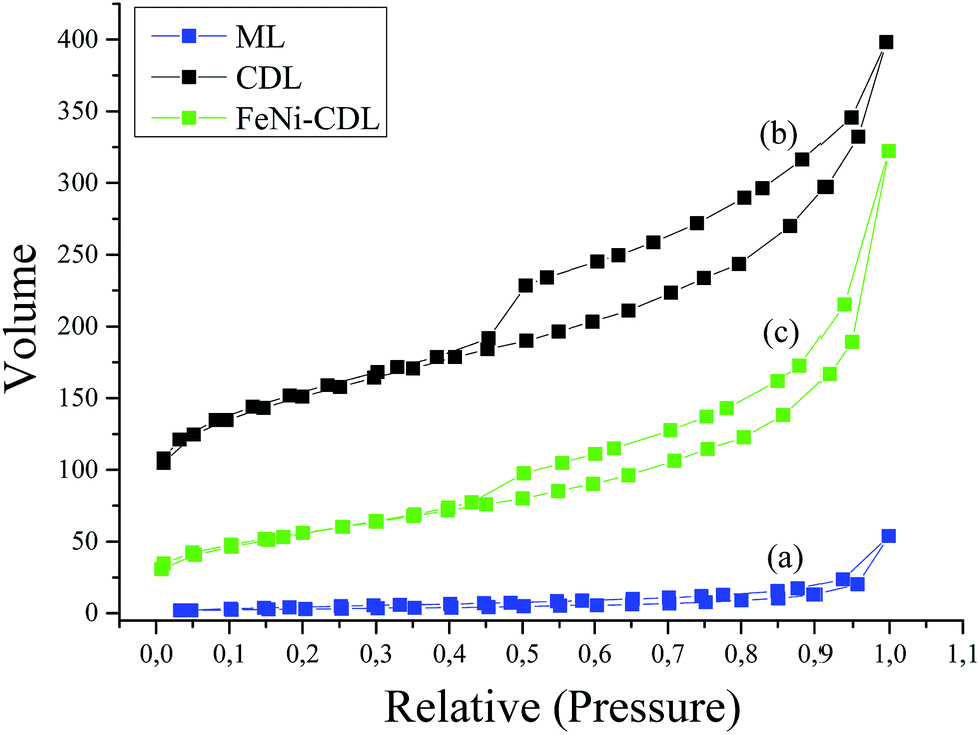 | ||
| Fig. 4 Nitrogen sorption isotherms of the ML (a), CDL (b) and FeNi–CDL (c). | ||
Here, the Brunauer–Emmett–Teller (BET) calculation for ML gave a negligible surface area of 11 m2 g−1 whereas the final carbonaceous material CDL showed an increased surface area of 539 m2 g−1 confirming the assumptions from the SEM images. Furthermore, the isotherm of CDL displayed high nitrogen uptake already at low relative pressures indicative for a reasonable amount of micropores. The isotherm of CDL shows a type IV behavior which is usually ascribed to the presence of mesopores. Average pore size was calculated from the nitrogen sorption isotherms using the NLDFT model for slit pores to be of 5 nm in diameter (see Fig. S2†). Here, it is to be mentioned that this size is much smaller than the barium carbonate crystallite size obtained from XRD measurements which is attributed to the deviating properties of the carbon described here from the model on which calculations are based as well as to cavitation effects.
Eventually, the direct heat treatment of ML followed by acid washing could be exploited for the synthesis of high surface area carbonaceous materials with micro- and mesoporosity. Such features are highly desirable for catalytic reactions where high mass transport is required.
With this is mind, the CDL was thus considered as a catalyst precursor and further functionalized with iron nickel nanoparticles (FeNi–CDL). This was achieved by impregnation of CDL with an alcoholic solution of the respective metal nitrate salts followed by heat-treatment at 800 °C for 2 h under N2 atmosphere (Scheme 1, step 3). This yielded a fine black powder and the XRD confirmed the formation of a fcc crystalline phase of a FeNi alloy with a NP crystallite size of 17 nm (Fig. 1c).31 Besides the main phase, an additional less intense peak found at 44.75θ revealed the presence of a concomitant bcc phase of the same alloy.32 The morphology of the FeNi–CDL resembled quite well the one of the original carbonaceous material, however, well visible bright metal nanoparticles with an average size of 48 nm in diameter could be detected (Fig. 3c and c′). The presence of iron and nickel was confirmed by EDX analysis (see Fig. S3 in the ESI†).
TEM analysis of FeNi–CDL revealed a relatively broad size distribution and the coexistence of metal particle conglomerates in the micrometer range, together with round shape nanoparticles embedded in the carbonaceous matrix (see Fig. 5a and b). The dispersed non-aggregated nanoparticles visible at high magnification showed an average size distribution of 48 nm (the size distribution graph is reported in the ESI in Fig. S4†). The fact that the mean particle size observed from both SEM and TEM images was larger than the average crystallite size calculated using the Scherrer equation from the XRD pattern (17 nm) confirmed that these nanoparticles were partly conglomerates.
 | ||
| Fig. 5 Low (a) and high (b) magnification TEM images of the carbonized/demineralized lignin impregnated with the salt precursors of Fe and Ni and then heat-treated at 800 °C for 2 h under N2. | ||
Also nitrogen sorption analysis still revealed an isotherm shape similar to the one of the initial CDL, i.e. particles are deposited on the outer surface, pointing to the successful functionalization of CDL under preservation of the porosity as well as morphology. The somewhat lowered surface area of 198 m2 g−1 could be ascribed to the increased mass of the additional metals (Fig. 4c).
Inductively coupled plasma-optical emission spectroscopy (ICP-OES) analysis of FeNi–CDL displayed the simultaneous presence of Fe (14 wt%) and Ni (22 wt%) in the initially employed ratio of the metal salt precursors as well as residues of Ba and Si from the ML (see Table S1†). As mentioned earlier, the focus of the present contribution was more the facile and sustainable valorization of the alkali hydrothermal process chain value. Therefore, a standardized washing procedure was applied without optimizing conditions for a full removal of inorganic impurities. Moreover, as it will be discussed in the following, the residual presence of Si and Ba was not detrimental for the catalytic performances of the FeNi–CDL, and possible additional effects caused by these inorganic impurities will be evaluated in future studies.
Having established the protocol for the preparation of FeNi–CDL from the alkali lignin, we assessed the catalytic performance of such material for hydrogenation reactions. For this purpose, the FeNi–carbonaceous composite powder was packed into a cartridge and directly employed as a catalyst within a bench-top continuous flow reactor (H-Cube Pro™) provided with an internal H2 source and equipped with a liquid feed.
Table 1 reports the conversion and selectivity for the hydrogenation of two different model substances. The flow rate and the molarity of the different substrates were kept constant (50 mM and 0.3 mL min−1), while temperature and hydrogen pressures were varied to optimize conversion and selectivity.
| Entry | Substrate | Product | Conversione (%) | Selectivitye (%) | T (°C) | p-H2 (bar) |
|---|---|---|---|---|---|---|
| a Conditions: 50 mM in ethanol, 0.3 mL min−1. b CDL was used as catalyst. c 100 mM, 0.5 mL min−1. d Lindlar catalyst. e Liquid feed compositions are expressed as uncorrected GC/MS peak area percentages. | ||||||
| 1a |
|
|
>99 | 95 | 125 | 20 |
| 2a,b |
|
|
8 | 99 | 125 | 20 |
| 3a |
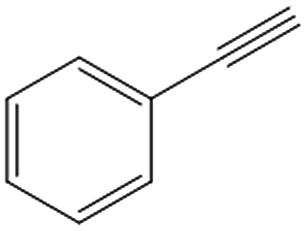
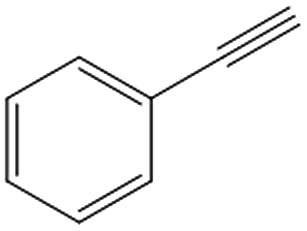
|
|
>99 | 95 | 150 | 50 |
| 4c |
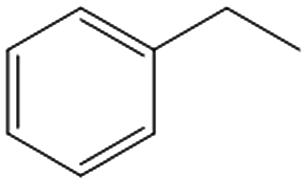
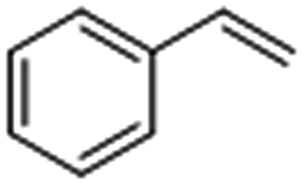
|
|
84 | 75 | 50 | 10 |
| 5c,d |
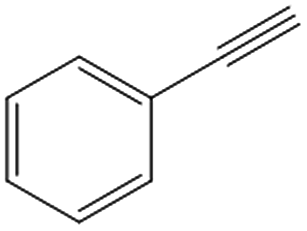
|
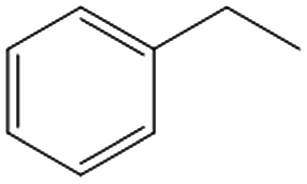
|
>99 | 89 | 50 | 10 |
| 6c,d |
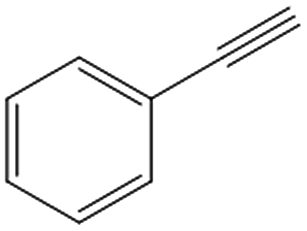
|
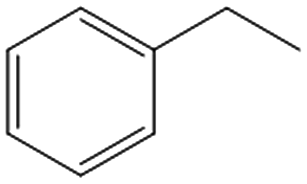
|
>99 | 82 | 25 | 1 |
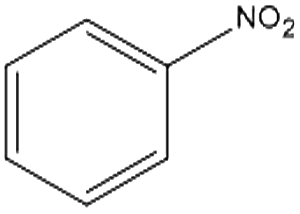
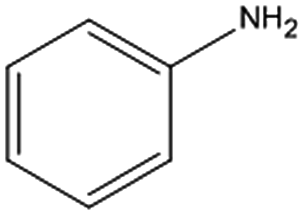
The catalytic hydrogenation of nitroaromatic compounds is a crucial reaction for the production of anilines, which represent an important intermediate for the synthesis of pharmaceuticals, polyurethanes, dyes and agricultural products.33,34 As a model for such transformation, the reduction of nitrobenzene was evaluated. The latter compound could be quantitatively converted into aniline (entry 1) at 125 °C and 20 bar of H2. The GC chromatograms and the corresponding mass spectra of the major products from Table 1 are reported in Fig. S5 and S6 in the ESI.†
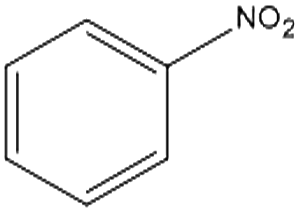
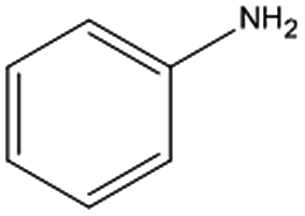
Control experiments performed with the non-functionalized CDL did not show effective hydrogenation of nitrobenzene under the same experimental conditions, confirming in a preliminary way that the presence of inorganic residues inherently present from the staring material was negligible with regard to the catalytic performance and underlined the active role of FeNi NPs in the catalytic cycle (entry 2).
Phenylacetylene was also tested as substrate for the continuous hydrogenation in order to prove the versatility of the FeNi–CDL for the reduction of additional functional groups (entry 3). The complete conversion of the starting material at high selectivity toward ethylbenzene was achieved at 150 °C and 50 bar of H2.
Interestingly, the selectivity of the reaction could be tuned towards styrene at milder conditions (entry 4) and increasing the molarity (100 mM) as well as the flow rate (0.5 mL min−1). Traditionally, similar processes are performed using commercial catalysts as the Lindlar, a palladium catalyst poisoned with traces of lead and quinoline.35 Nevertheless, using the latter under the same experimental conditions (entry 5), ethylbenzene was obtained. The selective hydrogenation to styrene could not be achieved even when the reaction was performed at room temperature and with 1 bar of H2 (entry 6), revealing our FeNi–CDL composite as an interesting candidate for selective hydrogenation processes.
Conclusions
In this work we showed how the integrated processes for the smart recycle and upcycle of biomass derived waste streams could be efficiently combined with strategies for the preparation of functional carbonaceous and composites materials based on cheap and abundant metal precursors of iron and nickel. Porous carbonaceous materials were obtained via the heat-treatment of mineralized lignin isolated from the waste stream of the alkaline hydrothermal digestion of rye straw. The BaCO3 contained in the “all-in-one” crude lignin carbonaceous precursor could be in this way upcycled and served as an efficient in situ activation and poration agent, leading to the formation of porous carbonaceous materials which were further functionalized with FeNi alloys nanoparticles. The resulting nanocomposite displayed remarkable catalytic activity for the flow hydrogenation of nitrobenzene and phenylacetylene to aniline and ethylbenzene, respectively. Moreover the FeNi–CDL was effective for the selective partial hydrogenation of phenylacetylene to styrene under mild conditions without the use of any additives, unlike the commercial Lindlar catalyst. Therefore, the approach described in this work demonstrates nicely the efficient valorization of a waste stream from an independent biomass conversion scheme and opens the way to a fully integrated biorefinery with increased chain value. Future studies will focus on the preparation of analogs of this novel catalytic system and the evaluation of their activity for additional catalytic transformations. Moreover, the possible synergistic effects of the inorganic residues contained in the waste biomass on the overall catalytic performance of the final composites will be further evaluated.Acknowledgements
The authors gratefully thank Hendrik Wetzel for ICP OES measurements (Fraunhofer Institute for Applied Polymer Research).Notes and references
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- J. Lora and W. Glasser, J. Polym. Environ., 2002, 10, 39–48 CrossRef CAS.
- L. da Costa Sousa, S. P. S. Chundawat, V. Balan and B. E. Dale, Curr. Opin. Biotechnol., 2009, 20, 339–347 CrossRef PubMed.
- Y. C. Park and J. S. Kim, Energy, 2012, 47, 31–35 CrossRef CAS PubMed.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- V. Fierro, V. Torné-Fernández and A. Celzard, Microporous Mesoporous Mater., 2007, 101, 419–431 CrossRef CAS PubMed.
- Y. Gao, Q. Yue, B. Gao, Y. Sun, W. Wang, Q. Li and Y. Wang, Chem. Eng. J., 2013, 217, 345–353 CrossRef CAS PubMed.
- J. Bedia, J. M. Rosas, J. Rodríguez-Mirasol and T. Cordero, Appl. Catal., B, 2010, 94, 8–18 CrossRef CAS PubMed.
- E. Guillén, R. Rico, J. M. López-Romero, J. Bedia, J. M. Rosas, J. Rodríguez-Mirasol and T. Cordero, Appl. Catal., A, 2009, 368, 113–120 CrossRef PubMed.
- J. A. Zazo, J. Bedia, C. M. Fierro, G. Pliego, J. A. Casas and J. J. Rodriguez, Catal. Today, 2012, 187, 115–121 CrossRef CAS PubMed.
- P. Gallezot, P. J. Cerino, B. Blanc, G. Flèche and P. Fuertes, J. Catal., 1994, 146, 93–102 CrossRef CAS.
- S. Sitthisa, W. An and D. E. Resasco, J. Catal., 2011, 284, 90–101 CrossRef CAS PubMed.
- G. Chieffi, C. Giordano, M. Antonietti and D. Esposito, J. Mater. Chem. A, 2014, 2, 11591–11596 CAS.
- D. Esposito and M. Antonietti, ChemSusChem, 2013, 6, 989–992 CrossRef CAS PubMed.
- G.-Q. Chen and M. K. Patel, Chem. Rev., 2012, 112, 2082–2099 CrossRef CAS PubMed.
- R. Luque, A. Pineda, J. C. Colmenares, J. M. Campelo, A. A. Romero, J. C. Serrano-Riz, L. F. Cabeza and J. Cot-Gores, J. Nat. Gas Chem., 2012, 21, 246–250 CrossRef CAS.
- M.-M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- R. J. White, V. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 3401–3418 RSC.
- P. J. M. Suhas, M. M. L. Carrott and R. Carrott, Lignin – from natural adsorbent to activated carbon: A review, Bioresour. Technol., 2007, 98(12), 2301–2312 CrossRef CAS PubMed.
- J. M. Dias, M. C. M. Alvim-Ferraz, M. F. Almeida, J. Rivera-Utrilla and M. Sánchez-Polo, J. Environ. Manage., 2007, 85, 833–846 CrossRef CAS PubMed.
- C. Zhang, M. Antonietti and T.-P. Fellinger, Adv. Funct. Mater., 2014, 24, 7655–7665 CrossRef CAS PubMed.
- N. Fechler, T.-P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75–79 CrossRef CAS PubMed.
- (a) S. Kataoka, Y. Takeuchi, A. Harada, T. Takagi, Y. Takenaka, N. Fukaya, H. Yasuda, T. Ohmori and A. Endo, Appl. Catal., A, 2012, 427–428, 119–124 CrossRef CAS PubMed; (b) J. Hu, Z. Zhou, R. Zhang, L. Li and Z. Cheng, J. Mol. Catal. A: Chem., 2014, 381, 61–69 CrossRef CAS PubMed.
- J. A. Onwudili and P. T. Williams, Int. J. Hydrogen Energy, 2009, 34, 5645–5656 CrossRef CAS PubMed.
- N. Tipcompor, T. Thongtem, A. Phuruangrat and S. Thongtem, Mater. Lett., 2012, 87, 153–156 CrossRef CAS PubMed.
- B. Xiao, X. F. Sun and R. Sun, Polym. Degrad. Stab., 2001, 74, 307–319 CrossRef CAS.
- S. Munir, S. S. Daood, W. Nimmo, A. M. Cunliffe and B. M. Gibbs, Bioresour. Technol., 2009, 100, 1413–1418 CrossRef CAS PubMed.
- A. E. Ghaly and A. Al-Taweel, Energy Sources, 1990, 12, 131–145 CrossRef CAS PubMed.
- G.-M. Gao, H.-F. Zou, S.-C. Gan, Z.-J. Liu, B.-C. An, J.-J. Xu and G.-H. Li, Powder Technol., 2009, 191, 47–51 CrossRef CAS PubMed.
- Y. Chen, X. Luo, G.-H. Yue, X. Luo and D.-L. Peng, Mater. Chem. Phys., 2009, 113, 412–416 CrossRef CAS PubMed.
- Y. Liu, Y. Chi, S. Shan, J. Yin, J. Luo and C.-J. Zhong, J. Alloys Compd., 2014, 587, 260–266 CrossRef CAS PubMed.
- T. Stemmler, A.-E. Surkus, M.-M. Pohl, K. Junge and M. Beller, ChemSusChem, 2014, 7, 3012–3016 CrossRef CAS PubMed.
- X. Meng, H. Cheng, Y. Akiyama, Y. Hao, W. Qiao, Y. Yu, F. Zhao, S.-I. Fujita and M. Arai, J. Catal., 2009, 264, 1–10 CrossRef CAS PubMed.
- H. Lindlar and R. Dubuis, Org. Synth., 1966, 46, 89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06635b |
| This journal is © The Royal Society of Chemistry 2015 |