
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Artificial metalloenzymes for the diastereoselective reduction of NAD+ to NAD2H†
Tommaso
Quinto
,
Daniel
Häussinger
,
Valentin
Köhler
and
Thomas R.
Ward
*
Department of Chemistry, University of Basel, Spitalstrasse 51, CH-4056 Basel, Switzerland. E-mail: thomas.ward@unibas.ch
First published on 7th November 2014
Abstract
Stereoselectively labelled isotopomers of NAD(P)H are highly relevant for mechanistic studies of enzymes which utilize them as redox equivalents. Whereas several methods are firmly established for their generation in high diastereomeric purity by enzymatic methods, alternative methods have so far not been investigated. The article presents the stereoselective deuteration of NAD+ at the 4-position (90% de) of the pyridinium-ring by means of an artificial metalloenzyme. The artificial metalloenzyme consists of a biotinylated iridium cofactor embedded in streptavidin isoforms and the resulting constructs have been previously shown to be compatible with natural enzymes. Alternative methods for stereoselective NAD(P)+ reduction are expected to be of high interest for the mechanistic study of enzymes that accept NAD(P)H mimics and for the synthesis of structurally related fine chemicals.
Introduction
The coenzymes NAD(P)+ and NAD(P)H play a crucial role in the redox machinery of living systems. This relates to their ability to either accept a hydride at the C4-position of the pyridine ring in their oxidized form or to donate the respective hydride in the reverse-reaction (Scheme 1). When the hydride is replaced by a deuteride in the reduction step, two diastereoisomers can be formed, due to the presence of the enantiopure adenine dinucleotide moiety. The diastereoisomers thus differ only in their configuration at C4. Stereoselective labelling of the coenzymes with deuterium or tritium yields precious mechanistic information for the corresponding enzymes. Methods for their preparation in high stereoisomeric purity are well established.1–3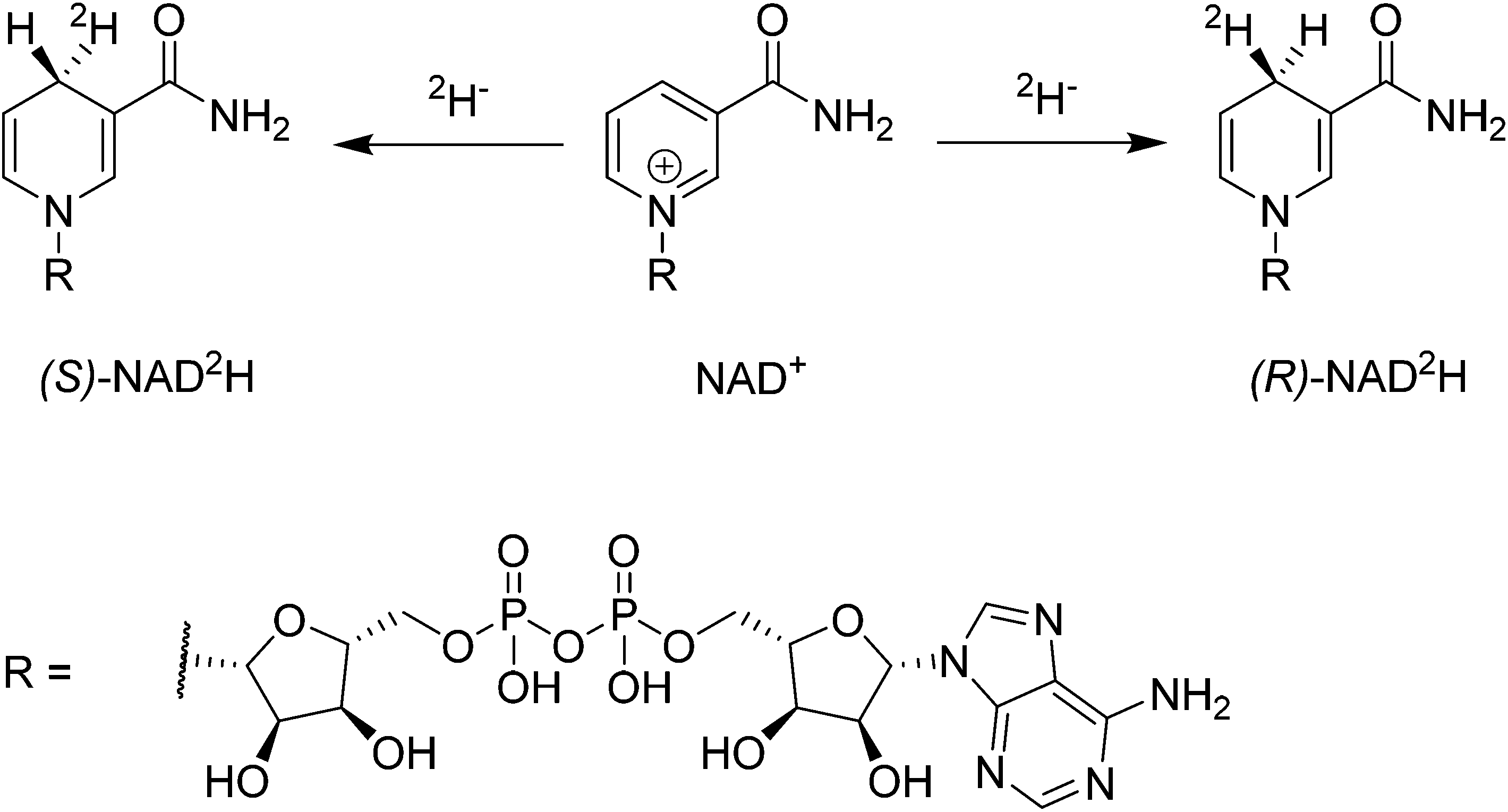 | ||
| Scheme 1 Deuteration of NAD+ at the C4 position yields diastereoisomers. | ||
Biocatalysis gains an ever increasing share in modern chemical manufacture, boosted by powerful protein engineering strategies.4 NAD(P)H or NAD(P)+ mol equivalents are required in various biotransformations, including ketoreductases, alcohol dehydrogenases, Baeyer Villiger monooxygenases, imine reductases, and P450s.5–8 These co-substrates are typically regenerated enzymatically in a catalytic concurrent fashion.9 However, alternative approaches employing e.g. chemocatalysis or electrochemical reduction have received increasing attention since they offer additional flexibility in process conditions and are readily transferrable to NAD(P)-mimics, which are substantially cheaper than their natural analogues.10
One complex which has been frequently used in non-enzymatic regeneration systems for the transfer of reducing equivalents to NAD(P)+ is the achiral [RhCp*(bipy)(H2O)]2+.11 At least one report exists where an enantiopure complex was utilized.12 However, no labelling studies were undertaken and accordingly, no diastereoselectivity for the reduction step studied. We recently reported on the application of an artificial transfer hydrogenase (ATHase) for the concurrent regeneration of NADH in a monooxygenase-coupled reaction.13 To generate the artificial metalloenzyme, a biotinylated iridium pianostool complex was incorporated into streptavidin-mutant S112A (Fig. 1). The biotinylated ligand for the IrCp* moiety is achiral in the vicinity of the metal center and the respective complex has been shown to induce only negligible stereocontrol in the reduction of prochiral imine substrates in the absence of streptavidin (Sav). However, upon incorporation into Sav, substantial stereocontrol can be achieved.14 Importantly, the concurrent process with the monooxygenase and other enzymes was only effective, when the complex was located inside Sav; otherwise dramatic deactivation of the monooxygenase and/or the artificial Ir-cofactor occurred.13
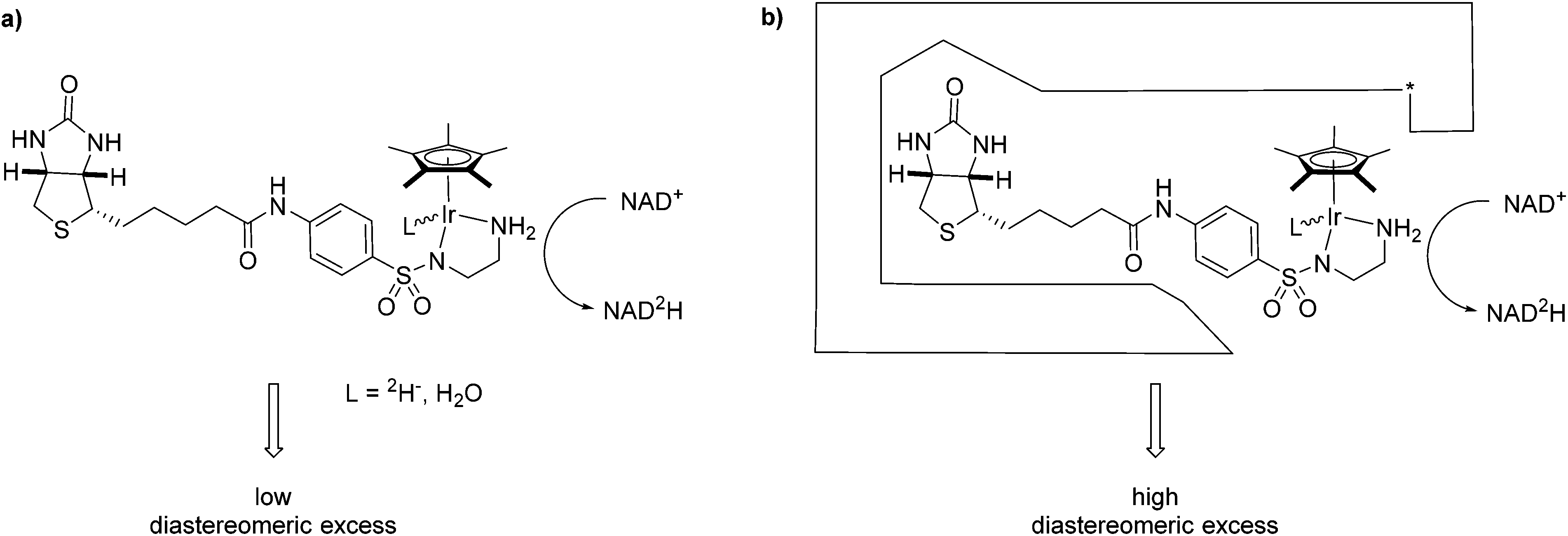 | ||
| Fig. 1 NAD+ reduction, (a) with a biotinylated Ir-complex and deuterated sodium formate in the absence of streptavidin mutants proceeds with low diastereoselectivity. (b) Upon incorporation into streptavidin mutants, the reduction proceeds with high diastereoselectivity. The star symbolizes a mutation in streptavidin. | ||
We have previously shown that substrate reduction with ATHases derived from streptavidin mutants S112A and S112K lead to R- and S-enantiomers for both ketones and imines, respectively.14 The corresponding X-ray structures of [Cp*Ir(Biot-p-L)Cl] ⊂ S112A14 and [Cp*Ir(Biot-p-L)Cl] ⊂ S112K15 were recently reported and analysed: for S112A, the absolute configuration at [Cp*Ir(Biot-p-L)Cl] is (S) and for S112K, the absolute configuration is (R). Based on modeling studies, we hypothesise that the absolute configuration at Ir by-and-large determines the absolute configuration of the alcohol and amine products: we term this phenomenon “induced lock-and-key” whereby the protein determines the configuration of the metal which in turn dictates which prochiral face of the substrate is reduced.15
Here we report on the diastereoselectivity of NAD deuteration employing the Ir-cofactor by itself and upon incorporation into two Sav-mutants (Fig. 1).
Results and discussion
To determine the diastereomeric ratio after reduction with deuteride, 1H-NMR-spectroscopy offers a convenient means.16,17 This technique enables the direct determination of the diastereoselectivity in the reaction mixture without requiring isolation of the products. The only noteworthy differences to potential preparative reactions lie in the presence of a small amount of deuterium oxide (5%), the dissolution of the biotinylated Ir-complex in DMF-d7 instead of DMF and a short centrifugation step. Based on catalysis results obtained in asymmetric imine reduction14 and NADH regeneration13 with artificial metalloenzymes, the selectivity with two representative mutants was investigated. Mutant S112A, employed previously for concurrent NADH regeneration, leads to superior enantiomeric excess and high turnover numbers in the reduction of 1-methyl-6,7-dimethoxy-3,4-tetrahydroisoquinoline yielding (R)-configured salsolidine (up to 96% ee).14 Reduction of the same substrate with the AME based on S112 K, in contrast, leads to a bias in favour of the (S)-enantiomer (up to 78% ee).14For the stereoselective NAD+ reduction experiments, deuterated sodium formate was used as a deuteride source in the presence of [Cp*Ir(biot-p-L)Cl]·S112A Sav and NAD+. Following centrifugation, the crude reaction mixtures were analyzed by 1H-NMR spectroscopy. While background signals of the protein and other components of the crude reaction mixture prevented unambiguous integration of 1D 1H-NMR data, a selective one-dimensional TOCSY NMR experiment using irradiation on H6 (5.89 ppm) yielded clean spectra with sufficient signal to noise ratio to reliably integrate the two resonances attributed to both diastereomers (Fig. 1). As the TOCSY transfer occurs with slightly different efficiency for (4R-2H)-NADH and (4S-2H)-NADH, a calibration TOCSY experiment with commercial, non-deuterated NADH was performed (Fig. 2a) and the resulting integrals (1.00![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.13 for H4S:H4R) were used to normalize the integrals of the deuterated species (Fig. 2b–d). Due to the collapse of the 2JHH coupling constant of 18.1 Hz into a 2JHD of 2.8 Hz, both H4 resonances appear as broad singlets in the TOCSY spectrum and the fine structure is lost. The integration yielded a de of 87% for mutant S112A and a de of 90% for mutant S112K, while a de of 38% was obtained for the sample without Sav (all with an error margin of ±1%), after 16 hours reaction time (Fig. 2).
:1.13 for H4S:H4R) were used to normalize the integrals of the deuterated species (Fig. 2b–d). Due to the collapse of the 2JHH coupling constant of 18.1 Hz into a 2JHD of 2.8 Hz, both H4 resonances appear as broad singlets in the TOCSY spectrum and the fine structure is lost. The integration yielded a de of 87% for mutant S112A and a de of 90% for mutant S112K, while a de of 38% was obtained for the sample without Sav (all with an error margin of ±1%), after 16 hours reaction time (Fig. 2).
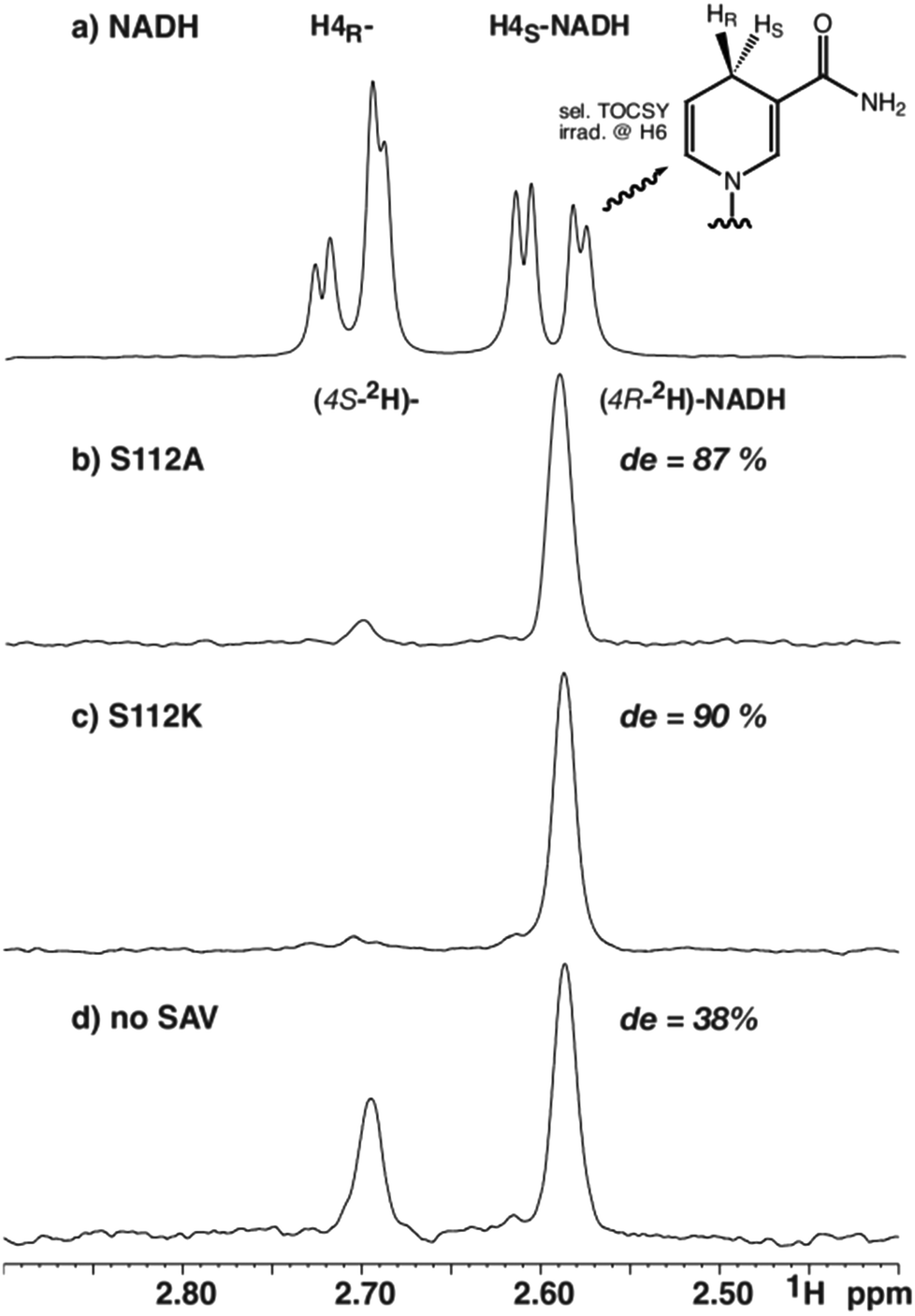 | ||
| Fig. 2 Selected region from selective 1H-TOCSY spectra of (a) commercial NADH, and catalytic reduction of NAD+ using 2HCOONa with, (b) S112A; (c) S112K; (d) no Sav. | ||
The modest, but significant diastereoselectivity observed in the absence of Sav, arises most likely from substrate control considering the formation of basically racemic product from achiral starting materials in related transformations.13,14 Both mutants led to a significant improvement in the diastereomeric excess compared to the result obtained in the absence of streptavidin (Fig. 2).
Interestingly, for the NAD+ reduction, both S112A and S112K ATHases afford the same diastereomer preferentially. This observation stands in stark contrast with results previously reported for ketone and imine reduction whereby S112A and S112K afford (R)- and (S)-reduction products respectively. In a matched-mismatched spirit, we speculate that the inherent chirality of the sugar moiety of NAD+ counterbalances the ATHase preference. This is supported by the inherent preference of the substrate in the absence of Sav. Alternatively, one NAD+ substrate could bind in the biotin vestibule in S112A, thus modifying the environment of the metal complex. Similar results were computed for the imine reductase with S112A.15 The preparation and screening of further mutants might provide guiding answers towards the design of ATHases with inverted selectivities for the reduction of NAD+.
Conclusions
The high chiral induction observed is promising for further applications such as the investigation of specificity in enzymes utilizing NAD(P)-mimics and the resulting information would be of interest for future enzyme engineering tasks. Furthermore, related prochiral structures can be envisaged, namely 3,4 substituted pyridinium ions, which would provide valuable building blocks upon asymmetric conversion.18Experimental part
Nicotinamide adenine dinucleotide hydrate (NAD+, contains 10% H2O) was purchased from Sigma. 2HCOONa was purchased from ABCR. Streptavidin was prepared as previously reported.13 [Cp*Ir(biot-p-L)Cl] was a kind gift from M. Dürrenberger and prepared as reported in ref. 19. NMR experiments were performed at 25 °C (MeOH calibration) on a Bruker Avance III NMR spectrometer operating at 600 MHz proton frequency, equipped with a direct detection dual channel, broadband probe-head with z-gradient. Chemical shifts were referenced against proton solvent peaks (4.773 ppm for H2O). The selective TOCSY experiment20,21 was performed using a MLEV17 spinlock sequence with a mixing time of 120 ms, trim pulses of 2.5 ms and a selective Gauss shaped refocusing pulse of 80 ms duration.Stock solutions: buffer 1: sodium phosphate 50 mM, pH 7.5; buffer 2: 2HCOONa 200 mM, sodium phosphate, pH 7.5 (2HCOONa (138 mg) was dissolved in H2O (5 ml), sodium phosphate buffer (0.5 ml, 1 M) was added and the pH adjusted to 7.5 by addition of aq. NaH2PO4. Water was added to a total volume of 10 ml); NAD+ stock solution: 10 mM in buffer 1 (NAD+ (29.9 mg, 405 μmol) was dissolved in buffer 1 (4.05 ml); Ir-stock solution: 10 mM [Cp*Ir(biot-p-L)Cl] (2.55 mg, 3.17 μmol) was dissolved in DMF-d7 (317 μl).
Preparation of ATHase: Sav-mutant S112A (2.53 mg, assuming 3 free binding sites) was dissolved in buffer 1 (1.144 mL), and Ir-stock solution was added (11.6 μl).
Sav-mutant S112K (2.65 mg assuming 3 free binding sites) was dissolved in buffer 1 (1.195 ml), and Ir-stock solution was added (12.1 μl).
Sample without SAV mutant (intermediate solutions): buffer 1 (0.495 ml) and Ir-stock solution was added (5 μl). Mixing was achieved by means of a vortex mixer. Reaction set up: To buffer 1 (125 μL) was added buffer 2 (250 μl; final concentration of 2HCOONa = 100 mM), ATHase (or intermediate solutions) (100 μl, final concentration of Ir = 20 μM) and NAD+-stock solution (25 μl, final concentration = 500 μM).
The reaction was incubated and agitated at 30 °C and 200 rpm for 24 h by means of a Thermomixer (HLC Biotech Model MHR 23).
Subsequently D2O (50 μl) was added and the NMR spectra recorded.
Abbreviations
| NADH | Nicotinamide adenine dinucleotide (reduced form) |
| NAD+ | Nicotinamide adenine dinucleotide |
| NAD(P)H | Nicotinamide adenine dinucleotide (phosphate) (reduced form) |
Acknowledgements
This research was made possible thanks to a grant from the European Union Seventh Framework Program (FP7/2007–2013) under grant agreement n° FP7-PEOPLE-ITN-2008-238531. Additional funding was provided by the Swiss National Science Foundation (200020_144354). TRW thanks Umicore for a generous loan of [Cp*IrCl2]2 and Prof. Cantor for the Sav gene.
References
- T. E. Benson, J. L. Marquardt, A. C. Marquardt, F. A. Etzkorn and C. T. Walsh, Biochemistry, 1993, 32, 2024–2030 CrossRef CAS PubMed.
- R. E. Viola, P. F. Cook and W. W. Cleland, Anal. Biochem., 1979, 96, 334–340 CrossRef CAS PubMed.
- G. Ottolina, S. Riva, G. Carrea, B. Danieli and A. F. Buckmann, Biochim. Biophys. Acta, Protein Struct., 1989, 998, 173–178 CrossRef CAS.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Buhler, Green Chem., 2011, 13, 226–265 RSC.
- F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2314 RSC.
- F. Leipold, S. Hussain, D. Ghislieri and N. J. Turner, ChemCatChem, 2013, 5, 3505–3508 CrossRef CAS.
- M. Rodríguez-Mata, A. Frank, E. Wells, F. Leipold, N. J. Turner, S. Hart, J. P. Turkenburg and G. Grogan, ChemBioChem, 2013, 14, 1372–1379 CrossRef PubMed.
- H. Wu, C. Tian, X. Song, C. Liu, D. Yang and Z. Jiang, Green Chem., 2013, 15, 1773–1789 RSC.
- C. E. Paul, I. W. C. E. Arends and F. Hollmann, ACS Catal., 2014, 4, 788–797 CrossRef CAS.
- R. Ruppert, S. Herrmann and E. Steckhan, Tetrahedron Lett., 1987, 28, 6583–6586 CrossRef CAS.
- M. de Torres, J. Dimroth, I. W. C. E. Arends, J. Keilitz and F. Hollmann, Molecules, 2012, 17, 9835–9841 CrossRef CAS PubMed.
- V. Köhler, Y. M. Wilson, M. Dürrenberger, D. Ghislieri, E. Churakova, T. Quinto, L. Knörr, D. Häussinger, F. Hollmann, N. J. Turner and T. R. Ward, Nat. Chem., 2013, 5, 93–99 CrossRef PubMed.
- M. Dürrenberger, T. Heinisch, Y. M. Wilson, T. Rossel, E. Nogueira, L. Knörr, A. Mutschler, K. Kersten, M. J. Zimbron, J. Pierron, T. Schirmer and T. R. Ward, Angew. Chem., Int. Ed., 2011, 50, 3026–3029 CrossRef PubMed.
- V. Muñoz Robles, M. Dürrenberger, T. Heinisch, A. Lledós, T. Schirmer, T. R. Ward and J.-D. Maréchal, J. Am. Chem. Soc., 2014, 136, 15676–15683 CrossRef PubMed.
- L. Brecker and D. W. Ribbons, Trends Biotechnol., 2000, 18, 197–202 CrossRef CAS PubMed.
- B. Morawski, G. Casy, C. Illaszewicz, H. Griengl and D. W. Ribbons, J. Bacteriol., 1997, 179, 4023–4029 CAS.
- J. Wu, W. Tang, A. Pettman and J. Xiao, Adv. Synth. Catal., 2013, 355, 35–40 CrossRef CAS.
- Y. M. Wilson, M. Dürrenberger and T. R. Ward, Organometallic chemistry in protein scaffolds, in Protein Engineering Handbook, ed. S. Lutz and U. T. Bornscheuer, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, vol. 3, pp. 215–241 Search PubMed.
- H. Kessler, H. Oschkinat, C. Griesinger and W. Bermel, J. Magn. Reson., 1986, 70, 106–133 CAS.
- A. Bax and D. G. Davis, J. Magn. Reson., 1985, 65, 355–360 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra with water suppression and selective 1H-TOCSY NMR spectra. See DOI: 10.1039/c4ob02071e |
| This journal is © The Royal Society of Chemistry 2015 |