
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface redox chemistry and mechanochemistry of insulating polystyrene nanospheres†
Thomas S.
Varley
,
Martin
Rosillo-Lopez
,
Sandeep
Sehmi
,
Nathan
Hollingsworth
and
Katherine B.
Holt
*
Department of Chemistry, University College London, 20, Gordon St., London WC1H 0AJ, UK. E-mail: k.b.holt@ucl.ac.uk
First published on 26th November 2014
Abstract
Cyclic voltammetry (CV) of polystyrene nanospheres was carried out after immobilisation onto boron-doped diamond electrodes. Although the polystyrene is insulating, a voltammetric response was obtained. This was attributed to the high surface area of the nanospheres, allowing the redox chemistry of the polystyrene surface to be probed despite the non-conducting nature of the bulk. The polystyrene redox response was found to be strongly dependent on prior mechanical agitation. Centrifuged, sonicated and vortexed polystyrene nanospheres all exhibited significantly higher oxidation currents than the non-agitated polystyrene. Mechanical treatment by sonication and centrifugation was found to bring about changes to surface chemistry of the polystyrene spheres, in particular the introduction of oxygen functionalities. For these samples the CV response is attributed to the presence of surface phenol functionalities. On the non-agitated and vortex treated polystyrene surfaces X-ray photoelectron spectroscopy revealed an absence of oxygen functionalities that could explain the redox response. Repetition of the CV experiment in the presence of a solution spin trap suggests that radical species play a role in the observed response. For the vortexed sample the increased oxidation currents were attributed to significant surface roughening and deformation, as revealed by Transmission Electron Microscopy.
1. Introduction
Non-conducting materials, such as wide band-gap polymers, are not typically associated with measurable electrochemistry. However, surface redox chemistry of insulating materials attributed to defect sites, dangling bonds and band-gap states may give rise to interesting electrochemistry. For nanoparticulate materials the large surface atom to bulk atom ratio allows the redox chemistry of surface states and functional groups to dominate over the insulating response of the bulk, enabling a voltammetric response to be obtained. We have previously reported this to be the case for insulating diamond nanoparticles, where surface states within the band gap are able to exchange electrons with their surroundings (solution redox probes, an underlying electrode).1 In this study we report the cyclic voltammetry (CV) response of electrode-immobilised polystyrene (PS) nanospheres (Scheme 1). We show here that, despite their insulating nature, PS nanobeads exhibit a redox response when immobilised on a boron-doped diamond (BDD) electrode. In addition, we subject the PS nanospheres to different forms of mechanical agitation and activation, namely centrifugation, sonication and vortex mixing, prior to immobilisation onto the electrode. We find that the redox response of the PS is strongly influenced by the nature of the previous mechanical treatment, indicating that mechanically-induced changes to the PS surface may be detected and quantified electrochemically.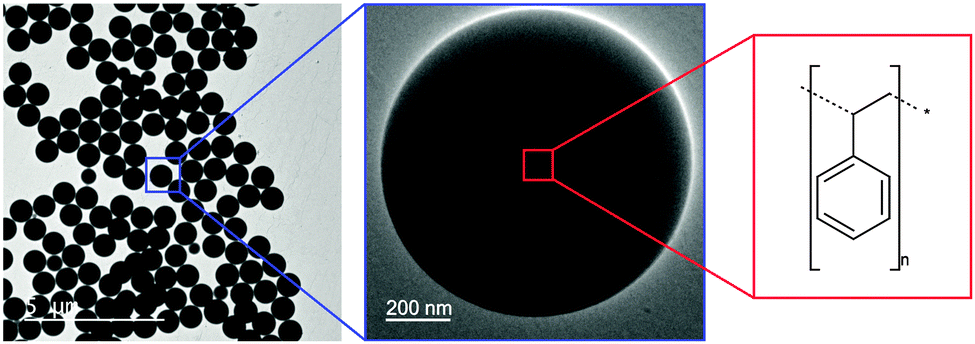 | ||
| Scheme 1 Monodisperse 800 nm diameter PS nanospheres used in this study. | ||
The effect of mechanical force upon the surface chemistry of insulating polymers has been previously studied with regards to understanding mechanical degradation mechanisms2 and the ‘mechanochemistry’ related to surface radical formation on polymers.3 Surface radicals (mechanoradicals) are formed via homolytic bond cleavage of the polymer chain under mechanical strain.3,4 Although polymer covalent bonds are very strong (typical C–C bond strength is ca. 350 kJ mol−1) experimental results from Electron Spin Resonance techniques show that these radicals can be formed as a result of mechanical work upon the surface and persist for some period of time.4 There is substantial technological relevance to studying this chemistry, as mastication (mechanical breakdown of polymers to achieve desired properties), recycling by pulverisation and lubrication processes can all generate mechanoradicals.5
Polymer mechanochemistry, well-established in the tribology and polymer literature, has recently been re-examined by researchers engaged in study of electrostatic charge accumulation.6–11 Electrostatic charging (tribocharging, contact electrification) is the separation of charge that occurs when two surfaces are contacted together and then separated.6 Although an everyday phenomenon that has been well studied, a unified theory explaining the mechanism of charge accumulation by wide band-gap polymers has not yet been achieved. The exact nature of the charge is not firmly established, with both electrons7 and ions8 suggested as charge carriers. Transfer of material fragments from one surface to another resulting in both positively and negatively charged ‘mosaics’ has also been observed.9 Regardless of the nature of the charge, the role of surface radicals has been shown to be important,10 with recent studies showing charge and radical co-localisation on polymer surfaces after contact.11 Moreover, treatment of a polymer surface with a radical spin trap molecule resulted in no charge accumulation, confirming the co-dependence of the two.11
Our primary motivation in this study was to establish whether surface redox chemistry for otherwise insulating materials could be observed using voltammetry, through the use of high surface area nanomaterials. However, given the proposed importance of surface radicals in charge accumulation and electrostatic charging10,11 we additionally probed the redox response of PS nanospheres after surface activation by mechanical means, to determine whether surface mechanoradicals or other reactive species thus formed showed different redox activity that could be used in their characterisation.
2. Experimental methods
Preparation of PS nanosphere suspensions
Sulfate-terminated PS beads of diameter 800 nm were purchased from Sigma-Aldrich (LB7, 10% solids in aqueous suspension). The suspension also contains 0.1–0.5% sodium dodecyl sulfate surfactant and 0.2% inorganic salts (potassium sulfate and sodium bicarbonate). To remove these extraneous agents the PS beads were cleaned by membrane dialysis against water. The membrane was a pre-treated standard grade regenerated cellulose dialysis membrane (Spectrum Laboratories), with a molecular weight cut off of 3500 Da and a flat width of 45 mm. Membranes were received in an aqueous solution containing sodium azide preservative (0.05%), which was removed by dialysis against distilled water for 2 h. Dialysis of the PS solution (diluted tenfold from the commercial stock solution) was subsequently performed against distilled water for 48 h, changing the water every 12 h. The resulting cleaned suspension was 1% PS solids in deionised water and was stored in a glass container throughout. Alternatively the PS nanospheres were separated from their native solution by centrifugation (with three cleaning and rinsing steps) followed by re-suspension in deionised water to give a final solution of 1% PS solids. These samples were both centrifuged and stored in polypropylene centrifuge tubes (Eppendorf 1.5 mL Flex tube microcentrifuge tubes); according to the manufacturers these tubes are “manufactured without the use of slip agents, plasticizers and biocides-substances that have been shown to leach from plastic consumables into the sample and negatively affect bio-assay results”.12For some experiments the PS suspensions were subjected to mechanical agitation, either by sonication for 1 hour in an ultrasonic bath or by vortex mixing. Sonication was performed using a Fisher Scientific Ultrasonic bath (FB 15049) filled with deionised water. PS suspensions were held in place via a clamp and positioned such that as much of the polypropylene centrifuge tube was submerged as possible. PS solutions were vortex mixed within their polypropylene centrifuge tubes using a Vortex Genie 2 (Scientific Industries Bohemia, N.Y., USA) on setting 3. Control experiments were also carried out by vortexing samples in a glass container, to ensure that material transfer from the plastic container walls was not responsible for the redox activity (see ESI†).
To further ensure that plasticizers or impurities did not leach from container walls and contaminate the suspension, the composition of the aqueous supernatant after storage and mechanical treatment was determined by Nuclear Magnetic Resonance (NMR). The PS nanosphere surface chemistry and morphology were also investigated, before and after mechanical treatment, using Transmission Electron Microscopy (TEM) and X-ray Photoelectron Spectroscopy (XPS). See ESI† for details.
Preparation and electrochemistry of PS-modified electrodes
Prior to modification with PS, the boron-doped diamond (BDD) working electrode was polished with 0.05 μm alumina slurry, rinsed and cycled repeatedly between 0 and 1 V vs. Ag/AgCl in the relevant buffer solution until a stable background was achieved. The PS suspensions were drop-coated onto a boron-doped diamond (BDD) electrode to form an adherent film with average coverage (5 ± 1) × 105 beads mm−2. Reproducibility of surface coverage of PS was evaluated by inspection of each modified electrode under an optical microscope and analysis of the resulting image using imaging software (see ESI† for details). The surface coverage of PS was the same regardless of suspension preparation method. CV experiments with the electrode-immobilised PS was carried out with a standard three electrode cell in 0.2 M phosphate buffer solutions (PBS) containing different proportions of K2HPO4 and KH2PO4 to adjust the pH. All solutions were made up with 18 MΩ cm deionised water. Spin trap experiments were performed using the same three electrode cell, but using 0.2 M PBS containing 10 μM N-tert-butyl-α-phenylnitrone (PBN) as the electrolyte solution.3. Results
CV response of electrode-immobilised PS nanospheres cleaned by centrifugation
In contrast to the featureless BDD background response, for the PS-modified electrode oxidation currents begin to flow at ca. 0.45 V, rising to a distinct peak centred at ca. 0.7 V (Fig. 1). There is no corresponding reduction peak on the reverse scan, indicating the oxidation process is irreversible. However a small reduction peak at about 0.25 V is noted on the reverse scan and on the second scan a corresponding oxidation peak is observed at 0.28 V. This reversible couple centred at ca. 0.26 V is stable to repeated cycling and results from the oxidation process at 0.7 V (Fig. 1 inset, ESI†). The peak currents exhibit a proportional relationship with voltammetric scan rate, confirming a surface-confined species. On the second and subsequent scans the oxidation peak at 0.7 V is no longer present, indicating that the species responsible is not regenerated following its oxidation during the first cycle. The reproducibility of the CV response was determined by evaluating the charge passed under the 0.7 V and 0.28 V oxidation peaks for ten repetitions with a freshly modified electrode (ESI†). It was determined that (28 ± 5) nC of charge are passed during the oxidation process centred at 0.7 V, while (2.5 ± 0.5) nC is passed during the oxidation process at 0.28 V. Inspection of the electrode under a microscope showed that coverage of PS was unchanged after the CV scans had been carried out. This observation indicates that PS remains immobilised on the electrode throughout the experiment. | ||
| Fig. 1 CV in 0.2 M pH 6 PBS of centrifuged PS immobilised on BDD electrode. Red: 1st scan; blue: 2nd scan; black: background response of unmodified BDD. Scan rate 0.02 V s−1. | ||
Effect of mechanical agitation on voltammetric response of PS nanospheres
When the PS nanospheres were cleaned by dialysis, rather than centrifugation, the form of the CV was unchanged from that in Fig. 1, but the magnitude of the oxidation currents was noticeably decreased, with charge passed under the 0.7 V oxidation peak being (13 ± 3) nC. Larger currents for the centrifuged beads could be attributed to mechanical ‘activation’ of the PS surface by shear forces. To investigate the effect of mechanical force on the beads, the dialysed suspension was subject to deliberate mechanical agitation, either by sonication or vortex mixing, prior to immobilisation onto the electrode. The form of the resulting CV for both methods was similar to Fig. 1 but higher oxidation currents were observed. Fig. 2a shows the mean charge obtained from integration of the 0.7 V oxidation peak for all samples, where a statistically significant difference between the data sets can be observed. In particular the vortexed PS shows greatly enhanced oxidation response, approximately ten times that of the dialysed samples. | ||
| Fig. 2 (a) Average charge passed under 0.7 V oxidation peak for different PS preparation methods; (b) time dependence of charge passed under 0.7 V oxidation peak for vortexed PS; (c) CVs in 0.2 M pH 6 PBS (scan rate 0.02 V s−1) comparing response of electrode-immobilised PS after prior sonication in air-saturated (black) and deoxygenated (red) solutions. | ||
Time and oxygen dependence of voltammetric response for mechanically agitated PS
The time-dependence of the enhanced redox response was investigated over a period of 14 days. The PS sample was vortex mixed and every day thereafter three repeat CVs were carried out with the same sample. The charge for the 0.7 V oxidation peak remained significantly higher than for the dialysed, centrifuged and sonicated beads, indicating a permanent change to the PS surface (Fig. 2b). Similarly PS suspensions having undergone centrifugation or sonication maintained their enhanced oxidation currents compared to the dialysed samples, although some decline in electrochemical activity was noted over a period of several months.The effect of dissolved oxygen present during the mechanical agitation process was explored by sonicating a deoxygenated PS suspension (previously cleaned by dialysis) prior to drop-coating onto the electrode. Sonication in the absence of oxygen gives rise to two peaks associated with oxidation of the PS layer, at ca. 0.63 V and ca. 0.77 V, in contrast to the single peak at 0.7 V observed after sonication in the presence of oxygen (Fig. 2c). Additionally the reduction peak at ca. 0.2 V on the reverse peak is much less prominent for the PS sonicated in the deoxygenated solution.
XPS analysis of the PS surface chemistry after mechanical treatment
The carbon 1s region spectrum for the dialysed PS (Fig. 3a) can be fit with three peaks, corresponding to sp3 carbon–carbon bonding of the backbone (284.9 eV) and sp2 carbon–carbon bonding of the phenyl ring (284.3 eV and 291.1 eV).13 There are no obvious peaks in the region 296–289 eV that could be attributed to carbon bonded to oxygen.14 Oxygen is present on the surface of the PS, as indicated by the oxygen 1s spectrum (ESI†) but this can be attributed to the sulfate terminating groups.15 The centrifuged PS C 1s spectrum (Fig. 3b) can be fit with an additional two peaks: at 286.6 eV for carbon bound to oxygen via a single bond and at 288.8 eV corresponding to carbon bound to more than one oxygen (or engaged in double bonding with one oxygen).14 The sonicated PS (Fig. 3c) shows a greater degree of oxidation than the centrifuged sample, with more prominent peaks observed in the regions for C–O (286.6 eV) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.8 eV) bonding. In contrast, the vortexed PS (Fig. 3d) shows similar oxygen content to the dialysed PS, with no evidence from the C 1s spectrum that carbon is engaged in bonding with oxygen.
O (288.8 eV) bonding. In contrast, the vortexed PS (Fig. 3d) shows similar oxygen content to the dialysed PS, with no evidence from the C 1s spectrum that carbon is engaged in bonding with oxygen.
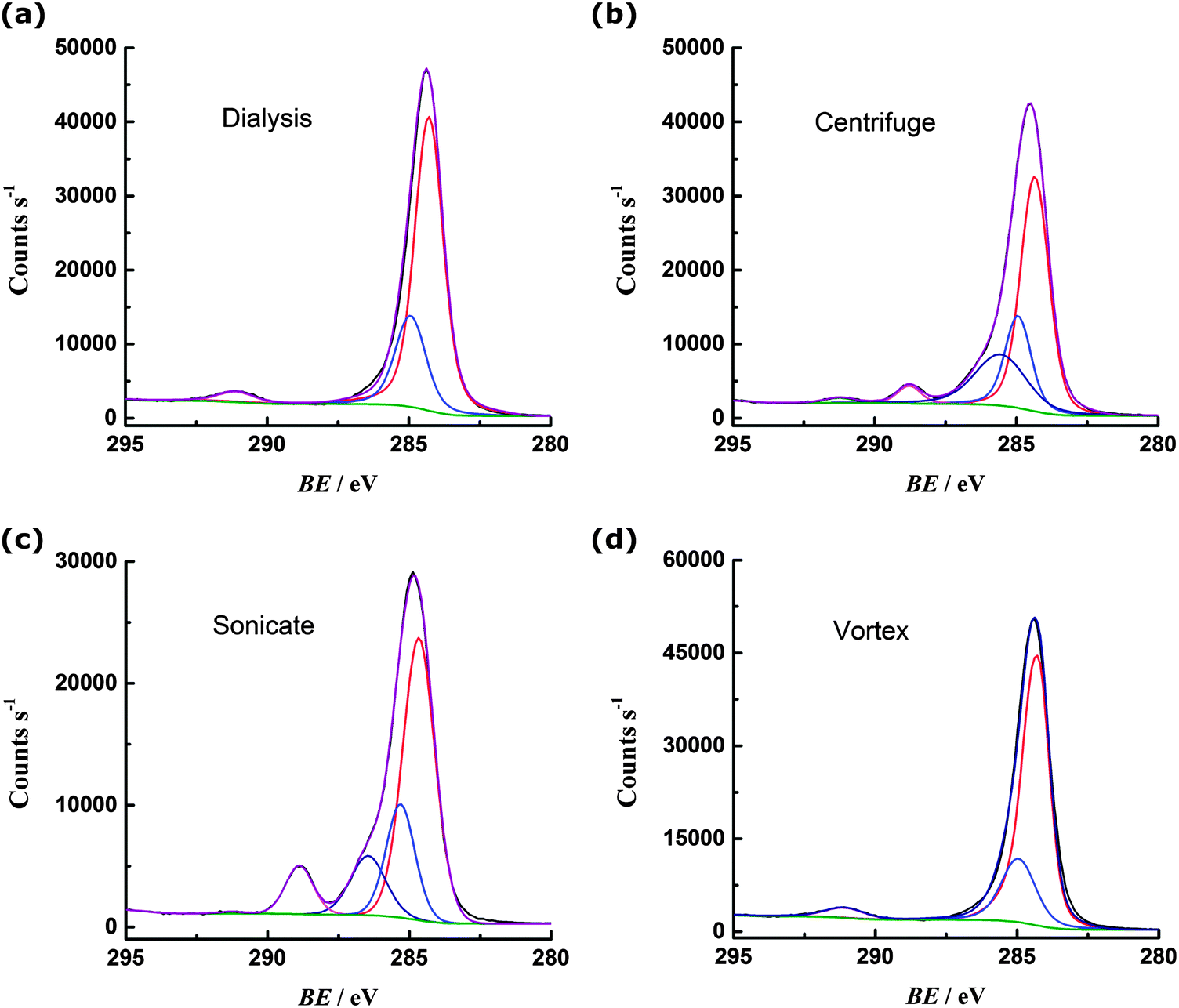 | ||
| Fig. 3 Carbon 1s region XPS spectra for PS (a) cleaned by dialysis; (b) cleaned by centrifugation; (c) subject to 1 hour sonication; (d) subject to 1 hour vortex mixing. | ||
Additional information about the nature of the carbon–oxygen bonding is obtained from the valence band XPS spectra for the samples (Fig. 4).14,16 An increase in intensity of the peak(s) at ca. 25 eV is noted for the centrifuged and sonicated samples, concomitant with a decrease in intensity of the peaks at 17 and 9 eV. The peaks at 25 eV are attributed to electrons with oxygen 2p character, i.e. originating from O(2p)–H(1s) and O(2p)–C(2p) molecular orbitals.14 The presence of these peaks for the centrifuged and sonicated PS is consistent with the carbon–oxygen bonding in the core level spectra and increased oxygen content for these samples. Valence band peaks at 17 and 9 eV are assigned to electrons of C 2s character, associated with the phenyl ring of PS.13 The loss of intensity of these peaks for the oxidised sonicated and centrifuged samples therefore indicates a change in carbon bonding in the phenyl ring, perhaps through insertion of singly or doubly bonded oxygen. The sample found to contain the highest oxygen content was PS sonicated in a deoxygenated solution. As noted above, the electrochemical response was different for this material also; however the increased oxygen content is somewhat surprising given the lack of dissolved oxygen available to react with the PS surface.
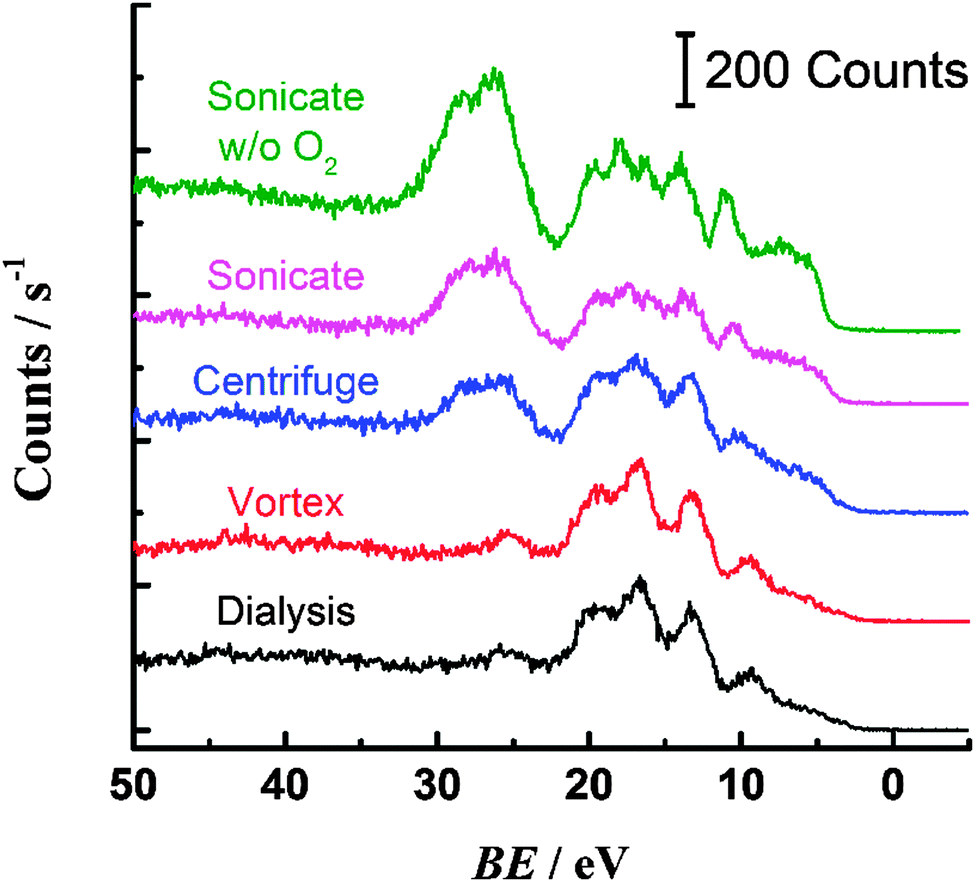 | ||
| Fig. 4 Valence band XPS spectra for polystyrene cleaned by dialysis (black); cleaned by centrifugation (blue); subject to 1 hour sonication in air-saturated solution (pink); subject to 1 hour sonication in deoxygenated solution (green) and subject to 1 hour vortex mixing (red). | ||
TEM images of the PS samples after mechanical treatment
Dialysed, sonicated and vortexed PS nanospheres were examined with TEM to observe changes in morphology associated with the mechanical treatment. Fig. 5a shows the dialysed PS beads are smooth and spherical with no evidence of surface deformation. The sonicated PS beads (Fig. 5b) are still spherical but the surface is roughened and black patches of transferred material are observable on the surface. In the regions where the sonicated beads touch, ‘strings’ of material can be seen extending between the beads, where material is shared. Under the heating effect of the TEM beam, occasionally the contacted beads are seen to move apart and as they do so they leave irregular dark ‘scars’ on the surface where the beads were touching and the surface has been roughened and material transferred (see ESI†). The dark patches on the beads have therefore been attributed to prior collisions between these beads during the sonication treatment. | ||
| Fig. 5 TEM images of (a) dialysed PS; (b) PS sonicated for 1 hour; (c) PS vortexed for 1 hour. | ||
Vortex treatment leads to even more dramatic deformation in morphology (Fig. 5c); here thick strings and lumps of polymer material can be seen extending between the beads, almost sticking them together. Many of the beads show significant deformation and loss of their smooth spherical morphology. In addition some amorphous grey matter can be seen occasionally in the images, attributed to polystyrene material that has been pulled loose from the surface of the beads during impact.
Dialysed PS: origin of the redox response
The phenyl rings of PS do not undergo direct oxidation in the potential range of this experiment, as benzene and toluene are oxidised above 2 V vs. Ag/AgCl at BDD.17 The terminating sulphate groups of the dialysed PS beads likewise oxidise at higher potentials (>2.5 V vs. Ag/AgCl).18 Substituted phenols undergo irreversible oxidation at 0.7 V (vs. Ag/AgCl) and above19,20 resulting in generation of redox couples in the region 0.0–0.4 V on subsequent scans. This behaviour is remarkably similar to that observed for the electrode-immobilised PS. However for the dialysed PS the presence of surface phenol moieties is inconsistent with the XPS data, which shows that little if any carbon is engaged in C–O bonding (Fig. 3a and 4); it is therefore important to consider possible origins for voltammetric response of the dialysed polystyrene.While adsorption of impurities onto the electrode, rather than the PS, may be responsible for the voltammetric response, we have taken great precautions to prevent this and have been unable to identify such a contaminant. The dialysed PS is stored exclusively in glass containers and the only brief contact with plastic is with a pipette tip during drop-coating. We ensure that all plastic used in our experiments is free from plasticizers and other possible contaminants (see Experimental). The electrode is carefully cleaned and a background response obtained in the absence of PS before every experiment. The same drop-coating procedure (volume, drying time, environment) is used for all samples and yet we see a significant difference between the data sets (Fig. 2a) so it seems unlikely that an airborne contaminant or a process taking place during deposition is responsible for the response. The supernatant is tested for dissolved redox species using NMR (see ESI†) and we have identified no species that could be responsible for the redox response. There is no detectable solid material other than PS beads observed in multiple TEM images. Other control experiments are reported in the ESI.† There may be an as yet unidentified contamination source that is responsible for the redox peaks, however in its apparent absence other explanations for the redox activity of the immobilised PS layer must be considered.
One explanation for the discrepancy between the observed electrochemistry and the absence of surface functionalities could be the difference in environment between the solution phase electrochemistry experiments and the ultra-high vacuum of the XPS experiments and the reorganisation of the polymer surface in response to the environment. It has been shown that PS surfaces are highly dynamic and reorganise in response to changes in hydrophobicity/hydrophilicity of the interface conditions.21,22 When exposed to water, polar groups are more likely to be pulled from the bulk to the surface to interact with the water and reduce the free energy at the interface.21 Conversely in a hydrophobic environment (e.g. in a nitrogen atmosphere) oxygen-containing polar groups migrate from the surface to the bulk of the polymer.22 If the UHV environment can be considered as hydrophobic, oxygen functionalities, such as phenols, may move into the bulk and so not be so readily detected with XPS. Another explanation could be presence of persistent and long lived radicals on the PS surface, where the unpaired electron is delocalised about the phenyl ring. This species is likely to be amenable to further oxidation at relatively mild potentials with a possible product being a phenolic species. Such a species would not readily be detected with XPS.
An alternate explanation for the observed redox response is that ‘phenol-type’ functionalities are formed on the electrode-immobilised PS surface in situ through reaction with electrogenerated hydroxyl radicals. It is well-established that hydroxyl radicals are generated at BDD electrodes at oxidative potentials as a result of water discharge23 however usually at more positive potentials than used in this study. However it has been suggested that non-diamond inclusions (e.g. sp2 impurities) in BDD films may support their formation within the potential range of this study24 and that electrogenerated OH plays a role in the oxidation mechanism of sulphides and disulphides at BDD at potentials well below that of water discharge.25 The CV of the unmodified BDD (Fig. 1) shows increases in current at potentials of 0.6 V and above, which may indicate water oxidation to form hydroxyl radicals at active sites on the BDD surface. These hydroxyl radicals would then react immediately with the PS immobilised at the electrode interface. The oxidation of benzene to phenol at different OH radical generating metal oxide electrodes has been reported to take place at similar applied potentials to those used in this study.26
To test this proposed mechanism the CV experiments were repeated with addition of 10 μM N-tert-butyl-α-phenylnitrone (PBN) to the solution (Fig. 6a). PBN is a radical spin trap molecule, which competitively reacts with the OH radicals as they are generated27 preventing them from reacting with the PS surface. Without PBN the oxidation peak is observed at ca. 0.7 V, however when the spin trap is present the oxidation currents are significantly diminished. This appears to support a hypothesis that OH radicals generated at the BDD electrode are necessary to observe a redox response for dialysed PS nanospheres and indicates that although PS itself is not redox active within the potential range investigated, redox active phenolic groups may be generated and then detected in situ at the BDD electrode. However it is also possible that addition of PBN has a different effect, for example it may react directly with the surface of the PS if carbon-centred radicals are present. This would prevent their further oxidation and hence suppress the voltammetric peak. Additionally the PBN may adsorb to the surface of the PS, preventing reorganisation and the migration of polar oxygen groups from within the bulk to the surface where they can be oxidised. Although we cannot definitively determine the origin of the redox response of the unmodified PS, the ability of PS spheres to reduce Ag+ ions (and hence themselves undergo oxidation) has previously been reported, although no PS surface functionality was specifically identified as being responsible for the redox activity.28
 | ||
| Fig. 6 CVs of first scan of electrode immobilised PS in 0.2 M pH 6 PBS (0.02 V s−1) in the absence (black) and presence (red) of 10 μM PBN spin trap: (a) dialysed PS; (b) centrifuged (PS); (c) sonicated PS; (d) vortexed PS. | ||
Mechanically agitated PS: origin of redox response
XPS data (Fig. 3 and 4) indicate that sonicated and centrifuged PS are oxidised prior to electrode-immobilisation, hence their redox response can be attributed to the presence of these surface oxygen functionalities. In contrast to the dialysed PS, there is no significant difference in the CV response of the sonicated PS in the presence or absence of PBN (Fig. 6c). Likewise the centrifuged PS shows only a small decrease in current with the spin trap present, although the oxidation peak becomes less well-defined (Fig. 6b). These experiments show that, if the PS surface is already oxidised, the CV response becomes independent of the presence of spin trap. XPS and CV data suggest surface phenol groups as a likely origin of the redox response. Additionally, the XPS data is consistent with the sonicated sample being more oxidised than the centrifuged and hence less sensitive to the presence of spin trap.The vortexed PS showed the most significant increase in oxidation charge, but XPS reveals that the surface oxygen content is similar to the dialysed PS (Fig. 3d) and so the response cannot be attributed to prior presence of oxygen functionalities. Addition of PBN spin trap during CV experiments (Fig. 6d) leads, as observed for the dialysed sample, to the absence of the distinct peak at 0.7 V. This indicates that the same PBN sensitive oxidation mechanism takes place for the vortexed and dialysed samples.
The charge related to oxidation of the centrifuged, sonicated and vortexed PS is greater that that for the dialysed PS by factors of two, three and ten respectively (Fig. 2a). Although the presence of more redox active functionalities could partly explain the enhanced electrochemical response for the oxidised samples, increased surface area due to roughening and deformation under mechanical agitation is also a likely explanation. For the case of the vortexed PS, the very significant deformation observed by TEM is supportive of the increase in surface area being the primary reason for the increased oxidation currents. In addition loose, amorphous polymer material can be seen in the TEM images of the vortexed sample and adsorption of this high surface area material to the electrode may be a cause of the significantly higher currents observed for this sample.
4. Discussion
CV mechanisms of immobilised PS
When phenol functionalities are present on the PS surface the oxidation mechanism could proceed as shown in Scheme 2, in an overall four electron/four proton oxidation at ca. 0.7 V with the final product being a quinone. Assuming uniform monolayer coverage of 5 × 105 beads mm−2 and transfer of ca. 13 nC of charge for the dialysed PS, the number of electrons transferred during the oxidation process at 0.7 V is of the order of 104 per bead (for detailed calculations see ESI†). Assuming smooth spherical particles, this translates very roughly to 3 or 4 electrons per styrene unit (i.e. per C6H5–C2H4-moiety) on the surface of the sphere and within tunnelling distance of the electrode, consistent with the mechanism in Scheme 2. | ||
| Scheme 2 Suggested mechanism for oxidation of phenol surface groups, as potential origin of voltammetric response of electrode-immobilised PS. | ||
The generated quinone species then undergoes a reversible reduction to the hydroquinone at potentials consistent with the small redox couple observed for oxidised PS at ca. 0.26 V. The noted charge discrepancy between the oxidation peaks at 0.7 V and 0.28 V for all samples (for the dialysed PS (28 ± 5) nC and (2.5 ± 0.5) nC respectively) indicates that this is not the only reaction pathway and other redox inactive products must be formed.
A proposed mechanism to explain the generation of phenolic species at a previously unoxidised PS surface is proposed in Scheme 3, here electrogenerated OH radicals abstract a hydrogen atom from the polymer backbone to produce a tertiary carbon radical. If dissolved oxygen is present a peroxy radical may be formed and this may be a terminating step. Alternatively the radical may be delocalised around the aromatic ring resulting in phenolic species after reaction with water or dissolved oxygen. In addition direct addition of OH radical to the aromatic ring will result in formation of phenolic moieties. The rate constants for OH reaction via H abstraction or by aromatic ring addition are comparable (of the order 109 M−1 s−1 in aqueous solution)29 hence the two processes take place in competition. The phenols generated via either route then undergo immediate oxidation at ca. 0.7 V, via the mechanism shown in Scheme 2, thereby explaining why a voltammetric response consistent with the presence of phenol is observed, despite no phenol functionality being present at the PS surface at the start of the experiment.
 | ||
| Scheme 3 Proposed mechanism for generation of redox-active phenolic groups on the PS surface through reaction with hydroxyl radicals. | ||
Polymer mechanochemistry: changes to the PS surface induced by mechanical forces
The XPS and electrochemical results show clearly that changes to PS surface chemistry are induced by different mechanical agitation techniques. The vortexed sample does not show any changes to surface termination as a result of the mechanical forces imposed, which is surprising given the degree of morphological deformation observed. This is perhaps a consequence of the types of forces experienced by the PS nanospheres. During vortex mixing the main forces are likely to be compressive, rather than shear forces. While this leads to substantial changes in surface morphology as the beads impact against each other, the collisions may have insufficient energy to allow homolytic cleavage of the polymer backbone.In contrast the centrifuged PS is likely to experience significant shear force, due to the direction in which the force is imposed relative to the surface, resulting in homolytic bond breakage, forming mechanoradical species.2–4 The sonicated samples will also experience shear force as the PS spheres rub against other, but additionally may be exposed to cavitation events.30 For both sonicated and centrifuged PS incorporation of oxygen functionality into the PS is observed, presumably due to mechanoradical formation (through cleavage of the backbone) followed by reaction with dissolved oxygen or with water.3 Cavitation during ultrasound treatment may also generate hydroxyl radicals that can react with the PS.30
The role of dissolved oxygen in the aqueous mechanochemistry of PS is clear from the different electrochemical response (Fig. 2c) and XPS spectra (Fig. 4) obtained for PS sonicated in air-saturated and argon-saturated solutions. Homolytic bond cleavage results in formation of alkyl radicals on the PS backbone, and these react rapidly with dissolved oxygen, resulting in unstable peroxy radicals3,4 that rapidly decompose to alcohols and other functionalities.31 In the absence of oxygen, the alkyl radical is more long-lived, resulting in propagation of chain reactions and radical delocalisation around the phenyl ring. Reaction of the resulting species with water or hydroxyl radicals produced through cavitation may lead to a wider range of oxidised species than obtained in the presence in oxygen, where formation of the peroxy radical is a terminating step.31 This may explain the surprising result that, according to the XPS analysis, PS sonicated in the absence of oxygen has a higher oxygen content than that sonicated with oxygen present. The difference in CV response for the two PS samples may provide insight into the identity of the redox active functionalities present and is the subject of further investigation.
Relevance to electrostatic charging of insulating materials
Insulating polymers such as Teflon, PMMA, nylon and polyethylene have been shown to perform redox chemistry when immersed in solution after electrostatic charging.7 These observations suggested that electron transfer between polymer surfaces could be responsible for the observed charge separation that takes place when two materials are rubbed together. However, the feasibility of electron transfer as a mechanism for electrostatic charging has been questioned due to the high energy required to transfer an electron from the HOMO of one wide band-gap insulator to the LUMO of another.8a In response, the concept of a ‘cryptoelectron’ was invoked to explain how electrons of mid band-gap energy may be available for transfer. ‘Cryptoelectrons’ were defined as ‘electrons residing in a material whatever their source (e.g. surface states, impurities, bulk defects) with energies significantly different from those expected from molecular states (or bands) in the material’.7c Recently at least some of the chemistry attributed to the transfer of ‘cryptoelectrons’ has been associated with the presence of surface mechanoradicals.10In the present study the mechanical activation of the PS nanospheres has been carried out in solution, rather than rubbing together two materials in an ambient environment, making a direct comparison to previous studies inappropriate. However we do see evidence for chemical modification of the PS surface after mechanical agitation in solution, where the shear forces and interfacial environment between two polymer surfaces may be comparable to that achieved by rubbing together two polymers in humid air. The surface modifications we observe are consistent with radical formation.3 The electrochemical response of the resulting surface species shows that after mechanical treatment surface groups and states are available in the energy gap between the frontier molecular orbitals of the polystyrene and could potentially facilitate electron transfer between polymer surfaces if the energies of the surface states was matched.
5. Conclusions
This relatively simple CV study has revealed an unexpected degree of insight into the reactivity and mechanochemistry of the PS surface in solution. Further work is planned to compare and contrast the behaviour of other polymers under similar conditions. One of the motivations of this study was to determine whether the redox chemistry of insulating polymers could be measured using voltammetric techniques. A complex, pH dependent CV response has previously been obtained for electrode-immobilised undoped (non-conductive) diamond nanoparticles.1 The response was attributed to the presence of surface functionalities (alcohols, quinones etc.) that could undergo direct electron transfer with the electrode and with dissolved redox species. The present study confirms that, if redox active surface functionalities are present, a CV response can also be obtained for polymer nanoparticles such as PS, despite the insulating nature of the bulk. This presents a limitless opportunity for further investigation of the surface chemistry of insulating materials, suggesting that many materials at the nanoscale may exhibit an electrochemical response.Acknowledgements
EPSRC Grant no EP/J0100061 is acknowledged for funding. The Electrostatics Discharge (ESD) Association is thanked for the award of a University Research Grant.References
-
(a) K. B. Holt, Phys. Chem. Chem. Phys., 2010, 12, 2048 RSC
; (b) K. B. Holt, D. J. Caruana and E. J. Millan-Barrios, J. Am. Chem. Soc., 2009, 131, 11272 CrossRef CAS PubMed
-
(a) T. Q. Nguyen and H. M. Kausch, Macromolecules, 1990, 23, 5137 CrossRef CAS
-
(a) M. K. Beyer and H. Clausen-Schaumann, Chem. Rev., 2005, 105, 2921 CrossRef CAS PubMed
-
(a) J. Sohma, Prog. Polym. Sci., 1989, 14, 451 CrossRef CAS
-
(a) G. Schmidt-Naake, A. Frendel, M. Drache and G. Janke, Chem. Eng. Technol., 2001, 24, 889 CrossRef CAS
- D. J. Lacks and R. M. Sankaran, J. Phys. D: Appl. Phys., 2011, 44, 453001 CrossRef
-
(a) C. Liu and A. J. Bard, Nat. Mater., 2008, 7, 505 CrossRef CAS PubMed
-
(a) L. S. McCarty and G. M. Whitesides, Angew. Chem., Int. Ed., 2008, 47, 2188 CrossRef CAS PubMed
-
(a) H. T. Baytekin, A. Z. Patashinski, M. Branicki, B. Baytekin, S. Soh and B. A. Grzybowski, Science, 2011, 333, 308 CrossRef CAS PubMed
- B. Baytekin, H. T. Baytekin and B. A. Grzybowski, J. Am. Chem. Soc., 2012, 134, 7223 CrossRef CAS PubMed
- H. T. Baytekin, B. Baytekin, T. M. Hermans, B. Kowalczyk and B. A. Grzybowski, Science, 2013, 341, 1368 CrossRef CAS PubMed
- http://eshop.eppendorfna.com/products/Eppendorf_Flex-Tube_microcentrifuge_tube .
- J. J. Pireaux, J. Riga, R. Caudano, J. J. Verbist, J. Delhalle, S. Delhalle, J. M. André and Y. Golillon, Phys. Scr., 1977, 16, 329 CrossRef CAS
- E. H. Lock, D. Y. Petrovykh, P. Mack, T. Carney, R. G. White, S. G. Walton and R. F. Fernsler, Langmuir, 2010, 26, 8857 CrossRef CAS PubMed
- M. M. Nasef, H. Saidi, H. M. Nor and M.
O. Yarmo, J. Appl. Polym. Sci., 2000, 76, 336 CrossRef CAS
- R. Foerch, G. Beamson and D. Briggs, Surf. Interface Anal., 1991, 17, 842 CrossRef CAS
- R. T. S. Oliviera, G. R. Salazar-Banda, M. C. Santos, M. L. Calegaro, D. W. Miwa, S. A. S. Machado and L. A. Avaca, Chemosphere, 2007, 66, 2152 CrossRef PubMed
- C. Provent, W. Haenni, E. Santoli and P. Rychen, Electrochim. Acta, 2004, 49, 3737 CrossRef CAS PubMed
- X. Zhu, S. Shi, J. Wei, F. Lv, H. Zhao, J. Kong, Q. Hi and J. Ni, Environ. Sci. Technol., 2007, 41, 6541 CrossRef CAS
- T. A. Enache and A. M. Oliveira-Brett, J. Electroanal. Chem., 2011, 655, 9 CrossRef CAS PubMed
- J. A. Wiles, M. Fialkowski, M. R. Radowski, G. M. Whitesides and B. A. Grzybowski, J. Phys. Chem. B, 2004, 108, 20296 CrossRef CAS
- T. Murakami, S. Kuroda and Z. Osawa, J. Colloid Interface Sci., 1998, 202, 37 CrossRef CAS
- A. Stefanova, S. Ayata, A. Erem, S. Ernst and H. Baltruschat, Electrochim. Acta, 2013, 110, 560 CrossRef CAS PubMed
- T. A. Enache, A.-M. Chiorcea-Paquim, O. Fatibello-Fihlo and A. M. Oliveira-Brett, Electrochem. Commun., 2009, 11, 1342 CrossRef CAS PubMed
- C. Terashima, T. N. Rao, B. V. Sarada, Y. Kubota and A. Fujishima, Anal. Chem., 2003, 75, 1564 CrossRef CAS
- B. Lee, H. Naito and T. Hibino, Angew. Chem., Int. Ed., 2012, 51, 440 CrossRef CAS PubMed
-
(a) Z. Ma, B. Zhao and Z. Yuan, Anal. Chim. Acta, 1999, 389, 213 CrossRef CAS
- L. Johnson and D. A. Walsh, J. Mater. Chem., 2011, 21, 7555 RSC
-
(a) L. M. Dorfman, I. A. Taub and D. A. Harter, J. Chem. Phys., 1964, 41, 2954 CrossRef CAS PubMed
- T. Q. Nguyen, Q. Z. Liang and H. H. Kausch, Polymer, 1997, 38, 3783 CrossRef CAS
- C. von Sonntag, E. Bothe, P. Ulanski and A. Adhikary, Radiat. Phys. Chem., 1999, 55, 599 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp03938f |
| This journal is © the Owner Societies 2015 |