
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular control of transition metal complexes in water by a hydrophobic cavity: a bio-inspired strategy
Olivia
Bistri
and
Olivia
Reinaud
*
Laboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, UMR CNRS 8601, Université Paris Descartes, Sorbonne Paris Cité, 45 rue des Saints Pères, 75006 Paris, France. E-mail: Olivia.Reinaud@parisdescartes.fr
First published on 7th January 2015
Abstract
Supramolecular chemistry in water is a very challenging research area. In biology, water is the universal solvent where transition metal ions play major roles in molecular recognition and catalysis. In enzymes, it participates in substrate binding and/or activation in the heart of a pocket defined by the folded protein. The association of a hydrophobic cavity with a transition metal ion is thus a very appealing strategy for controlling the metal ion properties in the very competitive water solvent. Various systems based on intrinsically water-soluble macrocyclic structures such as cyclodextrins, cucurbituryls, and metallo-cages have been reported. Others use calixarenes and resorcinarenes functionalized with hydrophilic substituents. One approach for connecting a metal complex to these cavities is to graft a ligand for metal ion binding at their edge. Early work with cyclodextrins has shown Michaelis-Menten like catalysis displaying enhanced kinetics and substrate-selectivity. Remarkable examples of regio- and stereo-selective transformation of substrates have been reported as well. Dynamic two-phase systems for transition metal catalysis have also been developed. They rely on either water-transfer of the metal complex through ligand embedment or synergistic coordination of a metal ion and substrate hosting. Another strategy consists in using metallo-cages, which provide a well-defined hydrophobic space, to stabilize metal complexes in water. When the cages can host simultaneously a substrate and a reactive metal complex, size- and regio-selective catalysis was obtained. Finally, construction of a polydentate coordination site closely interlocked with a calixarene or resorcinarene macrocycle has been shown to be a very fruitful strategy for obtaining metal complexes with remarkable hosting properties. For each of these systems, the synergism resulting from the biomimetic association of a hydrophobic cavity and a metal ion is discussed within the objective of developing new tools for either selective molecular recognition (with analytical perspectives) or performant catalysis, in water.
 Olivia Bistri | Olivia Bistri (Paris, France 1978) received her PhD from the Université Pierre et Marie Curie (Paris) in 2006 where she studied the regioselective functionalisation of α- and β-cyclodextrins under the supervision of Prof. Pierre Sinaÿ and Matthieu Sollogoub. She then joined the group of Prof. Carsten Bolm (Institut für Organische Chemie, RWTH Aachen University, Germany) as a Humboldt post-doctoral fellow. In 2008, she was appointed CNRS Researcher at Université Paris Descartes and she is currently working in the group of Prof. Olivia Reinaud in the field supramolecular bioinorganic chemistry. |
 Olivia Reinaud | Olivia Reinaud completed a PhD in organic chemistry (1987, Dr M. Maumy, ESPCI-UPMC) and after a one-year postdoc in biochemistry (Dr D. Mansuy, Paris Descartes University), she was appointed CNRS researcher and developed novel biomimetic copper catalyzed processes (ESPCI). She took a two year sabbatical in inorganic chemistry (Delaware University, USA, Prof. K. Theopold). Upon return to France, she started a new project associating organic, inorganic and supramolecular chemistry with some “bio-inspiration”. Since 2001, she has a full Professorship at Paris Descartes. Her current research interest lies in “Supramolecular Bio-Inorganic Chemistry”, dealing with biomimetic metal complexes based on calixarene derivatives. |
1. Introduction
Molecular recognition and catalysis in water are attractive goals for both economical and environmental reasons. On the one hand, water is a cheap, non toxic and readily available solvent. On the other hand, it is important to develop efficient analytical tools for identifying, quantifying or even extracting objects from water for environmental purpose or for biology and development of therapeutics. Hence, this topic faces problems of selectivity, affinity, and analytical reporting. For catalysis, water can be involved in the reactivity, which needs to be controlled.An infinite source of inspiration for the design of molecular receptors and catalysts is Nature. Interestingly, ca. 30% of enzymes present one or several metal ions in their active site.1 The metal ion participates either in the enzyme–substrate molecular recognition process or in the chemical transformation itself, or both. In the active site, neither the metal ion nor the substrate does sit in an aqueous environment. Instead, the folded protein provides an organic medium constituted of the amino-acid residues defining the hydrophobic pocket, which however remains accessible to a definite number of water molecules. This pocket confers well-defined and controlled first and second coordination spheres to the metal ion(s) and thus participates in the formation of reactive species. It also provides a third coordination sphere (and further…) that allows substrate binding and pre-organization next to the metal center and ensures product release. These two points are essential for selectivity (regio- and stereo-), efficiency, and catalysis with high turnovers.
Water is a highly competitive medium for both molecular recognition and transition metal complexation.2 Indeed, the water molecule is a hydrogen bond donor and acceptor, and provides a medium of high polarity. It strongly solvates polar guests and decreases host–guest electrostatic attractive interactions.
Water is also a competitive ligand for a transition metal complex leading to the difficult control of the coordination sphere of a transition metal. It readily binds metal ions, with a more pronounced affinity for hard Lewis acids. Upon coordination, the pKa of water decreases, which readily leads to hydroxide- and oxide-bound metal species that, if not controlled, can dimerize or polymerize and precipitate.
Water however presents some attractive aspects. In this solvent, molecular recognition can take advantage from the hydrophobic effect. Indeed, structuration of water molecules due to the presence of apolar compounds has an energy cost. As a consequence, desolvation of a guest associated to release of water molecules embedded in the host participates in the stability of the host–guest association.3 Catalysis can also benefit from pH control and proton accessibility, which are keys in processes involving acid–base interaction or electron transfer associated to proton transfer.
Hence, the association of a hydrophobic cavity with a transition metal ion (TM) appears to be a very appealing strategy for the development of new tools for molecular recognition (with analytical perspectives) and chemical transformation (catalysis). The hydrophobic cavity provides an organic microenvironment around or next to the metal center, which may contribute to the metal ion control and stabilization. It participates in the hosting process of the analyte/substrate and allows preorganization of the guest next to the metal ion. Conversely, the metal ion can strengthen the host–guest complex through a coodination bond. It may polarize the guest, thus exacerbating its reactivity, or activate a reactant in the vicinity of the guest-substrate, in a protected microenvironment.
Artificial systems presenting one or several synergistic aspects mentioned above are the subject of this review. Early studies report systems built on a readily available water-soluble cavity, namely cyclodextrins. Various strategies were developed: one consists in grafting a ligand (for metal ion binding) at the edge of the cavity; another relies on dynamic two-phase systems for transition metal catalysis. More recently, metallo-cages have been used as water-soluble receptors for hosting simultaneously a substrate and a TM catalyst, or stabilizing a complex in water. Efforts directed at the construction of a polydentate coodination site closely interlocked with a macrocyclic scaffold (calixarenes) led to metal complexes with very unusual properties. The most significant and representative efforts devoted to the development of such systems, associating a hydrophobic cavity to a transition metal ion, with the perspective of either selective molecular recognition or performant catalysis (ultimately both) in water will be presented.
2. A ligand connected at the edge of a water-soluble cavity
2.1. One cavity for one ligand and one metal ion
The covalent linkage of one donor ligand at the edge of a water-soluble cavity constituted the first step in the design of metal complexes in the proximity of a cavity. The first cavity used for this purpose was the enzymatically produced β-cyclodextrin macrocycle (β-CD) because of its natural availability, its solubility in water and its possible mono-functionalization. Indeed, this macrocycle displays a truncated cone shape open at both extremities to the solvent. The inside of the cavity is paved with C–H bonds, which makes it non-polar and prone to benefit from hydrophobic effect upon guest binding. In contrast, the edges present hydroxyl groups that ensure water-solubility. Thanks to the selective mono-acylation of a secondary hydroxyl group of the native β-CD, Breslow et al., in the early 70s, reported a water-soluble cavity substituted by a pyridine-2-carboxylic group, which can bind a metal ion (Fig. 1).4 The Ni(II) complex obtained in the presence of one equivalent of the CD-based ligand and one equivalent of pyridine-carboxaldoxime was used as a catalyst for the hydrolysis of p-nitrophenylacetate (pNPA). It was found that the first step of the reaction, corresponding to the nucleophilic attack of the ester substrate by the carboxaldoxime group, was four times faster than when an equivalent nickel complex deprived of cavity was used. This corresponds to a 103 rate increase over the uncatalyzed reaction. The reaction was competitively inhibited by the addition of cyclohexanol, which displays a good affinity for the CD cavity. With a substrate that does not fit into the CD cavity, no rate increase was observed. These results are consistent with a pre-association between the catalyst and the pNPA substrate thanks to the embedment of the p-PNP group into the β-CD cavity. As a result, the ester bond is preorganized next to the nucleophile bound to the metal ion. The analogous Cu(II) complex of the CD-ligand was also found to increase by a factor of six the rate of hydrolysis of the p-nitrophenylester of glycine compared to the same complex deprived of cavity. As a result, this system was coined “an artificial metallo-enzyme”.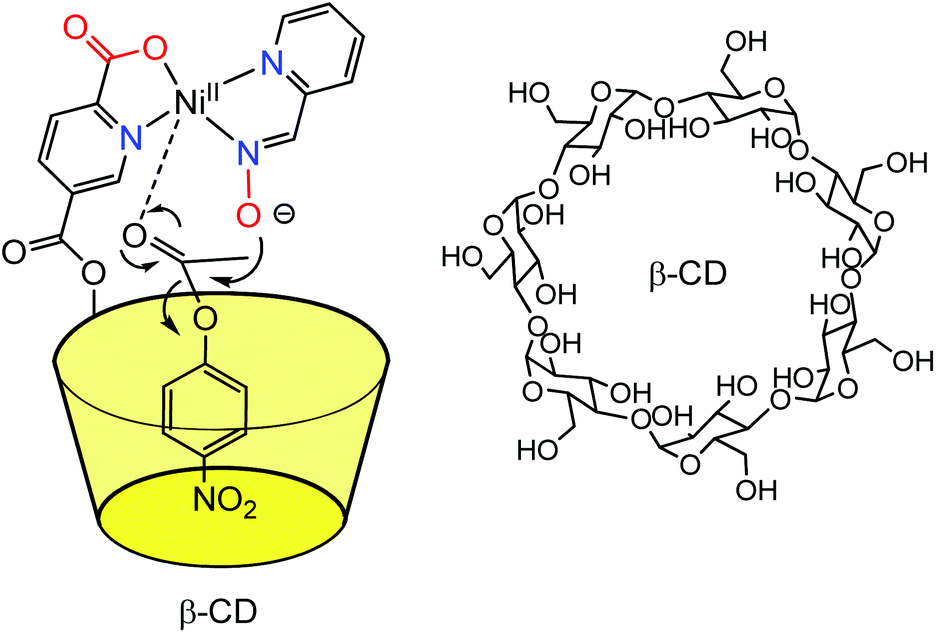 | ||
| Fig. 1 Early example of a M(II) complex in the proximity of a β-CD cavity reported by Breslow et al.4 | ||
Based on this strategy, many cyclodextrins incorporating functional groups have been synthesized and their catalytic properties evaluated. These works have already been carefully reviewed elsewhere.5,6 In 1999, Tabushi et al. reported a study describing a β-CD mono-functionalized by a polyethylene triamine, and coordinating various metal ions such as Zn(II), Cu(II) and Mg(II) in the vicinity of the hydrophobic cavity.7 The resulting metallo-hosts bind anionic guests containing a hydrophobic moiety (e.g. adamantan-2-one-1-carboxylate, 8-Anilinonaphthalene-1-sulfonate, and p-nitrophenate) more strongly than a simple β-CD deprived of metal complex at its edge. A representative example is displayed in Fig. 2, which depicts a Zn(II) complex that binds 2-oxo-1-adamantane carboxylate 330 times stronger than a simple β-CD. It emphasizes the positive cooperativity between metal–ligand interaction (at the level of the α-keto-carboxylate function) and cavity–guest interaction (with the adamantyl group), which benefits from the hydrophobic effect.
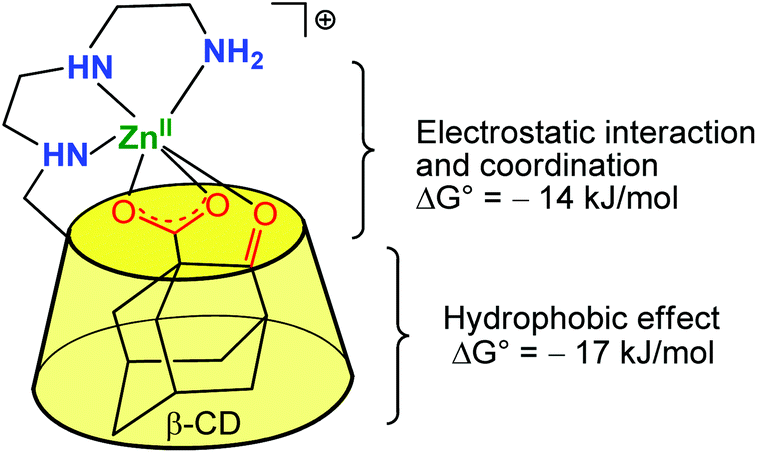 | ||
| Fig. 2 Early example of a β-CD Zn(II) complex associating the hydrophobic effect for guest inclusion and the interaction of the ligand with a metal center.7 | ||
A metal ligand with a stronger chelating property (triazacyclododecane, tacd) was also grafted to the small rim of the β-CD in order to strengthen Co(III)8 and Zn(II)9 coordination (Fig. 3). These complexes were used as mimics of zinc protease at pH = 7 and were shown to enhance the rate of hydrolysis of p-nitrophenylacetate by 900 and 300-fold compared to simple metal complexes based on the tacd ligand. Various studies drove Kim et al. to conclude that the ester substrate is CD-bound but not Zn(II)-coordinated. The rate increase is then proposed to be a consequence of substrate positioning in close proximity to the reactive metal center. In other words, the mere thermodynamic effect of substrate binding in the CD cavity has a kinetic impact on the activity of the metal catalyst.
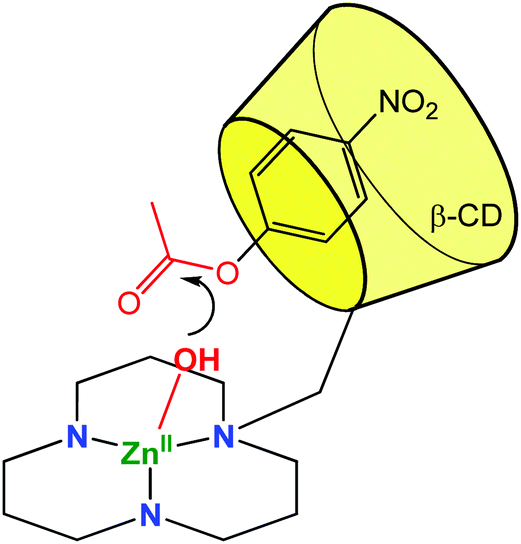 | ||
| Fig. 3 Biomimetic model using a CD cavity for substrate encapsulation in proximity of a (tacd)Zn(II) complex.9 | ||
The CD capacity of hosting, and thus recognizing a hydrophobic core has also been exploited for obtaining regioselective catalysts. An elegant example of selective peptide hydrolysis was reported by Kostić et al.10 They combined the ability of [Pd(H2O)4]2+ to residue-selectively hydrolyze X-Pro bonds in peptides at 6 ≤ pH ≤ 9, with the ability of β-CD to recognize aromatic side chains. The conjugate reagent presents a propanedithiol ligand grafted at the small rim of the CD (Fig. 4). The corresponding Pd(II) complex acts as a sequence-specific peptidase cleaving the Ser6-Pro7 amide bond in bradykinin at pH 7. ROESY 1H NMR spectra gave evidence for inclusion of the side chain of Phe8 in the β-CD cavity, the interaction that makes the cleavage sequence specific.
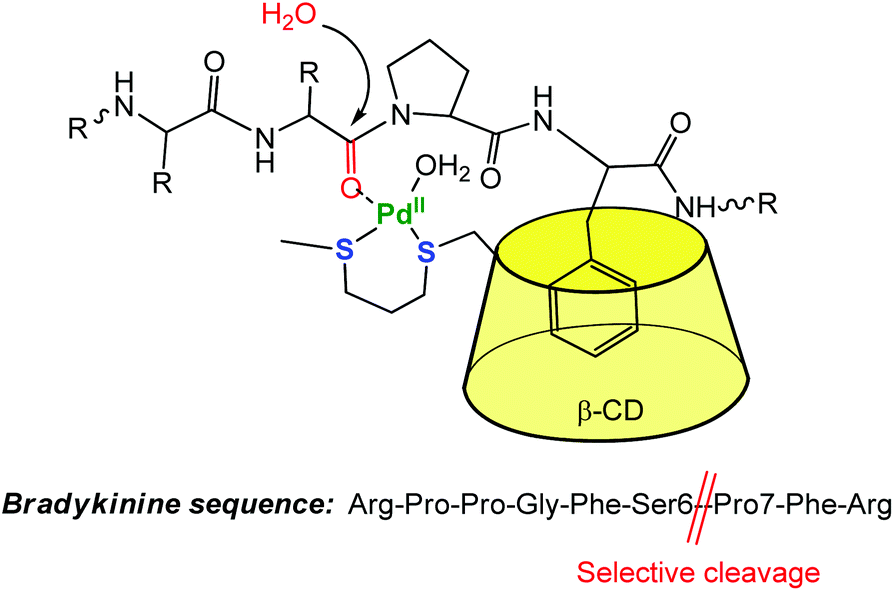 | ||
| Fig. 4 Selective cleavage of an amino-acid sequence by a β-CD palladium(II) complex in water.10 | ||
The last herein-selected example of transition metal complexes associated to a single CD illustrates this time a completely different chemistry. Breslow et al. reported in the 90s vitamin B-12 mimics. In a first study (Fig. 5),11 the CD itself is used as a ligand for a Co(III) ion through its primary methylene group. When it dissociates from B-12, it gives rise to a carbon radical that is able to abstract a phenylselenide group from a substrate bound in the CD cavity. Such a reaction mimics the deoxyadenosine cofactor abstracting an H atom from a substrate bound in the enzyme active site. In another study, the CD was attached to the propionic side chain of vitamin B-12. It was found that this species mediates some rearrangements related to those catalyzed by the enzyme and with a preference for substrates that bind in the CD cavity.12
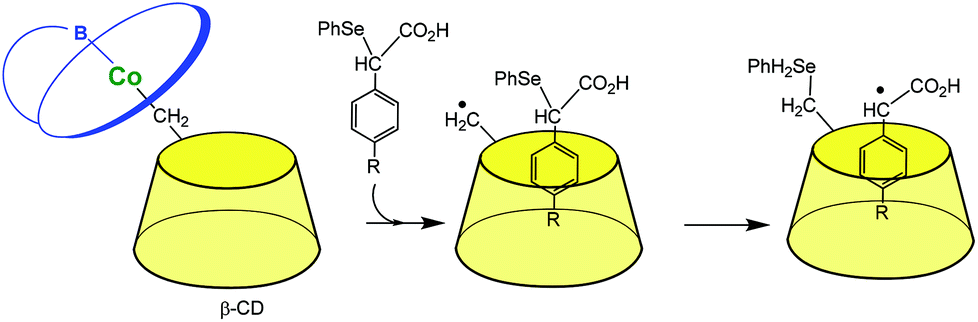 | ||
| Fig. 5 A mimic of vitamin B-12 (schematized in blue) enzymes.11 | ||
In summary, the selected examples described above have highlighted several important aspects that can be exploited when associating a transition metal complex to a host cavity in water:
● In water, the hydrophobic effect is at work for substrate binding, thus allowing its positioning in the proximity of the catalyst grafted at the edge of the cavity. This leads to faster kinetics.
● Substrate binding in the cavity can benefit from its coordination to the metal ion that strengthens the host–guest link and polarizes the substrate (in the case of metal-catalyzed hydrolysis).
● Host–guest recognition also allows substrate-selective transformations, and regio-selective reactions when using multifunctional substrates.
It is worth noting that in most of the above-mentioned examples, the metal complex was formed in situ in aqueous solution and, as a consequence, little is known about the exact nature of the metal environment. Finally, an important problem that most of these systems face is the associated low substrate conversion levels. This stems from the fact that substrate and product have often similar affinities for the CD cavity, which leads to product inhibition as the reaction progresses. Systems based on ditopic recognition of substrates with dimers of CDs rendered it possible to circumvent this obstacle in the case of hydrolytic catalysis, which will be presented below.
2.2. Several cavities for one ligand and one metal ion
CD monomers classically bind good guests with affinity constants of ca. 104 M−1 in water. In order to improve substrate binding and preorganization next to the catalyst, dimers of CDs connected through a ligand were synthetized. High affinity constants for ditopic substrates (having two moieties prone to CD embedment) were expected with both, enthalpic and entropic gains. The corollary of this strategy is that guests presenting a single binding group would display lower affinities for the dimer of CD. Hence, for reactions leading to the cleavage of ditopic substrates, high turnovers are expected.An early example, reported in 1992 by Breslow et al., consisted in two CD cavities connected through a bipyridine ligand (Fig. 6). In the presence of various metal ions (Cu2+, Ni2+, Zn2+), the hydrolysis of ester substrates presenting two hydrophobic groups prone to CD embedment was studied in aqueous media.13
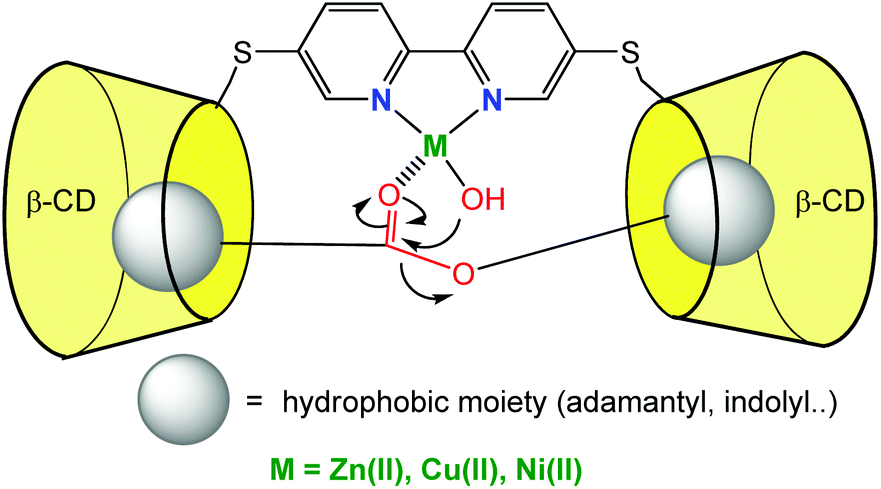 | ||
| Fig. 6 CD dimer with a catalytic binding group leading to fast ester hydrolysis.13 | ||
They observed a Michaelis-Menten kinetic behavior for the hydrolysis of esters [in 80% (v/v) DMSO/20 mM HEPES (pH 7) at 25 °C], with a Km value of 50 μM consistent with the strong double binding of the ditopic substrate. The kcat value of 0.45 s−1 corresponds to a rate enhancement (kcat/kuncat) of 1.5 × 107, which is ca. 100 times higher than that obtained with the monomer. This example further illustrates the impact of good substrate positioning in front of a catalytic site. But also importantly, thanks to a better affinity for the ditopic substrate compared to the monotopic reaction products, no sign of product inhibition was observed after 50 turnovers. The same system could be exploited for phosphoester cleavage at pH 7 and 25 °C in the presence of La3+ metal cation and hydrogen peroxide.14
More recently, Mao et al. described a dinuclear copper complex of a dimeric β-CD ligand (Fig. 7).15 Interestingly, they showed that, in addition to achieving substrate binding, the proximity of the CDs tunes the nature of the metal cofactor. Indeed, the dinuclear Cu(II) complex displays a hydroxo bridging ligand associated to a surprisingly low pKa value (4.3). As a result, the complex exhibits good phosphodiesterase activity at acidic pH, which is rare among synthetic complexes. The dramatic pKa decrease was related to the local non-polar sphere inside the CDs and hydrogen bonding to CD hydroxyl groups. They also showed that bound cavities considerably improved the water tolerance of cerium(IV) complexes, owing to steric and hydrophobic interactions with the CDs.16 As a result, the second-order rate constant reached 2.3 × 102 M−1 s−1 at pH 6.5, which is one of the highest non-enzymatic rate of bis(4-nitrophenyl)phosphate cleavage under mild conditions. These cases reveal that CDs can participate in the catalysis by controlling the micro-environment around the metal center.
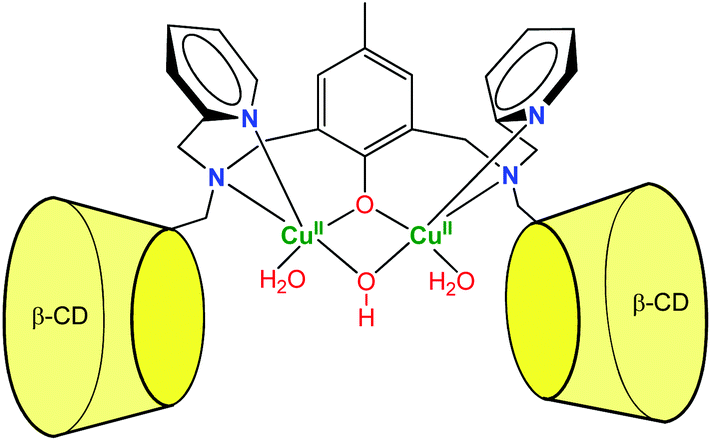 | ||
| Fig. 7 A dinuclear copper-β-cyclodextrin dimer complex exhibiting phosphodiesterase activity at acidic pH.15 | ||
Zhao et al. reported recently a photo-switchable version of the β-CDZn(II) catalyst (Fig. 8).17 They associated a dimethylaminopyridine-bridge β-CD dimer to gold nanoparticles presenting azobenzene units attached to their surfaces. Under visible light, the azobenzene units are in trans configuration and bind tightly to the CDs. This leads to reduced hydrolytic catalysis with p-nitrophenylacetate as a substrate. Under UV light, the azobenzene units are cis and bind loosely to the dimer. This allows substrates to get in and hydrolysis to occur with a 30-fold rate increase. With the ditopic substrate bis(p-nitrophenyl)carbonate, such a photo-switchable behavior was not observed because the binding affinity of the ditopic substrate for the CD dimer is too strong compared to the trans-azobenzene units of the gold nanoparticles.
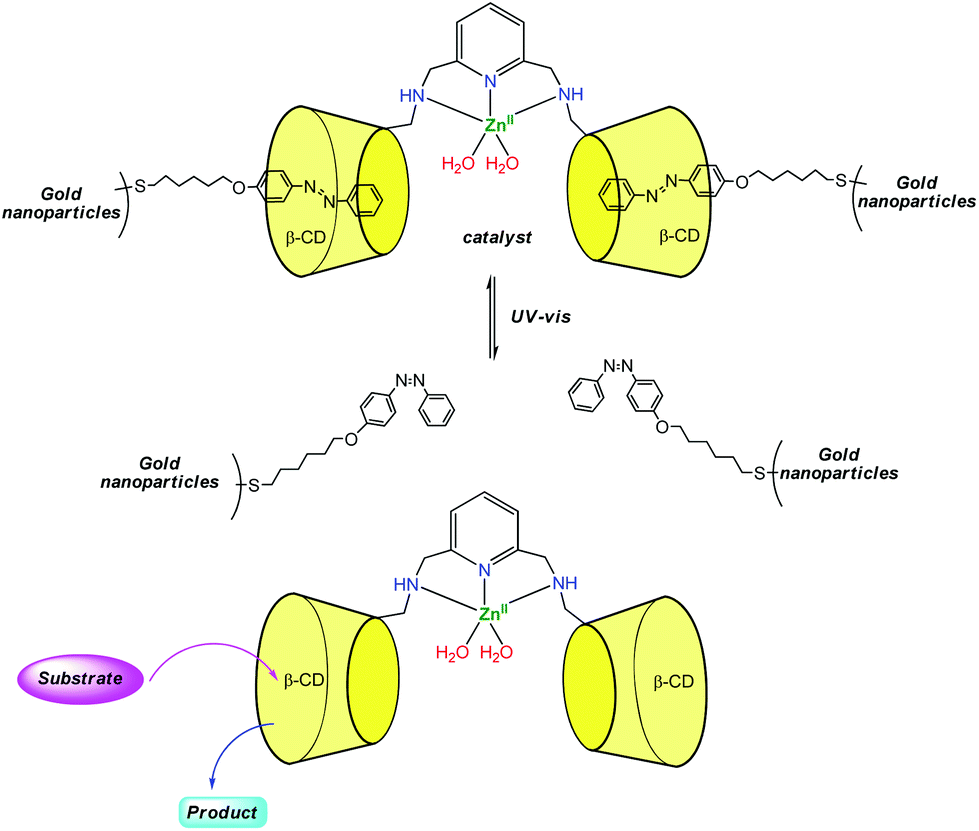 | ||
| Fig. 8 Design of a photo-switchable supramolecular catalyst.17 | ||
In the field of cytochrome P450 mimics, Kuroda et al. reported an iron-porphyrin complex sandwiched between two β-CD cavities. They compared its activity to a water-soluble porphyrin (P) deprived of cavity for alkene epoxidation in aqueous phosphate buffer in the presence of PhIO (Fig. 9).18 A 55% yield (based on consumed PhIO) was reported for the epoxidation of cyclohexene in the presence of the CD dimer, whereas less than 2% yield were obtained in the absence of cavity (i.e. with P). By contrast, with the much more reactive 2,3-dimethylbut-2-ene substrate, the same yield was obtained with or without cavity (ca. 30%). Here, the authors propose that the major role of the CDs is to protect the reactive oxene species, P·FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. With a good substrate, the iron oxo species reacts faster with the substrate than it decomposes, whatever the porphyrin. With a substrate of low reactivity, it self-decomposes before it reacts with the substrate, unless it is protected by CD cavities. Indeed, it is well-known that unprotected iron oxo species are readily trapped by the iron-porphyrin precursor in its reduced state through the formation of dinuclear species. Hence, the CDs prevent the trapping of the reactive oxo species by another iron complex while maintaining the access to the metal center open for substrate epoxidation.
O. With a good substrate, the iron oxo species reacts faster with the substrate than it decomposes, whatever the porphyrin. With a substrate of low reactivity, it self-decomposes before it reacts with the substrate, unless it is protected by CD cavities. Indeed, it is well-known that unprotected iron oxo species are readily trapped by the iron-porphyrin precursor in its reduced state through the formation of dinuclear species. Hence, the CDs prevent the trapping of the reactive oxo species by another iron complex while maintaining the access to the metal center open for substrate epoxidation.
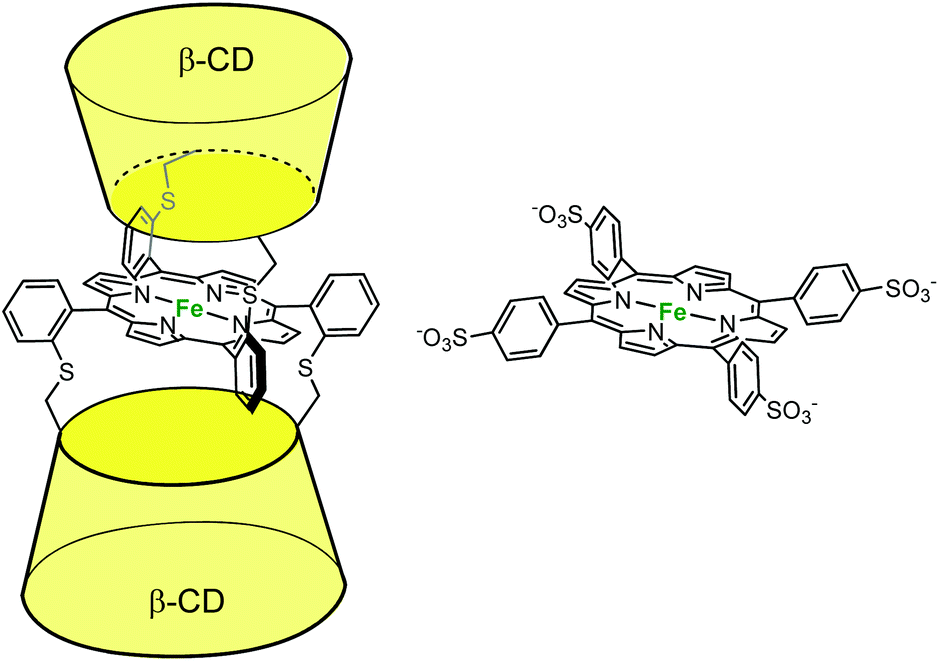 | ||
| Fig. 9 β-CD sandwiched iron-porphyrin catalyst (left) and its water-soluble analog deprived of cavity (right).18 | ||
An additional breakthrough in the field of oxidation chemistry was achieved with a catalyst capable of regio-selectively hydroxylating a polyfunctional substrate. Breslow et al. synthesized a Mn-porphyrin bearing at its meso position four β-CDs (Fig. 10). This P-450 mimic was tested for the selective hydroxylation of a steroid. Androstanediol was first converted to a diester derivative which presents tBu-phenyl binding groups and a water-solubilizing moiety. This diester was submitted to the action of iodosylbenzene in the presence of the catalyst. Quite remarkably, a single methylene group of the steroid was regio- and stereo-specifically hydroxylated with 3–5 turnovers before oxidative destruction of the catalyst.19 Two tBu groups of the substrate serve as guests for two diametrically opposed β-CDs. This double anchoring positions the steroid in such a way that one C–H bond is preferentially exposed to the MnO species and thus selectively hydroxylated. When changing the tBu guest substituents for alkyl chains with lower affinity for β-CDs, a mixture of six oxidation products was obtained.
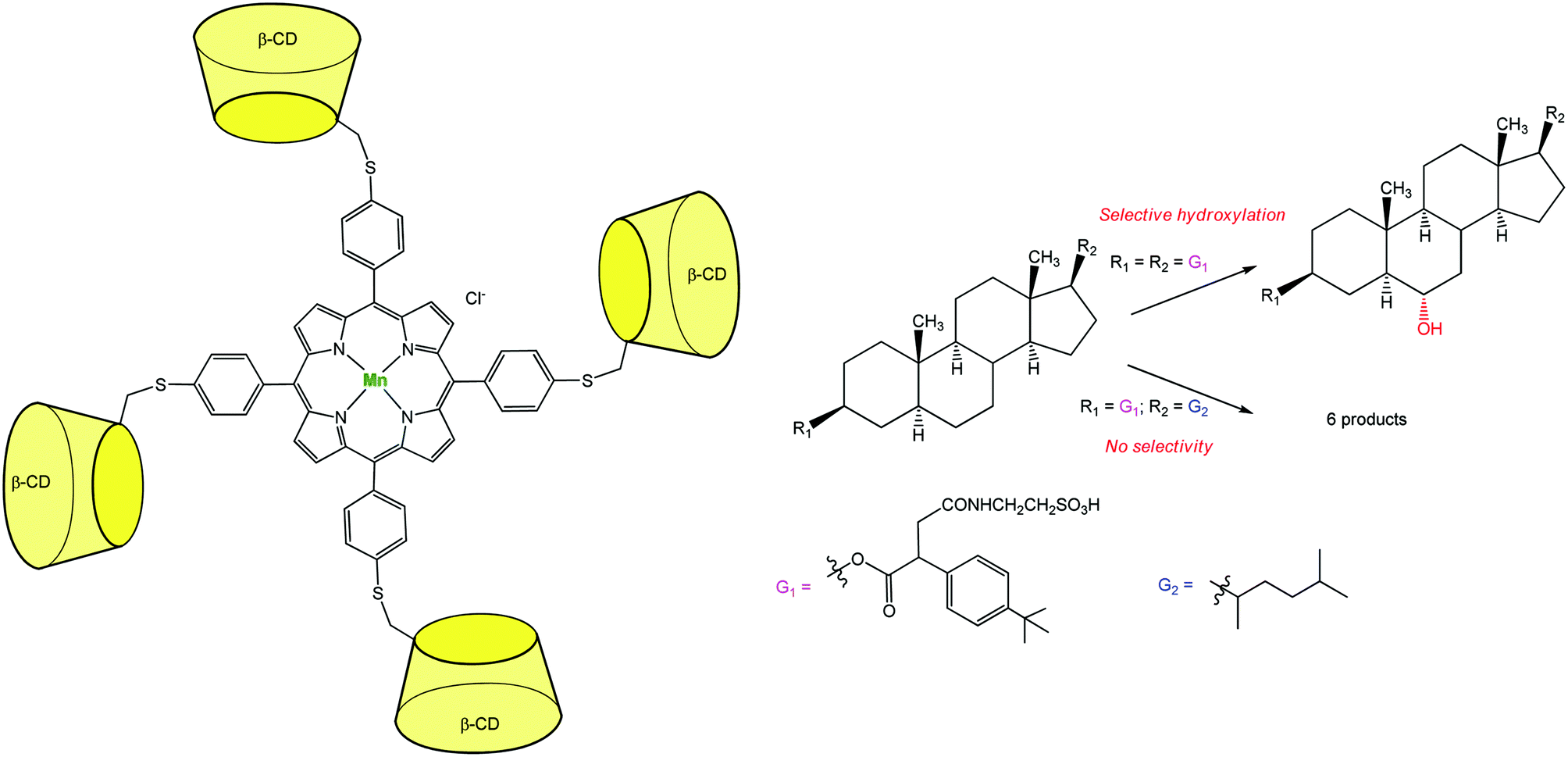 | ||
| Fig. 10 Highly selective Mn-porphyrin catalyst based on a β-CD tetramer for steroid hydroxylation.19 | ||
From the above-described studies involving polytopic recognition thanks to the association of several CDs to a metal complex, several conclusions can be drawn:
● Double embedment of the substrate strengthens its pre-association next to the metal complex and better preorganizes its reactive moiety in front of the catalyst. This yields faster kinetics.
● In the case of hydrolytic processes that split ditopic substrates into two pieces, monotopic products displaying lower affinities for the CD dimer are produced. This prevents product inhibition and gives rise to higher conversion rates.
● In the case of oxidative processes, multipoint recognition allows regio- and stereo-selectivities for the hydroxylation of the substrate.
● The CD cavities may also be used as a protective group around the metal center, which prevents its deactivation (e.g. through dimerization), while maintaining an access to the substrate.
In order to increase the variety of catalysts grafted to artificial cavities, the formation of polynuclear complexes was explored. The strategy consisted in the covalent grafting of several ligands to a macrocyclic scaffold for the coordination of several metal ions in the vicinity of a cavity.
2.3. One cavity for several ligands and several metal ions
One difficulty of employing CDs is the difficult control of their functionalization. While methods allowing their mono-functionalization were early developed, mastering their poly-functionalization appeared very difficult. Therefore, chemists turned to a different scaffold, namely calixarenes. These macrocycles are much easier to selectively functionalize. A drawback, however, lies in their high lipophilic character that makes them insoluble in water, unless they are decorated by several hydrophilic substituents. Gutsche, and later Reinhoudt, have taken advantage of the straightforward functionalization of the calix[4]arene macrocycle for the preorganization of multiple catalytic groups in the vicinity of each other.20 These systems mimic polynuclear metallo-enzyme active-sites. Thanks to the functionalization at the small rim of the calix[4]arene scaffold by four ethoxyethyl groups, the inversion of the aromatic units was prevented and the calixarene constrained in the cone conformation. Large rim functionalization by two or three 2,6-bis[(dimethylamino)methyl]pyridine ligands allowed the design and study of dinuclear and trinuclear phosphodiesterase models with Zn(II) cations. However, all studies had to be conducted in aqueous-organic solvents, due to two problems relative to water as a solvent: (i) the low solubility of the systems in pure water and (ii) low affinity of solvated Zn(II) ions for the pyridyl cores at neutral pH. Better ligands (1,5,9-triazacyclododecane) with higher affinity for transition metal ions in water were later introduced.21 Zn(II)-tacd-calix[4]arene complexes were unfortunately insoluble in water and hydrolytic studies were again run in 50% aqueous acetonitrile. Cu(II)-complexes, however, were fully water-soluble and used as catalysts for the hydrolysis of 2-hydroxypropyl p-nitrophenyl phosphate (HPNP) and diribonucleoside monophosphate at pH = 7 and 25 °C (Fig. 11).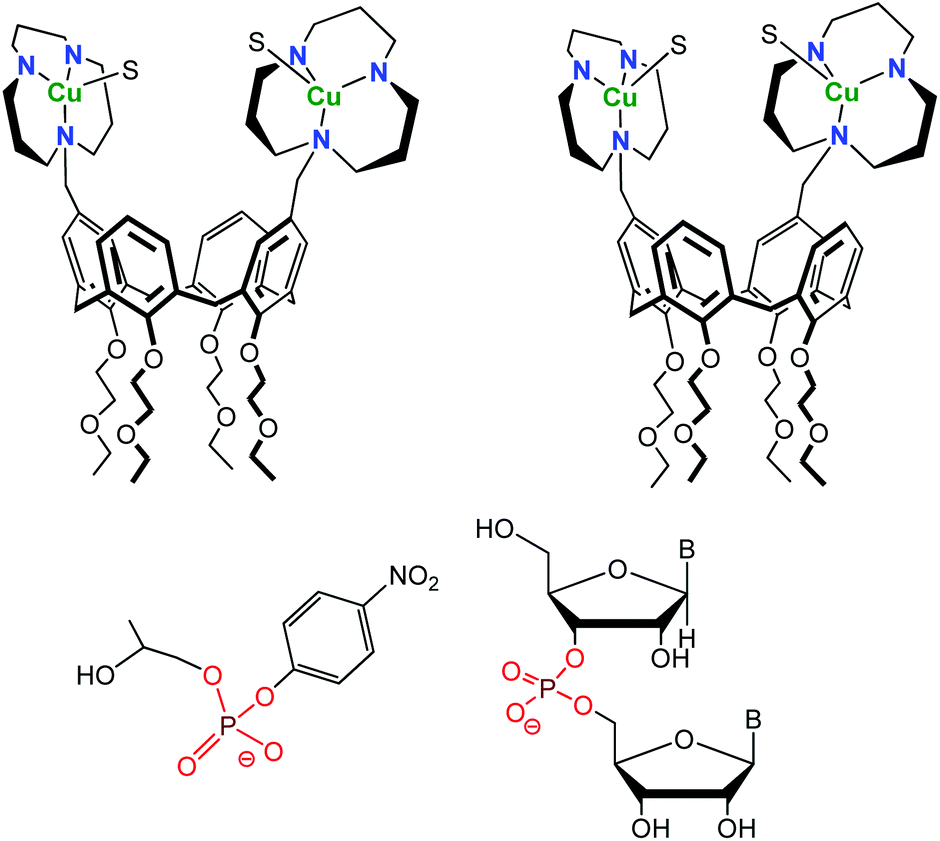 | ||
| Fig. 11 Structure of the 1,3-distal (left) and 1,2-vicinal (right) Cu(II) calix[4]arene (top) and substrates used for hydrolytic studies (HPNP, left and diribonucleoside monophosphate, right). S stands for a coordinated solvent molecule.21 | ||
Interestingly, the relative position of the metal complexes was varied (1,2-vicinal positions or 1,3-distal positions) and this modulation was shown to have a strong impact on the catalytic activity: the 1,2-vicinal Cu(II)-bimetallic catalyst revealed to be more efficient than the 1,3-distal one. Cooperativity between metal ions was observed in the case of the 1,2-vicinal bimetallic complex, as demonstrated by a 70-fold rate enhancement relative to a monometallic analogue [a tacd-Cu(II) complex] for HPNP cleavage and at least a 104 fold rate enhancement in the case of some diribonucleoside-3′,5′-monophosphates.
Hence, introducing several metal complexes instead of one appears as an interesting strategy for obtaining higher reactivity. However, this strategy faces new obstacles related to the more difficult functionalization of the macrocycle that needs to be substituted by hydrophilic substituents on one side, and by several ligands on the other side. Although these calix[4]arene-based systems provided some successful results, substrate embedment into the (too) small calix[4]arene cavity was not evidenced. In these systems, the macrocycle does not seem to play any role in the interaction between the substrate and the metal center. It acts essentially as a molecular platform that helps controlling the relative positioning of the metal ions in the polynuclear complexes.
3. Dynamic systems
3.1. Two-phase systems with substrate hosting
β-CD can also be used as a shuttle for mass-transfer in reactions catalyzed by transition metals under biphasic conditions. Early work in this field was reported in 1986 by Alper22 and Takahashi,23 who demonstrated that olefins were oxidized to ketones in the presence of oxygen, palladium chloride, copper chloride and native CDs. Later, the use of chemically modified cyclodextrins improved the catalytic activity of these mass-transfer additives in numerous reactions. Indeed, the introduction of a metal-binding group on the β-CD led to metal catalysts able to work in two-phase media (as schematized in Fig. 12) and this subject has already been reviewed.24 Reetz et al. pioneered this field: a diphosphine ligand, attached to the primary rim of the β-CD, was used in the presence of [Rh(cod)2]BF4 in a H2O–DMF (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30) mixture as hydrogenation and hydroformylation catalysts.25 Similarly, Karakhanov et al. have demonstrated that β-CD modified with nitrile-containing groups are good ligands for Pd(II) in two-phase Wacker oxidation of olefins.26 More recently, Woggon et al. have synthesized Ru complexes of amino-alcohol substituted β-CD and used them as catalysts for the reduction of ketones in the presence of sodium formate as hydrogen source in a two-phase system (lipophilic substrate and H2O–DMF, 3:1).27
:30) mixture as hydrogenation and hydroformylation catalysts.25 Similarly, Karakhanov et al. have demonstrated that β-CD modified with nitrile-containing groups are good ligands for Pd(II) in two-phase Wacker oxidation of olefins.26 More recently, Woggon et al. have synthesized Ru complexes of amino-alcohol substituted β-CD and used them as catalysts for the reduction of ketones in the presence of sodium formate as hydrogen source in a two-phase system (lipophilic substrate and H2O–DMF, 3:1).27
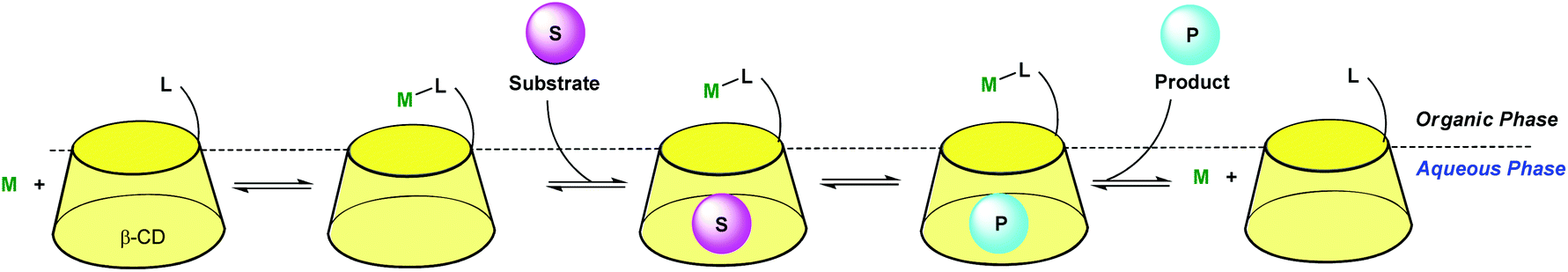 | ||
| Fig. 12 β-CD as a mass-transfer promoter in aqueous organometallic catalysis. | ||
A more recent example published by Harada et al. describes a mono-phosphine substituted β-CD ligand for ruthenium working as a supramolecular polymerization catalyst for ring-opening metathesis polymerization in pure water. The advantage of the cavity is evidenced by the lower polymerization activity of the catalyst in organic solvent. Its ability to complex the monomer unit 7-oxanorbornene dimethanol by hydrophobic effect was demonstrated by experiments conducted in the presence of a competitive guest molecule.28
3.2. Water-solubilization of a complex through ligand hosting in a cavity (CD)
In order to dissolve low-coordinated organometallic catalysts in water, Monflier et al. have developed a strategy based on the use of phosphine ligands embedded in a β-CD cavity.29,30 In these systems, the cavity only encapsulates an appropriate ligand and helps the metal ion to sit at its edge. As a consequence, low-coordinate palladium species were observed in water for the first time. The same group recently synthesized a CD dimer aimed at simultaneously hosting the substrate and the catalyst in each cavity (Fig. 13).31 The catalyst is a rhodium phosphine complex formed by complexation of the Rh(I) ion to a phosphine ligand bearing a biphenyl moiety prone to β-CD encapsulation. The proximity between the catalyst and the substrate leads to high catalytic activities.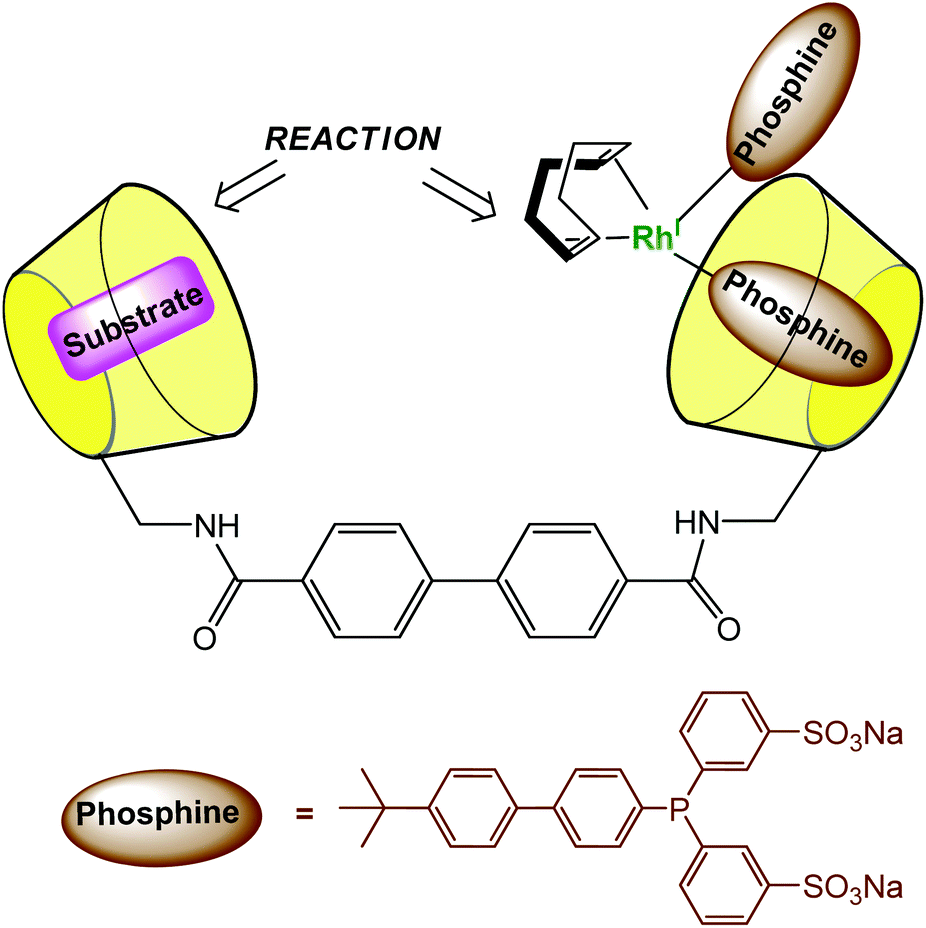 | ||
| Fig. 13 Cyclodextrin dimer as a platform for aqueous organometallic catalysis.31 | ||
3.3. Bimodal anchoring of a metal ion: one ligand encapsulated and one ligand covalently bound to the cavity
The above described strategies (i.e. the use of a β-CD functionalized by an aza ligand and a phosphine presenting a hydrophobic moiety prone to CD encapsulation) were combined to provide a supramolecular bidentate ligand offering a P,N mixed donor environment for the metal catalyst (Fig. 14). NMR and ITC studies confirmed the 1:1 stoichiometry of the metal complexes, with the phosphine ligand entering the CD cavity through its primary rim. Coordination of these P,N-bidentate ligands to platinum and rhodium in water was evidenced by NMR.32,33 The water-soluble platinum complex of this bidentate ligand was used as a catalyst for the hydrogenation of water-soluble allylic alcohols (2-methyl-3-buten-2-ol or 2-methylbutan-2-ol).
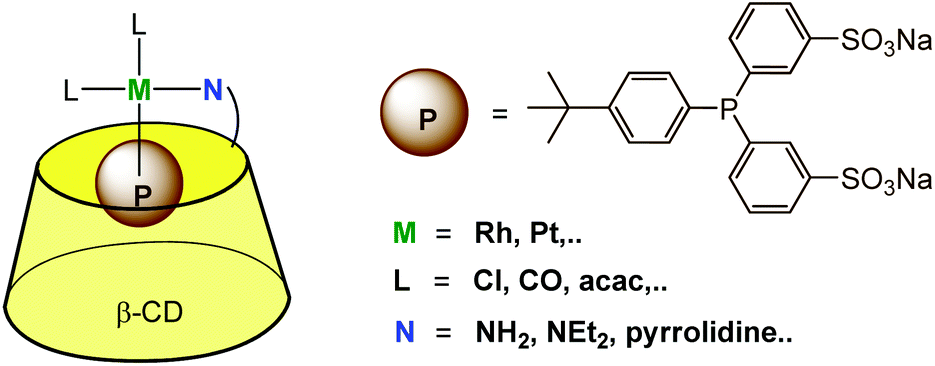 | ||
| Fig. 14 P,N-bidentate ligands used for the water-solubilization of low-coordinate metal complexes. | ||
In the above reported systems, efficient biphasic supramolecular catalysis was obtained with functionalized cyclodextrins. Three different strategies were explored: (i) the CD allows simultaneous shuttling of the substrate into water and binding of the catalyst through a grafted donor interacting with the metal ion, (ii) the host shuttles both the substrate and the catalyst through embedment of a hydrophobic group grafted onto the ligand that holds the metal complex (iii) supramolecular bidentate ligands for the metal catalyst are obtained in the water phase thanks to a CD functionalized covalently by one donor and hosting a second one the cavity.
Other dynamic systems have been obtained with other macrocycles, leading to different behaviors described below.
3.4. Ternary dynamic systems
In biochemistry, cooperative effects occur when the binding properties of the protein toward a ligand changes as a consequence of the interaction with another ligand. This cooperative effect has also been evidenced in an artificial system combining cavity, guest and metal ion, thus creating supramolecular metallo-enzyme models. Nau et al. have mixed p-sulfonatocalix[4]arene, Zn2+ or Co2+ and a bicyclic diazoalkane in water at pH = 7 (Fig. 15).34 The fluorescent guest is a very weak ligand for the metal ion [Kmet = 4 M−1 for Zn(II)] and presents an association constant with the cavity of Kcav = 1000 M−1. However, the metal cavity complex binds the guest with a higher association constant of K = 4000 M−1. In this synergistic system, the host assists the formation of a metal–ligand bond. The resulting ternary complex is further stabilized by a positive cooperative effect. The process is guest-selective as it was evidenced for 2,3-diazabicyclo[2.2.2]oct-2-ene and not with other diazoalkanes (Fig. 15).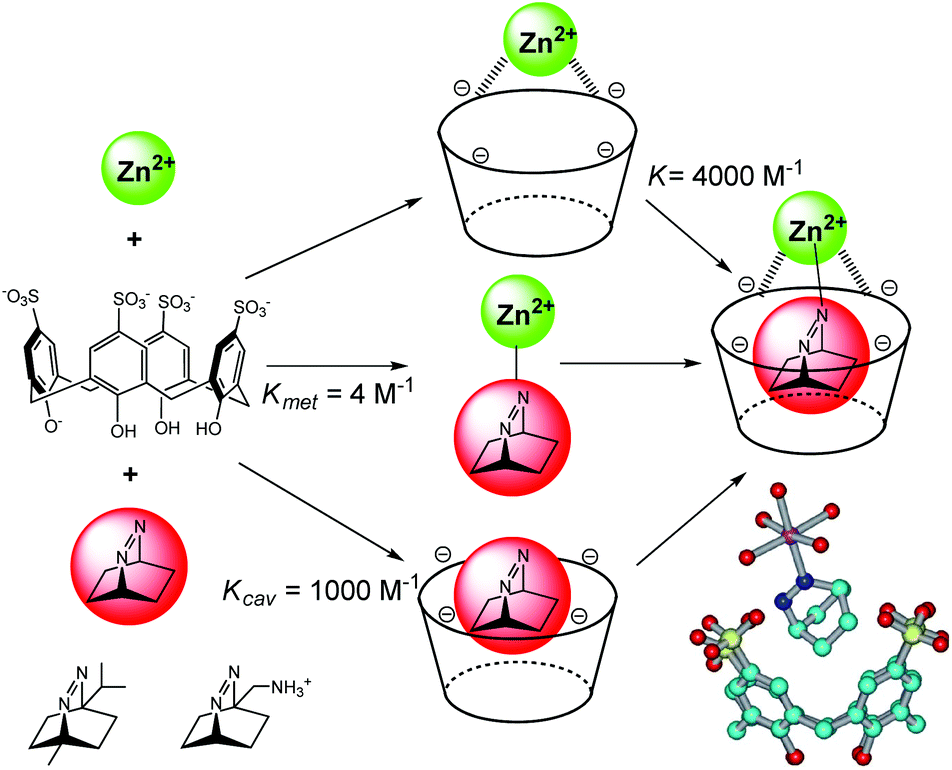 | ||
| Fig. 15 Selected binding constants in ternary complexes (left); other bicyclic diazoalkanes tested (bottom left); crystal structures of the Zn(II) complex (bottom right). (Reprinted with permission from ref. 34 Copyright 2006 John Wiley and Sons). | ||
The same strategy was applied to another type of cavity, a cucurbit[7]uril (CB[7]). This cavity is constituted of glycoluril units (Fig. 16, bottom). Beside a hydrophobic cavity, cucurbiturils also present a carbonyl-rich rim with good affinity for metal ions. In a two-phase system, a photoreactive substrate was encapsulated in the cavity in the aqueous layer, thereby favoring metal complexation to the carbonyl rim. The resulting ternary complex chemo-selectively produced a photoproduct after laser photolysis, which has a lower affinity for the cavity and accumulated in the organic layer.35
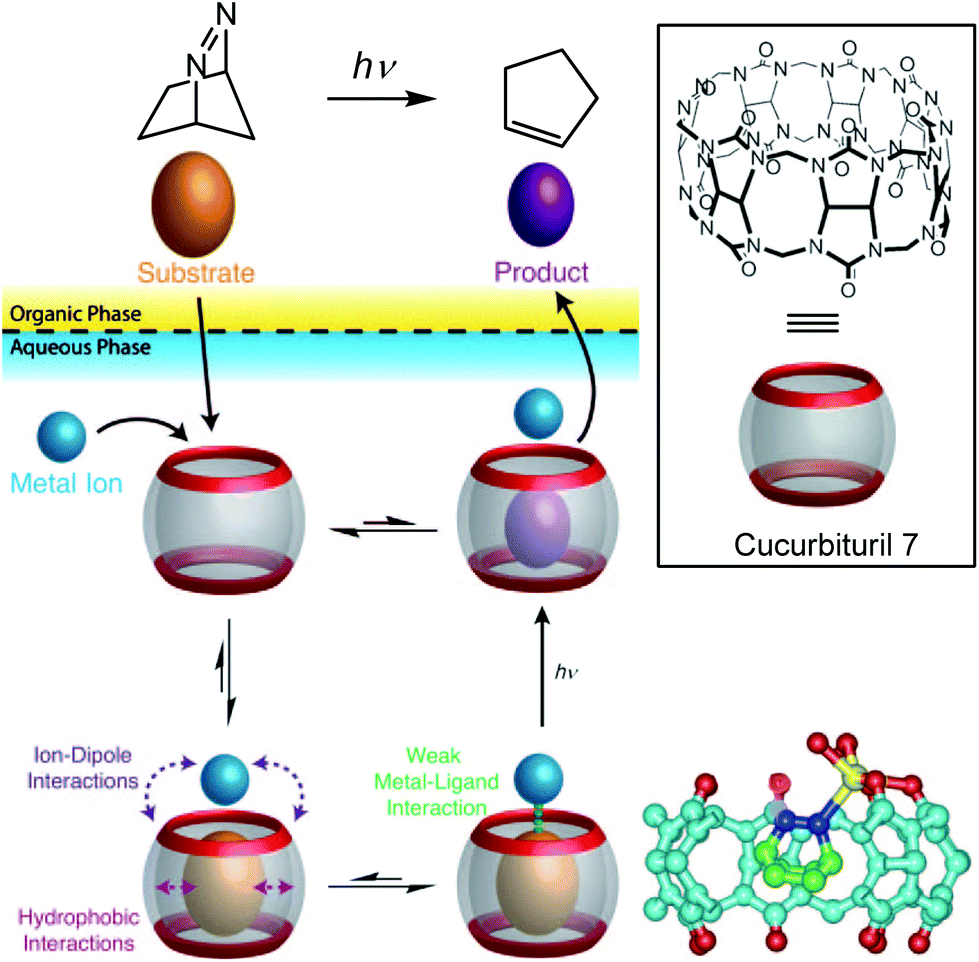 | ||
| Fig. 16 Top: Transition metal promoted chemoselective photoreaction through the dynamic formation of a ternary adduct in water. Bottom right: crystal structure of the ternary complex obtained with CB7, diazabicyclooctene and Ag+ (in yellow). (Reprinted with permission from ref. 35 Copyright 2011 John Wiley and Sons). | ||
Recently, Masson et al. reported a cucurbituril- and Ag(I)-based catalytic system to promote the desilylation of trimethylsilylalkynyl derivatives in water (Fig. 17).36 A 10% catalytic amount of CB[7] increased 4.3 times the desilylation rate. A ternary dynamic system is again proposed: a π-alkynyl silver intermediate stabilized by the carbonyl groups of the cavity.
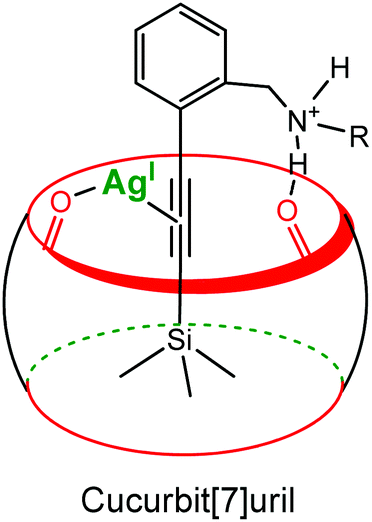 | ||
| Fig. 17 Schematic representation of a π-alkynyl silver intermediate stabilized by the carbonyl groups of the cucurbituril rim.36 | ||
In all of the above reported examples, the coordination of the metal ion occurs outside the cavity, or at best at its edge. Hence, the metal complex remains in an essentially aqueous medium, even though in interaction with the cavity through ligand/guest embedment. A completely different system is obtained when using water-soluble cage compounds. The idea here is to simultaneously host both, the metal catalyst and the substrate, in a hydrophobic environment, with water as a solvent. Representative examples are discussed below.
3.5. Catalysts in self-assembled cages
Self-assembled cages developed by Fujita et al. using the [enPd(II)]2+ (en = ethylenediamine) unit as a key building block present remarkable molecular recognition properties in water.37 Indeed, the presence of six cationic Pd(II) centers make the cage shown in Fig. 18 highly water-soluble, whereas the inside, defined by the poly-heteroaromatic walls, is hydrophobic. The resulting cavity is very large and capable of binding as many as four adamantane molecules in water thanks to the strong hydrophobic effect associated to the host–guest recognition process. These systems found impressive developments, acting as large molecular flasks and their capacities to mediate reactions has been reported. Surprisingly, only few examples involving a transition metal ion as a central actor in the cage-controlled reaction have been described.38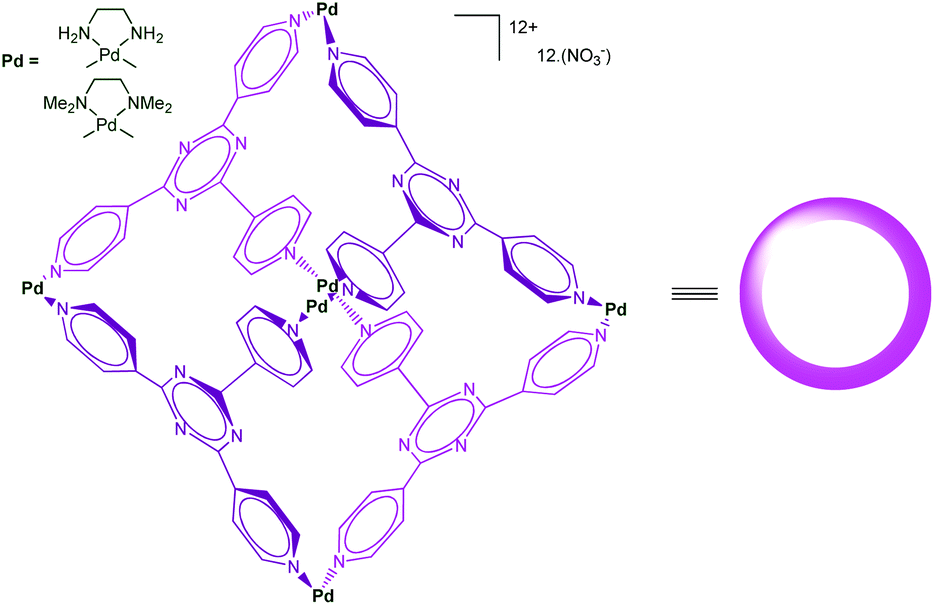 | ||
| Fig. 18 A self-assembled cage reported by Fujita et al.39 | ||
In early examples, this molecular container was used as a phase-transfer catalyst for Wacker oxidation: styrene is partially transferred into the aqueous solution, encapsulated in the cage and transformed into acetophenone in the presence of free Pd(II) catalyst [(en)Pd(NO3)2 10 mol%, 24 h at 80 °C, 82% yield]. Acetophenone, which is less hydrophobic than the substrate, is readily displaced by unreacted styrene, allowing turnovers. Both, the cage and the free catalyst [(en)Pd(NO3)2], are essential for catalysis and no catalytic activity was observed in the presence of a strongly competitive guest.40 The metallo-cage obtained with the capping tetramethylethylenediamine ligand was shown later to readily host two equivalents of linear ω-alkenol, the extremities of which sit next to the Pd(II) corners of the cage. Interestingly, upon heating, the alkenol was catalytically converted into the acetyl derivative in the absence of additional free Pd(II). This suggests that, in this case, the oxidation is promoted by the cage itself thanks to the favorable geometry of the guest substrate relative to the Pd(II) ions situated at the cage corners.41
Another interesting example recently reported by Fujita et al. is the cage-encapsulated [(Me4Cp)Ru(CO)2]2 complex (Fig. 19). This complex displays a much higher photo-stability in water in the presence of the cage. Under irradiation, it undergoes photo-substitution of one CO ligand by an alkyne without Ru–Ru bond cleavage usually observed. Here, the supramolecular assembly helps stabilizing the reactive metal center and improving the lifetime of the organometallic complex.
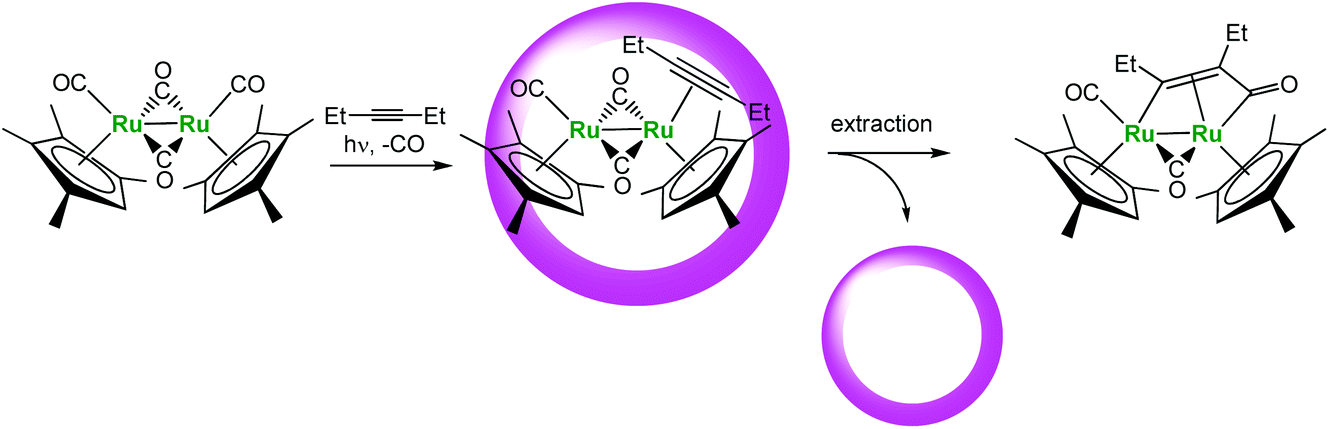 | ||
| Fig. 19 Photo-substitution reaction in a molecular vessel.42 | ||
Another water-soluble cage (Fig. 20) published by Raymond et al. has demonstrated its ability to encapsulate organometallic complexes and their subsequent catalytic activities have been studied.
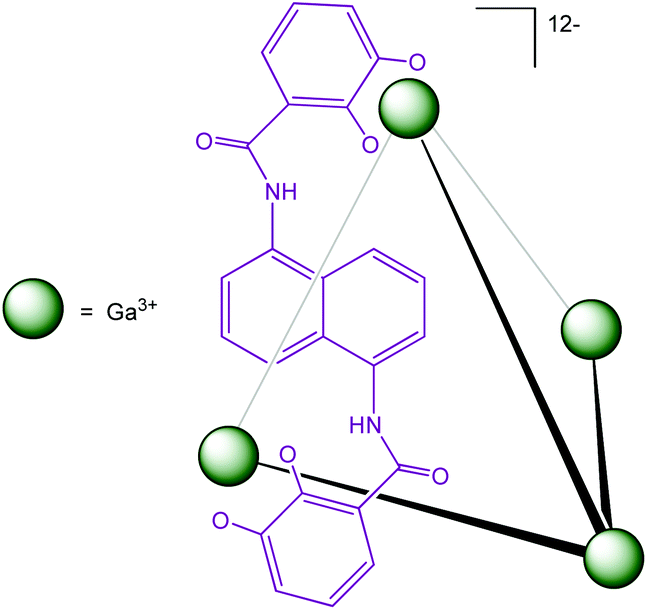 | ||
| Fig. 20 M4L6 tetrahedral assembly developed by Raymond et al.43 | ||
The encapsulation of complex [Cp(PMe3)Ir(Me)(C2H4)] in the Ga4L6 cage was observed by 1H and 31P NMR spectroscopy and electrospray mass spectrometry in D2O.44 This encapsulated complex was shown to stoechiometrically activate aldehydic CH-bonds in water. The cage imposes a size-controlled selectivity: acetaldehyde, propionaldehyde and butyraldehyde undergo CH-bond activation, whereas no reaction occurs with larger aldehydes such as valeraldehyde and benzaldehyde. Later, size- and shape-selectivities were observed in water for the catalytic isomerization of allylic alcohols by an encapsulated rhodium catalyst.45 Finally, gold_phosphine complexes were also encapsulated in this same cage in water. The embedment of Me3PAu+ led to an enhancement in the catalytic activity in the hydroalkoxylation of allenes in pure water and demonstrated a high selectivity.46 Interestingly, the supramolecular host is also able to stabilize reactive organometallic intermediates, whereas they usually decompose within minutes in water.47 The encapsulation of a Ru(II) catalyst in water by the same cage allowed the protection from decomposition of the latter and yielded high-turnover catalysis (TON = 1070) for allylic alcohol isomerization.48
Hence, the use of molecular cages allows creating a hydrophobic microenvironment in water around the transition metal complex. These cages can act as a protective envelop for stabilizing metal species that would otherwise be destroyed in water. They also have an insulating role preventing two reactive species to interact with each other or stabilizing unusual ligand/metal and metal/metal complexes. Most interestingly, when capable of hosting simultaneously a reactive metal complex and a substrate, very selective transformations are obtained. This is obviously due to the well-defined but restricted space provided by these hosts, which on the other side limits their scope of application.
The last strategy presented in this review concerns systems providing interlocked cavity and ligand. A macrocycle is used to create a ligand of specific geometry while exogenous ligand binding to the coordinated embedded metal ion, and/or redox properties, are under the control of the cavity itself.
4. Control of the micro-environment – interlocked cavity/ligand
4.1. Tuning the metal ion properties; protection of reactive species
Native cyclodextrins are able to encapsulate small organometallic complexes in their hydrophobic cavity in water. For example, ferrocene forms a (1:1) inclusion complex with β-CD in water and its association constant with β-CD was found to be 1.65 × 104 M−1 by solubility measurements.49 The resulting encapsulated complex presents modified chemical, electrochemical and photochemical properties.50 Likewise, Rebek and Kaifer reported the inclusion of ferrocene derivatives into a water-soluble “deep-cavitand”. As previously noticed with other cavities,51 the electrochemical behavior of this complex is different from that of the free ferrocene derivatives in aqueous solution. The host–guest complex, in that case, is not electroactive,52 which suggests that electron transfer from the inclusion complex is hindered.
Darensbourg et al. described a cyclodextrin host/guest approach to mimic a hydrogenase active site cavity.53 For this purpose, they prepared a 2Fe2S synthetic cluster mimicking the active site, outfitted with an aryl sulfonate moiety to promote inclusion into a β-CD. As schematized in Fig. 21, the self-assembly associates two β-CDs wrapping the diiron center into their hydrophobic cones and bridged by a sodium cation. The X-ray crystal structure of the self-assembled structure shows an increased torsion angle between the apical CO ligands, indicating that the supramolecular cage destabilizes the eclipsed geometry typical of diiron model complexes.
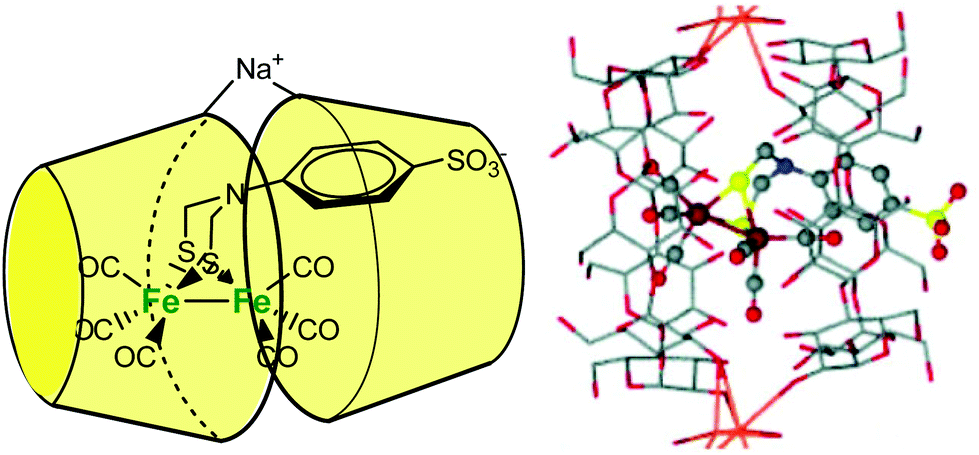 | ||
| Fig. 21 Schematic representation of the CD-encapsulated model of [FeFe]-dihydrogenase (left) and corresponding X-ray crystal structure of the clathrate (right).53 | ||
Based on NMR studies, the authors note that the complex is exchanging rapidly in and out of the cyclodextrin dimer. However, despite this fast exchange, the encapsulated model displays modified electrochemical properties compared to its free form. In the free form, the FeIFeI→FeIFe0 couple reduces at −1.2 V vs. Ag/AgCl (sat. KCl), whereas, in the presence of β-CD, the same reduction potential is shifted ca. 80 mV towards the more negative end. Proton reduction by the complex encapsulated in the β-CD dimer also occurs at a more negative potential (−1.4 V). These results demonstrate how the hydrophobic environment provided by the β-CD cavity has a strong impact on redox processes.
Likewise, Kano et al. have used two permethylated β-CD bridged by a pyridyl group to encapsulate an iron porphyrin (Fig. 22). In order to favor the encapsulation, the porphyrin was substituted by arylsulfonate groups at its meso positions. The pyridyl bridge serves also as an axial ligand for the iron center. The O2 adduct of this encapsulated porphyrin complex (FeIII–OO˙) was found to be stable for up to 30 h at 25 °C.54 By reaction between the encapsulated FeIII–OH and H2O2, the hydroperoxo adduct FeIII–OOH was observed for the first time, under physiological conditions. This adduct further evolved to yield a ferryl species (FeIVO coined compound II in heme biochemistry) that displayed also remarkable stability (7 h at 25 °C).55 It was proposed that the presence of the β-CD dimer creates a hydrophobic environment from which water molecules are excluded, thus protecting the reactive metal center.
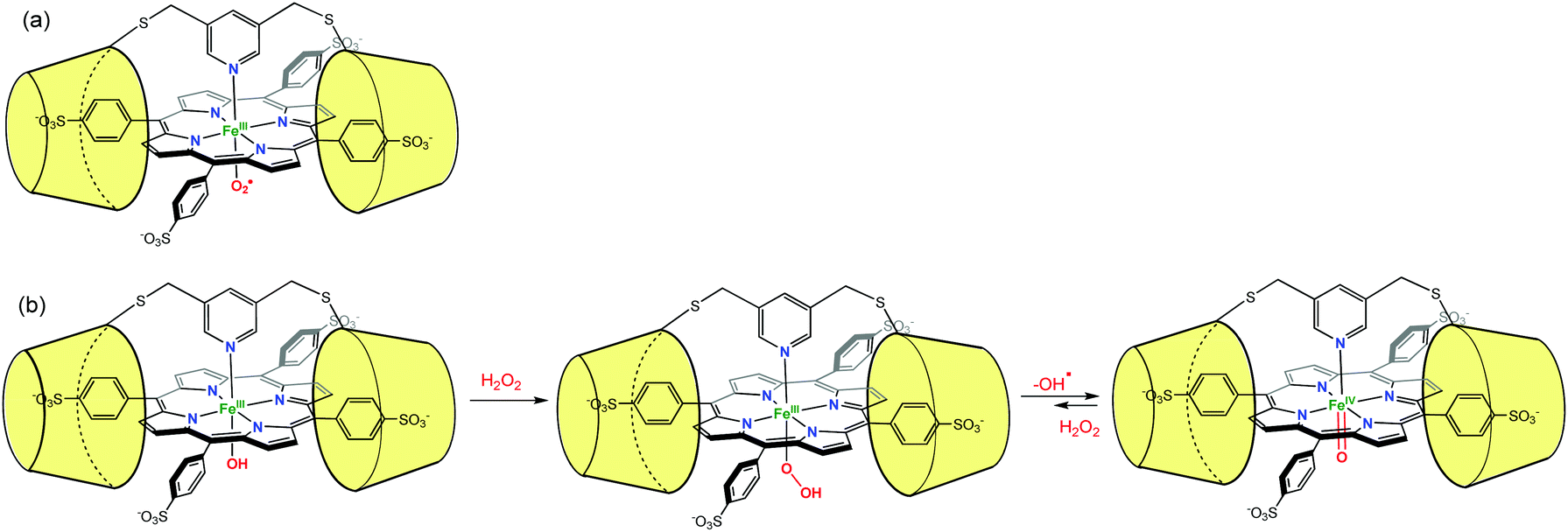 | ||
| Fig. 22 Protection of reactive species by their supramolecular environment: (a) porphyrin FeIII-superoxo species in a β-CD dimer environment; (b) formation and stabilization of FeIII–OOH and FeIVO species.54 | ||
4.2. The cavity participates in the ligand design – the ligand is “built-in” at the edge of the cavity
The systems described in this part get structurally closer to metallo-enzyme active sites. The cavity is considered as a real platform on which the first coordination sphere of the metal is constructed. Thanks to an adequate spacer length, several N-donors are positioned at the edge of the cavity, and this proximity allows the metal ion to sit in a controlled micro-environment.An early example was reported in 1984 by Tabushi et al., who functionalized a β-CD cavity at its primary rim by two histamine groups (Fig. 23).56 The complexation constant of ZnII to this bidentate ligand was determined to be 4.5 × 102 in water, at pH = 7.5. The corresponding ZnII complex was used as a carbonic anhydrase model. Kinetic analyses of CO2 hydration in imidazole buffer showed that a ZnII ion coordinated to two histidine ligands of the CD and to one exogenous imidazole from the buffered aqueous solution was the active species with a catalytic constant of ca. 103 M−1 s−1 at pH 7.5–7.8. It is proposed that the hydrophobic environment enhances the CO2 hydration activity since the analogous ZnII complex deprived of cavity was much less active. Dehydration, on the other hand, was not appreciably catalyzed by the complex, which is attributed to bicarbonate coordination inhibiting the catalyst.
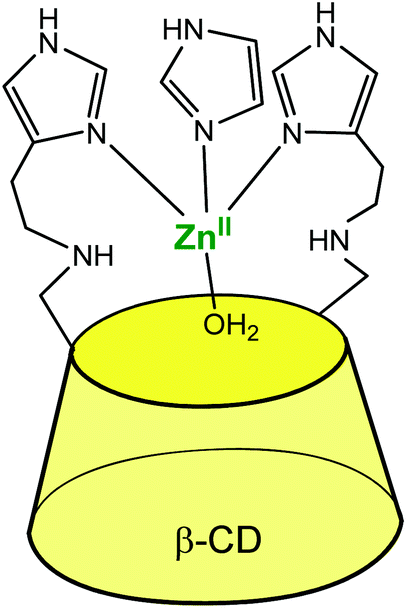 | ||
| Fig. 23 Bis(histamine)-β-CD ZnII complex.56 | ||
For the past decade, Reinaud et al. have been developing biomimetic receptors based on calix[6]arenes functionalized by three N-donors at the small rim.57,58 The resulting polydentate ligands mimic the first coordination sphere (a poly-histidine core) encountered in many metallo-enzyme active sites.59 Upon binding a metal ion, the conformational freedom of the calixarene is restricted and the cone conformation is stabilized. In these so-called “funnel complexes”, the geometry of the system favors endo-binding of a guest ligand and as a result behave as remarkable biomimetic receptors for neutral molecules in organic solvents.
For a few years, the water-solubility issue of these systems has been tackled. The strategy consists in the functionalization of the large rim of the calix cavity by hydrophilic substituents in order to maintain the same first coordination sphere around the metal center at the small rim. The first success was obtained with a tris(sulfonato)-tris(imidazolyl)-calixarene that provided the first example of a Cu(I) complex stable in pure water under air despite the presence of a free valence. (Fig. 24, top).60 In this system, the calixarene hydrophobic cone protects the metal center (preventing disproportionation and autoxidation), while allowing coordination of a small guest molecule such as CO in the concave area of a hydrophobic cavity. Hence, this complex reproduces not only the first coordination sphere encountered in many copper-enzymes and the frequently hydrophobic micro-environment of the active site, but also the aqueous macro-environment of a physiological medium.
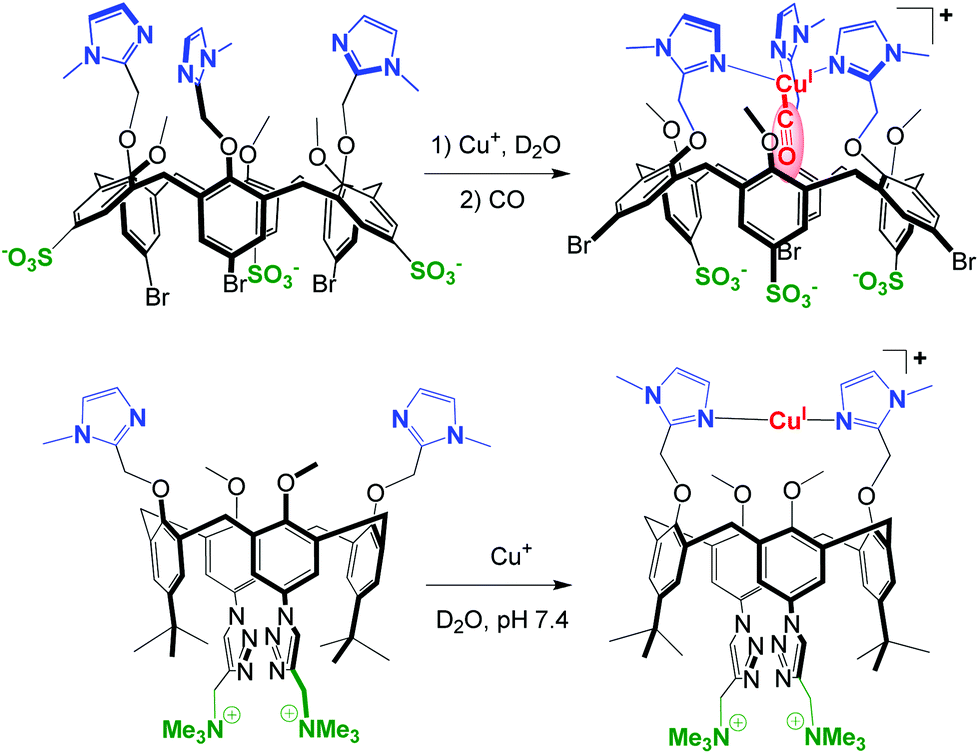 | ||
| Fig. 24 Water-soluble and air-stable CuI-calixarene complexes. Top: model of a Cu-enzyme active site with the selective binding of CO in the hydrophobic pocket.60 Bottom: CuI-selective ligand that ensures strong CuI binding at pH = 7.4.61 | ||
Very recently, a water-soluble copper(I)-selective ligand based on the calix[4]arene scaffold was obtained. The design of Cu(I)-selective chelators is of particular interest not only for the creation of therapeutic objects but also as useful tools to gain insights into the coordination of Cu(I) in a biological medium. In this system, the water-solubility of the calixarene macrocycle is ensured by the introduction of two ammonium groups at its large rim (Fig. 24, bottom). The linear bis-imidazole binding motif at the small rim allows strong coordination of Cu(I) in water at physiological pH (K = 1014 M−1 at pH = 7.4), and prevents autoxidation as well as disproportionation reactions. Furthermore, this ligand appears to be selective vs. other biologically relevant metal ions (Na+, K+, Mg2+, Ca2+, Zn2+), as well as vs. Cu2+.61 In this case however, the smaller calix[4]arene scaffold does not allow embedment of guest ligands as in the case of calix[6]arene-based complexes.
Coordination of Zn(II) to the above-mentioned water-soluble tris(sulfonato)-tris(imidazole)-calix[6] ligand (Fig. 24, top) led to precipitation of the complex in water. This was attributed to charge neutralization. Therefore, a tricationic version of this ligand was synthesized. However, no coordination was observed in water whatever the pH, although in organic solvents (MeCN, chloroform), stoechiometric coordination was observed at the same concentration. This was attributed to the competitive protonation of the ligand (favored under acidic conditions) and to the formation of Zn(II)-hydroxo species of lower Lewis acidity (favored under basic conditions). Quite remarkably however, the addition of a hydrophobic primary amine led to the formation of a ternary complex (calix/ZnII/amine) at physiological pH. This ternary complex is formed in a highly synergistic and allosteric manner and stabilizes the neutral form of the amino guest. It constitutes the first example of molecular receptor that efficiently and selectively binds primary amines in water, in spite of the high solvation energy associated to the corresponding ammonium ion present at this pH (Fig. 25).62 As previously observed in organic solvents,63 the tris-imidazolyl ligand coordinates ZnII in a tetrahedral environment, with the amino guest strongly embedded in the hydrophobic cavity of the macrocycle.
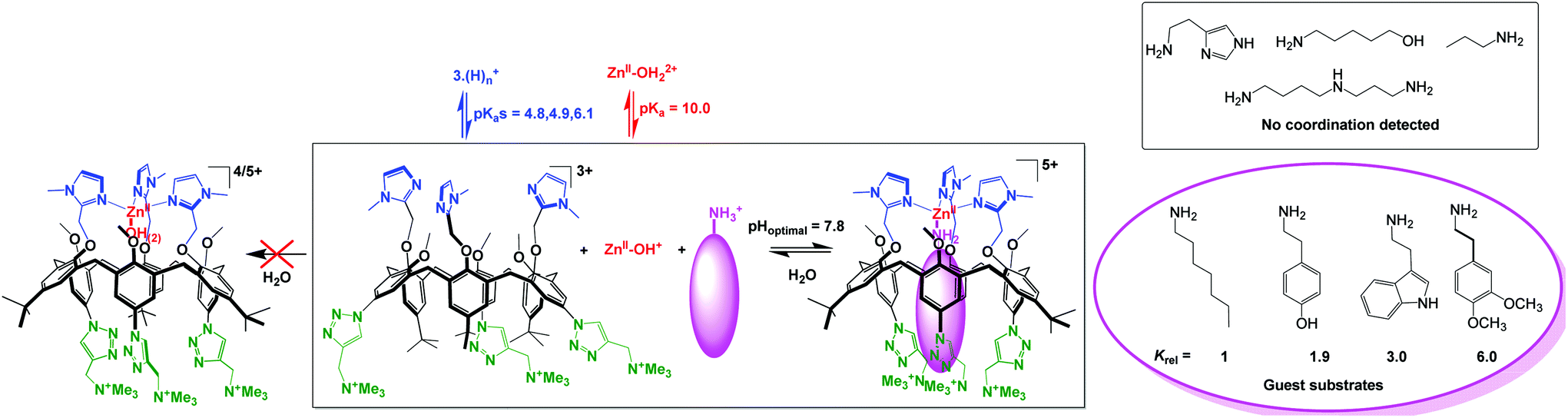 | ||
| Fig. 25 Water-soluble calix[6]arene ligand and ZnII coordination properties in water (left) and in the presence of hydrophobic primary amines (right).62 | ||
Hence, the molecular design of this ligand confers a micro-environment to the metal center that favors the formation of a mononuclear complex and prevents its precipitation. It also takes advantage of the hydrophobic effect as a key driving force for the formation of the ternary complex since the smaller propylamine guest is not recognized. The system thus behaves as an efficient and selective receptor for neutral lipophilic primary amines in water. The resulting pseudo-pKa shift of 7 units for the amine associated to the formation of the ternary complex is quite remarkable. Such an impressive stabilization of the basic form of the amine at neutral pH is obviously the result of both the coordination to Zn(II) and the micro-environment provided by the calixarene pocket.
Very recently, Reinaud et al. started developing another family of cavity-complexes. The so-called “bowl ligand” presents a resorcin[4]arene equipped with a tris(imidazole) core similar to those of the calix[6]arene-based funnel system. With M(II) ions, the bowl-ligand leads to 5-coordinate complexes presenting two different sites open for an interaction with exogenous donors: one is in endo position, under the supramolecular control of the bowl cavity, whereas the other is exposed to the solvent, in exo position relative to the cavity.64 The water-soluble version of this bowl ligand was obtained by introducing four quaternary ammonium groups at its “legs” (Fig. 26).65 Interestingly, and in strong contrast with the calix ligand, the bowl ligand readily binds Zn(II) ion in water within a relatively large pH window (pH = 6.5 to 9.5) in the absence of guest (other than water). The stronger ability to bind Zn(II) is attributed to the higher rigidity of the resorcinarene macrocycle (compared to calix[6]arene), which allows better pre-organization of the tris(imidazole) core for Zn(II) coordination. The Zn(II) complex is reported to bind acetate at an optimum pH value of 7.4 (K′ = 600 ± 100 mol−1 L). Guest binding further stabilizes the metal complex as evidenced by an increase of the pH window at which it can be detected (at ca. 5 mM, pH 3.7 to 10.6).
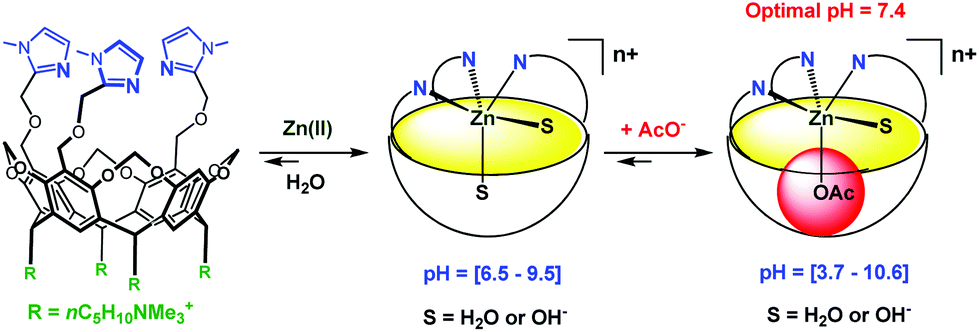 | ||
| Fig. 26 Schematic representation of the bowl ligand and its coordination properties in water.65 | ||
Hence, this study represents one step further in biomimetism: a biomimetic coordination core (three imidazole residues) able to coordinate a bio-relevant metal ion, Zn(II), presenting two labile sites in cis-position relative to each other. It is important to note that this feature is key for hydrolytic metallo-enzyme mimicry, since the mechanism is known to proceed via a five-coordinate intermediate.
4.3. Full control of the metal environment and guest binding
The substitution of three tBu groups of the large rim of a calix[6]azacryptand by three trimethylammonium groups enabled its water-solubilization. In this system, a TMPA unit [TMPA = tris(2-pyridylmethyl)amine, see Fig. 27] is covalently capping the calixarene cone. This provides a more rigid ligand with strong chelating properties. As a result, the corresponding copper complex is stable in water in its two oxidation states. The spectroscopic and electrochemical data recorded in pure water indicate that the host–guest properties of the funnel complex are retained at both oxidation states of the copper cation. The presence of a vacant coordination site shielded from the aqueous environment by the calixarene core, makes it highly reminiscent of the active site of copper monooxygenases.66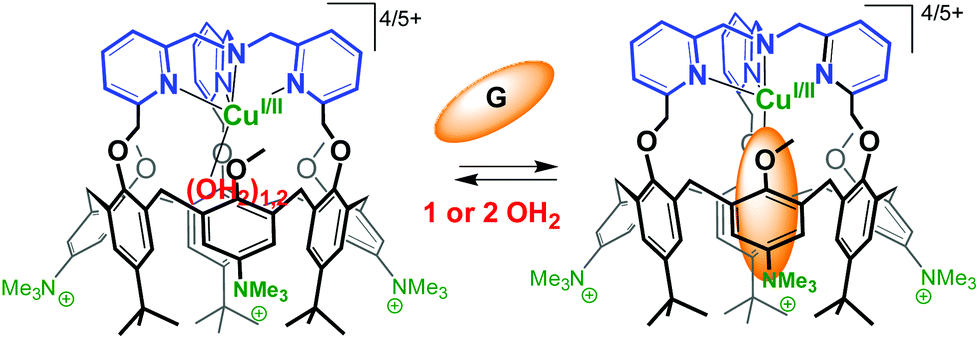 | ||
| Fig. 27 Water-soluble CuI and CuII complexes of TMPA-capped calix[6]arene. G stands for a guest molecule such as MeCN or PrCN.66 | ||
An interesting observation is the decrease of E1/2 when switching from an organic solvent (CH2Cl2 or CH3CN) to water (ΔE1/2 = 350 mV and 290 mV, respectively). It likely results from the better stabilization of a dicationic vs. monocationic state as the medium polarity increases. On the other hand, the values measured in water for the TMPA-capped calix[6]arene and the TMPA-Cu complex deprived of cavity differ by more than 500 mV (E1/2 = +0.53 V and −0.04 V, respectively). This nicely illustrates the insulating effect of the calixarene cavity from the media and highlights the spectacular ability of a hydrophobic embedment to tune the redox potential of metal ions in water. This water-soluble calix[6]arene-based ligand is a very rare example (if not the first) of an artificial structure capable of stabilizing in pure water a copper ion featuring a coordinative unsaturation in both +I and +II oxidation states. Remarkably, the major structural and functional properties observed in organic solvents are maintained: mononuclear embedment, exogenous ligand-binding, host–guest exchange, impressive affinity for small nitriles despite the highly competitive medium, protection from auto-oxidation.
Interestingly, this CuII-complex demonstrated a very strong and selective affinity for fluoride anion in buffered water at pH = 6–7 (Fig. 28).67 The binding constant at pH 5.9 is 85000, which corresponds to one of the highest values ever reported in water at this pH. Molecular modeling suggests that the species recognized by the host is a cluster [F·H2O·H2O]−, stabilized by OH–π interactions with the aromatic walls of the calixarene, which facilitates fluoride extraction from water by decreasing its dehydration cost.
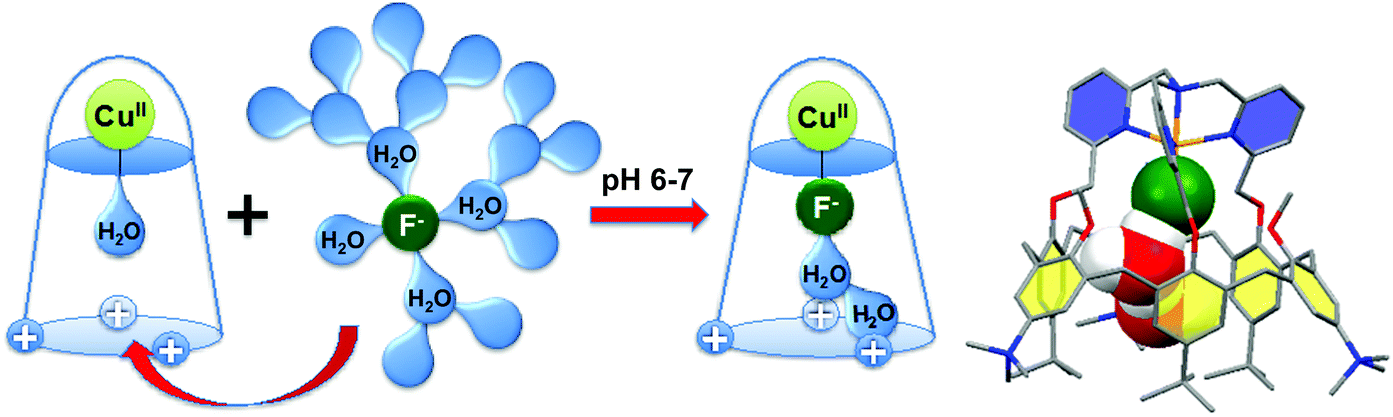 | ||
| Fig. 28 Fluoride binding in water by a water-soluble tmpa-capped calix[6]arene; DFT optimized structure of the solvated fluoride Cu(II) complex.67 | ||
Hence, while some examples of intricate cavity/ligand systems have been reported with cyclodextrins, the use of the more readily functionalizable calix[6]arene core allows the construction of a variety of polydentate donors. A careful design can give rise to the formation of metal complexes, the binding properties of which are under the supramolecular control of the cavity. The above reported examples show that these complexes behave as selective funnels for a variety of guest ligands, either lipophilic thanks to the hydrophobic effect associated to their embedment, or hydrophilic thanks to the hosting of water solvates. It is also shown that the redox properties of the metal ion can be tuned by the supramolecular environment provided by the cavity that participates in 2nd (and further) coordination sphere. Interestingly, as well, the resorcinarene core gives rise to bowl-shape complexes presenting two labile coordination sites in cis position relative to each other, thus prone to reactivity. One drawback of these systems is the associated important synthetic work since several donors must be regioselectively grafted on one edge of the macrocycle, while water—solubilizing groups must be introduced on the other side of the structure.
5. Conclusion
The design, synthesis and study of cavity-appended metal complexes is a rich research area where creativity and bio-inspiration play a major role.Different kinds of macrocyclic structures have been employed for playing the role of a cavity
Some of them are intrinsically water-soluble. The naturally available cyclodextrins were first exploited, whereas the synthetic cucurbiturils were more recently developed. Their respective truncated-cone and cylinder shape provides a hydrophobic space open to the solvent on both sides. Cages constituted by aromatic walls self-assembled by metal ions offer completely different systems, coined molecular flasks. Their communication with the guests relies on the size of the holes separating the organic units and on the lability of the coordination bonds that seal them together. One problem encountered with all these compounds is the difficult introduction of functional groups, namely a ligand, allowing the association of a well-defined metal complex with chemical activities in a controlled environment. Calixarenes and resorcinarenes offer an appealing alternative since synthetic methodologies for their selective functionalization have been largely developed and are now well-mastered. However, these macrocycles are not water-soluble and must be equipped with hydrophilic substituents. The double task of appending ligands for metal ion control and polar substituents for water-solubility makes the design and synthesis of the systems very challenging. Among them, calix[4]arenes appear too small to play the role of a host, whereas calix[6]arenes face problems related to their size: their increased conformational mobility must be mastered if the cone shape is desired to play the role of an efficient host. Resorcin[4]arenes offer a more rigid bowl-shape cavity, but are open to the solvent by one side only. Their small size also restricts their scope as receptors.The major outcomes of the herein-described studies of water-soluble cavity-complexes are very different when using one macrocyclic scaffold or another, and appear actually complementary.
● Early work with cyclodextrins evidenced Michaelis-Menten like catalysis in water with a transition metal ion as the reactive species. Like in enzymatic catalysis, the synergy of the association of a metal complex and a cavity is illustrated by enhanced kinetics, thanks to substrate embedment and preorganization of its reactive site next to the catalyst. It is associated to substrate selectivity based on host–guest relative affinities, which leads to substrate differentiation (i.e. competition), or regio-selective transformation of polyfunctional substrates. It also allows inhibition by competitive host binding with non-substrate guests, which opens doors for controlling the activity of the catalyst. Remarkable regio- and stereo-selectivity are obtained through multi-point recognition strategies involving several cavities associated to the catalyst. In the case of hydrolytic catalysis, ditopic recognition of substrates was exploited to circumvent problems related to product inhibition.● Cyclodextrins but also cucurbiturils have been used to perform biphasic catalysis with TMs. Strategies relying on either water-transfer of the metal complex through ligand embedment or synergistic coordination of a metal ion and substrate embedment, or both, lead to increased performance of TM catalysts. Interesting cases of host-assisted establishment of a metal–ligand bond also allows for the formation of ternary complexes displaying very high selectivity, a characteristic that is frequently encountered in biology.
● The presence of a cavity next to the metal ion also affects the properties of the metal ion itself. Indeed, the macrocycle provides a microenvironment that can protect reactive species against the solvent or any undesired process. This is illustrated by TM complexes formed at the edge of cyclodextrins or partially embedded into them. Even more spectacular, self-assembled metallo-cages provide a supramolecular hydrophobic environment that allows isolation of reactive species from water, and protection against decomposition. Interesting cases of co-encapsulation of a TM catalyst with its substrate give rise to efficient, size-selective transformations, very much like natural systems that work in water, while providing a hydrophobic environment around their active site.
● Functionalizing the calix[6]arene scaffold allowed building new ligand systems, that very much like proteins, fold around the metal ion to provide metal complexes with a labile site closely interlocked with the macrocyclic structure. This gives rise to induced-fit phenomena, highly cooperative formation of ternary systems leading to selective recognition of small molecules. In these cases, the multitopic recognition process benefits from: (i) coordination of the guest to the metal ion, (ii) further stabilization within the second coordination sphere providing hydrogen bonding sites, (iii) hydrophobic effect through embedment of a lipophilic guest residue, and (iv) flexibility of the host that adapts to its guest. The interests in these systems also lie in the possible characterization of the metal species in its microenvironment at the atomic level. This allows comprehensive analyses of the tuning of the metal properties by the surrounding non-aqueous, well-defined microenvironment. Hence, redox properties and ligand exchange can be adjusted, which is a fundamental aspect of biological systems.
Several difficulties in these bio-inspired quests remain to be overcome. Indeed, water-soluble cavities appended with the desired functionalities often represent a synthetic challenge. Moreover, the construction of a scaffold allowing the interlocking of a metal ion coordination site within a macrocycle open to the solvent is a real challenge from a design point of view. These are key aspects for controlling guest ligand binding (for recognition), substrate positioning and exchange (for catalysis). More biomimetism is desirable but it faces the difficult construction of a system with a specific first and second coordination sphere for the TM that are often ill-controlled, whereas they should be ideally tunable at will.
The major, middle and long term, perspectives are to obtain new receptors leading to new devices efficient in water, and new catalysts, selective, performant, with high turnovers and displaying new reactivity patterns leading to new reactions! This is a very challenging and exciting area of research.
Acknowledgements
The authors thank the CNRS (Institut de Chimie), the Ministère de l'Enseignement Supérieur et de la Recherche and the Agence Nationale pour la Recherche [Cavity-zyme(Cu) Project ANR-2010-BLAN-7141] for financial support. They also acknowledge the COST Action “Supramolecular Chemistry in Water” (CM 1005).References
- (a) I. Bertini, H. B. Gray, E. I. Stiefel and J. S. Valentine, Biological Inorganic Chemistry, Structure and Reactivity, University Science Books, Sausalito, CA, 2007 Search PubMed; (b) S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, Mill-Valley, CA, 1994 Search PubMed; (c) R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239 CrossRef CAS.
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648 RSC.
- F. Biedermann, W. M. Nau and H.-J. Schneider, Angew. Chem., Int. Ed., 2014, 53, 2 CrossRef.
- R. Breslow and L. E. Overman, J. Am. Chem. Soc., 1970, 92, 1075 CrossRef CAS.
- R. Breslow and S. V. Dong, Chem. Rev., 1998, 98, 1997 CrossRef CAS PubMed.
- F. Bellia, D. La Mendola, C. Pedone, E. Rizzarelli, M. Savian and G. Vecchio, Chem. Soc. Rev., 2009, 38, 2756 RSC.
- I. Tabushi, N. Shimizu, T. Sugimoto, M. Shiozuka and K. Yamamura, J. Am. Chem. Soc., 1977, 99, 7100 CrossRef CAS.
- E. U. Akkaya and A. W. Czarnik, J. Am. Chem. Soc., 1988, 110, 8553 CrossRef CAS.
- D. H. Kim and S. S. Lee, Bioorg. Med. Chem., 2000, 8, 647 CrossRef CAS.
- N. M. Milović, J. D. Badjić and N. M. Kostić, J. Am. Chem. Soc., 2004, 126, 696 CrossRef PubMed.
- R. Breslow, P. J. Duggan and J. P. Light, J. Am. Chem. Soc., 1992, 114, 3982 CrossRef CAS.
- M. Rezac and R. Breslow, Tetrahedron Lett., 1997, 38, 5763 CrossRef CAS.
- (a) R. Breslow and B. Zhang, J. Am. Chem. Soc., 1992, 114, 5882 CrossRef CAS; (b) B. Zhang and R. Breslow, J. Am. Chem. Soc., 1997, 119, 1676 CrossRef CAS.
- R. Breslow and B. Zhang, J. Am. Chem. Soc., 1994, 116, 7893 CrossRef CAS.
- M. Zhao, H. L. Wang, L. Zhang, C. Zhao, L. N. Ji and Z. W. Mao, Chem. Commun., 2011, 47, 7344 RSC.
- M. Zhao, C. Zhao, X.-Q. Jiang, L.-N. Ji and Z.-W. Mao, Dalton Trans., 2012, 41, 4469 RSC.
- L. Zhu, H. Yan, C. Y. Ang, K. T. Nguyen, M. Li and Y. Zhao, Chem. – Eur. J., 2012, 18, 13979 CrossRef CAS PubMed.
- (a) Y. Kuroda, T. Hiroshige, T. Sera, Y. Shiroiwa, H. Tanaka and H. Ogoshi, J. Am. Chem. Soc., 1989, 111, 1912 CrossRef CAS; (b) Y. Kuroda, T. Hiroshige and H. Ogoshi, J. Chem. Soc., Chem. Commun., 1990, 1594 RSC.
- (a) R. Breslow, X. Zhang, R. Xu, M. Maletic and R. Merger, J. Am. Chem. Soc., 1996, 118, 11678 CrossRef CAS; (b) R. Breslow, X. Zhang and Y. Huang, J. Am. Chem. Soc., 1997, 119, 4535 CrossRef CAS.
- P. Molenveld, J. F. J. Engbersen and D. N. Reinhoudt, Chem. Soc. Rev., 2000, 29, 75 RSC.
- R. Cacciapaglia, A. Casnati, L. Mandolini, D. N. Reinhoudt, R. Salvio, A. Sartori and R. Ungaro, J. Am. Chem. Soc., 2006, 128, 12322 CrossRef CAS PubMed.
- H. A. Zahalka, K. Januszkiewicz and H. Alper, J. Mol. Catal., 1986, 35, 249 CrossRef CAS.
- A. Harada, Y. Hu and S. Takahashi, Chem. Lett., 1986, 15, 2083 CrossRef.
- (a) H. Bricout, F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, Sustainability, 2009, 1, 924 CrossRef CAS PubMed; (b) F. Hapiot, H. Bricout, S. Tilloy and E. Monflier, Eur. J. Inorg. Chem., 2012, 1571 CrossRef CAS.
- M. T. Reetz and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 1997, 36, 865 CrossRef CAS.
- E. Karakhanov, A. Maximov and A. Kirillov, J. Mol. Catal. A: Chem., 2000, 157, 25 CrossRef CAS.
- A. Schlatter, M. K. Kundu and W. D. Woggon, Angew. Chem., Int. Ed., 2004, 43, 6731 CrossRef CAS PubMed.
- Y. Takashima, K. Uramatsu, D. Jomori, A. Harima, M. Otsubo, H. Yamaguchi and A. Harada, ACS Macro Lett., 2013, 2, 384 CrossRef CAS.
- L. Caron, M. Canipelle, S. Tilloy, H. Bricout and E. Monflier, Eur. J. Inorg. Chem., 2003, 595 CrossRef CAS.
- C. Machut-Binkowski, M. Canipelle, H. Bricout, S. Tilloy, F. Hapiot and E. Monflier, Eur. J. Inorg. Chem., 2006, 1611 CrossRef.
- C. Blaszkiewicz, H. Bricout, E. Léonard, C. Len, D. Landy, C. Cézard, F. Djedaïni-Pilard, E. Monflier and S. Tilloy, Chem. Commun., 2013, 49, 6989 RSC.
- C. Machut, J. Patrigeon, S. Tilloy, H. Bricout, F. Hapiot and E. Monflier, Angew. Chem., Int. Ed., 2007, 46, 3040 CrossRef CAS PubMed.
- J. Patrigeon, F. Hapiot, M. Canipelle, S. Menuel and E. Monflier, Organometallics, 2010, 29, 6668 CrossRef CAS.
- H. Bakirci, A. L. Koner, M. H. Dickman, U. Kortz and W. M. Nau, Angew. Chem., Int. Ed., 2006, 45, 7400 CrossRef CAS PubMed.
- A. L. Koner, C. Marquez, M. H. Dickman and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 545 CrossRef CAS PubMed.
- X. Lu and E. Masson, Org. Lett., 2010, 12, 2310 CrossRef CAS PubMed.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 371 CrossRef PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed.
- M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469 CrossRef CAS.
- H. Ito, T. Kusukawa and M. Fujita, Chem. Lett., 2000, 598 CrossRef CAS.
- M. Yoshizawa, N. Sato and M. Fujita, Chem. Lett., 2005, 34, 1392 CrossRef CAS.
- S. Horiuchi, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 12029 CrossRef CAS PubMed.
- D. L. Caulder, R. E. Powers, T. N. Parac and K. N. Raymond, Angew. Chem., Int. Ed., 1998, 37, 1840 CrossRef CAS.
- D. H. Leung, D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2004, 43, 963 CrossRef CAS PubMed.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746 CrossRef CAS PubMed.
- Z. J. Wang, C. J. Brown, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 7358 CrossRef CAS PubMed.
- D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745 CrossRef CAS PubMed.
- C. J. Brown, G. M. Miller, M. W. Johnson, R. G. Bergman and K. N. Bergman, J. Am. Chem. Soc., 2011, 133, 11964 CrossRef CAS PubMed.
- J. S. Wu, K. Toda, A. Tanaka and I. Sanemasa, Bull. Chem. Soc. Jpn., 1998, 71, 1615 CrossRef CAS.
- F. Hapiot, S. Tilloy and E. Monflier, Chem. Rev., 2006, 106, 767 CrossRef CAS PubMed.
- A. E. Kaifer, Acc. Chem. Res., 1999, 32, 62 CrossRef CAS.
- D. Podkoscielny, R. J. Hooley, J. Rebek and A. E. Kaifer, Org. Lett., 2008, 10, 1865 CrossRef PubMed.
- (a) M. L. Singleton, J. H. Reibenspies and M. Y. Darensbourg, J. Am. Chem. Soc., 2010, 132, 8870 CrossRef CAS PubMed; (b) M. L. Singleton, D. J. Crouthers, R. P. Duttweiler, J. H. Reibenspies and M. Y. Darensbourg, Inorg. Chem., 2011, 50, 5015 CrossRef CAS PubMed.
- K. Kano, H. Kitagishi, C. Dagallier, M. Kodera, T. Matsuo, T. Hayashi, Y. Hisaeda and S. Hirota, Inorg. Chem., 2006, 45, 4448 CrossRef CAS PubMed.
- H. Kitagishi, M. Tamaki, T. Ueda, S. Hirota, T. Ohta, Y. Naruta and K. Kano, J. Am. Chem. Soc., 2010, 132, 16730 CrossRef CAS PubMed.
- I. Tabushi and Y. Kuroda, J. Am. Chem. Soc., 1984, 106, 4580 CrossRef CAS.
- For organo-soluble host–guest systems based on tris(imidazolyl)-calix[6]arene Zn(II) complexes, see: (a) O. Sénèque, M.-N. Rager, M. Giorgi and O. Reinaud, J. Am. Chem. Soc., 2000, 122, 6183 CrossRef; (b) D. Coquière, S. Le Gac, U. Darbost, O. Sénèque, I. Jabin and O. Reinaud, Org. Biomol. Chem., 2009, 7, 2485 RSC (Perspective); and more recent developments: (c) D. Coquière, A. de la Lande, O. Parisel, T. Prangé and O. Reinaud, Chem. – Eur. J., 2009, 15, 11912 CrossRef PubMed; (d) D. Coquière, A. de la Lande, S. Martí, O. Parisel, T. Prangé and O. Reinaud, Proc. Natl. Acad. Sci., U. S. A., 2009, 106, 10449 CrossRef PubMed; (e) C. Monnereau, J.-N. Rebilly and O. Reinaud, Eur. J. Org. Chem., 2011, 166 CrossRef CAS.
- For organo-soluble host–guest complexes based on calix[6]aza-cryptands, see: (a) U. Darbost, M.-N. Rager, S. Petit, I. Jabin and O. Reinaud, J. Am. Chem. Soc., 2005, 127, 8517 CrossRef CAS PubMed; (b) X. Zeng, D. Coquière, A. Alenda, E. Garrier, T. Prangé, Y. Li, O. Reinaud and I. Jabin, Chem. – Eur. J., 2006, 12, 6393 CrossRef CAS PubMed; (c) D. Over, A. de la Lande, X. Zeng, O. Parisel and O. Reinaud, Inorg. Chem., 2009, 48, 4317 CrossRef CAS PubMed; (d) D. Over, X. Zeng, C. Bornholdt, J. Marrot and O. Reinaud, Inorg. Chem., 2013, 52, 14089 CrossRef CAS PubMed.
- Thematic issues on bioinorganic enzymology, Chem. Rev, 1996, 96, 2237–3042 Search PubMed; 2014, 114, 3367–4038 and 4039–4620.
- Y. Rondelez, G. Bertho and O. Reinaud, Angew. Chem., Int. Ed., 2002, 41, 1044 CrossRef CAS.
- A. Maurin, S. Varatharajan, B. Colasson and O. Reinaud, Org. Lett., 2014, 16, 5426 CrossRef CAS PubMed.
- O. Bistri, B. Colasson and O. Reinaud, Chem. Sci., 2012, 3, 811 RSC.
- Y. Rondelez, O. Sénèque, M.-N. Rager, A. F. Duprat and O. Reinaud, Chem. – Eur. J., 2000, 6, 4218 CrossRef CAS.
- (a) A. Višnjevac, J. Gout, N. Ingert, O. Bistri and O. Reinaud, Org. Lett., 2010, 12, 2044 CrossRef PubMed; (b) J. Gout, S. Rat, O. Bistri and O. Reinaud, Eur. J. Inorg. Chem., 2014, 2819 CrossRef CAS; (c) J. Gout, A. Višnjevac, S. Rat, A. Parrot, A. Hessani, O. Bistri, N. Le Poul, Y. Le Mest and O. Reinaud, Inorg. Chem., 2014, 53, 6224 CrossRef CAS PubMed.
- S. Rat, J. Gout, O. Bistri and O. Reinaud, submitted.
- G. Thiabaud, A. Brugnara, M. Carboni, N. Le Poul, B. Colasson, Y. Le Mest and O. Reinaud, Org. Lett., 2012, 14, 2500 CrossRef CAS PubMed.
- A. Brugnara, F. Topić, K. Rissanen, A. de la Lande, B. Colasson and O. Reinaud, Chem. Sci., 2014, 5, 3897 RSC.
| This journal is © The Royal Society of Chemistry 2015 |