
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAb initio calculations as a quantitative tool in the inelastic neutron scattering study of a single-molecule magnet analogue†
Michele
Vonci
 a,
Marcus J.
Giansiracusa
a,
Robert W.
Gable
a,
Willem
Van den Heuvel
a,
Kay
Latham
b,
Boujemaa
Moubaraki
c,
Keith S.
Murray
c,
Dehong
Yu
d,
Richard A.
Mole
*d,
Alessandro
Soncini
*a and
Colette
Boskovic
*a
a,
Marcus J.
Giansiracusa
a,
Robert W.
Gable
a,
Willem
Van den Heuvel
a,
Kay
Latham
b,
Boujemaa
Moubaraki
c,
Keith S.
Murray
c,
Dehong
Yu
d,
Richard A.
Mole
*d,
Alessandro
Soncini
*a and
Colette
Boskovic
*a
aSchool of Chemistry, University of Melbourne, VIC, 3010, Australia. E-mail: c.boskovic@unimelb.edu.au; asoncini@unimelb.edu.au
bSchool of Applied Science, RMIT University, Melbourne, VIC 3001, Australia
cSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia
dBragg Institute, Australian Nuclear Science and Technology Organisation, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia. E-mail: richardm@ansto.gov.au
First published on 3rd December 2015
Abstract
Ab initio calculations carried out on the Tb analogue of the single-molecule magnet family Na9[Ln(W5O18)2] (Ln = Nd, Gd, Ho and Er) have allowed interpretation of the inelastic neutron scattering spectra. The combined experimental and theoretical approach sheds new light on the sensitivity of the electronic structure of the Tb(III) ground and excited states to small structural distortions from axial symmetry, thus revealing the subtle relationship between molecular geometry and magnetic properties of the two isostructural species that comprise the sample.
With their signature energy barrier to magnetic relaxation and quantum tunnelling through this barrier, single-molecule magnets (SMMs) are important candidate molecules for future applications in molecular spintronics and quantum computation. Among the most promising SMMs are those based on trivalent lanthanoid ions (Ln-SMMs).1 Many future applications of SMMs will require their deposition on surfaces, which makes essential the investigation of the effect on the SMM properties of small structural changes that will likely arise from deposition.1e Thus the development of correlations between the magnetic behaviour, electronic structure and Ln(III) coordination geometry is crucial for achieving a better understanding of the range of structural parameters that will guarantee the retention of SMM properties. One of the most powerful experimental techniques for elucidating the electronic structure of SMMs is inelastic neutron scattering (INS), which provides a direct probe of the relevant energy levels.2 INS is very sensitive to the electronic structure of the lowest lying energy levels of Ln(III) ions, which are dominated by crystal field (CF) splitting effects. Despite these advantages, relatively few INS spectra with well-defined magnetic scattering have been reported for Ln-SMMs.1e,3 Complementing experimental measurements are theoretical approaches involving either (i) semi-empirical CF parameterisation of experimental magnetic susceptibility or spectroscopic data, or (ii) ab initio calculations, where the only input from experiments consists of atomic coordinates from crystal structure data. While CF models are very informative for high-symmetry structures, in low-symmetry cases they can suffer from overparameterisation. A useful description of the subtle, but at times crucial, influence of the low-symmetry harmonics and non-point charge effects associated with a realistic CF environment can only be gauged via suitable ab initio methodologies. However, recently the deviations of up to 20% between experimental data and the best available ab initio calculations for some Ln-SMMs has raised questions about this approach.4 Indeed, most experiments provide access only to ground state properties, while a more realistic benchmarking of ab initio methodologies should assess their ability to correctly determine the energetics of the CF states probed experimentally by techniques such as INS.
A family of complexes that has proved extremely useful for achieving the present understanding of Ln-SMMs are the [Ln(W5O18)2]9− polyanions,5 in part because of the relatively high local symmetry at the Ln center, which is sandwiched between two polyoxotungstate ligands. In recent investigations Coronado and co-workers have performed detailed magnetic susceptibility, heat capacity and EPR spectroscopy measurements, complemented by CF fitting of the data, finding SMM behaviour for the Ho, Er, Gd and Nd analogues.6 Their studies have contributed to the development of a semi-empirical approach that makes use of a radial effective charge model and experimental data for isostructural analogues to determine the CF splitting of Ln(III) ions. Although not a SMM due to an apparent MJ = 0 ground state, Na9[Tb(W5O18)2] (Tb) is an excellent model candidate for further study given the low neutron absorption cross section and the predicted small energy splittings of the J = 6 ground multiplet.6b In this communication we report a combined experimental and theoretical study of Tb, in which the application of the well-established ab initio CASSCF/RASSI computational strategy (see ESI†)7 provides key insights for interpretation of high quality INS spectra. In particular the ab initio calculations allowed us to assign the INS spectra to two distinct co-crystallised polymorphic phases Tb-a and Tb-b, which have not been recognised previously. Subtle differences in the local coordination environments of the two Tb centres in essentially isostructural polyanions (Fig. 1) contribute distinct INS signals, with ab initio calculations accounting precisely for the relationship between the differences in the coordination environment of the Tb(III) ions and the changes in the electronic structures of the two polymorphs.
 | ||
| Fig. 1 Molecular structure (left) and representations of the two distortion angles of the Tb coordination (right) for the [Tb(W5O18)2]9− polyanion in Tb; atom colour code: W (yellow), O (red) and Tb (violet). | ||
A bulk sample of Tb was prepared by modified literature procedures and characterised by single crystal X-ray diffraction, elemental analysis, infrared spectroscopy and magnetic susceptibility measurements (ESI,† Fig. S1 and S2).5,6a,b Recrystallisation from D2O afforded a sample for INS of Na9[Tb(W5O18)2]·20D2O (TbD). Samples of the diamagnetic Y analogues (Y and YD) were prepared similarly (ESI†) and employed to determine the phonon contributions to the INS spectra (Fig. S3, ESI†). The sample TbD was analysed on the time-of-flight INS spectrometer Pelican at the Australian Nuclear Science and Technology Organisation.8 The spectra collected at two neutron wavelengths, λ = 4.74 and λ = 2.37 Å, are shown in Fig. 2 (see also Fig. S4–S7, ESI†). At the base temperature of 5 K, four well resolved peaks are observed at 1.000 ± 0.001, 1.620 ± 0.001, 2.786 ± 0.002, 4.087 ± 0.009 meV across the two data sets (peaks Ia IIa, Ib, IIb, Fig. 2). The analysis of the temperature and momentum transfer vector (Q) dependence (Fig. S8, ESI†) classifies them as magnetic excitations from the ground state. Upon increasing temperature, three peaks become more evident at 5.00 ± 0.04, 6.25 ± 0.03, and 7.61 ± 0.04 meV (peaks IIIa, IVa, Va; Fig. 2) and are assigned as CF transitions from excited states through the analysis of the temperature dependence and comparison with the spectra of YD (Fig. S3, ESI†).
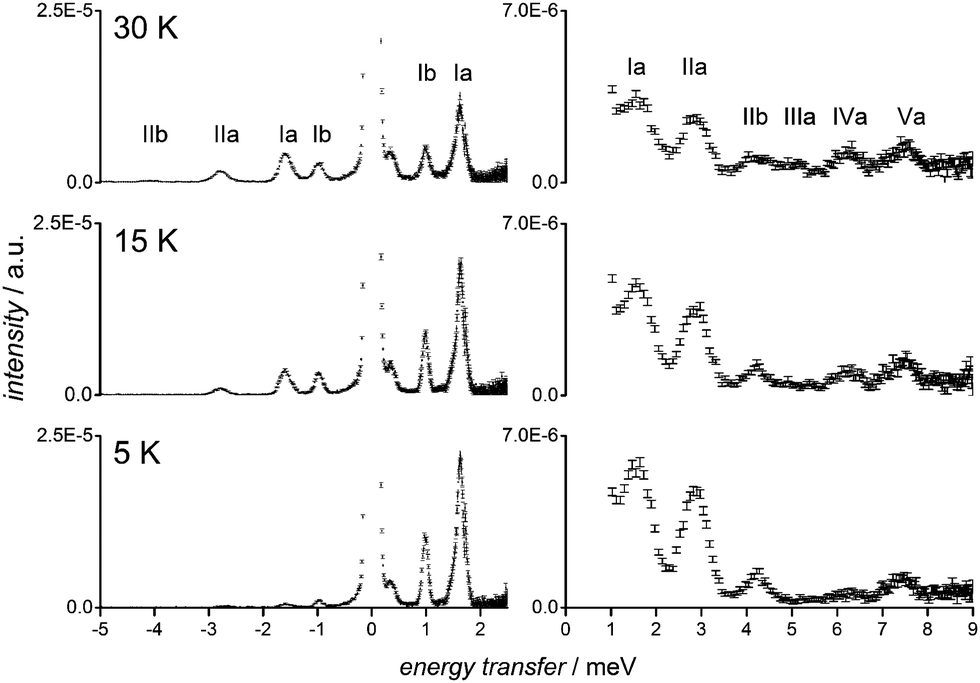 | ||
| Fig. 2 Variable temperature INS spectra of TbD. Left: λ = 4.74 Å, integration interval 0.8 Å−1 < Q < 1.2 Å−1 and right: λ = 2.37 Å, integration interval 1.15 Å−1 < Q < 1.65 Å−1. Positive numbers correspond to neutron energy loss. | ||
The literature CF models of the electronic structure for this system suggest an MJ = 0 ground state, and a first (second) excited pseudo-doublet MJ = ±1 (MJ = ±2.)4a,6b On this basis only one or two ground state transitions (MJ = 0 → MJ = ±1) are expected at 5 K. It is thus not possible to rationalise the four observed transitions without invoking either (i) a strong admixture of states, which would lead to matrix elements of the total angular momentum operator between ground and higher excited states large enough to justify the relative intensities of the observed peaks,4a,6b or (ii) the presence of multiple species in the sample, although it is analytically pure and literature studies of this compound imply a single polyanion. However, close scrutiny of the crystalline sample found the latter situation to indeed be the case, with two distinct polymorphic phases, Tb-a and Tb-b, comprising the sample analysed by INS. The two compounds both crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with only very small differences in the cell parameters and in the crystal habit: flat blade-shaped crystals for Tb-a and flat hexagonal prismatic crystals for Tb-b. Although a crystal structure has not been previously reported for Na9[Tb(W5O18)2], reported structures of other Ln analogues are isomorphous with Tb-a or Tb-b.5,6a,b Crystals of both polymorphs were characterised by single crystal X-ray diffraction (Table S1, ESI†), while powder X-ray diffraction data (Fig. S9, ESI†) are consistent with the INS sample TbD being comprised of a mixture of the two species. The single crystal analysis shows that Tb-a and Tb-b share the same connectivity of the isostructural [Tb(W5O18)2]9− polyanion and in both cases the pseudo C4 molecular axes are oriented along the same crystallographic direction. The most apparent difference between the two polymorphs is the arrangement of the water-coordinated sodium cations (Fig. S10, ESI†). In both structures the Tb(III) ion is sandwiched between two {W5O18} moieties with eight O donor atoms arranged in a distorted square antiprismatic (SAP) fashion, resulting in a distorted D4d molecular point symmetry. The distortions that the reciprocal position of the two square faces of the antiprism introduce into the first coordination sphere of Tb(III) can be evaluated using the average deviation from the ideal 45° value of the skew angles ϕ (AD(ϕ)45) and the angle φ between the normal vectors to the faces (Table 1, Fig. 1 and Fig. S11, ESI†). The values of these angles for the two polyanions reveal that the SAP local geometry of the Tb(III) ion is further from ideal D4d symmetry in Tb-b than in Tb-a in terms of both dihedral and axial distortion. Continuous shape measurements performed on the two polymorphs with the software SHAPE confirm the SAP geometry and larger distortion of the Tb(III) coordination sphere in Tb-b.9
, with only very small differences in the cell parameters and in the crystal habit: flat blade-shaped crystals for Tb-a and flat hexagonal prismatic crystals for Tb-b. Although a crystal structure has not been previously reported for Na9[Tb(W5O18)2], reported structures of other Ln analogues are isomorphous with Tb-a or Tb-b.5,6a,b Crystals of both polymorphs were characterised by single crystal X-ray diffraction (Table S1, ESI†), while powder X-ray diffraction data (Fig. S9, ESI†) are consistent with the INS sample TbD being comprised of a mixture of the two species. The single crystal analysis shows that Tb-a and Tb-b share the same connectivity of the isostructural [Tb(W5O18)2]9− polyanion and in both cases the pseudo C4 molecular axes are oriented along the same crystallographic direction. The most apparent difference between the two polymorphs is the arrangement of the water-coordinated sodium cations (Fig. S10, ESI†). In both structures the Tb(III) ion is sandwiched between two {W5O18} moieties with eight O donor atoms arranged in a distorted square antiprismatic (SAP) fashion, resulting in a distorted D4d molecular point symmetry. The distortions that the reciprocal position of the two square faces of the antiprism introduce into the first coordination sphere of Tb(III) can be evaluated using the average deviation from the ideal 45° value of the skew angles ϕ (AD(ϕ)45) and the angle φ between the normal vectors to the faces (Table 1, Fig. 1 and Fig. S11, ESI†). The values of these angles for the two polyanions reveal that the SAP local geometry of the Tb(III) ion is further from ideal D4d symmetry in Tb-b than in Tb-a in terms of both dihedral and axial distortion. Continuous shape measurements performed on the two polymorphs with the software SHAPE confirm the SAP geometry and larger distortion of the Tb(III) coordination sphere in Tb-b.9
| AD(ϕ)45a | φ | Tb–Oc | O–Od | D p−p | SHAPE indexf | |
|---|---|---|---|---|---|---|
| a Mean absolute deviation from the ideal value of 45° for the angles ϕ between the square faces of the SAP. b Dihedral angle between the square faces. c Average distance between the Tb(III) ion and the 8 bound O atoms. d Average distance in O–O pairs in the square faces. e Distance between the square faces. f The software SHAPE yields an index that accounts for distortion compared to an ideal geometry; the closer the index is to zero, the closer to the ideal geometry. | ||||||
| Tb-a | 2.3(6) | 1.1(1) | 2.39(2) | 2.88(2) | 2.497 | 0.059 |
| Tb-b | 4(1) | 2.2(1) | 2.40(2) | 2.88(1) | 2.512 | 0.138 |
Ab initio CASSCF/RASSI calculations were performed using Molcas 8.0 (see ESI†) to determine the effects on the electronic structure of the differences in the first coordination sphere of the Tb(III) centers in the two molecules. The structural inputs were the atomic positions obtained from single crystal X-ray diffraction data collected at 130 K for Tb-a and Tb-b. The calculations show that the energy spectra of the J = 6 ground state multiplet for the two compounds are qualitatively similar (Fig. S12 and Table S2, ESI†), with an almost pure MJ = 0 singlet as ground state, accounting for the lack of SMM behavior. The two doubly degenerate states expected as first and second excited levels in an ideal D4d symmetry are in fact manifestly split pseudo-doublets (Fig. 3 and 4c) the components of which are mainly a combination of MJ = ±1 and MJ = ±2 states, respectively (Tables S3 and S4, ESI†). The remaining excited states, although strongly mixed, are predicted to be closely spaced pseudo-doublets (Tables S3 and S4, ESI†). Ab initio calculations establish a direct correlation between the increase in structural distortions and the increment of the energy splitting between the components of the first two excited pseudo-doublets, offering a precise and simple explanation of the four ground state transitions observed at 5 K in TbD and suggesting the possible dynamics of the “hot” transitions (Fig. 3 and 4).
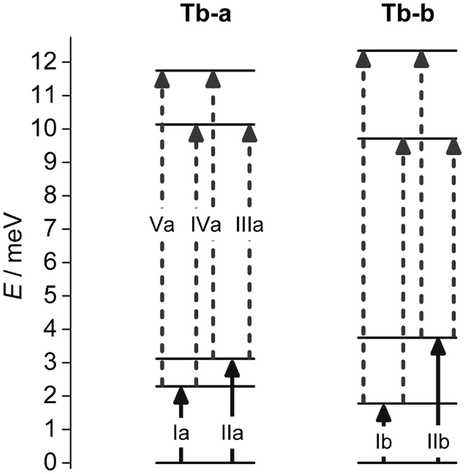 | ||
| Fig. 3 Low lying energy levels of the J = 6 ground multiplet for Tb-a and Tb-b calculated with ab initio methods. The arrows represent the INS transitions allowed from the ground state (solid) and from the first excited pseudo-doublet (dashed). | ||
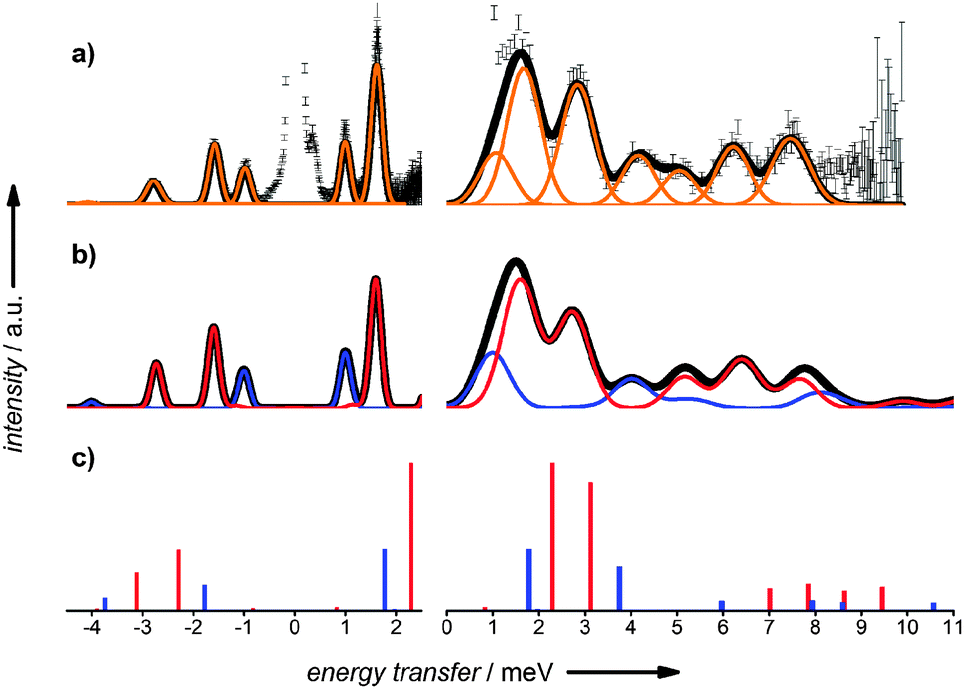 | ||
Fig. 4 INS spectra of TbD at λ = 4.74 (left) and λ = 2.37 Å (right) at 30 K. (a) Experimental spectra with Gaussian fitting (orange) and convolution of individual peak contributions (black). (b) Simulated INS spectra of Tb-a (red), Tb-b (blue) and combination (black) using the set of optimised CF parameters as described in the text. (c) Theoretical INS spectra arising from transition probabilities calculated from CASSCF/RASSI results for Tb-a (red) and Tb-b (blue). A 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 molar ratio of Tb-a:Tb-b is assumed for (b) and (c). :30 molar ratio of Tb-a:Tb-b is assumed for (b) and (c). | ||
We assign peaks Ia and IIa to ground state transitions to the two non-degenerate components of the first excited pseudo-doublet for Tb-a and peaks Ib and IIb to the equivalent transitions for Tb-b. On the basis of the relative intensities of peaks Ia and Ib the molar ratio between Tb-a and Tb-b is determined to be 70:30. After realising the existence of the two polymorphs, we were able to optimise the synthetic procedure to obtain pure samples of Tb-a and Tb-b (ESI†), although Tb-b is metastable, partially interconverting to Tb-a when wet. INS measurements confirmed the assignment of the ground state transitions for each phase and also allowed the assignment of “hot” transitions IIIa, IVa, and Va for Tb-a. The “hot” transitions of Tb-b are not apparent in the INS spectra of Tb, either because of low intensity due to the low concentration or because of partial overlap with Tb-a peaks.
Remarkably, the INS spectra arising from the transition probabilities calculated from CASSCF/RASSI results (Fig. 4c and Fig. S22a, ESI†), only fixing the relative ratio of Tb-a and Tb-b to the experimentally observed 70:30, are in good agreement with the experimental data, reproducing very well the relative intensities of the observed ground-state transitions and reasonably well the energies. They also qualitatively predict the position and relative intensities of “hot” transitions. Excellent simulations of the magnetic susceptibility data are also obtained (Fig. S2, ESI†).
The results of CASSCF/RASSI calculations can be presented more intuitively by projecting the lowest lying ab initio wave functions onto a (2J + 1)-dimensional pseudo-spin basis set (Tables S3 and S4, ESI†) and by computing the full set of CF parameters for the Tb(III) ions in Tb-a and Tb-b (Table S5, ESI†). The already mentioned admixture of the wavefunctions, particularly relevant for the first two pseudo doublets, is highlighted by the presence of off-diagonal CF parameters confirming that high-symmetry D4d approximations are inadequate to describe these molecules.6b,e
The limited number of peaks in the INS spectra prevents fitting the data to the full CF Hamiltonian. However, to elucidate the low symmetry effects suggested by ab initio calculations on the first and second excited pseudo doublets observed by INS, we employed extended Stevens operators10 to set up a simplified CF Hamiltonian containing a selected small number of terms (Table 2). The CF parameters included in the simplified model were chosen based on the following observations and symmetry arguments. The barycenter of the energy levels observed in the INS spectra could be satisfactorily fit employing B02 as the only diagonal parameter. The splitting of the first excited pseudo-doublet, observed for both Tb-a and Tb-b, was modeled by incorporating the off-diagonal parameter B22, allowing mixing of the MJ = ±1 components. Because the “hot” transitions to the second excited pseudo-doublet are only observed clearly for Tb-a, the off-diagonal parameter B44 was included for this polymorph only, allowing efficient mixing of the MJ = ±2 components. The experimental INS spectra were fit to the simplified CF Hamiltonian, using the values from the CASSCF/RASSI calculations (Table S5, ESI†), as initial values. The final values optimised for the two polymorphs (Table 2) afford accurate simulation of the INS spectra (Fig. 4b and Fig. S20–S22, ESI†).
| B 02 | B 22 | B 44 | |
|---|---|---|---|
| Tb-a | 0.705(1) | 0.0250(3) | −8.5(2) × 10−4 |
| Tb-b | 0.712(2) | 0.0729(4) |
The values obtained for the only diagonal parameter, B02, are very similar for Tb-a and Tb-b. The dihedral distortion, measured by the angle ϕ, preserves C4 rotational symmetry, and by introducing B44 and B46 parameters, can split the MJ = ±2 doublet only. Therefore the splitting of the MJ = ±1 doublet must arise from different symmetry-lowering structural distortions. Interestingly, the B22 parameter, which is almost three times larger for Tb-b than Tb-a (Table 2), correlates with the larger axial angle φ observed for Tb-b, which may thus be the main structural origin of the splitting of the MJ = ±1 doublet. More quantitative magnetostructural correlations may require additional crystal structure data collected at similar temperatures to the INS; although a recent study has concluded that, in the absence of a phase transition, the thermal effect of the molecular structure on the electronic structure of SMMs is negligible in practice.11
In summary we report herein how the first ab initio CASSCF/RASSI calculations performed on a Ln-polyoxotungstate play a heuristic role in the interpretation of otherwise inexplicable INS data measured on a co-crystallised mixture of two polymorphs of Na9[Tb(W5O18)2]. These calculations accurately account for the relationship between the increasingly distorted Tb(III) crystal fields in the two polymorphs and the energy shifts observed for the INS transitions. The observation that small changes in the Tb(III) coordination geometry significantly impacts the electronic structure has important implications for future surface deposition of SMMs and lays the foundation for a comprehensive investigation of the effect of environment-induced distortions on the magnetic behaviour of the SMM analogues in this family.
We thank the Australian Research Council for funding and AINSE Ltd for financial assistance to M. V. and M. J. G. We thank the Bragg Institute for beam time and technical support. W. V. d. H. thanks the University of Melbourne for a McKenzie Post-Doctoral Fellowship.
Notes and references
- (a) L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092 RSC; (b) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed; (c) J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Chem. Soc. Rev., 2012, 41, 7464 RSC; (d) M. Vonci and C. Boskovic, Aust. J. Chem., 2014, 67, 1542 CrossRef CAS; (e) K. S. Pedersen, L. Ungur, M. Sigrist, A. Sundt, M. Schau-Magnussen, V. Vieru, H. Mutka, S. Rols, H. Weihe, O. Waldmann, L. F. Chibotaru, J. Bendix and J. Dreiser, Chem. Sci., 2014, 5, 1650 RSC; (f) S. T. Liddle and J. van Slageren, Chem. Soc. Rev., 2015, 44, 6655 RSC.
- R. Basler, C. Boskovic, G. Chaboussant, H. U. Güdel, M. Murrie, S. T. Ochsenbein and A. Sieber, ChemPhysChem, 2003, 4, 910 CrossRef PubMed.
- (a) M. Kofu, O. Yamamuro, T. Kajiwara, Y. Yoshimura, M. Nakano, K. Nakajima and S. Ohira-Kawamura, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 064405 CrossRef; (b) F. Tuna, R. Valiente, L. H. Thomas, W. Wernsdorfer, S. T. Ochsenbein and M. Murrie, Inorg. Chem., 2014, 53, 8970 CrossRef PubMed.
- (a) J. J. Baldoví, J. M. Clemente-Juan, E. Coronado, Y. Duan, A. Gaita-Ariño and C. Giménez-Saiz, Inorg. Chem., 2014, 53, 9976 CrossRef PubMed; (b) J. van Leusen, M. Speldrich, H. Schilder and P. Kögerler, Coord. Chem. Rev., 2015, 289–290, 137 CrossRef CAS.
- (a) R. D. Peacock and T. J. R. Weakley, J. Chem. Soc. A, 1971, 1836 RSC; (b) T. Yamase, T. Ozeki and M. Tosaka, Acta Crystallogr., 1994, C50, 1849 CAS; (c) M. Barsukova, M. H. Dickman, E. Visser, S. Sankar Mal and U. Kortz, Z. Anorg. Allg. Chem., 2008, 634, 2423 CrossRef CAS.
- (a) M. A. Aldamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Ariño, J. Am. Chem. Soc., 2008, 130, 8874 CrossRef CAS PubMed; (b) M. A. AlDamen, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, C. Martí-Gastaldo, F. Luis and O. Montero, Inorg. Chem., 2009, 48, 3467 CrossRef CAS PubMed; (c) F. Luis, S. Cardona-Serra, C. Martí-Gastaldo, J. M. Clemente-Juan, J. Sesé, D. Drung and T. Schurig, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 060403 CrossRef; (d) J. J. Baldoví, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño and A. Palii, Inorg. Chem., 2012, 51, 12565 CrossRef PubMed; (e) M. J. Martínez-Pérez, S. Cardona-Serra, C. Schlegel, F. Moro, P. J. Alonso, H. Prima-García, J. M. Clemente-Juan, M. Evangelisti, A. Gaita-Ariño, J. Sesé, J. van Slageren, E. Coronado and F. Luis, Phys. Rev. Lett., 2012, 108, 247213 Search PubMed.
- (a) B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157 CrossRef CAS; (b) L. F. Chibotaru and L. Ungur, J. Chem. Phys., 2012, 137, 064112 CrossRef CAS PubMed.
- D. Yu, R. Mole, T. Noakes, S. Kennedy and R. Robinson, J. Phys. Soc. Jpn., 2013, 82, SA027 CrossRef.
- M. Llunell, D. Casanova, J. Cirera, J. M. Bofill, P. Alemany, S. Alvarez, M. Pinsky and D. Avnir, SHAPE 2.0, Universitat De Barcelona & The Hebrew University Of Jerusalem, Barcelona, 2003 Search PubMed.
- (a) C. Rudowicz and C. Y. Chung, J. Phys.: Condens. Matter, 2004, 16, 5825 CrossRef CAS; (b) C. Rudowicz, Coord. Chem. Rev., 2015, 287, 28 CrossRef CAS.
- K. Qian, J. J. Baldoví, S.-D. Jiang, A. Gaita-Ariño, Y.-Q. Zhang, J. Overgaard, B.-W. Wang, E. Coronado and S. Gao, Chem. Sci., 2015, 6, 4587 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for syntheses, X-ray crystallography, magnetic and INS measurements and ab initio calculations. ICSD 429951–429953. See DOI: 10.1039/c5cc07541f |
| This journal is © The Royal Society of Chemistry 2016 |