
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interplay between singlet and triplet excited states in a conformationally locked donor–acceptor dyad†
Mikhail A.
Filatov‡
*ab,
Fabian
Etzold
a,
Dominik
Gehrig
a,
Frédéric
Laquai
ac,
Dmitri
Busko
a,
Katharina
Landfester
a and
Stanislav
Baluschev
ade
aMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. E-mail: filatovm@tcd.ie
bInstitute of Polymers, Bulgarian Academy of Sciences, Acad. G. Bonchev Str., block 103-A, BG – 1113 Sofia, Bulgaria
cPhysical Sciences and Engineering Division (PSE), Material Science and Engineering (MSE), Solar and Photovoltaics Engineering Research Center (SPERC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia
dOptics and Spectroscopy Department, Faculty of Physics, Sofia University “St. Kliment Ochridski”, 5 James Bourchier, 1164 Sofia, Bulgaria
eFreiburg Institute for Advanced Studies (FRIAS), Albert-Ludwigs-Universität Freiburg, Albertstraße 19, D-79104 Freiburg, Germany
First published on 13th October 2015
Abstract
The synthesis and photophysical characterization of a palladium(II) porphyrin – anthracene dyad bridged via short and conformationally rigid bicyclo[2.2.2]octadiene spacer were achieved. A spectroscopic investigation of the prepared molecule in solution has been undertaken to study electronic energy transfer in excited singlet and triplet states between the anthracene and porphyrin units. By using steady-state and time-resolved photoluminescence spectroscopy it was shown that excitation of the singlet excited state of the anthracene leads to energy transfer to the lower-lying singlet state of porphyrin. Alternatively, excitation of the porphyrin followed by intersystem crossing to the triplet state leads to very fast energy transfer to the triplet state of anthracene. The rate of this energy transfer has been determined by transient absorption spectroscopy. Comparative studies of the dynamics of triplet excited states of the dyad and reference palladium octaethylporphyrin (PdOEP) have been performed.
Introduction
Chromophores producing long-lived triplet excited states under excitation with light have attracted much attention during the past decade due to great promise for application in diverse areas of research, particularly aimed at sunlight harvesting.1 Chemical modification is a key tool for the control of excited-state properties, and consequently emission and energy transfer parameters (efficiency and rates) of the chromophore.2 Particularly, introduction of intramolecular excited state donor–acceptor couplings prolongs the triplet lifetime by enabling a reversible intramolecular exciton transfer, the so called “energy reservoir” effect.3 Such modifications allow for improving the efficiencies of corresponding photonic applications.4 Phosphorescent donor-bridge-acceptor (D-b-A) dyes are important materials for a number of optoelectronic,5 sensing,6 bioimaging7 and applications based on triplet–triplet annihilation photon upconversion (TTA-UC).8In the TTA-UC process, D-b-A triplet sensitizers promise to provide a new generation of molecular photonic devices. Efficient implementation of the process requires two components in a mixture: a sensitizer (transition metal complex) and an emitter (aromatic hydrocarbon or BODIPY), which generate higher energy photons through a sequence of energy transfer processes.9 The development of a single molecular system combining the properties of sensitizer and emitter and capable of anti-Stokes emission is of fundamental importance and is desired for sensing applications. Attempts to incorporate 9,10-diphenylanthracene (DPA) into ruthenium(III) complex structures provided a delayed fluorescence (TTA-based upconversion emission) of covalently bound anthracene, however with low efficiency compared to the mixture.10 The key factors governing energy transfer properties in such dyads are to be elucidated in order to develop efficient single molecular upconversion systems.
A number of studies carried out with covalently/noncovalently linked porphyrin–porphyrin11 and porphyrin–nonporphyrinic chromophore12 assemblies have dealt with the effect of D-b-A distance on the rates of the triplet–triplet energy transfer (TTET) process, however, relatively little attention has been paid to the effects of mutual orientation between donor and acceptor.13 Giribabu and co-workers demonstrated striking orientation dependence of the energy transfer in a porphyrin-based D-b-A system, in which the donor subunits are linked both at the axial and peripheral sites of the porphyrin macrocycle (acceptor). Photoluminescence studies revealed that energy transfer (ET) from the porphyrin donor to the acceptor subunits bound at the peripheral positions is efficient, while no ET takes place to the acceptors bound at the axial positions.14
Another fundamental question arising from these studies is whether the electronic excitation leading to a long-lived triplet state is accompanied by a change in the molecular conformation, especially for porphyrins, known to possess various macrocycle distortion modes.15 Little is known about photophysical behaviour of conformationally rigid D-b-A porphyrin systems, specifically regarding the triplet state dynamics and phosphorescence energies. Although porphyrin D-b-A systems have been extensively studied,16 no examples of such system, which can be regarded as “rigid” meaning that no conformational changes in the excited state can take place, are known. In this work we studied a D-b-A system, capable of TTET, in which conformational changes are excluded due to the specific arrangements of donor and acceptor moieties along a bridging bicyclic subunit.
A molecular design concept of the target system is illustrated in Fig. 1. The assembly of porphyrin D-b-A dyads has commonly been based on the functionalization of meso-positions of the macrocycle, since corresponding synthetic methods are well developed.17 However, meso-substitution is known to cause non-planar distortion of the porphyrin macrocycle, adopting saddle, waved, ruffled or domed conformation.18 Systematic analysis of photophysical properties of meso-substituted porphyrins reveals that: (1) chromophores attached through meso-positions are in partial conjugation to the porphyrin's aromatic system and (2) rotational motions along Cmeso–Caryl bonds are possible.19 It results in lower quantum yields of emission and slows down excited state energy transfer processes. On the other hand, it was shown that β-substituted porphyrins with four bicyclo[2.2.2]octene-fused frameworks adopt a slightly waved, planar conformation.20 In all reported crystal structures of these porphyrins, the porphyrin chromophore was almost flat and showed only slight out-of-plane distortion.21 Separation of aromatic systems by a rigid bridges based on polycyclic hydrocarbon scaffolds has been shown to provide well-defined distances and orientations of the photoactive fragments.22 Based on these findings, we designed a porphyrin D-d-A dyad, in which the macrocycle is surrounded by four anthracene moieties connected via bicyclo[2.2.2]octene spacer (Fig. 1B) and studied its optical properties and excited state dynamics.
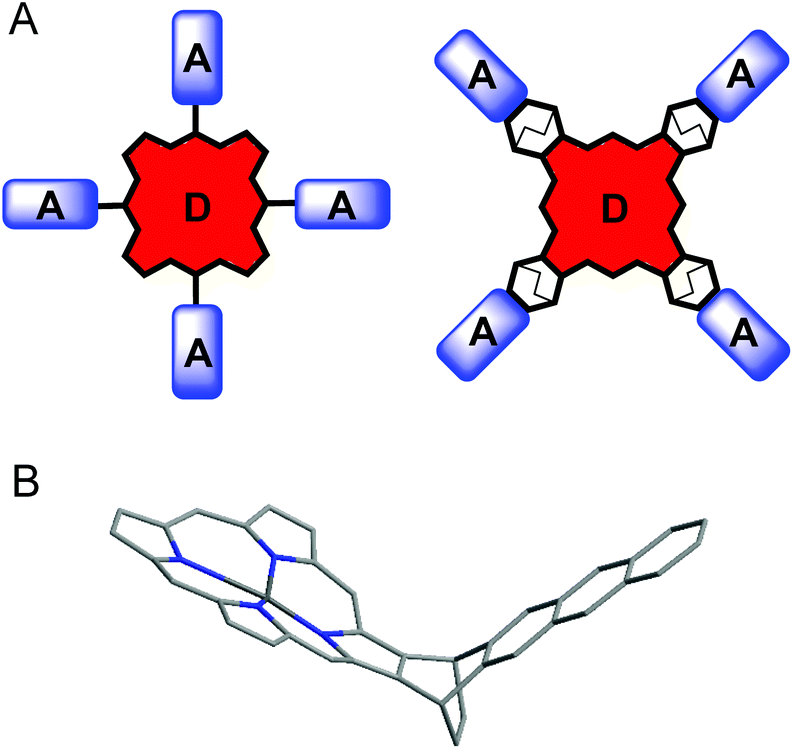 | ||
| Fig. 1 (A) Possible structures of porphyrin D-b-A system, (B) porphyrin-anthracene dyad with bicyclo[2.2.2]octane spacer. | ||
Results and discussion
Synthesis
The synthesis of the target molecule is shown in Scheme 1. Starting quinizarine was converted to 1,4-anthraquinone 1 by the reduction with NaBH4. Compound 1 was further subjected to Diels–Alder reaction with 1,3-cyclohexadiene in boiling chloroform which delivered product 2. It should be noted, that in the case of toluene as a solvent, heating of the reaction mixture above 100 °C resulted in the formation of tetracene-5,12-dione as a by-product via thermal retro-Diels–Alder reaction.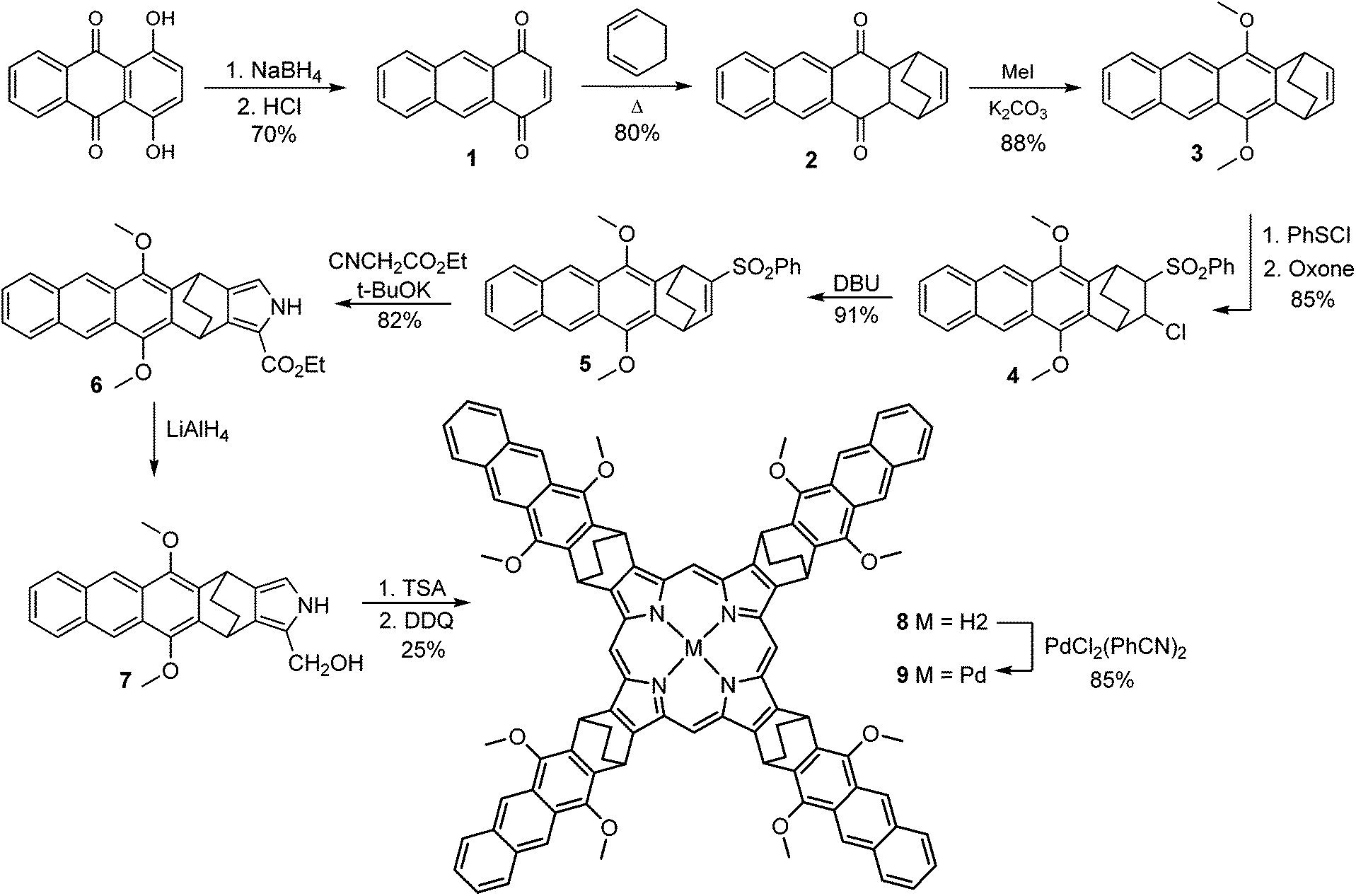 | ||
| Scheme 1 Synthesis of target molecule. | ||
Acenes are known to possess low solubility, requiring the introduction of alkyl, alkoxy or alkoxycarbonyl substituents. Introduction of such substituents affects excited states energies.23 The latter is of particular importance with respect to photophysical properties of a designed system, since an overlap of donor and acceptor triplet manifolds is to be achieved. Thus, we treated compound 2 with a base (DBU), to form deprotonated hydroquinone, which was then reacted with MeI to provide 1,4-dimethoxyanthracene derivative 3 in good overall yield. Anthracene 3 was subjected to a reaction with PhSCl that resulted in the corresponding chlorosulfide, which was further oxidized to give chlorosulfone 4. Subsequent HCl elimination under DBU treatment yielded the corresponding allylic sulfone 5, which was used for Barton–Zard reaction with isocyanacetate leading to pyrrole 6. Its reduction by LiAlH4 delivered pyrrole derivative 7 which was introduced into acid-catalyzed tetramerization24 without isolation to give corresponding porphyrin 8 in a good yield (25%).
Finally, the free-base porphyrin 8 was subjected to palladium(II) insertion reaction leading to the target complex 9. Careful control over the reaction temperature was critically important to prevent by-products formation. Palladium insertion into porphyrin macrocycles usually requires prolonged reflux in high boiling point solvents such as benzonitrile.25 In the case of porphyrin 9, heating over 150 °C was found to cause a retro-Diels–Alder reaction,26 leading to the corresponding tetracene-fused derivatives, as was evidenced by mass-spectroscopy.
In the course of isolation and purification of crude products 8 and 9 we noticed a formation of by-products upon their illumination with light under air. This probably results from porphyrin-sensitized singlet oxygen formation and its successive addition to the anthracene moiety, similarly to previously reported anthraporphyrins and anthracene-substituted porphyrins.27 However, protecting the corresponding solutions from light during purification we succeeded to isolate products 8 and 9 in pure form as evidenced by solution NMR and mass-spectroscopy data (see ESI‡).
It should be noted, that the obtained product 9 obviously consists of a mixture of stereoisomers, resulting from the fact that the attached anthracene fragment is not symmetrical with the respect to the porphyrin mean plane. However, taking into account that the geometry of the photoactive fragment (porphyrin-bridge-anthracene) is identical for each of the possible isomers, their photophysical properties are unlikely to show individual features.
Optical properties
Absorption and emission spectra of compound 3 (Fig. 2A) generally resembled those of a reference compound, namely 9,10-diphenylanthracene (DPA) in its maxima, band structure and lifetimes. On the other hand, a considerably lower fluorescence quantum yield was observed (0.51). In order to obtain its phosphorescence spectrum, we performed a photoluminescence measurement at 77 K in frozen methyl-cyclohexane solution. In this case a red-shifted emission with a considerably longer lifetime (∼300 ns) than the fluorescence (∼14.9 ns) was observed (see Fig. S18 and S19‡). However, a comparison with literature data shows, that the observed emission does not correspond to the anthracene triplet state, but rather to the emission of excimers.28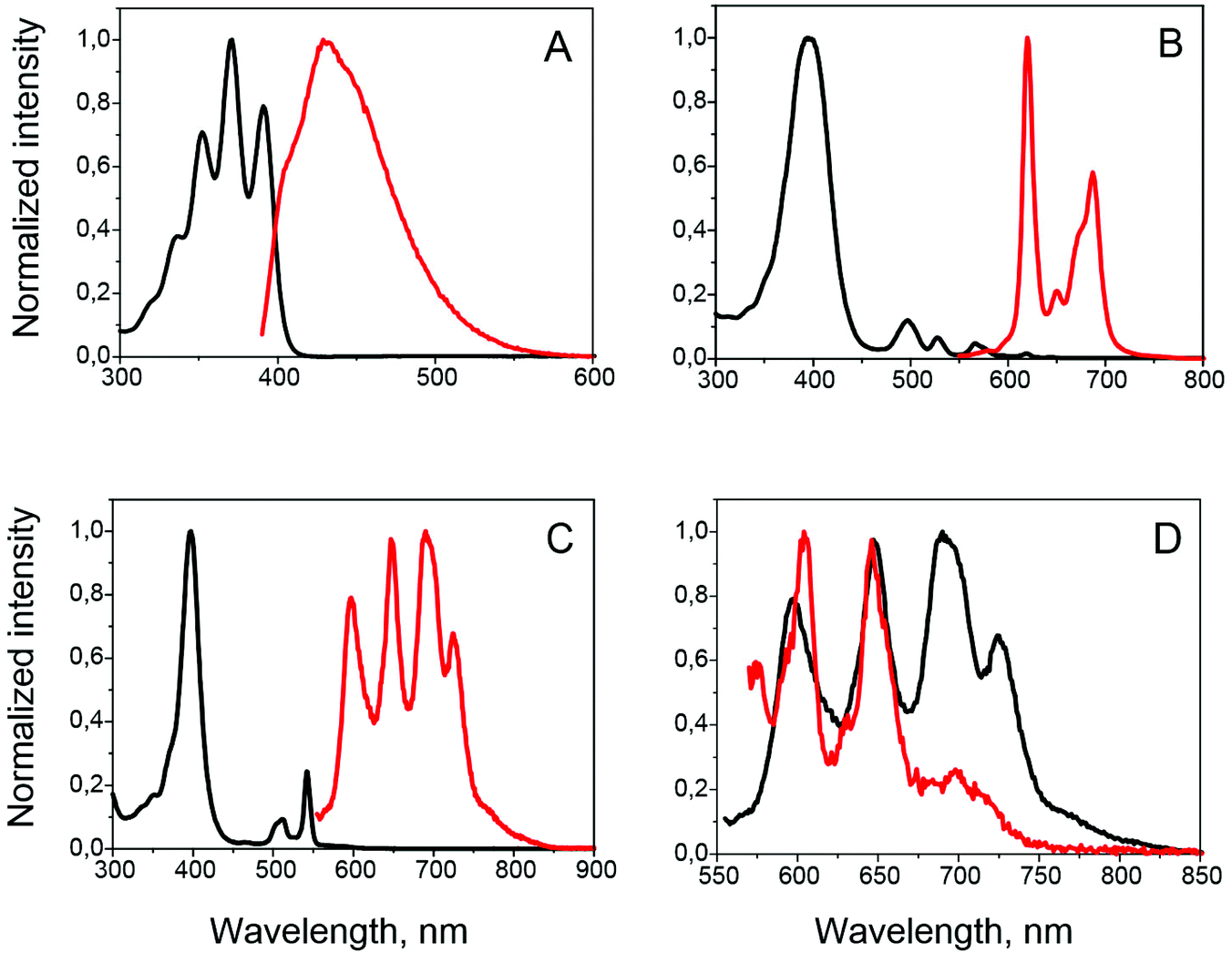 | ||
| Fig. 2 Absorption (black) and emission (red) spectra of anthracene 3 (A), porphyrins 8 (B) and 9 (C). (D) Comparison of porphyrin 9 (3 × 10−5 M) emission under inert athmosphere (black) and under air (red). | ||
UV/Vis and photoluminescence spectra of 8 and 9 are presented in Fig. 2. Similarity of the absorption features, i.e. maxima and vibrational structure, to those of octaethylporphyrin (OEP) and its Pd(II) complex (see Fig. S20 and S21‡ for comparison) confirms the absence of conjugation between porphyrin and anthracene. The only difference observed is a slight bandwidth broadening due to increased molecular size. Absorption of anthracene subunits is seen in the 300–400 nm region.
Upon excitation at the wavelength corresponding to the porphyrin Q-band absorption, compound 8 exhibits fluorescence consisting of two bands with maxima at 620 and 687 nm, typical of free base porphyrins, and virtually identical regarding peak maxima and vibronic structure to that obtained with the same excitation wavelength for OEP free-base. Fluorescence quantum yields were obtained as 0.17 and 0.13 for 8 and OEP, respectively (compared to tetraphenylporphyrin free-base as a reference). When excitation was done at 336 nm, in the region of the anthracene chromophore absorption, emission spectra showed predominantly emission from the porphyrin (Fig. S20‡). An excitation spectrum registered at different emission wavelengths (Fig. S20‡) closely resembles the absorption spectrum of 8.
Emission spectra of 9 solution (5 × 10−6 M), prepared in glove-box conditions (1 ppm O2), differ to those of the parent PdOEP (Fig. S21‡) and consist of four bands at 597, 648, 689 and 724 nm (Fig. 2c). Upon selective excitation in the region of anthracene absorption, similar emission spectra were observed. Excitation spectra measured at all emission maxima are similar to the absorption spectrum of 9 (Fig. S21‡). When the sample was left open to air, the emission beyond 670 nm was almost completely quenched (Fig. 2D), while the peaks at 597 and 648 nm were still observed, although with rather low intensity. Lifetimes of the emission at 597 and 648 nm were measured to be 602 and 648 μs, respectively (Fig. S23‡).
Excited state dynamics in PdOEP and porphyrin 9
In order to further clarify the excited state dynamics, porphyrin 9 was studied by ps–μs broadband transient absorption spectroscopy (TA); measurements on the pristine PdOEP were also performed and compared to those of 9.The measurements conducted on pristine PdOEP were done in toluene solution at a concentration of 10−4 M. After excitation into the Soret band at 400 nm, porphyrin singlet states are generated on a time scale faster than the TA instrument response. The respective sub-ps transient absorption spectrum is shown in Fig. 3. The prominent peaks between 500–550 nm are assigned to the bleaching of the ground state, as they perfectly well match the Q-band ground state absorption spectrum. The positive feature at 610 nm results from stimulated emission (SE) of the porphyrin S1 states (PS1) and can be used as a measure for the contribution of singlet excited states to the total excited state population. Between 850–1000 nm the photoinduced absorption (PAS1) of the singlets shows another prominent peak, which consequently can be used to monitor the dynamics of the excited singlet state population. After a few ps both the SE and the PAS1 of the singlets have vanished and another broad PA has appeared with a distinct peak between 740–790 nm. As this feature stayed constant throughout the time window of the TA experiment (4 ns), we assigned it to long-lived porphyrin T1 states (PT1). Thus the initially generated porphyrin singlet excitons undergo intersystem crossing to form porphyrin triplet states. Global fitting of the dataset with a single exponential function described the decay of the singlet excited states and concomitant rise of the triplet state population very well, allowing us to extract a rate constant for the intersystem crossing, as shown in the bottom of Fig. 3. The kinetics were extracted at the indicated spectral regions after the initial photogeneration of the singlet excitons (faster than the response time of the setup). The solid lines represent a monoexponential fit with a rate constant of kISC = 1/(7.97) ps−1 obtained from a global fit of the whole data set. The correlated decrease of porphyrin singlet PAS1 and increase of porphyrin triplet PAT1 allows to assign this rate constant to the ISC process. The obtained individual component spectra for the two states are shown in the ESI.‡
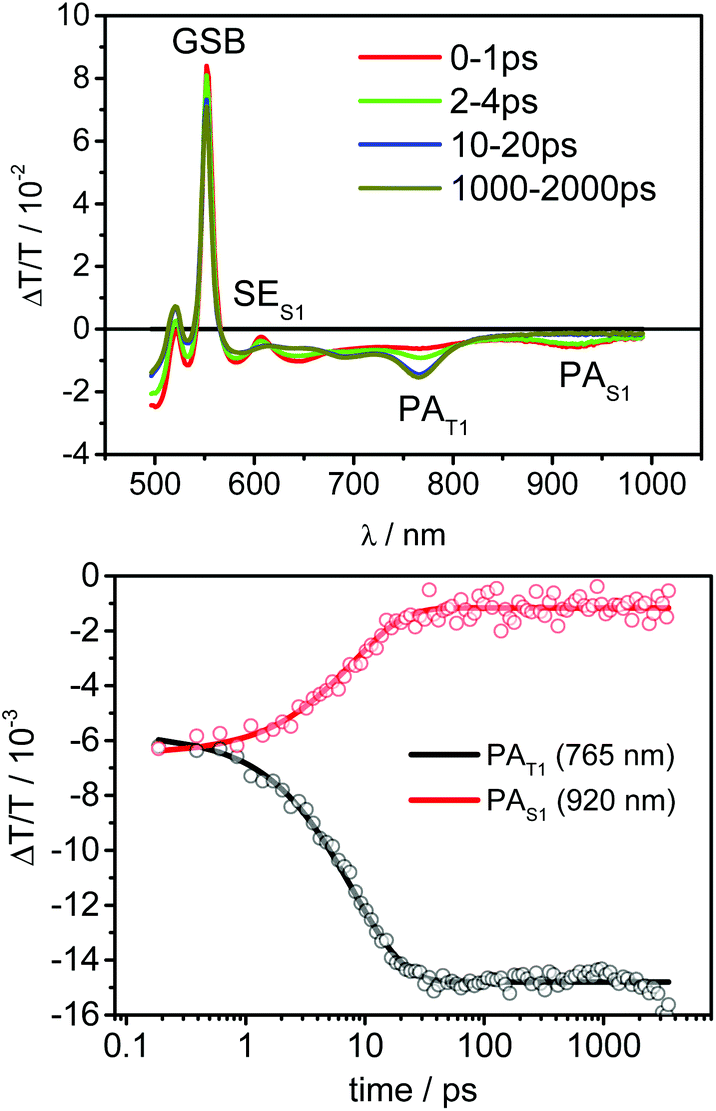 | ||
| Fig. 3 Transient absorption spectra of PdOEP after excitation at 400 nm and kinetics of the singlet and triplet population. | ||
With the knowledge of the singlet and triplet state transient absorption spectra, we interpreted the transient absorption data of compound 9 obtained in solution at a concentration of 10−4 M. Fig. 4 shows the respective transient absorption spectra.
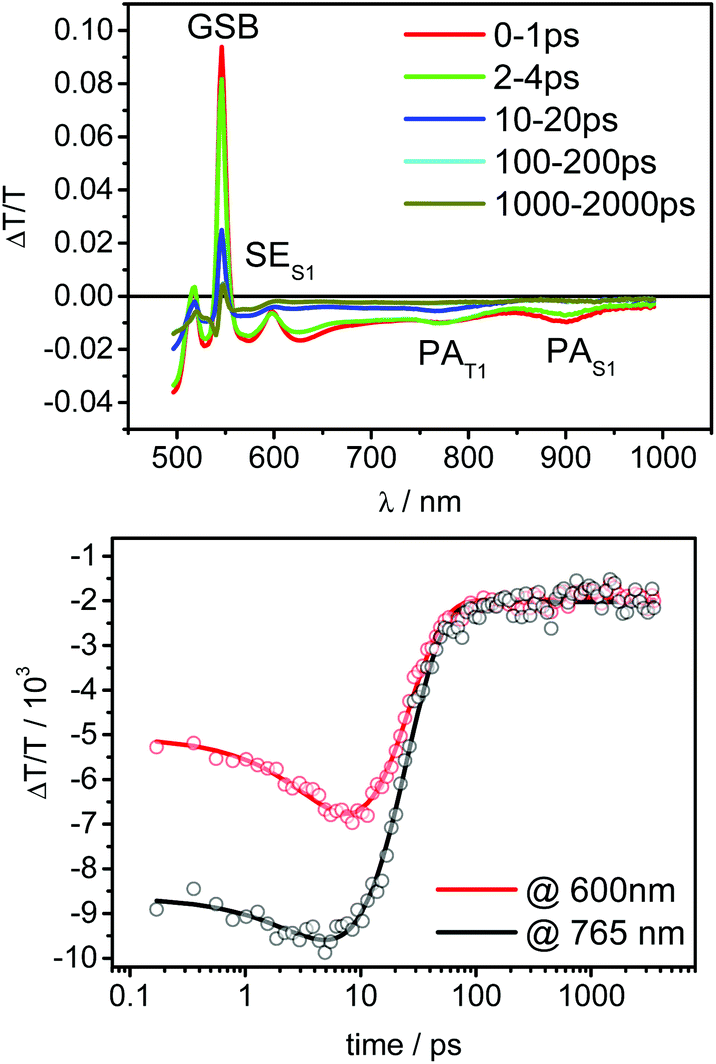 | ||
| Fig. 4 Transient absorption spectra of compound 9 and kinetics observed at the indicated wavelength regions. | ||
At early times, precisely 0–4 ps, porphyrin singlets can be identified by their characteristic SE peak around 600 nm and the photoinduced absorption feature at 850–950 nm. Both features disappear on the same time scale as observed for the PdOEP indicating triplet state formation on the porphyrin. However, the triplet signatures quickly decayed in contrast to the pristine PdOEP, and a very weak residual signal remained. The fast decay of the triplet signatures indicates efficient triplet energy transfer to the anthracene, which we modeled by a sequential model including two processes that follow single exponential dynamics, of which one describes the ISC and the other the triplet energy transfer from the porphyrin core to the adjacent anthracene.
Fig. 4 displays the kinetics in the region of SE of the porphyrin singlets (600 nm) and the PAT1 peak region of the porphyrin triplets (765 nm) together with the fit to the experimental data. Due to the superposition of the individual photoinduced absorption signatures, an initial signal rise can be observed, corresponding to the ISC process from porphyrin singlets to porphyrin triplets. After the ISC is completed the triplet population stays constant for more than 2 ns in the pristine PdOEP, while in compound 9 triplet energy transfer to the anthracene takes place and rapidly reduces the porphyrin triplet population. The rise of the signals on the sub-10 ps time scale corresponds to the population of the porphyrin triplet state, which is subsequently transferred to the anthracene, causing the signal to decay up to 100 ps. The remaining weak PA then stayed constant in signal height over the measured time frame. Due to its long-lived character and different shape compared to the triplet-induced absorption of the porphyrin, we assigned this feature to triplet states on anthracene. The global fit yielded an ISC rate of kISC = 1/8.97 ps−1 and a triplet energy transfer rate of ktrans = 1/14.1 ps−1. The spectra obtained for the porphyrin singlet and triplet state from the global fit are in good agreement with those obtained from the pristine porphyrin and thus further validate our data analysis (Fig. S26 and S27‡).
Excited state energy transfer processes in porphyrin 9
The energetic overlap of the porphyrin and anthracene triplet manifolds in molecule 9 was confirmed by measuring the emission of 3 in a mixture with PdOEP (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in toluene. Upon excitation at 542 nm, blue fluorescence (457 nm) was observed due to the TTA-UC process (Fig. 5). The efficiency of the process was found to be 7.5%, comparable with those of the PdOEP–DPA (12.5%).
:1) in toluene. Upon excitation at 542 nm, blue fluorescence (457 nm) was observed due to the TTA-UC process (Fig. 5). The efficiency of the process was found to be 7.5%, comparable with those of the PdOEP–DPA (12.5%).
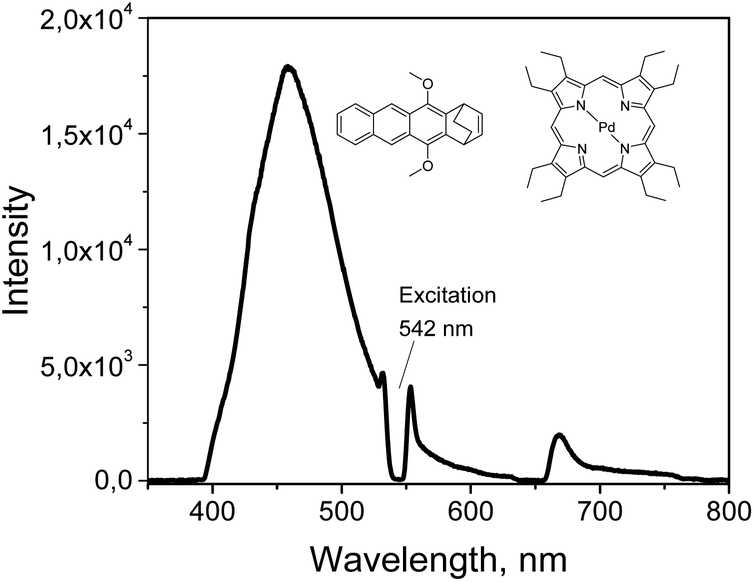 | ||
| Fig. 5 Upconversion spectrum of PdOEP (10−5 M) and 5,12-dimethoxy-1,4-dihydro-1,4-ethanotetracene (5 × 10−3 M) in air-free toluene. | ||
Thus, intersystem crossing giving the triplet state localized on the porphyrin subunit (1) is followed by excited state triplet transfer to the anthracene, as was shown by TA data (2):
| 1Por-b-Ant → 3Por-b-Ant | (1) |
| 3Por-b-Ant → Por-b-3Ant | (2) |
On the other hand, steady-state emission spectra and corresponding excitation spectra of 8 and 9 clearly indicate intramolecular excited state singlet energy transfer (3) from anthracene to the porphyrin core, since excitation in the region of anthracene absorption leads to the emission corresponding to the porphyrin chromophore. This is consistent with the excited-state behaviour of the previously reported porphyrin-anthracene dyads.29 Although direct excitation of both anthracene and porphyrin chromophores can't be excluded due to the overlap of the absorption, emission of the PdOEP–anthracene 3 mixture (1:4 ratio), coming predominantly from anthracene (Fig. S23‡) upon excitation in the same region, confirms energy transfer in dyad 9.
| Por-b-1Ant → 1Por-b-Ant | (3) |
Comparison of the emission spectra of palladium porphyrin 9 with the reported fluorescence and phosphorescence spectra of anthracene 3 and other anthracenes (9,10-dimethoxy or 9,10-diphenylanthracene) shows that there is no contribution from the anthracene subunits to the observed emission comprised by a series of peaks at 597, 648, 689 and 724 nm. Analytical data, suggesting reasonable purity, in conjunction with the good agreement of excitation and absorption spectra allow to rule out that some of these peaks are resulting from impurities. Alternatively, the fact that the emission peaks at 597 and 648 nm were observed in oxygen-saturated conditions, when the phosphorescence is completely quenched, allows to assign this emission as a fluorescence from the singlet state of the porphyrin,30 which is normally very weak for Pd and Pt porphyrins due to the high intersystem crossing efficiency.31
We conclude that the observed spectrum of 9 represents a combination of the emission from singlet and triplet excited states. Intensive singlet emission in degassed solution of 9 may result either from triplet–triplet annihilation or incomplete intersystem crossing.32 The latter can be however excluded in the case of molecule 9 based on the transient absorption studies results. We assigned the emission at 597 nm as a delayed fluorescence, while the emission at 648 nm as a superposition of the delayed fluorescence and phosphorescence. Indeed, similarly long-lived singlet emission was observed for PdOEP, although with much lower intensity compared to its phosphorescence.33
To prove the formation of porphyrin singlet states we performed long delay transient absorption measurement, i.e. at ns–μs timescale. TA spectra obtained after 1 ns are in agreement with the proposed process. After 1 μs the emergence of a clear positive signal between 550–600 nm (Fig. S28‡) is seen. Due to its spectral position this signal can only be assigned to the porphyrin's singlet state. The lifetime of this signal is in the range of hundreds of microseconds (Fig. S29‡), which is in line with the observation of long-lived photoluminescence from compound 9 in the same spectral region and supports our assignment. However, it remains unclear why the singlet states do not undergo rapid intersystem crossing to the triplet state as observed upon direct photoexcitation of the porphyrin's singlet state.
Intermolecular energy transfer processes, such as TTET, are concentration dependent. Indeed, registering the emission of 9 at different concentrations shows changes in the delayed fluorescence intensity, which almost completely disappears upon strong dilution. At 10−6 M concentration emission spectrum of 9 becomes nearly identical to those of PdOEP (Fig. 6), due to a loss of intermolecular TTA process efficiency.
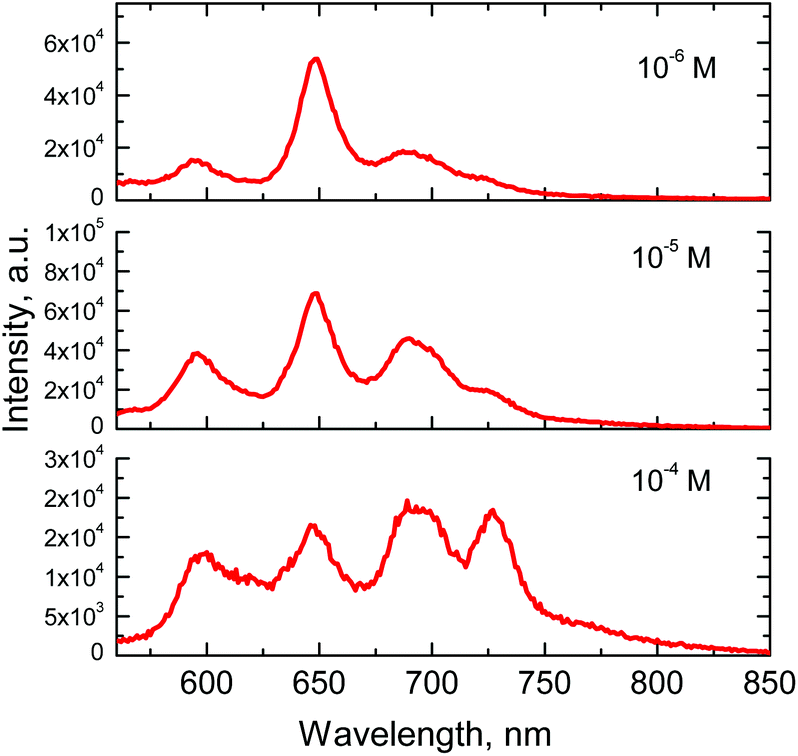 | ||
| Fig. 6 Emission spectra of porphyrin 9 at different concentrations in toluene. Excitation wavelength – 542 nm. | ||
Influence of the concentration on the emission spectra, i.e. intense delayed fluorescence at high concentration, evidences that intermolecular energy transfer between the molecules takes place in solution. Taking into account previous findings on triplet excited state relaxation channels,33 this confirms that triplet–triplet annihilation between two anthracene localized triplet states takes place, which results in the formation of a singlet excited state of anthracene:
| Por-b-3Ant + Por-b-3Ant → Por-b-1Ant + Por-b-Ant | (4) |
Such process is also consistent with the reported photophysical behavior of 9,10-diphenylanthracene appended ruthenium(III) complex structures, which undergo TTA process in the excited state.10
As shown in the steady-state emission experiments, singlet energy transfer from the anthracene to the porphyrin is very efficient. Thus TTA process (4) should be followed by the formation of porphyrin S1 states via intermolecular energy transfer process (2). Although the intersystem crossing is very fast, emission of the porphyrin singlet is observed due to recombination to the ground state. ns–μs TA experiments on compound 9 further corroborate porphyrin singlet excited state formation.
The observed processes are summarized in Fig. 7.
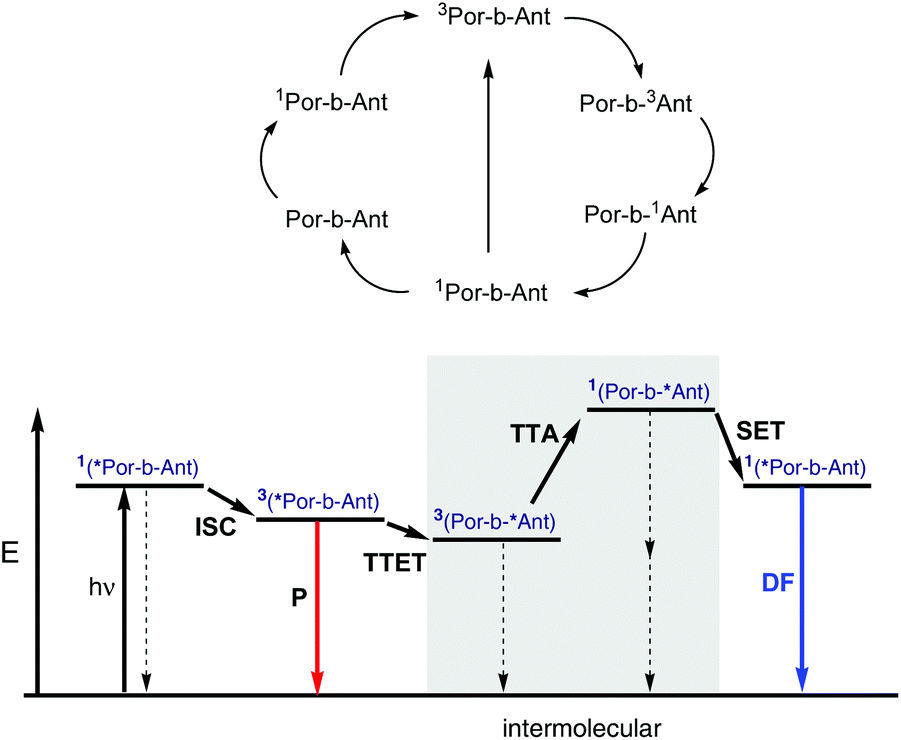 | ||
| Fig. 7 Transitions among excited states of 9 and qualitative Jablonski diagram. hv – photon excitation, ISC – intersystem crossing, P – phosphorescence, TTET – triplet–triplet energy transfer, TTA – triplet–triplet annihilation, SET – singlet energy transfer, DF – delayed fluorescence. | ||
An additional point is noteworthy: in the spectra of compound 9 no upconverted luminescence coming from the anthracene subunits was observed. This is in contrast with previous results reported for Ru(III) cyclometallated complexes, surrounded by diphenylanthracenes.10 A possible explanation could be that it is the short distance between donor and acceptor subunits in 9, which promotes fast energy back transfer in the singlet excited state formed in the course of TTA.
It was of particular interest to investigate whether molecule 9 can act as a triplet sensitizer for TTA-UC process in spite of the observed interplay between the singlet and triplet excited states. Another important question was if the intramolecular TTET from porphyrin to anthracene subunit, forming anthracene localized triplet state, can facilitate TTA-UC in a two-component system containing “free” anthracene 3 in the solution (emitter). Fig. 8 shows upconversion spectra for the samples containing 9 (3 × 10−5 M) as a sensitizer and 5, 10 or 20 equivalents of 3 as an emitter. The spectra were registered at different concentrations of “free” anthracene 3 while keeping the sensitizer concentration constant. Efficiency of the process reaches 3.8% at excitation intensities >1.5 W cm−2. However, in contrast to previously studied upconversion systems, which require excessive amounts of emitter with respect to amount of sensitizer used (usually 1:20–50), this couple shows strong emission even in 1:5 ratio (see Fig. S24‡ for comparison with PdOEP). Moreover, even in 1:1 ratio weak upconversion signal was still detected (Fig. S25‡). This suggests that dimethoxyanthracene antennas are enhancing the rate of TTET or triplet–triplet annihilation as well, or both rates. However, this is beyond the scope of the present manuscript and will be reported in a separate study.
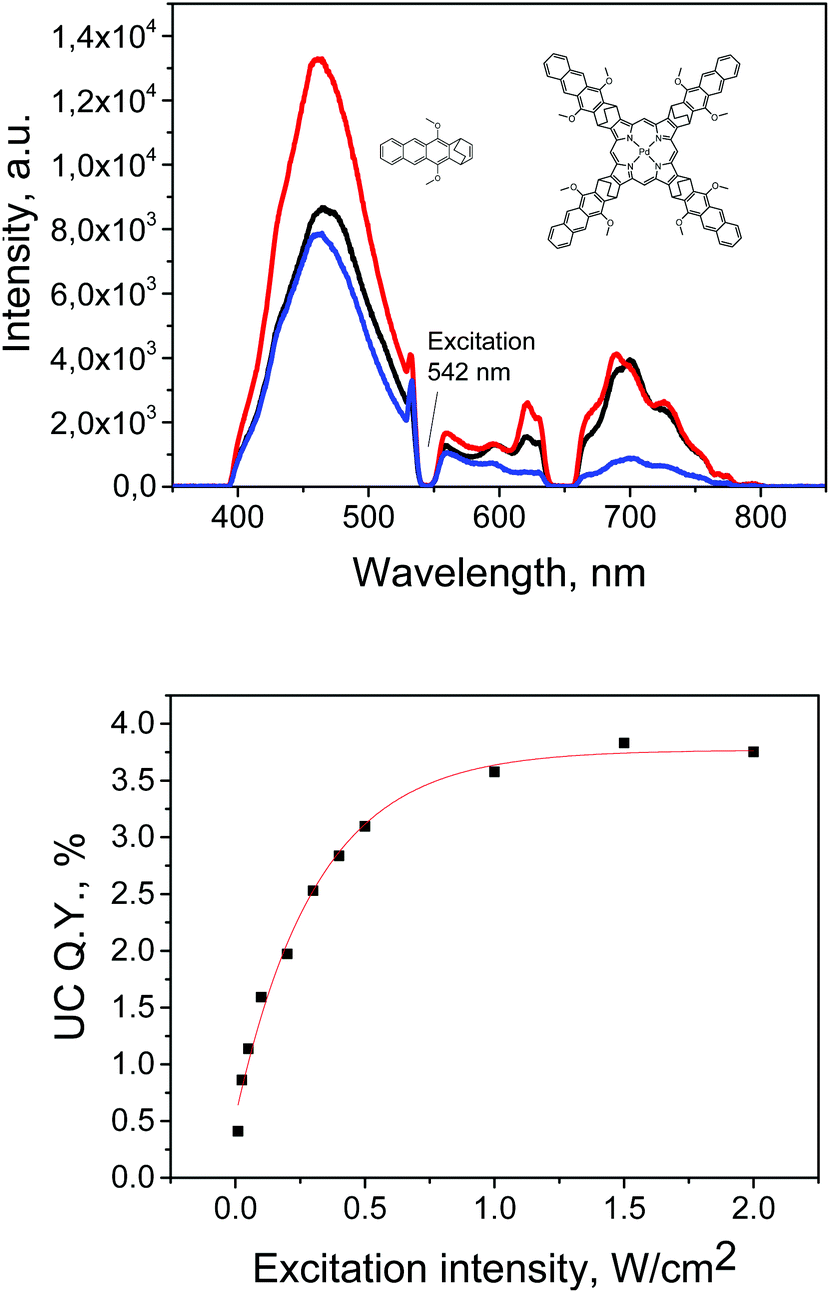 | ||
| Fig. 8 Upconversion luminescence of the mixture of 9 and 3 and quantum yield dependence on the excitation intensity (for the mixture of 6 × 10−4 M 3 and 3 × 10−5 M 9 respectively). Solvent: toluene. Samples were excited at λ = 542 nm (100 mW cm−2). | ||
Excited state behavior of 9 is intriguing from the viewpoint of chromophores design for photonic applications. Delayed fluorescence has been demonstrated as a tool for a real-time temperature probe with ratiometric response for a broad temperature interval.34 The main limitation of this approach is the relatively weak intensity of the delayed fluorescence of the metallated macrocycles, compared to phosphorescence signal. To increase the intensity of delayed fluorescence, encapsulation into polymer films (e.g. polystyrene), was applied.33 Although steric effects of the triplet–triplet annihilation process have not been adequately studied so far, experimental observations suggest that the TTA efficiency is critically dependent on the mutual orientation of two annihilating triplet excited state molecules. In particular, simulations showed that the magnetic field effect on TTA is very sensitive to mutual chromophore alignment, and the direction of the effect is consistent with a local ordering of excited state molecules.35 The fact that molecule 9 possesses strong delayed fluorescence in solution, may imply involvement of the factor of mutual orientation between annihilating excited states in the intermolecular TTA process. For further investigation of these steric factors, more dyad molecules with distinct geometries are to be prepared and investigated.
Conclusions
In conclusion, we have synthesized and characterized a conformationally rigid porphyrin-anthracene dyad that exhibits strong delayed fluorescence and phosphorescence in solution. Using broadband transient absorption spectroscopy, we have elucidated the formation of anthracene-localized triplet states via intramolecular energy transfer from the porphyrin core with an inverse rate constant of 14 ps and its further transformation into the porphyrin singlet state at μs – timescale. Excited state dynamics along with other spectroscopy data suggest that the porphyrin delayed fluorescence arises as a result of intermolecular triplet–triplet annihilation process. Observed properties of the prepared molecule demonstrate an example of singlet and triplet excitation management in donor–acceptor dyad and are of interest with respect to photon energy conversion systems.Experimental
Equipments and methods
Anthracene-1,4-dione was prepared according to published procedure.36 NMR spectra were recorded with the solvent proton or carbon signal as an internal standard. Emission quantum yields of the porphyrin compounds were measured relative to the fluorescence of free-base tetraphenylporphyrin (ϕf = 0.11)37 in deoxygenated toluene. For quantum yields measurements, sample concentrations were chosen to obtain an absorbance of 0.03–0.07 at the excitation wavelength, at least three measurements were performed for each sample. Emission of the diluted (1 × 10−6 M) rubrene solution in toluene under 542 nm excitation was used as a reference for UC quantum yield calculations. UC measurements were performed using previously described set-up.38 All the measurements were performed using deoxygenated samples prepared in glovebox (1 ppm oxygen).Transient absorption measurements were performed with a home-built pump–probe setup. For measurements in the time range of 1–3 ns with a resolution of ∼100 fs the output of a commercial titanium:sapphire amplifier (3.5 mJ, 1 kHz, 100 fs) was split with one portion used to generate a 400 nm excitation pulse (second harmonic generation in a BBO crystal cut at theta = 29.2°) and another used to generate a white light probe using a home-built two-stage broadband (480–850 nm) non-collinear optical parametric amplifier for white light generation and amplification in the visible spectral range. The variable delay of up to ∼3 ns between pump and probe was introduced by a broadband retroreflector mounted on a mechanical delay stage. Only reflective optics were used to guide the probe beam to the sample to minimize chirp. The excitation pulse was chopped at 500 Hz, while the white light pulses were dispersed onto a linear photodiode array which was read out at 1 kHz. Adjacent diode readings corresponding to the transmission of the sample after an excitation pulse and without an excitation pulse were used to calculate ΔT/T.
Preparation and characterization of the compounds
ε): 391 (3.67), 371 (3.77), 352 (3.62), 337 (3.35). Anal. Calcd for C22H20O2: C, 83.51; H, 6.37. Found: C, 83.59; H, 6.61.
:1) mixture (300 mL), and a suspension of Oxone (39.3 g, 64 mmol) in water (100 mL) was added under vigorous stirring. The mixture was stirred at room temperature for 3 days, diluted with water (200 mL), and extracted with CH2Cl2. The combined organic layers were dried with Na2SO4 and evaporated to dryness. Solid residue was recrystallized from ethanol. 4: 13.41 g (85%), yellow semisolid froth. 1H NMR (250 MHz, CD2Cl2) δ 8.69 (d, J = 5.0 Hz, 2H), 8.08 (dt, J = 6.0, 3.5 Hz, 2H), 7.93–7.80 (m, 2H), 7.61–7.41 (m, 5H), 4.36–4.29 (m, 1H), 4.23 (dd, J = 4.8, 2.9 Hz, 1H), 4.10 (s, 3H), 3.99 (s, 3H), 3.82 (dd, J = 5.3, 2.9 Hz, 1H), 3.64 (dd, J = 5.6, 1.8 Hz, 1H), 2.43 (m, 1H), 2.13–2.00 (m, 1H), 1.79–1.59 (m, 1H), 1.56–1.43 (m, 1H). 13C NMR (63 MHz, CD2Cl2) δ 147.8, 147.3, 138.3, 134.5, 132.1, 131.9, 129.6, 129.6, 128.9, 128.9, 127.5, 127.4, 127.2, 126.3, 126.3, 126.0, 122.0, 121.8, 72.8, 63.6, 63.4, 57.0, 37.0, 29.3, 27.3, 18.7. Anal. Calcd for C28H25ClO4S: C, 68.21; H, 5.11. Found: C, 68.63; H, 5.55.
ε): 394 (5.3), 497 (4.37), 528 (4.1), 566 (3.96), 619 (3.45). HRMS (ESI-TOF): m/z found 1463.5874, calcd for [M+] C100H78N4O8 1463.5853.
ε): 397 (5.3), 512 (4.26), 542 (4.68). HRMS (ESI-TOF): m/z found 1567.4713, calcd for [M+] C100H76N4O8Pd 1567.4731.
Acknowledgements
M. Filatov acknowledges the European Commission for Marie Curie Individual Fellowship (CONSORT, Grant No. 655142). S. Baluschev acknowledges the financial support from the Bulgarian National Science Fund (DFNI E 02/11 – SunStore-project “Molecular solar thermal systems, enhanced by annihilation upconversion”) and from the FCFP FRIAS COFUND Fellowship Programme (FP7-MCA-609305). F. Laquai thanks the Max Planck Society for funding the Max Planck Research Group. F. Etzold acknowledges financial support in the framework of the SPP1355 “Elementary Processes in Organic Photovoltaics”. D.W. Gehrig thanks the Fonds der Chemischen Industrie (FCI) for a Kekulé scholarship.Notes and references
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS PubMed; H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef PubMed; P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887 CrossRef PubMed; F. N. Castellano, Acc. Chem. Res., 2015, 48, 828 CrossRef PubMed; M. Filatov, S. Ritz, I. Ilieva, V. Mailänder, K. Landfester and S. Baluschev, SPIE Newsroom. DOI: DOI:10.1117/2.1201403.005378. Published Online: 7 April 2014; S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef.
- J. Zhao, W. Wu, J. Suna and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC; S. Guo, W. Wu, H. Guo and J. Zhao, J. Org. Chem., 2012, 77, 3933 CrossRef CAS PubMed; X. Cui, J. Zhao, Z. Lou, S. Li, H. Wu and K. Han, J. Org. Chem., 2015, 80, 568 CrossRef PubMed; D. Huang, J. Sun, L. Ma, C. Zhang and J. Zhao, Photochem. Photobiol. Sci., 2013, 12, 872 Search PubMed; P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847 RSC; S. Y. Takizawa, K. Shimada, Y. Sato and S. Murata, Inorg. Chem., 2014, 53, 2983 CrossRef PubMed; T. Takahashi, K. Shizu, T. Yasuda, K. Togashi and C. Adachi, Sci. Technol. Adv. Mater., 2014, 15, 034202 CrossRef.
- A. Harriman, M. Hissler, A. Khatyr and R. Ziessel, Chem. Commun., 1999, 735 RSC; N. D. McClenaghan, F. Barigelletti, B. Maubert and S. Campagna, Chem. Commun., 2002, 602 RSC; D. S. Tyson, J. Bialecki and F. N. Castellano, Chem. Commun., 2000, 2355 RSC; A. F. Morales, G. Accorsi, N. Armaroli, F. Barigelletti, S. J. A. Pope and M. D. Ward, Inorg. Chem., 2002, 41, 6711 CrossRef CAS PubMed; B. Maubert, N. D. McClenaghan, M. T. Indelli and S. Campagna, J. Phys. Chem. A, 2003, 107, 447 CrossRef; D. S. Tyson and F. N. Castellano, J. Phys. Chem. A, 1999, 103, 10955 CrossRef; S. G. Camera and H. E. Toma, J. Photochem. Photobiol., A, 2002, 151, 57 CrossRef.
- P. Duan, N. Yanai and N. Kimizuka, Chem. Commun., 2014, 50, 13111 RSC; J. Peng, X. Jiang, X. Guo, D. Zhao and Y. Ma, Chem. Commun., 2014, 50, 7828 RSC.
- A.-M. Stadler, F. Puntoriero, F. Nastasi, S. Campagna and J.-M. Lehn, Chem. – Eur. J., 2010, 16, 5645 CrossRef CAS PubMed; F. Laquai, Y.-S. Park, J.-J. Kim and T. Basche, Macromol. Rapid Commun., 2009, 30, 1203 CrossRef PubMed; B. Geiß and C. Lambert, Chem. Commun., 2009, 1670 RSC; M. O. Senge, M. Fazekas, M. Pintea, M. Zawadzka and W. J. Blau, Eur. J. Org. Chem., 2011, 5797 CrossRef PubMed.
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568 RSC; Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007 RSC; O. S. Finikova, P. Chen, Z. Ou, K. M. Kadish and S. A. Vinogradov, J. Photochem. Photobiol., A, 2008, 198, 75 CrossRef CAS PubMed.
- Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC; C. Wohnhaas, V. Mailänder, M. Droge, M. A. Filatov, D. Busko, Y. Avlasevich, S. Baluschev, T. Miteva, K. Landfester and A. Turshatov, Macromol. Biosci., 2013, 13, 1422 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395 CrossRef CAS PubMed.
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103 Search PubMed; V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345 RSC; T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef CAS PubMed.
- D. V. Kozlov and F. N. Castellano, Chem. Commun., 2004, 2860 RSC; G. Bergamini, P. Ceroni, P. Fabbrizi and S. Cicchi, Chem. Commun., 2011, 47, 12780 RSC.
- A. Ambroise, J. Li, L. Yu and J. S. Lindsey, Org. Lett., 2000, 2, 2563 CrossRef CAS; E. K. L. Yeow, P. J. Sintic, N. M. Cabral, J. N. H. Reek, M. J. Crossley and K. P. Ghiggino, Phys. Chem. Chem. Phys., 2000, 2, 4281 RSC; A. Osuka, J.-Y. Shin, R. Yoneshima, H. Shiratori, T. Ohno, K. Nozaki, Y. Nishimura, I. Yamazaki, S. Taniguchi, T. Shimizu and T. Okada, J. Porphyrins Phthalocyanines, 1999, 3, 729 CrossRef; P. Brodard, S. Matzinger, E. Vauthey, O. Mongin, C. Papamicaeel and A. Gossauer, J. Phys. Chem. A, 1999, 103, 5858 CrossRef; L. Flamigni, F. Barigelletti, N. Armaroli, B. Ventura, J.-P. Collin, J.-P. Sauvage and J. A. G. Williams, Inorg. Chem., 1999, 38, 661 CrossRef; K. Kils, J. Kajanus, J. Mùrtensson and B. Albinsson, J. Phys. Chem. B, 1999, 103, 7329 CrossRef.
- S. Kawabata, I. Yamazaki and Y. Nishimura, Bull. Chem. Soc. Jpn., 1997, 70, 1125 CrossRef CAS; R. Hermant, P. A. Liddell, S. Lin, R. G. Alden, H. K. Kang, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1993, 115, 2080 CrossRef; A. Osuka, H. Yamada, K. Maruyama, N. Mataga, T. Asahi, M. T. Ohkouchi, T. Okada, I. Yamazaki and Y. Nishimura, J. Am. Chem. Soc., 1993, 115, 9439 CrossRef; J. S. Lindsey, P. A. Brown and D. A. Siesel, Tetrahedron, 1989, 45, 4845 CrossRef; F. Effenberger, H. Schlosser, P. Bäuerle, S. Maier, H. Port and H. C. Wolf, Angew. Chem., Int. Ed. Engl., 1988, 27, 281 CrossRef PubMed; J. D. Megiatto Jr., D. I. Schuster, S. Abwandner, G. de Miguel and D. M. Guldi, J. Am. Chem. Soc., 2010, 132, 3847 CrossRef PubMed.
- I. Andreu, I. Morera, F. Palumbo, G. Sastre, F. Boscá and M. Miranda, Chem. Sci., 2015, 6, 4035 RSC.
- L. Giribabu, A. Ashok Kumar, V. Neeraja and B. G. Maiya, Angew. Chem., Int. Ed., 2001, 40, 3733 CrossRef.
- M. O. Senge, Chem. Commun., 2006, 243 RSC.
- M. Sirish and B. G. Maiya, J. Porphyrins Phthalocyanines, 1998, 2, 237 CrossRef CAS; M. Sirish and B. G. Maiya, J. Photochem. Photobiol., A, 1995, 85, 127 CrossRef; J. Seixas de Melo, A. J. F. N. Sobral, A. M. Rocha Gonsalves and H. D. Burrows, J. Photochem. Photobiol., A, 2005, 172, 151 CrossRef PubMed.
- M. O. Senge, Chem. Commun., 2011, 47, 1943 RSC; P. D. Harvey, M. A. Filatov and R. Guilard, J. Porphyrins Phthalocyanines, 2011, 15, 1150 CrossRef CAS; P. D. Harvey, A. Langlois, M. Filatov, D. Fortin, K. Ohkubo, S. Fukuzumi and R. Guilard, J. Porphyrins Phthalocyanines, 2012, 16, 685 CrossRef.
- R. E. Haddad, S. G. Azeau, J. Pecaut, J.-C. Marchon, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2003, 125, 1253 CrossRef CAS PubMed; C. Muzzi, C. J. Medforth, K. M. Smith, S.-L. Jia and J. A. Shelnutt, Chem. Commun., 2000, 131 RSC; J. A. Shelnutt, in The PorphyrinHandbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, Burlington, MA, 2000, vol. 7, ch. 50, p. 167 Search PubMed.
- L. Noss, P. A. Liddell, A. L. Moore, T. A. Moore and D. Gust, J. Phys. Chem. B, 1997, 101, 458 CrossRef CAS; A. Rosa, G. Ricciardi and E. J. Baerends, J. Phys. Chem. A, 2006, 110, 5180 CrossRef PubMed; A. Y. Lebedev, M. A. Filatov, A. V. Cheprakov and S. A. Vinogradov, J. Phys. Chem. A, 2008, 112, 7723 CrossRef PubMed.
- S. Ito, H. Uno, T. Murashima and N. Ono, Chem. Commun., 1999, 2275 RSC.
- H. Uno, H. Watanabe, Y. Yamashita and N. Ono, Org. Biomol. Chem., 2005, 3, 448 CAS.
- G. D. Scholes, K. P. Ghiggino, A. M. Oliver and M. N. Paddon-Row, J. Am. Chem. Soc., 1993, 115, 4345 CrossRef CAS; S. Plunkett, K. J. Flanagan, B. Twamley and M. O. Senge, Organometallics, 2015, 34, 1408 CrossRef.
- J. Sanning, P. R. Ewen, L. Stegemann, J. Schmidt, C. G. Daniliuc, T. Koch, N. L. Doltsinis, D. Wegner and C. A. Strassert, Angew. Chem., Int. Ed., 2015, 54, 786 CrossRef CAS PubMed.
- N. Ono, H. Kawamura, M. Bougauchi and K. Maruyama, Tetrahedron, 1990, 46, 7483 CrossRef CAS.
- O. S. Finikova, S. Y. Chernov, A. V. Cheprakov, M. A. Filatov, S. A. Vinogradov and I. P. Beletskaya, Dokl. Chem., 2003, 391, 222 CrossRef CAS; M. A. Filatov, A. Y. Lebedev, S. A. Vinogradov and A. V. Cheprakov, J. Org. Chem., 2008, 73, 4175 CrossRef PubMed; A. V. Cheprakov and M. A. Filatov, J. Porphyrins Phthalocyanines, 2009, 13, 291 CrossRef.
- S. Ito, N. Ochi, T. Murashima, N. Ono and H. Uno, Chem. Commun., 2000, 893 RSC; T. Okujima, T. Kikkawa, H. Nakano, H. Kubota, N. Fukugami, N. Ono, H. Yamada and H. Uno, Chem. – Eur. J., 2012, 18, 12854 CrossRef CAS PubMed; Y. Tomimori, T. Okujima, T. Yano, S. Mori, N. Ono, H. Yamada and H. Uno, Tetrahedron, 2011, 67, 3187 CrossRef PubMed; H. Uno, H. Uoyama, C. Chenxin, H. Tahara, Y. Shimizu, H. Hagiwara, Y. Hanasaki, H. Yamada and T. Okujima, Heterocycles, 2010, 80 Search PubMed; M. A. Filatov, E. Heinrich, K. Landfester and S. Baluschev, Org. Biomol. Chem., 2015, 13, 6977 Search PubMed.
- M. A. Filatov, S. Baluschev, I. Z. Ilieva, V. Enkelmann, T. Miteva, K. Landfester, S. E. Aleshchenkov and A. V. Cheprakov, J. Org. Chem., 2012, 77, 11119 CrossRef CAS PubMed; M. A. Filatov, E. Heinrich, D. Busko, I. Z. Ilieva, K. Landfester and S. Baluschev, Phys. Chem. Chem. Phys., 2015, 17, 6501 RSC.
- T. Seko, K. Ogura, Y. Kawakami, H. Sugino, H. Toyotama and J. Tanaka, Chem. Phys. Lett., 1998, 291, 438 CrossRef CAS.
- M. Ezoe, T. Minami, Y. Ogawa, S. Yagi, H. Nakazumi, T. Matsuyama, K. Wada and H. Horinaka, Photochem. Photobiol. Sci., 2005, 4, 641–646 Search PubMed; E. J. Shin, Bull. Korean Chem. Soc., 2006, 27, 751 CrossRef CAS; L. Giribabu and B. G. Maiya, Res. Chem. Intermed., 1999, 25, 769 CrossRef PubMed; L.-P. Long, M.-X. You, H. Wang, Y.-X. Wang and R.-H. Yang, Sci. China Ser. B: Chem., 2009, 52, 793 CrossRef.
- Y. Y. Cheng, T. Khoury, R. G. Clady, M. J. Tayebjee, N. J. Ekins-Daukes, M. J. Crossley and T. W. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 66 RSC.
- D. Madge, M. W. Windsor, D. Holten and M. Gouterman, Chem. Phys. Lett., 1974, 29, 183 CrossRef.
- J. B. Birks, Photophysics of Organic Molecules, Wiley, New York, 1970, p. 372 Search PubMed.
- A. Turshatov and S. Baluschev, Triplet–Triplet Annihilation-Assisted Upconversion: All-Optical Tools for Probing the Physical Parameter of Soft Matter Handbook of Coherent-Domain Optical Methods, Springer Science + Business Media, New York, 2013, ISBN-13: 978-1461451754 Search PubMed.
- T. Miteva, G. Nelles, A. Yasuda, S. Balouchev and J. Lupton, U.S. Pat, 7517144B2, Apr. 14, 2009 RSC; G. A. Baker, S. N. Baker and T. M. McCleskey, Chem. Commun., 2003, 2932 RSC; D. N. Messias, M. V. D. Vermelho, A. S. Gouveia-Neto and J. S. Aitchison, Rev. Sci. Instrum., 2002, 73, 476 CrossRef CAS PubMed.
- G. B. Piland, J. J. Burdett, D. Kurunthu and C. J. Bardeen, J. Phys. Chem. C, 2013, 117, 1224 CAS.
- D. H. Hua, M. Tamura, X. Huang, H. A. Stephany, B. A. Helfrich, E. M. Perchellet, B. J. Sperfslage, J.-P. Perchellet, S. Jiang, D. E. Kyle and P. K. Chiang, J. Org. Chem., 2002, 67, 2907 CrossRef CAS PubMed.
- P. G. Seybold and M. Gouterman, J. Mol. Spectrosc., 1969, 31, 1 CrossRef CAS.
- A. Turshatov, D. Busko, Y. Avlasevich, T. Miteva, K. Landfester and S. Baluschev, ChemPhysChem, 2012, 13, 3112 CrossRef CAS PubMed.
- P. B. Hopkins and P. L. Fuchs, J. Org. Chem., 1978, 43, 1208 CrossRef CAS.
- O. S. Finikova, A. V. Cheprakov, P. J. Carroll and S. A. Vinogradov, J. Org. Chem., 2003, 68, 7517 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Optical spectra, NMR and mass-spectra of newly synthesized compounds. See DOI: 10.1039/c5dt03784k |
| ‡ Current address: Trinity Biomedical Science Institute, Trinity College Dublin, 152-160 Pearse Street, Dublin 2, Ireland. |
| This journal is © The Royal Society of Chemistry 2015 |